

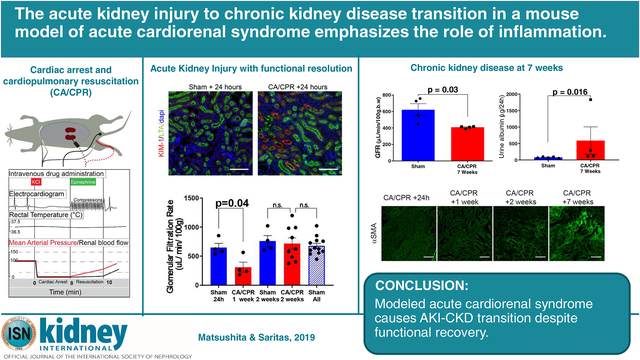
Abstract
Acute cardiorenal syndrome is a common complication of acute cardiovascular disease. Studies of acute kidney injury (AKI) to chronic kidney disease (CKD) transition, including patients suffering acute cardiovascular disease, report high rates of CKD development. Therefore, acute cardiorenal syndrome associates with CKD, but no study has established causation. To define this we used a murine cardiac arrest (CA) and cardiopulmonary resuscitation (CPR) model or sham procedure on male mice. CA was induced with potassium chloride while CPR consisted of chest compressions and epinephrine eight minutes later. Two weeks after AKI induced by CA/CPR, the measured glomerular filtration rate (GFR) was not different from sham. However, after seven weeks the mice developed CKD, recapitulating clinical observations. One day, and one, two, and seven weeks after CA/CPR, the GFR was measured, and renal tissue sections evaluated for various indices of injury and inflammation. One day after CA/CPR, acute cardiorenal syndrome was indicated by a significant reduction of the mean GFR (649 in sham, vs. 25 μL/min/100g in CA/CPR animals), KIM-1 positive tubules, and acute tubular necrosis. Renal inflammation developed, with F4/80 positive and CD3-positive cells infiltrating the kidney one day and one week after CA/CPR, respectively. Although there was functional recovery with normalization of GFR two weeks after CA/CPR, deposition of tubulointerstitial matrix proteins α-smooth muscle actin and fibrillin-1 progressed, along with a significantly reduced mean GFR (623 in sham vs. 409 μL/min/100g in CA/CPR animals), proteinuria, increased tissue transforming growth factor-β, and fibrosis establishing the development of CKD seven weeks after CA/CPR. Thus, murine CA/CPR, a model of acute cardiorenal syndrome, causes an AKI-CKD transition likely due to prolonged renal inflammation.
Keywords: Cardiac arrest, cardiorenal, acute kidney injury, chronic kidney disease
Graphical Abstract
Introduction
Acute kidney injury (AKI) is among the most common and expensive complications of severe illness and is a risk factor for the development of chronic kidney disease (CKD).1–3 Acute cardiorenal syndrome, AKI secondary to acute cardiac dysfunction, frequently complicates the course of cardiovascular illness and its treatment. For example, cardiac arrest, which affects 6 million persons yearly,4 causes acute cardiorenal syndrome in up to 50% of survivors.5–9 Studies of AKI-CKD transition which include patients suffering acute cardiovascular disease report high rates of development of CKD.1, 10–17 Therefore, acute cardiorenal syndrome may cause CKD. Since preventing CKD is highly desirable, extensive study has focused on mechanisms of AKI-CKD transition. However, mechanistic insights have been largely limited to models which do not replicate the most common human causes of AKI, and there is considerable scientific interest in novel translational AKI models.18–20 Further, the frequency of cardiovascular disease and the burden of CKD mandates mechanistic study of their association.
We previously reported that murine cardiac arrest and cardiopulmonary resuscitation (CA/CPR) causes elevated serum creatinine and reduction in glomerular filtration rate 24h later.21–24 However, whether there is tubular injury and repair, renal inflammation, recovery of function (as in humans), and whether CKD develops, are unanswered and critical translational questions. Characterization of AKI in acute cardiorenal syndrome and discovery of progression to CKD is expected to generate critical translational insight in this model, which replicates a cause of up to 3 million cases of AKI yearly, and thus may lead to prevention or therapy for CKD.
Here we report that acute cardiorenal syndrome caused by CA/CPR is characterized by acute, reversible loss of GFR and accompanied by robust tubular KIM-1 expression, extensive tubulopathy, and renal inflammation. Critically, 2 weeks following AKI due to CA/CPR, glomerular filtration rate (GFR) normalizes – but despite this, fibrosis progresses and mice develop CKD by 7 weeks after CA/CPR. Our results demonstrate that CA/CPR is a translational model of acute cardiorenal syndrome which can provide critical insight into AKI-CKD transition due to a very common condition, leading to clinically-relevant interventions.
Results
CA/CPR model and characteristics
Critical events of the CA/CPR model are summarized in figure 1A. Intravenous potassium chloride caused cardiac arrest in all mice which were assigned to CA/CPR. Return of spontaneous circulation occurred in 87% of mice. Preoperative body weight and survival data are depicted in figure 1B–E. Neither preoperative nor endpoint body weight was different between sham and CA/CPR animals. CA/CPR caused 40–63% 3 day mortality; all mice which survived to day 5 survived to the predetermined endpoint. Binary logistic regression indicated that for each additional second of resuscitation time, the OR for survival to predetermined endpoint was decreased by 0.4% (95% CI 0.1–0.7%, p=0.00003). However there was no difference in resuscitation time between groups (1F). Although generally negative, the correlation between resuscitation time and GFR at the experimental endpoint was not significant in either the 24h experiment or the 7 week experiment.
Figure 1:

Characteristics of cardiac arrest and cardiopulmonary resuscitation (CA/CPR) model and experiments. A. Summary of events and physiology during the cardiac arrest and cardiopulmonary resuscitation model in the mouse. During preparation the mouse is instrumented with an endotracheal tube, 3-lead electrocardiogram, right jugular PE-10 catheter, and rectal temperature probe in all experiments. Dotted lines denote renal cortex laser Doppler, and femoral arterial pressure catheter observations made in prior investigations (Hutchens et al 2010 and 2015, schematically reproduced, with approximate quantities, with permission). At (1) cardiac arrest is induced by administering potassium chloride (KCl), and the electrocardiogram demonstrates a sinusoidal, agonal rhythm. Immediately on cardiac arrest, mean arterial pressure drops to ~10% of baseline, and renal cortical blood flow to near zero. At (2), 8 minutes after KCl administration, chest compressions begin at a rate of 300/min, and epinephrine 8–16 μg administered intravenously over 30–60 seconds. Compression artifact is seen in the electrocardiogram. At (3) return of spontaneous circulation (ROSC) is observed in 90% of mice within 120 seconds. Hypertension (to ~125% baseline MAP) occurs in the 2 minutes following ROSC. Following resumption of adequate spontaneous respiration, by 15 minutes after ROSC, the endotracheal tube and other monitors are removed and the mouse is placed in a recovery cage. B-D Body weight and survival in independent experiments. B. Body weight was not different between sham and CA/CPR mice in the 24h experiment. C. Survival in the 7d CA/CPR cohort (this cohort does not have a respective sham group). Inset: body weight at start and end of the 7d experiment. D. Body weight and survival of CA/CPR and sham mice in the 2 week experiment. E. Body weight and survival of CA/CPR and sham mice in the 7 week experiment. F. The time to return of spontaneous circulation varies between mice, but not between groups. G: Scatter plots relate time to ROSC with GFR. Although the relationship is generally inverse, there is not significant correlation at 24h (note that GFR is near-zero 24h after CA/CPR) or at 7 weeks.
Acute Kidney Injury 24 hours after CA/CPR
CA/CPR resulted in AKI as shown in figure 2A: near-zero GFR (649±70 μL/min/100g in sham, vs. 25±11 μL/min/100g after CA/CPR, p<0.0001 n=4–5, see also supplemental figure S1) and oliguria (24h urine output 4.1 ± 0.5 mL in sham vs. 0.6 ± 0.3 mL after CA/CPR, p=0.002, n=4–7). Kidney injury molecule-1 (KIM-1) immunofluorescence exhibited a heterogeneous pattern (figure 2B) and was greatly upregulated in CA/CPR mice compared with rare positivity in sham (74.8±6.1 %+ tubules/HPF in CA/CPR vs. 0.06±0.06 in sham, p<0.0001, figure 2C), demonstrating tubular cell injury. Apoptotic cleaved caspase-3 positive cells were absent in sham and scarce 24h after CA/CPR mice, however luminal casts in CA/CPR mice were positive for caspase-3 (figure 2D, arrows). Since we previously found tubular epithelial caspase-3 upregulated 6h after CA/CPR,21 this may suggest apoptosis occurring earlier than 24h. Periodic-acid Schiff (PAS) stained sections demonstrated CA/CPR-induced proximal tubular brush border effacement, intracellular vacuoles within tubular epithelial cells, cell casts, and necrosis, concentrated at the corticomedullary junction (figure 2E).
Figure 2:

Histopathologic characterization of acute cardiorenal syndrome 24 hours after cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Glomerular filtration (A) and urine output (B) are near zero 24h after CA/CPR. B: Coimmunostaining for Kidney Injury Molecule-1 (KIM-1) and Lotus tetragonolobus lectin (LTA, a marker marker specific for brush borders of proximal tubules) demonstrates heterogeneously distributed injury to proximal tubule cells after CA/CPR, with tubules demonstrating the most KIM-1 expression also containing luminal LTA and brush-border loss (open arrow, example), while other tubular sections demonstrate faint or absent KIM-1 expression. KIM-1 expression, absent from sham, is significant 24h after CA/CPR (C). D: staining for the apoptosis marker cleaved caspase-3 demonstrates caspase-3 positive casts within tubular lumens and scattered tubular cells after CA/CPR and none in sham, suggesting that caspase-mediated apoptotic cell death occurs before 24h after CA/CPR. E: Brush border effacement, tubular cell swelling and vacuolization (closed arrowheads), tubular cell casts (open arrowheads), and tubular proteinaceous casts (star) are present after CA/CPR compared with normal architecture in sham. All scale bars are 100 μm. All column graph bars represent mean±S.E.M.
Renal inflammation and recovery of GFR
Recovery and progression of renal dysfunction may coexist in AKI-CKD transition.25 We therefore investigated whether these processes occur in the kidney following CA/CPR. GFR remained diminished at 1 week, but 2 weeks after CA/CPR had normalized (figure 3A). Tubular injury scoring (figure 3B) revealed that intraepithelial vacuolization, present at 24h after CA/CPR, declined rapidly thereafter, while epithelial cell flattening/tubular atrophy, increased progressively, and brush border loss remained elevated through 7 weeks, indicating that despite normalization of GFR, pathologic change evolves in tubular epithelium through 7 weeks. Figure 3C confirms that tubular epithelial cell injury continued through functional recovery, with KIM-1 positivity in renal cortex 1 week after CA/CPR and less severe but still present 7 weeks after CA/CPR. In some mice, KIM-1 positivity appears to shift over time from primarily cortical at 24h to primarily outer medullary at 1 week, however quantification of KIM-1 positive area ration did not substantiate this impression (figure 3D). Cellular proliferation, evidenced by Ki-67 immunopositivity, accompanied injury, peaking 1 week after CA/CPR (figure 3E), and shifting from the pertubular interstitium (in sham) to within tubular cells 24h after CA/CPR (3F, representative images 3G).
Figure 3:

Functional recovery from acute cardiorenal syndrome is accompanied by continued histopathologic injury. A. GFR recovers to normal by 2 weeks after CA/CPR. Although still reduced compared to sham 1 week after CA/CPR, 2 weeks after CA/CPR, GFR is identical to that of time-matched sham. 24h, 2 week, and 7 week groups were pooled to increase statistical power after analysis demonstrated they were not different (“sham all”); this was also not different from 2 week GFR. Bars are mean±SEM. Each dot represents one animal. B. Tubular injury scoring shows that while acute changes, such as epithelial vacuolization decline, epithelial atrophy increases through 7 weeks after CA/CPR. C-D: Kidney injury molecule-1 (KIM-1) signal remains elevated 1 week after CA/CPR, but is not different than sham 7 weeks after CA/CPR. KIM-1 signal is heterogeneously distributed between cortex and outer medulla over time after CA/CPR (shams not shown because KIM-1 is not present in sham). E: Quantification of Ki-67-positive cells. F: Tubular:interstitial ratio of Ki-67-positive cells demonstrates redistribution of proliferating cells from interstitital to tubular compartment 24 h after CA/CPR. G Representative low power renal sections with costaining for KIM-1, and the proliferative cell marker Ki-67 demonstrate corticomedullary junctional KIM-1 at 1 week (dotted white lines demarcate the border between inner and outer stripe of the medulla (ISOM, OSOM respectively). Although KIM-1 positive injury does not extend to the inner medulla, Ki-67 positive cells are present in the inner medulla. KIM-1 expression persists at 60 days after CA/CPR, indicating chronic injury in dilated proximal tubules. Scale bars are 1000 μM. KIM-1-delineated injury is most prominent 7 days following CA/CPR. Proliferating cells are most evident at this time point as well, consistent with tubular repair, which continues through 7 weeks C: Quantification of KIM-1. For C&E, bars represent mean, error lines SEM, and each dot is one animal. Six high-power fields were counted per animal.
The presence in renal cortex of the macrophage marker F4/80 and the T-cell marker CD3, strongly support an inflammatory response to AKI after CA/CPR. F4/80-positive cells were present in cortex at all time points, but sharply increased 24 h after CA/CPR (figure 4A), and were distributed in a perivascular and peritubular pattern. Inducible nitric oxide (iNOS, a marker for M1-type macrophages)-positive cells also increased after CA/CPR in the peritubular interstitium (figure 4B). CD3+ cells, not present in sham, followed a similar distribution to F4/80 positive cells, but were delayed and sustained relative to F4/80, present not at 24 h but at 1 and 7 weeks (figure 4C, representative images in figures 4F). Compared to sham mice, TGFβ concentration tended to be higher in renal tissue at 24h after CA/CPR, but this and other cytokines did not reach statistical significance (supplemental figure S2A). However, we found that tgfb1 mRNA is increased 24h after CA/CPR (supplemental figure S2B). These observations support recovery of renal function after CA/CPR accompanied by acute sustained and concurrent tubular epithelial cell injury and renal inflammation, likely starting at 24h, as well as tubular repair.
Figure 4:

Inflammatory cell infiltration following CA/CPR. A: F4/80-positive cells are present in low levels in sham, maximally present 24 h after CA/CPR, and remain present at 1 week and 7 weeks. B: Inducible nitric oxide (iNOS)-positivity increasingly characterizes perivascular cells, suggesting increasing contribution of m1-phenotype macrophages. C: CD3-positive cells, not present in sham or immediately after CA/CPR, infiltrate by 1 week, and persist to 7 weeks after CA/CPR. D: Representative F4/80-stained renal cortex high-power images. Representative iNOS-stained renal cortex high-power images. Representative CD3-stained renal cortex high-power images. Bar graph columns are mean±SEM. Scale bars are 100μm.
Development of CKD
Next, we determined whether CKD occurred after recovery from CA/CPR. Compared with sham mice survived for an identical period, CA/CPR mice demonstrated 30% reduced GFR (623±73 μL/min/100g in sham vs. 409 ± 5 μL/min/100g after CA/CPR, p=0.016, n=4/group) and increased serum urea nitrogen (33±4 vs. 49±3 mg/dL, p=0.013, n=9, figure 5A). PAS-stained kidney sections of CA/CPR-treated mice demonstrated morphologic changes of CKD: peritubular cellular infiltrates, tubulointerstitial protein deposition, atubular glomeruli, and protein casts indicative of proteinuria (figure 5B). Proteinuria suggested by histologic proteinaceous deposits was confirmed in 24h urine collections from 7 weeks, with CA/CPR mice demonstrating elevated total protein and urine albumin (figure 5C and D). Because inflammatory cytokines may mediate chronic renal injury, we repeated cytokine array analysis on renal tissue in mice 7 weeks after sham or CA/CPR. Renal TGFβ was significantly elevated in CA/CPR-treated mice (24.8 vs. 16.6 pg/mL, p=0.006). Other cytokines were not significantly different between sham and CA/CPR mice, although MCP-1 tended to be higher in CA/CPR mice.
Figure 5:

Chronic kidney disease (CKD) results from CA/CPR. A: 7 weeks after CA/CPR, renal function is impaired, demonstrated here by reduced GFR and increased serum urea nitrogen. B: The tubulointerstitial extracellular matrix is lastingly altered after acute cardiorenal syndrome, Periodic-acid Schiff (PAS)-stained sections demonstrate findings of CKD in CA/CPR-treated mice compared with sham. In the cortex, fibrosed areas coincide with cellular infiltrates. While some tubules demonstrate normal morphology, many are dilated. Atubular glomeruli (arrow) are present. In the medulla, interstitial fibrosis is also present, and tubular proteinaceous stain reveals proteinuria. C and D: Both 24h urine protein and 24h urine albumin are elevated in mice subjected to CA/CPR. E: Inflammation persists and TGFβ is elevated in renal tissue 7 weeks after CA/CPR. F: Progressive fibrosis occurs. The renal fibrosis marker α-smooth muscle actin (αSMA) is not detectable in the tubulointerstitium 24 h after CA/CPR except in vascular smooth muscle (bottom right). Thereafter, cortical interstitial αSMA signal increases; it is first visible at 1 week. By 2 weeks following CA/CPR some areas of interstitium are largely replaced with αSMA, and surrounding tubules appear dilated, and by 7 weeks there are areas of interstitium which appear entirely composed of αSMA. The difference between sham and CA/CPR at 7 weeks is quantified at right. G: Immunostaining for fibrillin-1 revealed its presence in the cortical and medullary interstitium after sham procedure. 7 weeks after CA/CPR, fibrillin-1 staining is greatly increased, forming linear peritubular bands in both the cortex and the medulla suggesting a role for this functional matrix protein in fibrotic renal injury. Scale bars are 100 μM. All column graph bars represent mean±S.E.M.
Tubular injury and repair continued indicated by KIM-1 positive tubules with dense surrounding Ki-67 (supplemental figure S3). In CA/CPR mice there was progression in tubulointerstitial deposition of the fibrosis marker α-smooth muscle actin (αSMA); by 7 weeks there was substantial αSMA signal (0.18±0.06 in sham vs. 1.6±0.43 in CA/CPR-treated mice, volume %, p=0.009, n=4/group, figure 5F). Picrosirius red stain demonstrated collagen accumulation (sham 8.4±0.4 vs. 26.6±1.5 volume % after CA/CPR, p<0.0001, supplemental figure S4). The status of the matrix protein Fibrillin-1, which plays a role in dermal fibrosis and may connect inflammation and fibrosis, is unknown in renal injury. Fibrillin-1 was present in sham mice, but was two-fold higher 7 weeks after CA/CPR (5.9±1.1 vs 13.9±3.3, volume %, p=0.04, n=4/group, figure 5G). Therefore, 7 weeks after CA/CPR, mice develop CKD characterized by loss of renal function, proteinuria, and accumulation of renal extracellular matrix.
Discussion
We provide the first report of acute cardiorenal syndrome-induced AKI-CKD transition. Despite normalization of GFR (mimicking recovery from human AKI), progressive deposition of extracellular matrix and continued inflammation occurs. By 7 weeks after CA/CPR, CKD is demonstrated by tubulointerstitial fibrosis, reduced GFR, and proteinuria. These findings are in accordance with results from other mouse AKI-CKD transition models: inflammation, loss of function, and delayed fibrosis occur after unilateral ureteral obstruction,26 ischemia-reperfusion injury,27–31 and proximal tubule-directed injury.32, 33 Our results are both novel and significant because CA/CPR mimics a common human disease state and may therefore provide a means to test mechanisms in a translational model. We note progression of tubulointerstitial αSMA (present 1 week following CA/CPR) while GFR, inflammatory markers, and KIM-1 labeled injury demonstrate rise and fall. This suggests that αSMA deposition is initiated at the time of CA/CPR, or within the first week, and may guide further mechanistic studies by suggesting the timing of fibrosis initiation.
We found that 24h after CA/CPR, mice demonstrate AKI characterized by oliguria, negligible glomerular filtration, tubular epithelial cell injury/death, inflammatory infiltration, and morphologic signs of AKI. These findings are significant additions to the limited prior characterization of this unique acute cardionrenal syndrome model, the full body of knowledge of which comes from two groups. The Rabb and Traystman groups demonstrated t-cell mediated injury and elevated intrarenal cytokines and serum elevated creatinine 24h after normothermic cardiac arrest in mice in a seminal 2003 investigation.34 Our group has previously demonstrated tubular necrosis, elevated neutrophil gelatinase-associated lipocalin, urea nitrogen and creatinine, and estrogen-mediated sex differences in the 24-hour AKI caused by CA/CPR. The current investigation adds evidence of elevated KIM-1, renal recovery demonstrated by cellular proliferation, and renal inflammation demonstrated by macrophage infiltration, and acute upregulation of tubulointerstitial aSMA. Together with the finding of tubular epithelial vacuolization, these findings confirm and extend the observation of Burne-Taney et al that compared with renal ischemia/reperfusion induced by vascular occlusion, acute cardiorenal syndrome induces surprisingly severe renal injury. We speculate this may implicate extrarenal contributions to injury which induce additional damage on top of stop-flow alone.
This is also the first report that renal fibrillin-1 is increased during renal injury. Fibrillin-1 is associated with dermal fibrosis in mice (the tight skin, tsk, mouse harbors a causative mutation in Fbn1)35 and in humans (Stiff Skin Syndrome is due to mutations in FBN1)36. Fibrillin-1 targets the large latent TGFβ complex to the extracellular matrix,37 and together with αSMA positive myofibroblasts, may be required for activation of TGFβ signaling during some types of fibrosis. As we found that tgfb1 mRNA is increased 24h after CA/CPR and TGFβ tissue concentration is increased 7 weeks after CA/CPR, renal fibrosis due to CA/CPR may therefore involve fibrillin-1 regulated TGFβ signaling. Whether this occurs clinically or in other models remains to be determined. TGFβ has been implicated as a driver of AKI-CKD transition,38–40 and our finding that early renal macrophage infiltration persists into the fibrotic stage and TGFβ associates with development of CKD after CA/CPR suggests that inflammation mediated by TGFβ may be a mechanism of AKI-CKD transition after CA/CPR. It is important to note that CA/CPR is distinct from other renal injury models in that after CA/CPR the kidney is exposed to endocrine and other signaling from injured organs outside the kidney, such as the heart. We recently reported that the cardiac transcription factor cardiac LIM protein, released into plasma after CA/CPR, induces renal fibrosis.41 Therefore the mechanism by which renal fibrosis is induced after CA/CPR may be distinct from that of other models, involving extrarenal signals. Future investigation in our laboratory is focused on this connection.
Our study has limitations. First, CA/CPR is a severe insult, with significant mortality. Interestingly, CA/CPR mortality in all cohorts was entirely prior to 7 days post-arrest. This early mortality in our translational model is consistent with clinical observations after CA in which the majority of mortality is within days of resuscitation.42, 43 37% 5-day mortality in the 7-week survival cohort may have led to survival bias, a hypothesis which is supported by the observation of a trend toward shorter resuscitation times in the 7-week cohort. Therefore the long-term survivors in these experiments may be animals which suffered less severe acute kidney injury because more severely injured animals died before 7 weeks. The possibility that less severe AKI may lead to CKD after CA/CPR is tantalizing and deserves further investigation.
Second, we measured GFR in anesthetized mice to reduce movement artifact, which has disproportionate impact on very low GFRs as measured in our 24h CA/CPR cohort. Therefore as a result, GFRs are generally lower than those reported in awake mice, but are in line with previously reported GFR in anesthetized mice in our lab and others21, 44. All mice underwent identical procedures for GFR monitoring, and therefore we are confident in the relative differences in GFR between groups.
Lastly, we were not able to quantify the M1/M2 macrophage balance. Literature supports that M2-macrophages have anti-inflammatory properties and may appear in advanced stages of injury, therefore potentially influencing CKD progression.45–47 Nevertheless, we observed sustained tubule injury and progression of renal fibrosis, suggesting no dominant effects of M2 macrophages in our model.
In summary, we report that AKI-CKD transition occurs due to murine CA/CPR, a model of acute cardiorenal syndrome. Therefore, CA/CPR is a translational model of AKI-CKD transition which may support critical investigation and important insights into a common and burdensome human disease.
Methods
Animals
Animal procedures, performed on 8–12 week-old male C57BL/6 mice which were purchase from Jackson Laboratories, were approved by the Oregon Health & Science University Institutional Animal Care and Use Committee (#IP0000035). There were 4 a priori-defined experiments involving CA/CPR in this investigation. We compared matched sham-treated and CA/CPR-treated mice at 24h, 2 weeks, and 7 weeks after treatment. A separate group of mice, without sham control, underwent CA/CPR and outcome measurement 7 days after CA/CPR.
Cardiac arrest and cardiopulmonary resuscitation (CA/CPR)
CA/CPR was performed as previously described and summarized in figure 1.21–24 Briefly, after induction of general anesthesia with isoflurane (2–4%) by mask, endotracheal intubation, jugular cannulation, rectal temperature probe placement, and subcutaneous electrocardiography electrode placement were carried out. Anesthesia was maintained with isoflurane 1.2–1.5% in 30% oxygen/air mixture and normothermia was maintained using a heating lamp and temperature controller. Cardiac arrest was induced by intravenous administration of 50 μL 0.5M potassium chloride. The endotracheal tube was disconnected, isoflurane discontinued, and the mouse covered with an insulating blanket. After 7.5 min, the endotracheal tube was reconnected to the ventilator. 8 minutes after potassium chloride administration, chest compressions were initiated at a rate of 300 per minute, and epinephrine 8–16 μg administered intravenously over 30–60 seconds. Chest compressions were discontinued after 180 seconds or upon observing sustained spontaneous electrocardiographic activity (return of spontaneous circulation, ROSC). When the spontaneous respiratory rate was >40, usually 10–15 min after ROSC, the endotracheal tube, rectal probe, and jugular catheter were removed and the mouse placed in a recovery cage which was warmed to 37° for 2h. Sham-treated mice received 15 minute isoflurane anesthesia with endotracheal intubation, temperature, and electrocardiography monitoring, but did not undergo intravenous catheterization, potassium chloride injection, or chest compression.
Immunofluorescence and Immunohistochemistry
A list of antibodies and reagents is in Table S1. Kidneys were perfusion-fixed via the cardiac left ventricular apex with 4% paraformaldehyde, and 5 μm thick, paraffin-embedded sections were stained using Periodic acid-Schiff (PAS) stain, α smooth muscle actin-fluorescein isothiocyanate (αSMA-FITC) conjugate, and picrosirius red. For immunofluorescence, sections were incubated for 2h at room temperature with primary antibodies in 1% bovine serum albumin/phosphate buffered saline, followed by Cy2, Cy3 or Cy5-conjugated secondary antibodies (all 1:500, Thermo Fisher Scientific, Carlsbad CA) for 1h at room temperature, and stained with diaminopyridine (DAPI) in the mounting medium. Immunohistochemistry was performed using the Vectastain ABC kit, (Vector Labs, Burlingame CA) according to manufacturer instructions. Antigens were unmasked with citrate buffer, and sections were blocked with 10% normal goat serum for 20 minutes, followed by an incubation with primary antibody in 10% normal goat serum for 1h. Then sections were incubated with biotinylated secondary antibody (Vector Labs, 1:200, 30 minutes), Vectastain ABC and DAB substrate, and mounted.
Glomerular filtration rate and measurement of urine volume
GFR was measured by determining elimination of fluorescein isothiocyanate (FITC)-sinistrin transcutaneously as described.48 Under brief isoflurane anesthesia a fluorescence detector (NIC-Kidney; Mannheim Pharma & Diagnostics GmbH, Germany) was placed on a depilated region of the back and FITC-sinistrin (75 mg/kg body weight) was injected retro-orbitally. Data were acquired in anesthetized (isoflurane, 1%) mice for 90 min and GFR calculated using the half-life (t1/2).48 24h urine volume was collected in metabolic cages.
Imaging
Fluorescence images were captured an epifluorescence microscope (Axio Imager M2, Zeiss, Jena Germany). Light microscopy images were acquired using an auto-imaging system (Evos, Thermo-Fisher Scientific, Carlsbad CA). 6 images (200× field or 400× high-power-field, hpf) were randomly taken per kidney cortex and outer medulla. 6 images (200× field or 400× high-power-field, HPF) were randomly taken per kidney cortex and outer medulla. Cell numbers/HPF (% positive tubular sections/HPF and cortical versus outer medullary area% ratio for KIM-1) were calculated using ImageJ. To ensure unbiased quantification of extracellular matrix components αSMA and fibrillin-1, slides containing 5 sagittal kidney sections, cut at 160 μm intervals starting at random distance from the caudal renal pole, were scanned using a slide scanner (Axioscan, Zeiss, Jena Germany) and semiautomated unbiased stereology was performed using a custom ImageJ macro as previously described.49 Injury scoring was performed on 80–100 random HPF from four consecutive scanned PAS-stained sections. Loss of brush border or sloughing off of tubular epithelial cells, flattening of tubular epithelium (tubular atrophy), and vacuolization within tubular epithelial cells was scored in each section and weighted by severity to express a summative score for each pathologic finding.50
Multiplex cytokine array
Kidney homogenates were prepared using a rotary tissue disruptor in radioimmunoprecipitation assay buffer and sent to Quansys biosciences where the enzyme-lined immunosorbent assay array was performed. Concentrations for interleukin 4 (IL-4), interleukin 10 (IL-10), interleukin 13 (IL-13), interleukin 17 (IL-17), murine chemoattractant protein 1 (MCP-1), interferon 4 (IFN-γ), tumor necrosis factor α (TNF-α), macrophage inflammatory protein-1 (MIP-1), granulocyte-monocyte colony stimulating factor (GM-CSF), and transforming growth factor β (TGFβ) were quantified, and normalized to sample total protein concentration.
Quantitative Polymerase Chain Reaction (qPCR)
Kidneys used for qPCR were removed after saline perfusion but prior to paraformaldehyde perfusion at the time of euthanasia. Ribonucleic acid extraction was performed using a commercial kit (Qiagen, Germantown MD). Reverse transcription of was performed with SuperScript VILO (Invitrogen, Calsbad, CA). qPCR (using NE Biolabs Master Mix, part #M3004S with cycle parameters per manufacturer instructions) was then performed using the tgfb1 Taqman Gene Expression Assay Mm_01178820, normalized to GAPDH (Mm_99999915). Relative gene expression was expressed using the delta-delta CT method.
Statistics
Statistical analysis was performed using Prism 7.0 (GraphPad, LaJolla CA) with the exception of binary logistic regression, which was performed in R (v3.5.1) using package glm. Two-group comparisons were performed using Student’s t-test, or repeated measures ANOVA in the case of analysis of weight at multiple time points. Comparisons of survival were conducted using the log-rank test. Multiple-group comparisons were performed with ANOVA with Holm-Sidak’s test as appropriate. Statistical significance was inferred from p <0.05. Mean and standard error are shown in the figures and text.
Supplementary Material
Supplementary Table S1: List of reagents and resources used in this study.
Supplementary Figure S1: CA/CPR markedly reduced FITC-sinistrin transcutaneous fluorescence decay. FITC-sinistrin fluorescence decay in mice 24h after sham or CA/CPR. Shown are mean±SD (n=4/group). CA/CPR-treated mice demonstrate no appreciable reduction in FITC-sinistrin transcutaneous fluorescence during 90 minutes after bolus injection, while sham mice demonstrate normal reduction in fluorescence, due to indicator clearance. This data was used to derive GFR depicted in figure 2A.
Supplementary Figure S2: A. Results of inflammatory cytokine assay 24h after CA/CPR or sham. CA/CPR did not mediate differential cytokine tissue concentration (mean±SEM shown, p=0.18, ANOVA) B. Quantitative polymerase chain reaction perfomed at the same time point demonstrates increase in tgfb1 mediated by CA/CPR relative to sham.
Supplementary Figure S3: High power renal cortical sections demonstrate acute injury and progression of KIM-1 signal and cell proliferation after sham and CA/CPR. 24h after CA/CPR, lotus tetragonolobus lectin, which specifically marks the proximal tubular brush border, is seen within lumens of injured tubules. KIM-1 signal is sparse (see also figure 3B). 7 days after CA/CPR, KIM-1 and Ki-67 signal are widespread in the cortex, particularly in areas denuded of brush border. By 7 weeks after CA/CPR, Ki-67 and KIM-1 signal colocate surrounding tubular lumens in the renal cortex.
Supplementary Figure S4: Collagen deposition in the tubulointerstitial is dramatically increased 7 weeks after CA/CPR as compared with sham. Using a polarizing microscope, random cortical high-power fields (9 per section, 4 sections per mouse) were imaged. Using imageJ, an 80-point grid was superimposed on each image. Each grid point was determined to be either birefringence positive or negative, and the fraction of postive points expressed as % positive.
Translational Statement.
Sufferers of acute cardiovascular illness often develop renal injury, termed acute cardiorenal syndrome. Although acute cardiorenal syndrome is usually self-resolving, recent studies indicate that it confers high risk for early development of chronic kidney disease. In this study, we describe development of chronic kidney disease after cardiac arrest and acute cardiorenal syndrome in mice. This investigation provides mechanistic insight into the transition between acute cardiorenal syndrome and chronic kidney disease; our long-term goal is to enable development of tools to interrupt the development of chronic kidney disease following acute cardiovascular illness.
Acknowledgements
NIDDK K08DK090754 to MPH, NIDDK R01 DK098141 to JAM, DFG German Research Foundation 332853055 and Else Kröner-Fresenius Stiftung 2015_A197 to TS. This material is the result of work (by MPH) which was supported with resources and the use of facilities at the Portland Veterans Affairs Medical Center. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
The authors have no conflicts of interest to disclose.
References
- 1.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney international 2012; 81: 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chawla LS, Amdur RL, Shaw AD, et al. Association between AKI and long-term renal and cardiovascular outcomes in United States veterans. Clinical journal of the American Society of Nephrology : CJASN 2014; 9: 448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bucaloiu ID, Kirchner HL, Norfolk ER, et al. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney International 2012; 81: 477–485. [DOI] [PubMed] [Google Scholar]
- 4.Mehra R Global public health problem of sudden cardiac death. J Electrocardiol 2007; 40: S118–122. [DOI] [PubMed] [Google Scholar]
- 5.Ronco C, McCullough PA, Anker SD, et al. Cardiorenal syndromes: an executive summary from the consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol 2010; 165: 54–67. [DOI] [PubMed] [Google Scholar]
- 6.Pickering JW, James MT, Palmer SC. Acute kidney injury and prognosis after cardiopulmonary bypass: a meta-analysis of cohort studies. American Journal of Kidney Diseases : The Official Journal of the National Kidney Foundation 2015; 65: 283–293. [DOI] [PubMed] [Google Scholar]
- 7.Englberger L, Suri RM, Li Z, et al. Clinical accuracy of RIFLE and Acute Kidney Injury Network (AKIN) criteria for acute kidney injury in patients undergoing cardiac surgery. Critical Care (London, England) 2011; 15: R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigues FB, Bruetto RG, Torres US, et al. Incidence and mortality of acute kidney injury after myocardial infarction: a comparison between KDIGO and RIFLE criteria. PLoS One 2013; 8: e69998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tujjar O, Mineo G, Dell’Anna A, et al. Acute kidney injury after cardiac arrest. Critical Care (London, England) 2015; 19: 169-015-0900-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishani A, Nelson D, Clothier B, et al. The magnitude of acute serum creatinine increase after cardiac surgery and the risk of chronic kidney disease, progression of kidney disease, and death. Arch Intern Med 2011; 171: 226–233. [DOI] [PubMed] [Google Scholar]
- 11.James MT, Ghali WA, Knudtson ML, et al. Associations between acute kidney injury and cardiovascular and renal outcomes after coronary angiography. Circulation 2011; 123: 409–416. [DOI] [PubMed] [Google Scholar]
- 12.Ishani A, Xue JL, Himmelfarb J, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 2009; 20: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newsome BB, Warnock DG, McClellan WM, et al. Long-term risk of mortality and end-stage renal disease among the elderly after small increases in serum creatinine level during hospitalization for acute myocardial infarction. Arch Intern Med 2008; 168: 609–616. [DOI] [PubMed] [Google Scholar]
- 14.James MT, Hemmelgarn BR, Wiebe N, et al. Glomerular filtration rate, proteinuria, and the incidence and consequences of acute kidney injury: a cohort study. Lancet 2010; 376: 2096–2103. [DOI] [PubMed] [Google Scholar]
- 15.Brown JR, Solomon RJ, Robey RB, et al. Chronic Kidney Disease Progression and Cardiovascular Outcomes Following Cardiac Catheterization-A Population-Controlled Study. J Am Heart Assoc 2016; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gammelager H, Christiansen CF, Johansen MB, et al. Five-year risk of end-stage renal disease among intensive care patients surviving dialysis-requiring acute kidney injury: a nationwide cohort study. Crit Care 2013; 17: R145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Legouis D, Galichon P, Bataille A, et al. Rapid Occurrence of Chronic Kidney Disease in Patients Experiencing Reversible Acute Kidney Injury after Cardiac Surgery. Anesthesiology 2017; 126: 39–46. [DOI] [PubMed] [Google Scholar]
- 18.de Caestecker M, Humphreys BD, Liu KD, et al. Bridging Translation by Improving Preclinical Study Design in AKI. Journal of the American Society of Nephrology : JASN 2015; 26: 2905–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nath KA. Models of Human AKI: Resemblance, Reproducibility, and Return on Investment. Journal of the American Society of Nephrology : JASN 2015; 26: 2891–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuk A, Palevsky PM, Fried L, et al. Overcoming Translational Barriers in Acute Kidney Injury: A Report from an NIDDK Workshop. Clin J Am Soc Nephrol 2018; 13: 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeda M, Wakasaki R, Schenning KJ, et al. Determination of renal function and injury using near-infrared fluorimetry in experimental cardiorenal syndrome. Am J Physiol Renal Physiol 2017; 312: F629–F639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda M, Swide T, Vayl A, et al. Estrogen administered after cardiac arrest and cardiopulmonary resuscitation ameliorates acute kidney injury in a sex- and age-specific manner. Critical Care (London, England) 2015; 19: 332-015-1049-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutchens MP, Kosaka Y, Zhang W, et al. Estrogen-Mediated Renoprotection following Cardiac Arrest and Cardiopulmonary Resuscitation Is Robust to GPR30 Gene Deletion. PloS one 2014; 9: e99910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutchens MP, Fujiyoshi T, Komers R, et al. Estrogen protects renal endothelial barrier function from ischemia-reperfusion in vitro and in vivo. Am J Physiol Renal Physiol 2012; 303: F377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basile DP, Bonventre JV, Mehta R, et al. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J Am Soc Nephrol 2016; 27: 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He W, Dai C, Li Y, et al. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 2009; 20: 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao L, Zhou D, Tan RJ, et al. Sustained Activation of Wnt/beta-Catenin Signaling Drives AKI to CKD Progression. J Am Soc Nephrol 2016; 27: 1727–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai CF, Lin SL, Chiang WC, et al. Blockade of cysteine-rich protein 61 attenuates renal inflammation and fibrosis after ischemic kidney injury. Am J Physiol Renal Physiol 2014; 307: F581–592. [DOI] [PubMed] [Google Scholar]
- 29.Ahmed A, Huang L, Raftery AT, et al. Cyclosporine A sensitizes the kidney to tubulointerstitial fibrosis induced by renal warm ischemia. Transplantation 2004; 77: 686–692. [DOI] [PubMed] [Google Scholar]
- 30.Barrera-Chimal J, Perez-Villalva R, Ortega JA, et al. Mild ischemic injury leads to long-term alterations in the kidney: amelioration by spironolactone administration. Int J Biol Sci 2015; 11: 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weng X, Shen H, Kuang Y, et al. Ischemic postconditioning inhibits the renal fibrosis induced by ischemia-reperfusion injury in rats. Urology 2012; 80: 484 e481–487. [DOI] [PubMed] [Google Scholar]
- 32.Grgic I, Campanholle G, Bijol V, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 2012; 82: 172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takaori K, Nakamura J, Yamamoto S, et al. Severity and Frequency of Proximal Tubule Injury Determines Renal Prognosis. J Am Soc Nephrol 2016; 27: 2393–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burne-Taney MJ, Kofler J, Yokota N, et al. Acute renal failure after whole body ischemia is characterized by inflammation and T cell-mediated injury. American journal of physiologyRenal physiology 2003; 285: F87–94. [DOI] [PubMed] [Google Scholar]
- 35.Siracusa LD, McGrath R, Ma Q, et al. A tandem duplication within the fibrillin 1 gene is associated with the mouse tight skin mutation. Genome Res 1996; 6: 300–313. [DOI] [PubMed] [Google Scholar]
- 36.Tug E, Loeys B, De Paepe A, et al. A Turkish patient of typical Loeys-Dietz syndrome with a TGFBR2 mutation. Genet Couns 2010; 21: 225–232. [PubMed] [Google Scholar]
- 37.Isogai Z, Ono RN, Ushiro S, et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem 2003; 278: 2750–2757. [DOI] [PubMed] [Google Scholar]
- 38.Doi S, Zou Y, Togao O, et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem 2011; 286: 8655–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lan R, Geng H, Polichnowski AJ, et al. PTEN loss defines a TGF-beta-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol Renal Physiol 2012; 302: F1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geng H, Lan R, Wang G, et al. Inhibition of autoregulated TGFbeta signaling simultaneously enhances proliferation and differentiation of kidney epithelium and promotes repair following renal ischemia. Am J Pathol 2009; 174: 1291–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wakasaki R, Matsushita K, Golgotiu K, et al. Glomerular filtrate proteins in acute cardiorenal syndrome. JCI Insight 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hypothermia after Cardiac Arrest Study G. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. The New England journal of medicine 2002; 346: 549–556. [DOI] [PubMed] [Google Scholar]
- 43.Lemiale V, Dumas F, Mongardon N, et al. Intensive care unit mortality after cardiac arrest: the relative contribution of shock and brain injury in a large cohort. Intensive Care Medicine 2013; 39: 1972–1980. [DOI] [PubMed] [Google Scholar]
- 44.Schock-Kusch D, Sadick M, Henninger N, et al. Transcutaneous measurement of glomerular filtration rate using FITC-sinistrin in rats. Nephrol Dial Transplant 2009; 24: 2997–3001. [DOI] [PubMed] [Google Scholar]
- 45.Lee S, Huen S, Nishio H, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 2011; 22: 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belliere J, Casemayou A, Ducasse L, et al. Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J Am Soc Nephrol 2015; 26: 1363–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guiteras R, Flaquer M, Cruzado JM. Macrophage in chronic kidney disease. Clin Kidney J 2016; 9: 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schock-Kusch D, Geraci S, Ermeling E, et al. Reliability of transcutaneous measurement of renal function in various strains of conscious mice. PLoS One 2013; 8: e71519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wakasaki R, Eiwaz M, McClellan N, et al. Automated systematic random sampling and Cavalieri stereology of histologic sections demonstrating acute tubular necrosis after cardiac arrest and cardiopulmonary resuscitation in the mouse. Histol Histopathol 2018: 18012. [DOI] [PubMed] [Google Scholar]
- 50.Saritas T, Cuevas CA, Ferdaus MZ, et al. Disruption of CUL3-mediated ubiquitination causes proximal tubule injury and kidney fibrosis. Sci Rep 2019; 9: 4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: List of reagents and resources used in this study.
Supplementary Figure S1: CA/CPR markedly reduced FITC-sinistrin transcutaneous fluorescence decay. FITC-sinistrin fluorescence decay in mice 24h after sham or CA/CPR. Shown are mean±SD (n=4/group). CA/CPR-treated mice demonstrate no appreciable reduction in FITC-sinistrin transcutaneous fluorescence during 90 minutes after bolus injection, while sham mice demonstrate normal reduction in fluorescence, due to indicator clearance. This data was used to derive GFR depicted in figure 2A.
Supplementary Figure S2: A. Results of inflammatory cytokine assay 24h after CA/CPR or sham. CA/CPR did not mediate differential cytokine tissue concentration (mean±SEM shown, p=0.18, ANOVA) B. Quantitative polymerase chain reaction perfomed at the same time point demonstrates increase in tgfb1 mediated by CA/CPR relative to sham.
Supplementary Figure S3: High power renal cortical sections demonstrate acute injury and progression of KIM-1 signal and cell proliferation after sham and CA/CPR. 24h after CA/CPR, lotus tetragonolobus lectin, which specifically marks the proximal tubular brush border, is seen within lumens of injured tubules. KIM-1 signal is sparse (see also figure 3B). 7 days after CA/CPR, KIM-1 and Ki-67 signal are widespread in the cortex, particularly in areas denuded of brush border. By 7 weeks after CA/CPR, Ki-67 and KIM-1 signal colocate surrounding tubular lumens in the renal cortex.
Supplementary Figure S4: Collagen deposition in the tubulointerstitial is dramatically increased 7 weeks after CA/CPR as compared with sham. Using a polarizing microscope, random cortical high-power fields (9 per section, 4 sections per mouse) were imaged. Using imageJ, an 80-point grid was superimposed on each image. Each grid point was determined to be either birefringence positive or negative, and the fraction of postive points expressed as % positive.