

Acinetobacter baumannii undecaprenyl pyrophosphate synthase, a key enzyme in bacterial cell-wall biosynthesis, has been shown via X-ray structures to bind three fragments identified from direct crystal soaking of a cocktailed screening library.
Keywords: undecaprenyl pyrophosphate synthase, fragment screening, Acinetobacter baumannii
Abstract
Direct soaking of protein crystals with small-molecule fragments grouped into complementary clusters is a useful technique when assessing the potential of a new crystal system to support structure-guided drug discovery. It provides a robustness check prior to any extensive crystal screening, a double check for assay binding cutoffs and structural data for binding pockets that may or may not be picked out in assay measurements. The structural output from this technique for three novel fragment molecules identified to bind to the antibacterial target Acinetobacter baumannii undecaprenyl pyrophosphate synthase are reported, and the different physicochemical requirements of a successful antibiotic are compared with traditional medicines.
1. Introduction
A broad-spectrum antibiotic should ideally target an essential feature or process that is highly conserved amongst bacterial species and is absent in humans. The target of investigation here is Acinetobacter baumannii undecaprenyl pyrophosphate synthase (Ab-UppS), a key enzyme in bacterial cell-wall biosynthesis (Apfel et al., 1999 ▸) that catalyses cis-double-bond formation during the sequential condensation of isopentenyl pyrophosphate (IPP) with farnesyl pyrophosphate (FPP) in the generation of C55 undecaprenyl pyrophosphate, the lipid carrier for the precursors of various cell-wall structures (Ogura & Koyama, 1998 ▸). This enzyme is not expressed in human cells; therefore, blocking its function will induce only bacterial cell death and it is predicted that any inhibitor would synergize with established cell-wall biosynthesis inhibitors, such as the β-lactam class of antibiotics (Demain & Elander, 1999 ▸; Zhu et al., 2013 ▸).
The strategic combination of fragments into subsets based upon various selection criteria such as shape, rotatable bonds or pK a, with subsequent computational concatenation into diverse groups of easily distinguishable molecules, has been carried out in a multitude of ways (Beresini et al., 2014 ▸; Hann et al., 1999 ▸; Keserű et al., 2016 ▸; Menard et al., 1998 ▸; Verlinde et al., 2009 ▸), with the primary goal of producing a well defined set of non-cross-reactive mixed fragments covering as much chemical space as possible (Beresini et al., 2014 ▸; Hann et al., 1999 ▸; Verlinde et al., 2009 ▸). When applied in X-ray crystallography, this technique is often described as ‘fragment cocktailing’ (Caliandro et al., 2013 ▸; Davies & Tickle, 2012 ▸; Hartshorn et al., 2005 ▸; Verlinde et al., 2009 ▸; Xue et al., 2016 ▸).
Here, we present details of the creation of a cocktailed direct crystal-screening fragment library at the GlaxoSmithKline laboratories and three fragment structures derived from it for the antibacterial target Ab-UppS by X-ray crystallography as potential starting points for antibiotic drug discovery. Moreover, we discuss the physicochemical requirements for the development of potential antibiotic leads compared with traditional medicines.
2. Materials and methods
2.1. Protein expression and purification
A PCR reaction was carried out to amplify an N-terminally 6×His-thrombin-tagged ORF sequence of A. baumannii BM4454 flanked by CACC at the 5′ end for TOPO cloning into the Gateway entry vector pENTR-SD-TOPO. A Gateway TOPO reaction was carried out with ∼12 ng of PCR product purified using the Qiagen PCR purification kit, and the reaction was incubated at room temperature for ∼15 min before transformation into chemically competent Top10 cells. Subsequent plasmid DNA Midiprep purification was carried out on a few colonies from the TOPO reaction, and a correct pENTR-SD-TOPO/His-Thr-Ab-UppS clone was identified through sequence confirmation. A Gateway LR reaction was carried out using pENTR-SD-TOPO/His-Thr-Ab-UppS and the E. coli destination vector pDESTT7-ST and was incubated at room temperature for ∼1 h. Following the LR reaction, 2 µl of proteinase K was added and the reaction was incubated at 310 K for ∼10 min before transformation into chemically competent Top10 cells. Subsequent plasmid DNA Midiprep purification was carried out on a few colonies from the LR reaction and a correct pDESTT7-ST/His-Thr-Ab-UppS clone was identified through sequence confirmation.
Inoculation of 2 × 1 l LB broth with 75 µg ml−1 ampicillin was carried out in a 4 l glass flask with 25 ml (1:40 dilution) of overnight starter culture. The flask was then incubated at 307 K with shaking at 220 rev min−1 for approximately 3 h until the optical density (OD) reached 0.495. 0.5 mM IPTG was then added to induce expression and the flask was incubated at 303 K for 3 h. The cells were harvested by centrifugation 3 h post-induction at an OD of 1.81.
The cell pellet was homogenized in a lysis buffer consisting of 50 mM sodium phosphate pH 7.5, 500 mM NaCl, 10 mM imidazole, lysed by sonication on ice and centrifuged at 30 000g for 45 min to remove cell debris. Clarified supernatant was affinity-purified using Ni–NTA Superflow (Qiagen) via batch capture at 277 K, washed with 50 mM imidazole in lysis buffer and eluted with 250 mM imidazole in lysis buffer. The eluate was dialyzed into 50 mM Tris–HCl pH 7.5, 200 mM NaCl, 1 mM EDTA. The purified Ab-UppS was concentrated to 14 mg ml−1 as determined by the Bradford method (Bradford, 1976 ▸) and confirmed by UV absorbance at 280 nm (Layne, 1957 ▸). Protein identity was confirmed by intact mass analysis via LC/MS. Macromolecule-production information is summarized in Table 1 ▸.
Table 1. Macromolecule-production information.
| Source organism | A. baumannii BM4454 |
| DNA source | Synthetic |
| Cloning vector | pENTR-SD-TOPO |
| Expression vector | pDESTT7-ST |
| Expression host | E. coli BL21(DE3)pRR692 |
| Complete amino-acid sequence of the construct produced | MGSSHHHHHHSSGLVPRGSHMTDSEEYHLPQHVAIIMDGNNRFAKKNQMQKGDGHREGKNVLDPIVEHCVKTGVRALTVFAFSSENWNRPQYEVDLLMKLLEETIHEQIPRMKKFNIALRFIGDRSRLPSHLVALMEDAEQQTAHHDAMTLTIAVSYGGMWDIANAAKQVAQAVSRGEIDADQINVDLFEKYVSLNDLPAVDLLIRTGGDFRISNFLLWQAAYAELYFTDTLWPEFTVEEFDHALNVFSGRERRFGKTSEQIQQEKIEKL |
2.2. Crystallization
Diffraction-quality crystals were obtained by microseeding, in which small seed crystals of Ab-UppS were grown and crushed by vortexing with a Hampton Research seed bead into a 1 ml solution of 200 mM calcium acetate, 20%(w/v) PEG 3350 and used as a seeding solution to generate large diffraction-quality crystals (Table 2 ▸). Crystals grew to a maximum size over 10–14 days at 293 K. All drops were dispensed by a Mosquito nanolitre dispenser, incubated and imaged in a Formulatrix automated microscope. The resulting crystals were soaked overnight at 293 K in 9.5 µl of the above well solution and 0.5 µl of the fragment-cocktail solution to give a 25 mM concentration of each molecule in the cocktail, a total organic load of 100 mM and 5% DMSO. Crystals were captured in SPINE-standard cryo-loops and were immediately flash-cooled into a bath of liquid nitrogen.
Table 2. Crystallization.
| Seed crystals | Diffracting crystals | |
|---|---|---|
| Method | Vapour diffusion | Vapour diffusion |
| Plate type | Sitting drop | Sitting drop |
| Temperature (K) | 293 | 293 |
| Protein concentration (mg ml−1) | 14 | 14 |
| Buffer composition of protein solution | 50 mM Tris pH 7.5, 200 mM NaCl, 1 mM EDTA | 50 mM Tris pH 7.5, 200 mM NaCl, 1 mM EDTA |
| Composition of reservoir solution | 200 mM calcium acetate, 5–40%(w/v) PEG 3350 | 200 mM calcium acetate, 20%(w/v) PEG 3350, 20% glycerol |
| Volume and ratio of drop | 200 nl, 1:1 | 400 nl, 1:1 (+50 nl seeds) |
| Volume of reservoir (µl) | 80 | 80 |
2.3. Data collection and processing
Data were collected either in-house using a Rigaku FR-E generator with robotic sample handling and a Saturn A200 detector or at the Diamond Light Source (DLS) synchrotron using a PILATUS 6M-F detector. Data were processed with the Global Phasing program autoPROC (Vonrhein et al., 2011 ▸) using the integration software XDS (Kabsch, 2010 ▸), the isotropic scaling software AIMLESS (Evans & Murshudov, 2013 ▸) and the Global Phasing anisotropic scaling software STARANISO (Tickle et al., 2018 ▸). Data-collection and processing statistics are summarized in Table 3 ▸.
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| GR839/GSK513 | GW197 | |
|---|---|---|
| Diffraction source | Rigaku FR-E | I02, DLS |
| Wavelength (Å) | 1.54178 | 0.97950 |
| Temperature (K) | 100 | 100 |
| Detector | Saturn A200 | PILATUS 6M-F |
| Crystal-to-detector distance (mm) | 80 | 345.75 |
| Rotation range per image (°) | 0.5 | 0.5 |
| Total rotation range (°) | 153 | 180 |
| Exposure time per image (s) | 60 | 0.2 |
| Space group | C2 | C2 |
| a, b, c (Å) | 119.796, 63.630, 71.430 | 120.303, 64.117, 71.600 |
| α, β, γ (°) | 90.00, 92.84, 90.00 | 90.000, 92.696, 90.000 |
| Resolution range (Å) | 71.342–1.835 (1.958–1.835) | 71.521–1.647 (1.736–1.647) |
| Total No. of reflections | 122457 (6505) | 171154 (7541) |
| No. of unique reflections | 37882 (1896) | 52406 (2618) |
| Completeness† (%) | 89.8 (49.0) | 85.1 (32.4) |
| Multiplicity | 3.2 (3.4) | 3.3 (2.9) |
| 〈I/σ(I)〉 | 7.9 (1.4)‡ | 21.6 (1.6)§ |
| R meas | 0.110 (0.793) | 0.033 (0.582) |
| Overall B factor from Wilson plot (Å2) | 20 | 27 |
Resolution cutoff applied based on ellipsoidal diffraction limits.
The overall estimate of the ellipsoidal resolution limit from Mn〈I/σ(I)〉 > 1.2 is 1.835 Å. From Mn〈I/σ(I)〉 ≥ 2.00, R p.i.m. ≤ 0.6, CC1/2 ≥ 0.30, resolution limit = 2.089 Å and outer shell completeness (2.096–2.089 Å) = 95.9%. Anisotropic resolution limits as determined by STARANISO: 1.820, 1.891 and 2.015 Å. Delta-B tensor: B 11 = −6.40, B 22 = 4.54, B 33 = 1.86, B 31 = 2.33.
The overall estimate of the ellipsoidal resolution limit from Mn〈I/σ(I)〉 > 1.2 is 1.647 Å. From Mn〈I/σ(I)〉 ≥ 2.00, R p.i.m. ≤ 0.6, CC1/2 ≥ 0.30, resolution limit = 1.751 Å and outer shell completeness (1.757–1.751 Å) = 89.2%. Anisotropic resolution limits as determined by STARANISO: 1.636, 1.688 and 1.795 Å. Delta-B tensor: B 11 = −6.28, B 22 = 3.49, B 33 = 2.79, B 31 = 3.39.
2.4. Structure solution and refinement
Refinements were carried out with BUSTER (Bricogne et al., 2019 ▸; Smart et al., 2012 ▸). Ligand libraries were generated with phenix.elbow (Moriarty et al., 2009 ▸) and were modified by Mogul (Bruno et al., 2004 ▸). Model building was carried out with Coot (Emsley et al., 2010 ▸). Refinement statistics are summarized in Table 4 ▸.
Table 4. Structure refinement.
Values in parentheses are for the outer shell.
| GR839 | GSK513 | GW197 | |
|---|---|---|---|
| Fragment structure |
|
|
|
| Resolution range (Å) | 20.09–1.84 (1.91–1.84) | 23.87–1.65 (1.67–1.65) | |
| Completeness (%) | 80.5 (14.1) | 79.5 (30.7) | |
| No. of reflections, working set | 35922 (757) | 49773 (1048) | |
| No. of reflections, test set | 1911 (32) | 2625 (37) | |
| Final R cryst | 0.171 (0.2097) | 0.183 (0.2197) | |
| Final R free | 0.204 (0.2914) | 0.212 (0.2420) | |
| Cruickshank DPI | 0.143 | 0.108 | |
| No. of non-H atoms | |||
| Protein | 3898 | 3870 | |
| Ion | 1 | 1 | |
| Ligand | 28 | 9 | |
| Water | 368 | 324 | |
| Total | 4295 | 4204 | |
| R.m.s. deviations | |||
| Bonds (Å) | 0.01 | 0.01 | |
| Angles (°) | 1.0 | 1.6 | |
| Average B factors (Å2) | |||
| Protein | 23 | 31 | |
| Ion | 23 | 26 | |
| Ligand | 51 | 30 | |
| Water | 32 | 39 | |
| Ramachandran plot† | |||
| Favoured regions (%) | 98.60 | 99.08 | |
| Additionally allowed (%) | 1.40 | 0.69 | |
| Outliers (%) | 0 | 0.23 | |
| PDB code | 6szg | 6szh | |
Values determined by MolProbity (Chen et al., 2010 ▸).
3. Results and discussion
3.1. Cocktail set creation
The set used here took selected fragments from a broad-profile library of more than 1000 fragment molecules regularly screened as singletons against targets both biochemically and biophysically (Keserű et al., 2016 ▸). Shape-scoring profiles were calculated for each fragment using OMEGA (Hawkins et al., 2010 ▸) and ROCS (Hawkins et al., 2007 ▸) and, in an attempt to account for experimental X-ray data, through automated fitting (Emsley et al., 2010 ▸) of each fragment into calculated electron-density envelopes at a resolution of 3.5 Å (Winn et al., 2011 ▸) for all other fragments, with the resulting correlation coefficients used to gauge their fit (Adams et al., 2010 ▸).
Fragments were then grouped into categories of monocyclic, bicyclic, chlorine-containing and sulfur-containing molecules, with one of each selected for a cocktail based upon its shape and electron-density diversity scores against other members of the same cocktail to yield 48 solutions containing four shape-diverse molecules. Each cocktail was further scrutinized by a panel of medicinal chemists to remove any potential cross-reactive molecules. The final selection showed good coverage of the full library (Fig. 1 ▸).
Figure 1.

Scatter plot comparing the range of molecular weight versus clogP (derived using ChemAxon) observed across a broad GSK fragment library (blue) and the 192 fragments selected for cocktailing (red).
3.2. Ab-UppS structure and fragment binding
PDB entry 6acs (Ko et al., 2018 ▸) describes the structural features of Ab-UppS with bound citrate in a higher symmetry form than that observed here. Both structures presented here show the physiological dimer as the asymmetric unit, but the active-site pocket of protein chain B has significantly less well defined electron density than that of chain A, lacking residues 33–49, and no fragments were fitted into chain B. Considering the C2 symmetry of the crystal in the CCP4 program CONTACT (Winn et al., 2011 ▸) run against the superposition of chain A on chain B focused on the residue range 30–63 illustrates this region in chain A to primarily contact a single symmetry-related chain B residue Lys41 (−x + 1/2, y + 1/2, −z). For chain B this same symmetry operator yields contacts with chain A through Glu57, His58, Lys61 and Lys104 and by the additional operator −x, y, −z to Asn37 of chain B. The closer crystal packing in chain B in the context of a cocktailed fragment-soaking experiment seems to have a disruptive effect on the α-helical structure in this region.
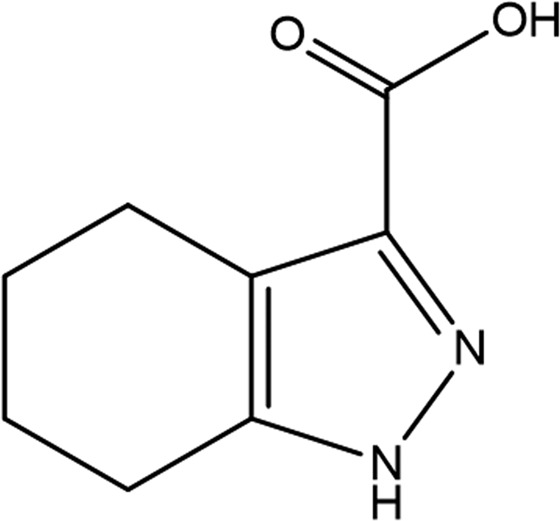
4,5,6,7-Tetrahydro-1H-indazole-3-carboxylate (GR839 in Tables 3 ▸ and 4 ▸) is bound to protein chain A only, showing clear electron density at 1 r.m.s.d., but with no evidence of electron density in the same region of protein chain B. Comparison with E. coli UppS containing farnesyl S-thiolodiphosphate (FSPP; PDB entry 1x06; Guo et al., 2005 ▸) in Fig. 2 ▸(a) shows the carboxylic acid of GR839 to mimic the sulfur-linked phosphate (PA) bridging the Arg79 side chain and the backbone N atom of Asn31 (a glycine in PDB entry 1x06). GR839 extends along the path of the poly(alkene) chain of FSPP, with the ring N atoms hydrogen-bonding to the backbones of Met27 and Asn30.
Figure 2.

(a) Protein subunit A illustrating the binding pocket as mixed ribbons and electrostatic surface and the binding positions of GR839 and GSK513 compared with those of FSPP (carbons as yellow spheres) and FPS902 (carbons as cyan spheres) overlaid from PDB entry 1x06. (b) The binding pocket and interactions of GR839 and GSK513 with the associated 2F o − F c electron density at 1 r.m.s.d. for the fragments and the electrostatic surface of the protein. (c) Dimer structure of Ab-UppS (protein chain A in white, protein chain B in orange) illustrating the binding position of GW197 at the dimer interface alongside superposed molecules of FSPP taken from PDB entry 1x06 to illustrate the catalytic pocket positions. (d) The binding pocket and interactions of GW197 with the associated 2F o − F c electron density at 1 r.m.s.d. for the fragment and the electrostatic surface of the protein. Residues in protein chain A have white C atoms and those in chain B have orange C atoms. Figures were generated with CCPmg (McNicholas et al., 2011 ▸).
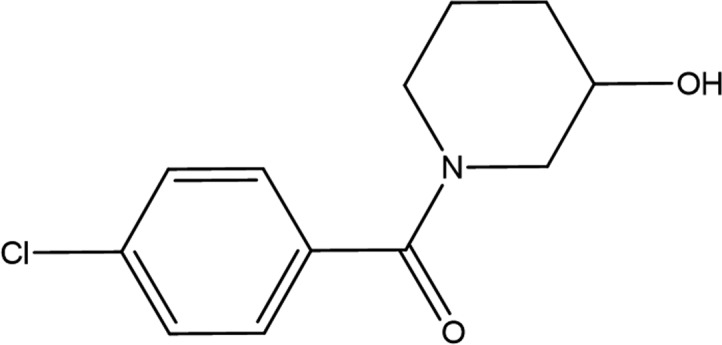
(4-Chlorophenyl)-(3-hydroxypiperidin-1-yl)methanone (GSK513 in Tables 3 ▸ and 4 ▸) comes from the same fragment cocktail as GR839 and is bound in the same structure ∼4 Å deeper in the pocket of protein chain A (Figs. 2 ▸ a and 2 ▸ b). The fragment electron density is more diffuse, with atomic B factors that are approximately twice as large as those of GR839. The piperidin-3-ol ring has been fitted in a higher energy boat conformation to allow a better fit to the electron density for the linking amide. The lower energy chair conformation leads to a series of close contacts to the protein and to the amide O atom rotating away from the water interactions bridging Gly48 and Leu52. The chlorophenyl group binds in a similar position to the geranyl moiety (FPS902) in the E. coli structure (PDB entry 1x06).
The binding modes of these two fragments were not determined separately at the time of this work and their proximity may influence the orientation that each adopts within the active-site pocket. However, throughout the course of this research a propensity was observed for carboxylic acids to be bound in positions similar to GR839 and halogenated aromatics to be bound in positions similar to GSK513.
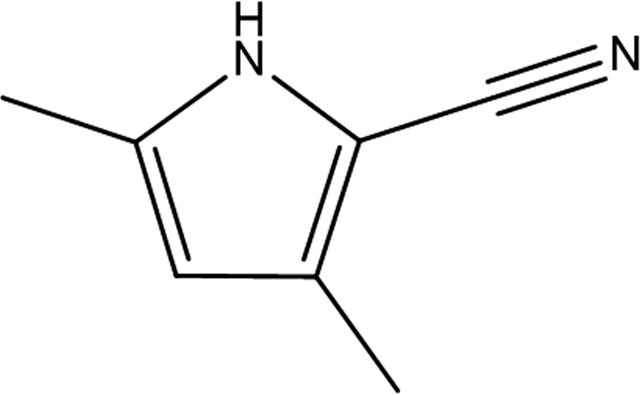
3,5-Dimethyl-1H-pyrrole-2-carbonitrile (GW197 in Tables 3 ▸ and 4 ▸) was bound remotely from the catalytic site (Fig. 2 ▸ c) in an induced pocket at the dimer interface formed by a shift in the position of the chain B residue Ile203, with the peptide carbonyl turned out of the pocket and the peptide carbonyl of Phe206 turned into the pocket. The pyrrole N atom hydrogen-bonds to the backbone carbonyl of Phe206 and the nitrile to the backbone N atom of Ser204 (Fig. 2 ▸ d). The pocket is formed near the centre of the protein dimer and is primarily composed of residues from protein chain B, with a few hydrophobic residues from chain A (Ile203, Leu208 and Leu216). Schrödinger SiteMap (Halgren, 2009 ▸) calculated a cavity volume of 332 Å3 and a ligand volume of 148 Å3.
3.3. Molecular properties
When considering the fragment structures presented here as potential starting points for the synthesis of new antibiotics, it is important to consider the molecular-property space that any potential medicine is likely to need to minimize development risks (Hann, 2011 ▸; Leeson & Young, 2015 ▸). Marketed antibiotics do not follow the same trends as traditional oral medicines, with higher molecular weights and lower lipophilicity being common features (Leeson & Davis, 2004 ▸). The different bacterial cell architecture and the significant impact of cell permeability and efflux means that antibacterial drugs tend to occupy a different property space compared with traditional small-molecule therapeutics (O’Shea & Moser, 2008 ▸).
Fig. 3 ▸ shows the calculated properties total polar surface area (tpsa) and logP for several classes of antibiotic, with the majority residing outside the traditional small-molecule therapeutic range. For this reason, traditional high-throughput screening (HTS) libraries may not be well suited to antibacterial drug discovery (Payne et al., 2007 ▸). Whilst fragment libraries are traditionally biased towards this same therapeutic window, as low-molecular-weight highly efficient starting points they have a greater potential to be developed towards favourable molecular-property space, increasing molecular weight and target affinity, whilst ensuring that key molecular-property indicators are travelling in a direction most likely to be successful in a bacterial setting.
Figure 3.

ChemAxon-calculated values of chromatographic logP versus the total polar surface area (tpsa), comparing the distribution between fragment hits identified in this work and known antibiotics.
4. Conclusions
The experiments here used direct X-ray screening of a set of cocktailed fragments, revealing a hit rate of 8.3% for the antibacterial enzyme Ab-UppS. Of the 16 hits identified, 13 fragments were observed to be bound along the length of the active-site pocket, two in the induced pocket at the dimer interface and one to the surface of the protein, all being determined without any further deconvolution through singleton soaks (although this may have yielded more structural results, it was not attempted). In our hands, this methodology can be a powerful tool to rapidly identify binding fragments for systems where crystallization, soaking and X-ray data collection are routine and provides a useful validation tool that helps to assess the predictive nature of pre-screening assay techniques in identifying successful fragments for crystallization studies.
Supplementary Material
PDB reference: A. baumannii undecaprenyl pyrophosphate synthase, complex with GR839/GSK513, 6szg
PDB reference: complex with GW197, 6szh
Acknowledgments
The authors thank the following GSK scientists for their contributions: Jessica Schneck (UppS program leader), Lisa A. Sloan, Yoshiaki Washio and Robert J. Young (medicinal chemistry), David P. Dixon and Claus E. P. Spitzfaden (biophysics), Stephen D. Picket (computational chemistry).
References
- Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L.-W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. & Zwart, P. H. (2010). Acta Cryst. D66, 213–221. [DOI] [PMC free article] [PubMed]
- Apfel, C. M., Takács, B., Fountoulakis, M., Stieger, M. & Keck, W. (1999). J. Bacteriol. 181, 483–492. [DOI] [PMC free article] [PubMed]
- Beresini, M. H., Liu, Y., Dawes, T. D., Clark, K. R., Orren, L., Schmidt, S., Turincio, R., Jones, S. W., Rodriguez, R. A., Thana, P., Hascall, D., Gross, D. P. & Skelton, N. J. (2014). J. Biomol. Screen. 19, 758–770. [DOI] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem. 72, 248–254. [DOI] [PubMed]
- Bricogne, G., Blanc, E., Brandl, M., Flensburg, C., Keller, P., Paciorek, W., Roversi, P., Smart, O., Vonrhein, C. & Womack, T. (2019). BUSTER-TNT v.2.11.7. Global Phasing Ltd., Cambridge, UK.
- Bruno, I. J., Cole, J. C., Kessler, M., Luo, J., Motherwell, W. D., Purkis, L. H., Smith, B. R., Taylor, R., Cooper, R. I., Harris, S. E. & Orpen, A. G. (2004). J. Chem. Inf. Comput. Sci. 44, 2133–2144. [DOI] [PubMed]
- Caliandro, R., Belviso, D. B., Aresta, B. M., de Candia, M. & Altomare, C. D. (2013). Future Med. Chem. 5, 1121–1140. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Davies, T. G. & Tickle, I. J. (2012). Top. Curr. Chem. 317, 33–59. [DOI] [PubMed]
- Demain, A. L. & Elander, R. P. (1999). Antonie Van Leeuwenhoek, 75, 5–19. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Guo, R.-T., Ko, T.-P., Chen, A. P.-C., Kuo, C.-J., Wang, A. H.-J. & Liang, P.-H. (2005). J. Biol. Chem. 280, 20762–20774. [DOI] [PubMed]
- Halgren, T. A. (2009). J. Chem. Inf. Model. 49, 377–389. [DOI] [PubMed]
- Hann, M. M. (2011). Med. Chem. Commun. 2, 349–355.
- Hann, M., Hudson, B., Lewell, X., Lifely, R., Miller, L. & Ramsden, N. (1999). J. Chem. Inf. Comput. Sci. 39, 897–902. [DOI] [PubMed]
- Hartshorn, M. J., Murray, C. W., Cleasby, A., Frederickson, M., Tickle, I. J. & Jhoti, H. (2005). J. Med. Chem. 48, 403–413. [DOI] [PubMed]
- Hawkins, P. C., Skillman, A. G. & Nicholls, A. (2007). J. Med. Chem. 50, 74–82. [DOI] [PubMed]
- Hawkins, P. C., Skillman, A. G., Warren, G. L., Ellingson, B. A. & Stahl, M. T. (2010). J. Chem. Inf. Model. 50, 572–584. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Keserű, G. M., Erlanson, D. A., Ferenczy, G. G., Hann, M. M., Murray, C. W. & Pickett, S. D. (2016). J. Med. Chem. 59, 8189–8206. [DOI] [PubMed]
- Ko, T.-P., Huang, C.-H., Lai, S.-J. & Chen, Y. (2018). Acta Cryst. F74, 765–769. [DOI] [PMC free article] [PubMed]
- Layne, E. (1957). Methods Enzymol. 3, 447–455.
- Leeson, P. D. & Davis, A. M. (2004). J. Med. Chem. 47, 6338–6348. [DOI] [PubMed]
- Leeson, P. D. & Young, R. J. (2015). ACS Med. Chem. Lett. 6, 722–725. [DOI] [PMC free article] [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- Menard, P. R., Mason, J. S., Morize, I. & Bauerschmidt, S. (1998). J. Chem. Inf. Comput. Sci. 38, 1204–1213.
- Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. (2009). Acta Cryst. D65, 1074–1080. [DOI] [PMC free article] [PubMed]
- Ogura, K. & Koyama, T. (1998). Chem. Rev. 98, 1263–1276. [DOI] [PubMed]
- O’Shea, R. & Moser, H. E. (2008). J. Med. Chem. 51, 2871–2878. [DOI] [PubMed]
- Payne, D. J., Gwynn, M. N., Holmes, D. J. & Pompliano, D. L. (2007). Nat. Rev. Drug Discov. 6, 29–40. [DOI] [PubMed]
- Smart, O. S., Womack, T. O., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., Vonrhein, C. & Bricogne, G. (2012). Acta Cryst. D68, 368–380. [DOI] [PMC free article] [PubMed]
- Tickle, I. J., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., Vonrhein, C. & Bricogne, G. (2018). STARANISO. Global Phasing Ltd., Cambridge, UK.
- Verlinde, C. L. M. J., Fan, E., Shibata, S., Zhang, Z., Sun, Z., Deng, W., Ross, J., Kim, J., Xiao, L., Arakaki, T. L., Bosch, J., Caruthers, J. M., Larson, E. T., Letrong, I., Napuli, A., Kelly, A., Mueller, N., Zucker, F., Van Voorhis, W. C., Buckner, F. S., Merritt, E. A. & Hol, W. G. J. (2009). Curr. Top. Med. Chem. 9, 1678–1687. [DOI] [PMC free article] [PubMed]
- Vonrhein, C., Flensburg, C., Keller, P., Sharff, A., Smart, O., Paciorek, W., Womack, T. & Bricogne, G. (2011). Acta Cryst. D67, 293–302. [DOI] [PMC free article] [PubMed]
- Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A. & Wilson, K. S. (2011). Acta Cryst. D67, 235–242. [DOI] [PMC free article] [PubMed]
- Xue, Y., Guo, H. & Hillertz, P. (2016). ChemMedChem, 11, 1881–1885. [DOI] [PubMed]
- Zhu, W., Zhang, Y., Sinko, W., Hensler, M. E., Olson, J., Molohon, K. J., Lindert, S., Cao, R., Li, K., Wang, K., Wang, Y., Liu, Y.-L., Sankovsky, A., de Oliveira, C. A., Mitchell, D. A., Nizet, V., McCammon, J. A. & Oldfield, E. (2013). Proc. Natl Acad. Sci. USA, 110, 123–128. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: A. baumannii undecaprenyl pyrophosphate synthase, complex with GR839/GSK513, 6szg
PDB reference: complex with GW197, 6szh