

SUMMARY
Generation of protective immunity to infections and vaccinations declines with age. Studies in healthy individuals have implicated reduced miR-181a expression in T cells as contributing to this defect. To understand the impact of miR-181a expression on antiviral responses, we examined LCMV infection in mice with miR-181ab1-deficient T cells. We found that miR-181a deficiency delays viral clearance, thereby biasing the immune response in favor of CD4 over CD8 T cells. Antigen-specific CD4 T cells in mice with miR-181a-deficient T cells expand more and have a broader TCR repertoire with preferential expansion of high-affinity T cells than in wild-type mice. Importantly, generation of antigen-specific miR-181a-deficient CD8 effector T cells is particularly impaired, resulting in lower frequencies of CD8 T cells in the liver even at time points when the infection has been cleared. Consistent with the mouse model, CD4 memory T cells in individuals infected with West Nile virus at older ages tend to be more frequent and of higher affinity.
Graphical Abstract
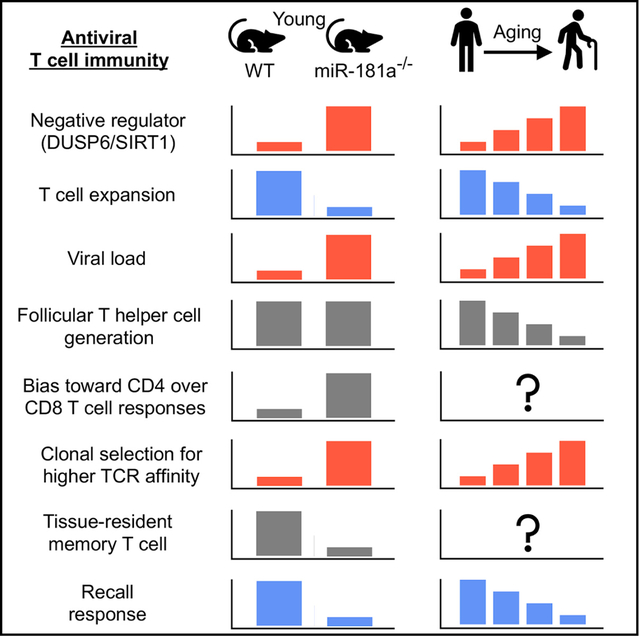
In Brief
T cell aging in humans is associated with progressive loss in miR-181a, the implications of which for antiviral immunity are unknown. Using mouse models, Kim et al. find that miR-181a deficiency in T cells reproduces many aging features including impaired effector T cell expansion, viral clearance, generation of tissue-residing T cells, and recall responses.
INTRODUCTION
With increasing age, the ability of the immune system to protect against infections erodes (Goronzy and Weyand, 2017; Nikolich-Žugich, 2018). Incidence and severity of viral infections increase. More than 90% of all influenza-related deaths in the United States occur in older individuals (Targonski et al., 2007; Thompson et al., 2003). Immune responses to influenza variants are usually a mixture of primary and recall responses in adults, and it is therefore undetermined whether the increased susceptibility is due to defective immune memory. However, mortality and morbidity with newly arising infections are at least equally increased. The risk of neuroinvasive disease from West Nile virus (WNV) increases with age, with the highest incidence, hospitalization, and case-fatality rate in persons aged ≥70 years (Lindsey et al., 2010). Similarly, defects in primary immune responses to several vaccines have been described, including tick-borne encephalitis, Japanese encephalitis, hepatitis A, and pandemic influenza strains (Cramer et al., 2016; D’Acremont et al., 2006; Jílková et al, 2009; Langley et al., 2011). For yellow fever vaccination, development of seroprotection is significantly delayed in older individuals (Roukens et al., 2011). A similar observation was made for the hepatitis B vaccine (Weinberger et al., 2018), where more booster vaccinations were required to achieve seroprotection in non-immune older adults. Interestingly, recall responses in immune individuals were not affected by age in this study.
Studies over the last decade have explored the mechanisms that could account for these defects (Goronzy and Weyand, 2019). In most old individuals, homeostatic mechanisms are able to maintain a sufficiently large and diverse naive CD4 T cell repertoire to respond to the variety of antigens (Qi et al., 2014). Naive CD8 T cells are less well preserved, which may in part explain the defective antiviral responses (Czesnikiewicz-Guzik et al., 2008; Nikolich-Žugich et al., 2012). Alternatively, age-associated T cell-intrinsic defects in cell signaling and differentiation may contribute to the finding of impaired adaptive immunity (Kim et al., 2017). In in vitro studies, we had initially observed that naive CD4 T cells from older individuals have impaired ERK phosphorylation upon T cell receptor (TCR) stimulation due to reduced expression of miR-181a (Li et al., 2012). Transcription of pri-miR-181a is regulated by a transcription factor network including YY1 and TCF1; the expression of these transcription factors and consequently the expression of miR-181a in naive T cells declines with age (Ye et al., 2018). An age-associated decline in miR-181a expression is also seen in mice (Figure S1), suggesting that this decline is a hallmark of T cell aging.
miR-181a was first described in mouse thymocytes and T cells as the master regulator of the TCR activation threshold by controlling the expression of the cytoplasmic DUSP6 and other negative-feedback pathways including PTPN22, SHP2, DUSP5, and SIRT1 (Li et al., 2007; Zhou et al., 2012, 2016). miR-181a is highly expressed in double-positive (DP) thymocytes; expression declines with differentiation to single-positive (SP) thymocytes and peripheral T cells (Li et al., 2007). It has been postulated that the high expression facilitates positive selection through the recognition of autoantigen, while the lower expression in peripheral T cells prevents autoimmunity (Ebert et al., 2009).
Here, we used a mouse model to determine the impact of miR-181a deficiency in T cells upon anti-viral responses in vivo and to infer the implications for the age-associated decline of adaptive immunity. We observed that the T cell response after lymphocytic choriomeningitis virus (LCMV) infection is skewed toward the expansion of antigen-specific CD4 T cells. Generation of CD8 effector T cells is subdued, which leads to delayed viral clearance and reduced numbers of liver-residing CD8 memory T cells, while generation of central memory CD4 and CD8 T cells is preserved. Consistent with the mouse data, viral clearance of WNV is delayed in older adults, while at later time points WNV-specific memory T cells are more frequent and show evidence of affinity selection.
RESULTS
Conditional Deletion of miR-181ab1 in Mature T Cells
To study the miR-181a function in mature T cells, we used mice expressing Cre recombinase under control of the distal Lck promoter (dLck-Cre), which initiates Cre expression after thymic positive selection (Zhang et al., 2005). We crossed dLck-Cre mice with Rosa26-YFP reporter mice (Rosa26YFP), which contain a loxP-flanked transcription termination site upstream of YFP under control of the ubiquitously expressed Rosa26 locus. dLck-Cre+ Rosa26YFP mice did not express YFP reporter in DP thymocytes and started to express YFP in SP thymocytes. Delayed Cre expression resulted in YFP expression in over 90% of CD8 T cells and 70% of CD4 T cells in the spleen (Figure S2A). We then crossed dLck-Cre+ Rosa26YFP mice with miR-181ab1fl/fl mice to generate dLck-Cre+ Rosa26YFP miR-181ab1fl/fl mice (referred to as miR-181a−/− mice) and dLck-Cre+ Rosa26YFP miR-181ab1+/+ mice (referred to as wild-type [WT] mice). miR-181a in YFP+ splenic T cells from miR-181a−/− mice was markedly reduced, with some minimal expression possibly deriving from the miR-181ab2 gene (Figure S2B). Deletion of miR-181a expression in mature T cells did not affect thymic T cell development, and miR-181a−/− mice had similar number of splenic CD4 and CD8 T cells as WT mice (Figures S2C and S2D). Importantly, T cells in WT and miR-181a−/− mice expressed similar levels of surface TCR without aberrant activation phenotypes (Figures S2E and S2F).
We screened T cells from miR-181a−/− mice for the expression of known miR-181a targets that are involved in regulation of T cell responses. SIRT1, previously shown to be targeted by miR-181a in mouse hepatocytes (Zhou et al., 2012) and human T cells (Ye et al., 2018), was increased in miR-181a−/− CD4 T cells (Figure S3A). Also, we observed higher DUSP6 expression (Figure S3B) and reduced ERK phosphorylation after TCR stimulation (Figures S3C and S3D). In contrast, AKT phosphorylation was equally induced in WT and miR-181a-deficient T cells, consistent with previous reports that PTEN was not upregulated in the miR-181a-deficient mice used here (Schaffert et al., 2015), in contrast to the mice reported by Henao-Mejia et al. (2013).
miR-181a Deficiency Impairs CD8 T Cell Expansion and Viral Clearance
We infected WT and miR-181a−/− mice with LCMV-Armstrong and assessed LCMV-specific CD8 T cell responses on day 8 using MHC class I tetramers. We observed a 2- to 3-fold lower number of activated CD62L– CD44+ CD8 T cells from miR-181a−/− mice relative to WT mice in the spleen (Figure 1A). Relative frequencies of CD8 T cells specific to three viral peptides were similar in spleen and liver (Figures 1B and S4A); reductions for all three peptide responses were seen in lymph nodes (Figure S4A). These data are consistent with the notion that miR-181a−/− mice globally generated fewer LCMV-specific CD8 T cells than WT mice, but without bias for the epitope recognized (Figure 1C). The reduction was not due to altered tissue migration, as we observed reductions in LCMV-specific CD8 T cells in lymph nodes and liver (Figure 1D).
Figure 1. miR-181a Deficiency Impairs Antigen-Specific CD8 T Cell Expansion and Viral Clearance after LCMV Infection.

dLck-Cre+ Rosa26YFP miR-181ab1+/+ (WT) and dLck-Cre+ Rosa26YFP miR-181ab1fl/fl (miR-181a−/−) mice were infected with LCMV-Armstrong; CD8 T cell responses were analyzed on day 8.
(A) Representative flow plots of CD44+ splenic CD8 T cells (left) and summary data of the total numbers of YFP+ CD44+ CD62L– CD8 T cells in the spleen from one experiment (right).
(B) Representative flow plots of Db LCMV GP33–41 (GP33), Db GP276–286 (GP276), and Db NP396–404 (NP396) tetramer+ cells gated on YFP+ CD8 T cells in the spleen of WT and miR-181a−/− mice.
(C and D) YFP+ CD8 T cells specific for the indicated epitopes in the spleen (C) and inguinal lymph nodes and liver (D).
(E) Splenocytes were restimulated with GP33, GP276, or NP396 peptides. Stacked bar graphs show the numbers of cytokine-producing YFP+ CD8 T cells (mean ± SEM).
(F) Viral titers in the spleen and kidney on day 6 after LCMV infection.
Data are representative of three independent experiments with 4–5 mice per group (A–C and E), one experiment with 4–5 mice per group (D), or pooled from two independent experiments with 8 mice per group (F). Unless stated otherwise, data are presented as means. Statistical significance by two-tailed unpaired t test (A, C, D, and F) or two-way ANOVA followed by Tukey’s post-test comparison (E). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. See also Figure S4.
To examine the ability of virus-specific CD8 T cells to produce effector cytokines on day 8, we restimulated whole splenocytes with cognate peptides ex vivo and measured intracellular cytokine production. Consistent with tetramer staining, we observed a 2- to 3-fold decrease in the number of LCMV-specific interferon gamma (IFNγ)-, tumor necrosis factor alpha (TNFα)- or interleukin 2 (IL-2)-producing CD8 T cells in the spleen from miR-181a−/− compared to WT mice (Figure 1E). While reduced in frequencies, virus-specific CD8 T cells were fully functional in producing cytokines upon ex vivo restimulation, indicating that miR-181a deficiency did not impair acquisition of effector functions (Figures S4B and S4C). On the contrary, miR-181adeficient virus-specific CD8 T cells had higher granzyme B expression and CD107a (LAMP1) cell surface staining on a per-cell basis (Figures S4D and S4E).
Consistent with reduced frequencies of CD8 effector T cells, miR-181a−/− mice showed significant impairment in their ability to clear LCMV (Figure 1F). Taken together, these data demonstrate that miR-181a expression in mature CD8 T cells is functionally important for antigen-specific T cells to expand and clear the virus.
Delayed Viral Clearance in miR-181a−/− Mice Leads to Increased CD4 T Cell Responses
We next examined whether miR-181a deficiency also impairs antiviral CD4 T cell responses. Surprisingly, in contrast to reduced CD8 T cell responses, we observed an increase in LCMV glycoprotein 61–80 epitope (GP61)-specific tetramer+ CD4 T cells in miR-181a−/− compared to WT mice on day 8 after LCMV infection (Figure 2A). Likewise, the frequency of CD4 T cells producing cytokines upon peptide restimulation was increased (Figure 2B). Like CD8 T cells, miR-181a-deficient CD4 T cells were on a per-cell basis equally able as WT cells to produce effector cytokines upon ex vivo restimulation (Figure 2C). The absence of a defect in T cell activation at the effector stage is consistent with the observation that miR-181a was very low in WT effector T cells. A kinetics of miR-181a expression in SMARTA cells, which are specific for the immunodominant CD4 epitope of the LCMV glycoprotein 61–80, showed a rapid decline of miR-181a after activation to minimal levels in effector cells; a partial recovery of miR-181a was seen in memory cells (Figure 2D).
Figure 2. Antiviral CD4 T Cell Responses in miR-181a−/− Mice.

(A–C) WT and miR-181a−/− mice were infected with LCMV; CD4 T cell responses were analyzed on day 8. (A) Representative flow plots for IAb GP66 tetramer+ cells gated on YFP+ CD4 T cells (left) and summary data of the frequencies and numbers of IAb GP66 tetramer+ YFP+ CD4 T cells in the spleen (right). (B) Representative flow plots of IFNγ and IFNα production by YFP+ CD4 T cells after restimulation with GP61 peptide (left) and summary data of cytokine-producing YFP+ CD4 T cells in the spleen (right). (C) Summary data of cytokine expression on a per-cell basis in day 8 effector CD4 T cells.
(D) Naive SMARTA CD4 T cells (CD45.1+) were adoptively transferred into B6 hosts (CD45.2+), followed by LCMV infection 1 day later. Graph shows the number of splenic SMARTA CD4 T cells and miR-181a levels on sorted SMARTA cells at indicated time points (mean ± SEM).
(E–G) Equal numbers of congenically marked WT and miR-181a−/−YFP+ SMARTA CD4 T cells were co-transferred into B6 mice that were infected with LCMV 1 day later. (E) Schematic diagram (left) and representative flow scatter plots of the relative proportion of WT and miR-181a−/− SMARTA CD4 T cells in the spleen (right). (F) Summary graph of the number of splenic WT and miR-181a−/− SMARTA CD4 T cells on day 8 from one experiment. (G) Ratios of miR-181a−/− to WT SMARTA CD4 T cells in the spleen, mesenteric lymph nodes (mLNs), inguinal lymph nodes (iLNs), and liver.
(H) Equal numbers of WT SMARTA cells (CD45.1+) were transferred into WT mice and mice with miR-181a-deficient T cells (schematic diagram, left), followed by LCMV infection 1 day later. Representative contour plots of expanded SMARTA cells gated on total CD4 T cells in WT and miR-181a−/− recipients (middle) and frequencies from four mice in each group on day 8 (right).
Data are pooled from two independent experiments with 8–10 mice per group (A and B), representative of two independent experiments with 4–5 mice per group (C and E–G) or one experiment with 3–4 mice per group (D and H). Unless stated otherwise, data are presented as means. Statistical significance by two-tailed unpaired (A–C and H) or paired (F) t test. *p < 0.05; **p < 0.01; ***p < 0.001; NS, not significant. See also Figure S5.
To determine whether expansion of antiviral CD4 T cells is resistant to miR-181a deficiency and CD4 responses are therefore increased in miR-181a−/− mice, TCR transgenic SMARTA mice were crossed to miR-181a−/− mice to generate dLck-Cre+ Rosa26YFP miR-181ab1fl/fl SMARTA mice (referred to as miR-181a−/− SMARTA mice) and dLck-Cre+ Rosa26YFP miR-181ab1+/+ SMARTA mice (referred to as WT SMARTA mice). We co-transferred congenically marked naive WT and miR-181a−/− SMARTA CD4 T cells at a 1:1 ratio into B6 recipient mice and infected them with LCMV 1 day later (Figure 2E). Similar to miR-181a-deficient CD8 T cells, miR-181a−/− SMARTA CD4 T cells showed a significant reduction in expansion on day 8 in the spleen as well as lymph nodes and liver (Figures 2E–2G). Silencing of DUSP6 in miR-181a-deficient SMARTA cells before adoptive transfer was not able to restore clonal expansion (Figure S5), consistent with the interpretation that a single miR-181a target did not account for the observed defect. Moreover, we observed a 2-fold higher expansion of WT SMARTA CD4 T cells, when adoptively transferred into miR-181a−/− compared to WT mice, suggesting that the observed increased CD4 T cell expansion is induced by exogenous stimuli (Figure 2H). In conclusion, miR-181a deficiency intrinsically impairs expansion of CD8 as well as CD4 T cells, thereby delaying viral clearance. The longer persistence of viral stimulation appears to lead to a compensatorily increased antiviral CD4 T cell response.
miR-181a Deficiency Selects a More Diverse and Higher Affinity Virus-Specific CD4 T Cell Repertoire
Given the ability of miR-181a in regulating TCR activation thresholds, we explored whether miR-181a deficiency contracts the TCR repertoire responding to LCMV. On day 8 after LCMV infection, we sorted LCMV glycoprotein 33–41 epitope (GP33)-specific CD8 T cells from the spleens of WT and miR-181a−/− mice. cDNA was generated by 5′ rapid amplification of cDNA end (5′-RACE) PCR, and TRB sequences were amplified and sequenced. WT and miR-181a−/− mice harbored a similar number of 160 distinct GP33-specific CD8 T cell clones (Figure 3A). Clonal size distribution was also similar, with 50% of the entire antigen-specific compartment made up by the ~18 top frequent clonotypes (Figure 3B). Accordingly, repertoire diversity, as determined by the Shannon entropy index, was not different (Figure 3C). Functionally, miR-181a-deficient CD8 effector T cells required similar amounts of GP33 peptide antigen to induce half-maximal responses compared to WT cells, indicating similar antigen sensitivity (Figure 3D). In summary, miR-181a deficiency affected the expansion but did not contract the repertoire of the responding CD8 T cell population.
Figure 3. miR-181a Deficiency Selects a More Diverse and Higher-Affinity Virus-Specific CD4 T Cell Repertoire.

WT and miR-181a−/− mice were infected with LCMV. (A–C) TRB genes from splenic Db GP33 tetramer+ YFP+ CD8 T cells on day 8 were sequenced. Total number of unique TRB sequences (A), the number of top frequent clones making up 50% of the entire repertoire (B), and the Shannon diversity index (C) are shown.
(D) Splenocytes were restimulated with a range of GP33 peptide concentrations. Dot plot shows the effective GP33 peptide concentration required to elicit a half-maximal IFNγ production.
(E) Representative histogram of tetramer binding intensity of IAb GP66 tetramer+ YFP+ CD4 T cells (left) and summary data of tetramer mean fluorescence intensity (MFI) from one experiment (right). Filled gray in histogram indicates naive CD4 T cells.
(F) Dot plot shows the effective GP61 peptide concentration required to elicit a half-maximal IFNγ production after in vitro restimulation.
(G and H) WT and miR-181a-deficient SMARTA cells were co-transferred into WT mice that were then infected with LCMV. MFI expression of the SMARTA TCR as histogram (G, left), summary plot from 5 mice in each group (G, right), and half-maximal effective concentration (EC50) of IFNγ production by SMARTA cells after peptide restimulation in vitro (H) on day 8. (I–K) TRB sequences from splenic IAb GP66 tetramer+ YFP+ CD4 T cells were determined on day 8. Total number of unique TRB sequences (I), the number of top frequent clones making up 50% of the entire repertoire (J), and the Shannon diversity index (K) are shown.
Data are from one experiment with 4–5 mice per group (A–C), representative of two independent experiments with 4–5 mice per group (D, E, and G–K) or pooled from two independent experiments with 7–8 mice per group(F). Unless stated otherwise, data are presented as means. Statistical significance by two-tailed unpaired (A–F and I–K) or paired (G and H) t test. *p < 0.05; **p <0.01; ***p < 0.001; NS, not significant.
In contrast, miR-181a deficiency influenced the repertoire of responding CD4 T cells. miR-181a-deficient GP61-specific CD4 effector T cells lacked a population of cells that weakly bound the tetramer (Figure 3E). The stronger tetramer binding corresponded to higher antigen sensitivity, as indicated by 20% less antigenic peptide needed to elicit a half-maximal response (Figure 3F). In contrast, TCR-transgenic WT and miR-181a−/− SMARTA CD4 T cells had similar TCR expression and peptide antigen sensitivity at the peak of LCMV infection (Figures 3G and 3H), consistent with the interpretation that the observed differences were caused by clonal selection within a polyclonal population. In spite of the more stringent selection, a higher number of CD4 T cells with unique TRB sequences were recruited by LCMV in miR-181a-deficient than WT mice, resulting in increased repertoire diversity (Figures 3I–3K). These data indicate that not all CD4 T cell clones are recruited in the response to an acute infection; in the presence of a reduced CD8 T cell response, the size as well as the clonal diversity of the CD4 T cell responses is increased, presumably due to the higher persistence of antigen.
Defective Generation of miR-181a-Deficient Short-Lived Effector and Long-Lived Liver-Residing Memory CD8 T Cells
In antiviral T cell responses, CD8 T cells differentiate into memory precursor and short-lived effector T cells that can be distinguished based on variable expression of CD127 (IL-7Rα) and KLRG1 (Joshi et al., 2007; Sarkar et al., 2008). The impaired expansion of miR-181a-deficient CD8 T cells reflected a selective defect in the accumulation of terminally differentiated KLRG1high CD127low effector cells, whereas the number of KLRG1low CD127high memory precursor cells was not altered (Figures 4A and S6A). Consistent with the relative increase in memory precursors, LCMV-specific miR-181a-deficient CD8 effector cells exhibited increased frequencies of IL-2 producers (Figure S6B). The difference in the frequencies of KLRG1+ cells even widened while frequencies were declining subsequent to the peak response (Figure 4B). In contrast, frequencies of CD127high long-lived memory CD8 T cells were not different in both mice, arguing against a kinetic difference in antiviral CD8 T cell responses (Figure 4B).
Figure 4. miR-181a Is Required for the Generation of Short-Lived Effector and Long-Lived Liver-Residing Memory CD8 T Cells.

(A) WT and miR-181a−/− mice were infected with LCMV, and their spleens were harvested on day 8. Stacked bar graphs of the numbers of short-lived effector (KLRG1high CD127low, left) and memory precursor (KLRG1low CD127high, right) Db tetramer+ YFP+ CD8 T cells for indicated epitopes (mean ± SEM).
(B) Frequencies of Db GP33 tetramer+ YFP+ CD8 T cells with a short-lived effector (left) or a long-lived memory phenotype (right) in peripheral blood mononuclear cells (PBMCs) at indicated time points.
(C–F) Antigen-specific memory CD8 T cells on day 85 after infection. Number of Db GP33 tetramer+ YFP+ CD8 T cells in the spleen (C), and representative histograms and dot plots of CD62L and CD27 expression by Db GP33 tetramer+ YFP+ CD8 T cells (D). Frequencies and numbers of Db GP33 tetramer+ YFP+ CD8 T cells in inguinal lymph nodes (E) and liver (F).
(G) CD69 and CXCR6 expression on liver-resident GP33-specific CD8 T cells on day 50.
(H) GP33-specific YFP+ memory CD8 T cells from WT and miR-181a−/− mice were transferred into B6 hosts infected with LCMV 1 day later. Representative flow plots of the frequency of donor-derived Db LCMV GP33 tetramer+ cells gated on total CD8 T cells in the spleen (left) and dot plots of the total numbers of Db LCMV GP33 tetramer+ CD8 T cells in the spleen, lymph nodes, and liver (right).
Data are representative of three independent experiments with 4–5 mice per group (A), pooled from two independent experiments with 6–7 mice per group (B–F) or one experiment with 5 mice per group (G and H). Unless stated otherwise, data are presented as means. Statistical significance by two-way ANOVA followed by Tukey’s post-test comparison (A) or two-tailed unpaired t test (B–H). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; NS, not significant. See also Figures S6 and S7.
On days 80 to 90 after infection, LCMV-specific memory CD8 T cell numbers in the spleens of WT and miR-181a−/− mice were similar; however, central memory cells were relatively enriched in miR-181a-deficient mice, as shown by increased CD62L and CD27 expression (Figures 4C, 4D, and S6C–S6E). Consistent with this functional bias, we saw increased frequencies of antigen-specific T cells in lymph nodes (Figures 4E and S6F). In contrast, miR-181a−/− mice had notably fewer LCMV-specific tissue-residing CD8 T cells in liver compared to WT mice (Figure 4F). This reduction equally affected CD69- and CXCR6-expressing cells (Figure 4G). Together, these data suggest that miR-181a deficiency impairs generation of effector CD8 T cells, resulting in lower frequencies of long-lived virus-specific CD8 T cells in tissues after the infection has been cleared.
Memory CD8 T cells developed in miR-181a−/− mice differed in functionality compared to WT CD8 T cells. Tetramer staining intensity was increased, suggesting more stringent selection for affinity (Figure S7A). In parallel, IFNγ and IFNα production on a per-cell basis was increased, and a higher proportion of cells co-produced IFNγ, IFNα, and IL-2, indicating polyfunctionality (Figures S7B and S7C). To examine their function in vivo, WT and miR-181a-deficient memory CD8 T cells were isolated from the spleens of WT and miR-181a−/− mice on day 50 after LCMV infection. Identical numbers of GP33-specific memory CD8 T cells were then adoptively transferred into naive B6 recipients, followed by LCMV infection 1 day later. Similar to the primary response, miR-181adeficient memory CD8 T cells had significantly reduced recall expansion in all tissues examined (Figure 4H). In summary, increased central memory phenotypes of miR-181a-deficient memory CD8 T cells did not translate into increased secondary responses.
Viral Infection Induces Increased Number of Functional miR-181a-Deficient Memory CD4 T Cells
To examine whether miR-181a deficiency influenced CD4 T cell differentiation, we analyzed SMARTA effector cells after co-transfer of WT and miR-181a−/− naive SMARTA cells into B6 recipient mice and LCMV infection. On day 8, miR-181a-deficient SMARTA cells included proportionally more T follicular helper (Tfh) cells and less Th1 cells than their WT counterparts (Figure 5A). When taking into account the reduced expansion of miR-181a-deficient SMARTA cells (Figures 2E–2G), we observed fewer Th1 cells lacking miR-181a, whereas the number of Tfh cells was not significantly altered (Figure 5A). A similar relative bias toward Tfh cell differentiation in miR-181a deficient cells was observed in polyclonal CD4 T cell responses (Figure 5B). These data are consistent with the model that strong TCR signals promote the expansion of Th1 over Tfh cells, while Tfh cells are efficiently developed when TCR signals are weaker (Krishnamoorthy et al., 2017; Snook et al., 2018). Also, memory CD4 T cells that are similar to Tfh cells in their differentiation pathways were relatively increased in miR-181a−/− mice in the spleen as well as lymph nodes (Figures 5C and 5D). Moreover, memory CD4 T cells derived from miR-181a-deficient cells had enhanced functions, as shown by stronger binding to tetramers, increased amounts of cytokine production on a per-cell basis, increased frequency of cells producing multiple cytokines, and heightened sensitivity to peptide antigen (Figures 5E–5H).
Figure 5. Viral Infection Induces Increased Number of miR-181a-Deficient Memory CD4 T Cells.

(A) Equal numbers of congenically marked WT and miR-181a−/−YFP+ SMARTA CD4 T cells were co-transferred into B6 mice subsequently infected with LCMV 1 day later. Representative flow plots of SLAM and CXCR5 expression by WT and miR-181a−/− SMARTA CD4 T cells (left) and dot plots of the total numbers of SLAM+ Th1 and CXCR5+ Tfh SMARTA CD4 T cells in the spleen (right) on day 8 after infection.
(B) Representative flow plots of SLAM and CXCR5 expression by IAb GP66 tetramer+ YFP+ CD4 T cells from LCMV-infected WT and miR-181a−/− mice (left) and dot plots of the total numbers of Th1 and Tfh tetramer+ CD4 T cells in the spleen (right) on day 8.
(C–E) Numbers of IAb GP66 tetramer+ YFP+ CD4 T cells in the spleen at indicated time points (C) and in mesenteric and inguinal lymph nodes on day 85 (D). Representative flow plots of IAb GP66 tetramer+ cells gated on YFP+ CD4 T cells (E, left), representative histogram of tetramer binding intensity (middle), and dot plot of tetramer MFI (right). Filled gray in the histogram indicates naive CD4 T cells.
(F–H) Dot plots show MFI of indicated cytokines (F), the proportion of cells co-producing IFNγ, IFNα, and IL-2 (G) and the effective GP61 peptide concentration required to elicit a half-maximal IFNγ production (H) by GP61-specific YFP+ CD4 T cells in the spleen on day 85 after LCMV.
Unless stated otherwise, data are representative of two independent experiments with 4–5 mice per group (A and B) or pooled from two independent experiments with 6–9 mice per group (C–H). Data are presented as means. Statistical significance by two-tailed paired (A) or unpaired (B–H) t test. *p < 0.05; **p < 0.01; ***p < 0.001; NS, not significant.
To determine whether the findings in the mouse model of miR-181a-deficient T cells accurately recapitulate human immune aging, we examined antigen-specific T cells in individuals who had had an acute infection with WNV at young or old age. WNV-specific memory T cells in the blood were examined by using the HLA-A2 restricted Env430–438 tetramer for CD8 T cells(Figures 6A–6C) and the DRB1*01:01 or DRB1*04:01 restricted NS1205–220 tetramers for CD4 T cells (Figures 6E–6G). We screened 48 individuals for the appropriate HLA types; due to the nature of tetramer studies, the final study population was small precluding definite conclusions, but showed suggestive trends. Phenotypes of virus-specific memory T cells were not influenced by age. WNV-specific CD8 T cells expressed CD45RA and in part CCR7, phenotypically resembling naive cells as described for yellow-fever-specific cells (Akondy et al., 2017), while all WNV-specific CD4 T cells expressed CD45RO, consistent with memory cells (Figures 6B and 6F). Frequencies of WNV-specific memory CD8 as well as CD4 T cells were generally higher in older than young individuals. Importantly, WNV-specific memory T cells in older subjects bound their cognate tetramers more strongly than cells of young individuals, mirroring the data in the mouse model (Figures 6C and 6G). This increased tetramer staining of WNV-specific memory T cells inversely correlated with the miR-181a expression in naive T cells for both the CD8 and the CD4 subsets (Figures 6D and 6H).
Figure 6. Characterization of West Nile Virus-Specific CD4 and CD8 Memory T Cells in Young and Old Adults.

Tetramer staining for West Nile virus (WNV)-specific T cells in PBMC from individuals who acquired infection at young or old age.
(A) Representative scatter plot of HLA-A2-restricted Env430–438 tetramer+ CD8 T cells gated on CD3+ T cells.
(B) CD45RA and CCR7 expression by WNV-specific CD8 T cells (red) and total CD8 T cells (gray).
(C) Frequencies of tetramer+ CD8 T cells among CD3+ T cells and MFI of tetramer relative to CD3 staining from four 20- to ~50-year-old and four 70- to ~85-year-old individuals. Horizontal lines in dot plots indicate the means.
(D) miR-181a levels in naive CD8 T cells (young closed circle, old open circle) are plotted versus tetramer MFI from (C). Dotted line indicates the best fit by linear regression.
(E) Representative scatter plot of DRB1*01:01 restricted NS1205–220 tetramer+ CD4 T cells gated on CD3+ T cells.
(F) CD45RA and CCR7 expression of WNV-specific CD4 T cells (red) and total CD4 T cells (gray).
(G) Frequencies of DRB1*01:01 or DRB1*04:01 tetramer+ cells among CD3+ T cells and MFI of tetramer relative to CD3 staining from two young and three older individuals. Horizontal lines in dot plots indicate the means.
(H) miR-181a levels in naive CD4 T cells (young closed circle, old open circle) are plotted versus tetramer MFI from (G). Dotted line indicates the best fit by linear regression.
Repertoire Contraction in Recall Responses of miR-181a-Deficient Memory CD4 T Cells
To examine recall function, LCMV-immune mice that had previously received WT and miR-181a−/− SMARTA cells and were infected with LCMV, were challenged with recombinant Listeria monocytogenes expressing the LCMV glycoprotein 61–80 epitope (Lm-gp61). Similar to the primary response, miR-181a-deficient SMARTA memory cells had a defect in their ability to generate responses, resulting in a progressive under-representation of miR-181a-deficient cells upon secondary infection with preferential diminution of non-Tfh cells (Figure 7A). Similar results were observed during recall responses of polyclonal memory CD4 T cells by challenging LCMV-immune WT and miR-181a−/− mice with Lm-gp61 (Figures 7B and 7C). A closer analysis of tetramer+ CD4 T cells at the peak of recall responses demonstrated strong binding to tetramers and higher antigen sensitivity to antigen stimulation by miR-181a-deficient than WT secondary effectors (Figures 7D and 7E).
Figure 7. miR-181a Is Required for Secondary Expansion of Memory CD4 T Cells.

(A) Equal numbers of congenically marked WT and miR-181a−/−YFP+ SMARTA CD4 T cells were co-transferred into B6 mice infected with LCMV 1 day later. On day 50 after infection, immune mice were rechallenged with Lm-gp61. Representative flow plots show YFP+ SMARTA cells gated on CD4 T cells (top left) and the relative frequencies of WT and miR-181a−/− SMARTA cells (bottom left) in the spleen at indicated time points after infection and SLAM and CXCR5 expression on WT and miR-181a−/− SMARTA CD4 T cells on day 7 after recall (middle). Dot plots on the right show the ratios of miR-181a−/− to WT SMARTA CD4 T cells.
(B–F) WT and miR-181a−/− mice were infected with LCMV. On day 80 after infection, immune mice were rechallenged with Lm-gp61 and cells were harvested on day 7 at the peak of the recall response. (B) Representative flow plots of IAb GP66 tetramer+ cells gated on YFP+ CD4 T cells. (C) Fold expansion of IAb GP66 tetramer+ memory CD4 T cells on day 7 after rechallenge compared to before infection. (D) Representative histogram of tetramer binding intensity of IAb GP66 tetramer+ YFP+ CD4 T cells (left) and dot plot of tetramer MFI (right). Filled gray in the histogram indicates naive CD4 T cells. (E) Effective GP61 peptide concentration required to elicit a half-maximal IFNγ production. (F) TRB genes from IAb GP66 tetramer+ YFP+ CD4 T cells in the spleen were sequenced. Dot plots show the total number of unique TRB sequences (left) and the Shannon diversity index (right).
Data are pooled from two independent experiments with 5–9 mice per group (A–E) or are from one experiment with 3 mice per group (F). Unless stated otherwise, data are presented as means. Statistical significance by two-tailed unpaired t test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; NS, not significant.
We next determined whether the reduced expansion miR-181a-deficient memory T cells in the recall response is associated with repertoire selection. Comparison of TRB sequences of GP61-specific CD4 T cells at the memory state and the peak of the recall response revealed that the number of unique T cell clones responding in the recall challenge was significantly lower than that of the unstimulated memory repertoire in miR-181a−/− mice. Accordingly, the Shannon diversity index declined (Figure 7F). In contrast, all memory CD4 T cell clones in WT mice equally contributed to the secondary responses without skewing toward particular clones (Figure 7F). Together, these data suggest that not all miR-181a-deficient memory CD4 T cells are capable of responding in a secondary challenge.
DISCUSSION
Previous studies have shown that T cell aging in humans is associated with a decline in miR-181a expression, in particular in naive T cells (Li et al., 2012). Since miR-181a targets several phosphatases that attenuate TCR signaling, the age-associated decline has been implicated in rendering aged T cells less responsive to antigenic stimulation (Goronzy and Weyand, 2013). Here, we used a mouse model with conditional deletion of miR-181ab1 in mature T cells to examine the impact on antiviral immune responses and delineate similarities to human immune aging.
miR-181a deficiency in CD4 or CD8 T cells impaired the expansion of antigen-specific cells after LCMV infection, consistent with defective TCR signaling. In contrast to its role in thymic selection, miR-181a deficiency did not exclude lower-affinity T cells from responding; however, impaired their expansion. This reduced expansion was not solely due to the overexpression of DUSP6 and the associated selective inhibition of ERK phosphorylation, as DUSP6 silencing alone did not restore expansion. Evidence of repertoire selection during clonal expansion was observed for CD4 but not CD8 T cells, with selection of effector as well as memory T cells binding the tetramer with higher affinity and responding to lower antigen concentrations. A similar constraint in repertoire selection appears to also occur with age in humans. Memory T cells from individuals who acquired WNV infection at older age showed higher tetramer binding than young individuals.
Impaired antiviral CD8 T cell responses in miR-181a−/− mice delayed viral clearance, which is characteristic for the aged immune system (Bender et al., 1991; Brien et al., 2009; Kapasi et al., 2002; Schulz et al., 2015; Smithey et al., 2011). Unexpectedly, CD4 T cell responses in miR-181a−/− mice were enhanced, both in terms of clonal expansion as well as repertoire diversity. miR181a-deficient SMARTA CD4 T cells expanded less than WT SMARTA in the same host, similar to miR-181a-deficient CD8 T cells. Moreover, WT SMARTA cells expanded more in miR-181a−/− than WT mice. Taken together, the increased expansion of miR-181a-deficient CD4 T cells appears to be caused by viral persistence. In concordance with this observation, studies have shown that a brief encounter with antigen is sufficient to activate CD8 T cells (Kaech and Ahmed, 2001), whereas CD4 T cells require prolonged TCR stimulation for full effector and memory differentiation (Obst et al., 2005; Williams and Bevan, 2004; Wil liams et al., 2008). In a mouse model of influenza infection, memory CD4 T cell generation was promoted by recognition of antigen at later stages (Jelley-Gibbs et al., 2005; McKinstry et al., 2014).
The observations of a skewing in the murine T cell response are pertinent for understanding the age-associated defects in human T cell responses to infections or vaccinations. Vaccine studies with live-attenuated yellow fever virus (YFV) have shown that older individuals had fewer YFV-specific CD8 T cells at the peak response and prolonged viremia than did young individuals (Roukens et al., 2011; Schulz et al., 2015). In parallel, YFV-specific CD4 T cells in these older vaccinees underwent prolonged expansion after YFV vaccination (Schulz et al., 2015). Increased frequencies of WNV-specific memory CD4 T cells in older individuals who had prior WNV infection were reported in a previous study as well as confirmed here (James et al., 2016). In conclusion, the response patterns of antiviral CD8 and CD4 T cells and the kinetics of viral clearance are interlinked and at least in part determined by the miR-181a expression in peripheral T cells. Of particular interest, we propose that the age-associated defect in viral clearance is due to defective CD8 T cell effector generation and the antiviral immune response in elderly individuals has to rely on the recruitment of a broadening repertoire of antigen-specific CD4 T cells.
Along with costimulatory molecules and cytokines, TCR signal strength controls effector versus memory T cell differentiation (Daniels and Teixeiro, 2015). Strong activation signals are required for terminal effector cell differentiation, while memory precursor cells are generated by a relatively weak stimulation. In concordance with this concept, reduced expansion of miR-181a-deficient CD8 T cells largely resulted from severe impairment of short-lived effector cell generation. Most strikingly, we found that miR-181a deficiency reduced the frequency of tissue-residing CD8 T cells in the liver not only during acute infection, but also at later memory time points. It is possible that the size of effector CD8 T cell responses during primary infection determines the number of more long-lived memory T cells in the tissue or that miR-181a support their generation and survival by some other mechanism. Recent human studies have identified tissue-residing memory T cells in the liver that are distinct from circulating T cells and persist not only in chronic infection but also after viral clearance (Pallett et al., 2017). Whether miR-181a deficiency impairs the generation of such a distinct tissue-resident memory T cell population remains to be proven, but would have obvious implications for immune health. Reduced frequencies of tissue-resident memory T cells could indeed explain in part the increased susceptibility in older individuals to respiratory infections. Cutaneous infections and malignancies are also more frequent in older humans (Vukmanovic-Stejic et al., 2011). Quantitative studies of age-associated changes in tissue-resident memory T cells are only in the beginning (Kumar et al., 2018). Recall responses to varicella zoster virus in older individuals are reduced in the skin, but normal in the circulation, which may reflect defective tissue-resident memory T cell function (Agius et al., 2009).
Corresponding to the model that strong TCR signaling in CD8 T cells favors generation of effector over memory precursor cells, weaker signals also bias CD4 T cell differentiation to Tfh cells over Th1 cells (Krishnamoorthy et al., 2017; Snook et al., 2018). Indeed, we observed a selective effect on Th1 cells with miR-181a-deficient SMARTA cells, while Tfh cell generation was not altered. Moreover, we observed enhanced polyfunctionality, such as co-production of multiple cytokines with higher amounts on a per-cell basis in miR-181a−/− mice. In spite of the polyfunctionality, we did not see a robust recall response of miR-181a-deficient memory T cells. Instead, the secondary expansion was again impaired, with narrowing of the antigen-specific CD4 T cell repertoire recruited by recall antigen. Therefore, the increased frequency of polyfunctional memory CD4 T cells did not translate into improved recall responses.
How well do these findings reflect the immune response in older individuals? Viral clearance is delayed in human immune aging, consistent with the reduced expansion of effector T cells. Surprisingly, several reports have found polyfunctionality in T cell responses of older individuals. The ability of human T cells with superantigen staphylococcal enterotoxin B to produce multiple cytokines increased with age (Van Epps et al., 2014). Also, polyfunctionality of CD8 T cells is increased in individuals with latent cytomegalovirus (CMV) infection (Pera et al., 2014) and does not decline with age for WNV, CMV, and Epstein-Barr virus (EBV)-specific CD8 T cells (Lelic et al., 2012). Moreover, the broadening of the CD4 TCR repertoire in miR-181a−/− mice with defective CD8 T cell responses is reminiscent of the more diverse T cell response to new infection in old mice chronically infected with murine CMV (Smithey et al., 2018).
In contrast, impaired Tfh generation is not apparent in the miR-181a-deficient mouse model. Circulating Tfh are increased with age; however, after influenza vaccination, frequencies correlate with humoral vaccine responses only in young but not old (Herati et al., 2014). Several microRNAs change expression level with aging (Gustafson et al., 2019); many of these age-associated changes mirror those seen with differentiation; also, age-associated epigenetic changes such as increased chromatin accessibility to bZIP transcription factors document that aged cells are more differentiated (Moskowitz et al., 2017). Some of the changes in microRNA expression may counteract those observed with miR-181a deficiency as recently shown for the age-associated increased production of miR-21 that favors Th1 effector cell generation (Kim et al., 2018).
In conclusion, we have developed a murine model based on a molecular defect that is correlated with human T cell aging. This model of mice with miR-181a-deficient T cells resembled several facets of the antiviral T cell response in older individuals including the failure to generate effector CD8 T cells resulting in delayed viral clearance and increased reliance on a larger and more diverse CD4 T cell response. In contrast to conventional models using aged mice, this approach is not dependent on the assumption that the process of T cell aging is analogous in spite of the large difference in life span and in the mechanisms of T cell homeostasis. Moreover, it allows studying the consequences of one defect in isolation reducing the complexity of aging studies and enabling the identification of pathways to be targeted.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jörg J. Goronzy (jgoronzy@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal
C57BL/6J (B6), distal Lck-Cre and Rosa26-YFP reporter mice were purchased from the Jackson Laboratory. miR-181ab1fl/fl mice were obtained from C.Z. Chen (Stanford University) (Fragoso et al., 2012), currently available at Jackson Laboratory (Stock No: 025872), and crossed to Rosa26YFP dLck-Cre+ mice for conditional knockout of miR-181a in mature T cells. SMARTA TCR transgenic mice, with TCR specific for the LCMV glycoprotein 61–80 epitope presented by IAb, were crossed to dLck-Cre+ Rosa26YFP miR-181ab1fl/fl mice to generate miR-181a-deficient SMARTA mice and fully backcrossed to B6 mice. dLck-Cre+ Rosa26YFP miR-181ab1+/+ mice were used as wild-type control mice for all experiments to exclude effects resulting from the Cre recombinase and YFP expression. Both sexes were included in the study. All animal experiments were approved by the Stanford University Institutional Animal Care and Use Committee.
Human study population
A cohort of 48 individuals previously recovered from WNV infection were HLA genotyped; six 22 to 50 year-old (all female) and five 70 to 79 year-old (4 male and 1 female) individuals who typed HLA-A*02:01, HLA-DRB1*01:01 and/or HLA-DRB1*04:01 were selected for tetramer staining. Blood samples were obtained between 1 and 6 years (young/middle-aged adults) and 3 and 7 years (old adults) after infection. The studies were approved by the University of Arizona and the Stanford University Institutional Review Boards.
Cell culture
BHK (ATCC CCL-10) hamster kidney fibroblast cells, Vero (ATCC CCL-81) monkey kidney epithelial cells and Plat-E (Cell Biolabs, Inc.) based on 293T human embryonic kidney cells were propagated in DMEM supplemented with 10% fetal bovine serum and 100 U/ml penicillin and streptomycin (Thermo Fisher Scientific). Cells were maintained at 37°C with 5% CO2.
METHOD DETAILS
Infections
LCMV-Armstrong was grown in BHK cells and titered in Vero cells as described previously (Ahmed et al., 1984). Mice were infected i.p. at a dose of 2 × 105 plaque-forming units (PFU). Organs were homogenized, and LCMV titers were determined by plaque assay on Vero cells. For recall responses of CD4 memory T cells, recombinant Listeria monocytogenes expressing the LCMV glycoprotein 61–80 epitope (Lm-gp61) were grown to log phase in BHI broth and concentration determined by measuring the O.D. at 600 nm (O.D. of 1 = 1 × 109 CFU/ml). LCMV-immune mice were injected i.v. with 2 × 105 colony forming units (CFU) for recall responses. All mice analyzed were infected at 8–10 weeks of age, and both sexes were included.
Adoptive transfer
For adoptive transfer of SMARTA CD4 T cells, naive CD4 T cells were negatively isolated from the spleen of wild-type SMARTA mice (CD45.1+) using a naive CD4 T cell isolation kit (Miltenyi Biotec). 104 naive SMARTA CD4 T cells were then adoptively transferred i.v. into WT or miR-181a−/− hosts (CD45.2+) before LCMV infection. For adoptive co-transfer experiments of WT and miR-181a−/− SMARTA cells, naive CD4 T cells were isolated from the spleens of WT (CD45.1+) and miR-181a−/−(CD45.2+) SMARTA mice. Cells were then mixed at a 1:1 ratio, and a total of 1×104 YFP+ SMARTA CD4 T cells were injected i.v. into B6 recipient mice 1 day prior to LCMV infection. For recall response of memory CD8 T cells, WT and miR-181a−/− mice were infected with LCMV. On day 50, CD8 T cells were negatively isolated from the spleen using a CD8 T cell isolation kit (Miltenyi Biotec), and CD44+ memory CD8 T cells were then positively enriched using anti-CD44-biotin and anti-biotin microbeads (Miltenyi Biotech). ~8×104 YFP+ CD44+ GP33-speicifc memory CD8 T cells from WT and miR-181a−/− mice were transferred into B6 hosts, followed by LCMV infection one day later.
Cell preparations and flow cytometry
Single-cell suspensions of spleen, lymph nodes and liver were placed in DMEM supplemented with 10% fetal bovine serum and 100 U/ml penicillin and streptomycin (Thermo Fisher Scientific). For cell surface staining, cells were incubated with fluorescently conjugated antibodies at 4°C in antibody staining buffer (PBS with 1% FBS). IAb LCMV GP66–77 DIYKGVYQFKSV (GP66), Db GP33–41 KAVYNFATC (GP33), Db GP276–286 SGVENPGGYCL (GP276) and Db NP396–404 FQPQNGQFI (NP396) tetramers were obtained from the NIH tetramer core facility (Atlanta, GA). IAb GP66 tetramers were incubated with cells in RPMI1640 containing 2% FBS and 0.1% sodium azide at 37°C for 2 hours, followed by cell surface staining. To stain LCMV-specific CD8 T cells, cells were incubated with Db CD8 tetramers along with cell surface antibodies at 4°C for 30 minutes in antibody staining buffer. For intracellular cytokine assays, cells were restimulated for 4 hours with 10 uM GP61–80 peptide (GLKGPDIYKGVYQFKSVEFD),0.1 uM GP33–41 peptide, 0.1 uM GP276–286 peptide or 0.1 uM NP396–404 peptide at 37°C in the presence of brefeldin A (GolgiPlug, BD Biosciences). To assess degranulation of CD8 T cells, fluorescently labeled anti-CD107a and monensin (GolgiStop, BD Biosciences) were added during ex vivo restimulation. Cells were then stained with cell surface antigen-specific antibodies, permeabilized with Cytofix/Cytoperm kit (BD Biosciences) and stained with fluorescently labeled antibodies specific to the indicated cytokines. To determine antigen sensitivity of cells in functional responses, splenocytes were restimulated with a range of peptide concentrations. The percentage of maximal response was determined by calculating the frequency of IFNγ-producing cells at any given concentration as a percentage of the frequency of IFNγ-producing cells at the highest peptide concentration. The effective peptide concentration required to elicit a half-maximal IFNγ production was then calculated. For staining of phosphorylated signaling proteins, total T cells negatively isolated using a Pan T cell isolation kit (Miltenyi Biotec) were stimulated by CD3 cross-linking at 37°C, fixed with Cytofix buffer (BD Biosciences) for 10 minutes at 37°C and permeabilized with Perm buffer III (BD Biosciences) for 30 minutes on ice. Cells were then incubated with fluorescently labeled antibodies for 60 minutes at room temperature. The following fluorochrome-conjugated antibodies were used for flow cytometry: anti-CD4 (RM4.5; BioLegend), anti-CD8 (53–6.7; BioLegend), anti-CD44 (IM7; BioLegend), anti-CD62L (MEL-14; BioLegend), anti-CD45.1 (A20; BioLegend), anti-CD45.2 (104; BioLegend), anti-Va2 (B20.1; BioLegend), anti-TCRβ (H57–597; BioLegend), anti-B220 (RA3–6B2; BioLegend), anti-KLRG1 (2F1; SouthernBiotech), anti-IL7Rα (A7R34; BioLegend), anti-CXCR5 (2G8; BD Biosciences), anti-SLAM (TC15–12F12.2; BioLegend), anti-CD27 (LG.3A10; BioLegend), anti-CD69 (H1.2F3; BioLegend), anti-CXCR6 (SA051D1; BioLegend), anti-CD107a (1D4B; BioLegend), LIVE/DEAD Fixable Aqua (Thermo Fisher Scientific), anti-IFNγ (XMG1.2; BioLegend), anti-IFNα (MP6-XT22; BioLegend), anti-IL-2 (JES6–5H4; BioLegend), anti-Granzyme B (GB11; BDBiosciences), anti-p-ERK (T202/Y204; 20A; BD Biosciences) and anti-p-AKT (S473; M89–61; BD Biosciences). Cells were analyzed on an LSRII or LSR Fortessa (BD Biosciences), and flow cytometry data was analyzed using FlowJo (TreeStar).
Retroviral production and transduction
Dusp6 shRNA sequence (5′-TGCTGTTGACAGTGAGCGAACACATCGAATCTGCCATTAATAGTG AAGCCACAGATGTATTAATGGCAGATTCGATGTGTGTGCCTACTGCCTCGGA-3′) was cloned into pLMPd-Ametrine vector, an MSCV-based retroviral vector for the expression of mir30-flanked shRNA (Choi and Crotty, 2015). The pLMPd-Ametrine vector with mouse shCD19 (shCtrl) was used as a control. The methods of retroviral production and transduction have been described in details (Choi and Crotty, 2015). Briefly, virion was produced by transfection of a vector into Plat-E cells. Culture supernatants were collected 48 and 72 hours after transfection, filtered through a 0.45-mm syringe filter (Milipore) and saved at 4 C until use. For retroviral transduction, miR-181a-deficient naive SMARTA CD4 T cells were activated with anti-CD3 (145–2C11; Thermo Fisher Scientific) and anti-CD28 (37.51; Thermo Fisher Scientific) adsorbed on plates. Cell were transduced with retrovirus at 24 and 36 hours after stimulation. After 72 hours, cells were washed and rested for another 3 days. Retrovirally transduced miR-181a-deficient SMARTA CD4 T cells (Ametrine+ YFP+ CD4+) were sorted and 104 shRNA+ SMARTA cells were intravenously transferred into B6 hosts, followed by LCMV infection three days later.
Analysis of West Nile virus-specific T cells
HLA-A*02 restricted Env430–438 (SVGGVFTSV), DRB1*01:01 restricted NS1205–220 (RLNDTWKLERAVLGEVK) or DRB1*04:01 restrictedNS1205–220 (RLNDTWKLERAVLGEVK) tetramers were described previously (James et al., 2016; Piazza et al., 2010) and obtained from the NIH tetramer core facility (Atlanta, GA). PBMC from individuals who acquired WNV infection previously were incubated with indicated tetramers for 1 hour at room temperature. Cells were then washed and stained with fluorochrome-conjugated surface antibodies including anti-CD3 (HIT3a; BD Biosciences), anti-CD4 (RPA-T4; BD Biosciences), anti-CD8 (RPA-T8; BD Biosciences), anti-CCR7 (G043H7; BD Biosciences), anti-CD45RA (HI100; BD Biosciences) and LIVE/DEAD Fixable Aqua (Thermo Fisher Scientific).
Generation of TRB gene libraries and high-throughput sequencing
After LCMV infection of WT and miR-181a−/− mice, splenic Db GP33 tetramer+ YFP+ CD8 T cells and IAb GP66 tetramer+ YFP+ CD4 T cells were sorted at indicated time points to > 97% purity on FACSAria (BD Biosciences). Total RNA was prepared using the RNeasy Micro Kit (QIAGEN), and first strand cDNA was generated by 5′ rapid amplification of cDNA end (5′ RACE) PCR using a SMARTer® RACE 5′/3′ kit (Clontech). TRB genes were amplified with the forward primer specific to the SMARTer II A oligonucleotide (5′-GGCATTCCTGCTGAACC GCTCTTCCGATCTNNNNACTAGGTAAAGCAGTGGTATCAACGCAGAGT-3′) and the reverse primer specific to constant region of TRB (5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT NNNNxxxxxxxxTGGCTCAAACAAGGA GACCT-3′). xxxxxxxx in the reverse primer represents barcode sequence. Illumina sequencing adaptors were added in a second PCR, amplicons were separated on an agarose gel using a QIAquick gel extraction kit (QIAGEN), pooled in equal amounts and sequenced with an Illumina Miseq sequencer (2 × 350-bp with paired-end reads).
Analysis of TCR Repertoires
The reads obtained from Miseq were de-barcoded after trimming adaptors, then aligned to the mouse TRB reference sequence library from IMGT (Jan-2017) (Lefranc and Lefranc, 2001) and assembled into TCR sequences using MiXCR v2.1.2 (Bolotin et al., 2015). The TCR sequences were filtered to exclude sequences contacting stop codons and out of frames along with those having CDR3 amino-acid length greater than 30. Identified TCR sequences were again filtered to include only those that occupy 97% of the total clonal space in each replicate in order to minimize contamination with erroneous sequences due to technical and clustering errors. Resulting clones were used for downstream analyses. Shannon diversity index, which reflects both abundance and richness, was used for evaluation of TRB sequence diversity (Stewart et al., 1997). Shannon diversity index was calculated as H’ = −∑[pi × ln(pi)], where pi is the proportion of TCR sequence i.
Western blotting
FACS-sorted cells were lysed in RIPA buffer containing PMSF and protease and phosphatase inhibitors (Santa Cruz Biotechnology) for 30 minutes on ice. Proteins were separated on denaturing 4%–15% SDS-PAGE (Bio-Rad), transferred onto PVDF membrane (Millipore) and probed with antibodies to SIRT1 (D60E1; Cell Signaling Technology), DUSP6 (ab76310; Abcam) and β-actin (13E5;Cell Signaling Technology). Membranes were developed using HRP-conjugated secondary antibodies and Pierce ECL western blotting substrate (Thermo Fisher Scientific).
miRNA quantification
Total RNA was isolated with a miRNeasy Micro kit (QIAGEN) or Trizol reagent (Thermo Fisher Scientific). RNA was reverse-transcribed using the miRCURY LNA Universal RT microRNA cDNA synthesis kit (Exiqon). Mature miR-181a expression levels were assessed by quantitative RT-PCR using the miRCURY LNA UniRT PCR primer for miR-181a-5p (Exiqon) and Power SYBR® Green PCR Master Mix (Thermo Fisher Scientific). U6 or RNU48 (Exiqon) were used as internal control to normalize miR-181a expression. In some experiments, a TaqMan MicroRNA Reverse Transcription kit and TaqMan MicroRNA Assays (Thermo Fisher Scientific) were used for reverse transcription and quantitative RT-PCR of miR-181a and sno202. Expression levels were displayed as 2−ΔΔCt.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Prism (GraphPad). Unless stated otherwise, data are presented as mean, and error bars indicate the standard error of the mean. Paired or unpaired two-tailed Student’s t tests were used for comparing two groups. Two-way ANOVA with Tukey’s post hoc test was used for multi-group comparisons. p < 0.05 was considered statistically significant. Statistical details and significance can be found in the figure legends.
DATA AND CODE AVAILABILITY
The accession number for TRB sequencing data reported in this paper is SRA: PRJNA558782.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD3 (145-2C11) | Thermo Fisher Scientific | Cat# 16-0031-82; RRID: AB_468847 |
| anti-CD28 (37.51) | Thermo Fisher Scientific | Cat# 16-0281-82; RRID: AB_468921 |
| anti-Armenian and Syrian Hamster IgG | BD Biosciences | Cat# 554009; RRID: AB_395205 |
| anti-CD4 (RM4.5) | BioLegend | Cat# 100536; RRID: AB_493701 |
| anti-CD8 (53-6.7) | BioLegend | Cat# 100730; RRID: AB_493703 |
| anti-CD44 (IM7) | BioLegend | Cat# 103020; RRID: AB_493683 |
| anti-CD62L (MEL-14) | BioLegend | Cat# 104418; RRID: AB_313103 |
| anti-CD45.1 (A20) | BioLegend | Cat# 110728; RRID: AB_893346 |
| anti-CD45.2 (104) | BioLegend | Cat# 109808; RRID: AB_313445 |
| anti-Vα2 (B20.1) | BioLegend | Cat# 127810; RRID: AB_1089250 |
| anti-TCRβ (H57-597) | BioLegend | Cat# 109220; RRID: AB_893624 |
| anti-B220 (RA3-6B2) | BioLegend | Cat# 103212; RRID: AB_312997 |
| anti-KLRG1 (2F1) | SouthernBiotech | Cat# 1807-09; RRID: AB_2795369 |
| anti-IL7Rα (A7R34) | BioLegend | Cat# 135022; RRID: AB_1937273 |
| anti-CXCR5 (2G8) | BD Biosciences | Cat# 551960; RRID: AB_394301 |
| Cat# 561988; RRID: AB_10893355 | ||
| anti-CD27 (LG.3A10) | BioLegend | Cat# 124233; RRID: AB_2687192 |
| anti-CD69 (H1.2F3) | BioLegend | Cat# 104522; RRID: AB_2260065 |
| anti-CXCR6 (SA051D1) | BioLegend | Cat# 151109; RRID: AB_2616760 |
| anti-SLAM (TC15-12F12.2) | BioLegend | Cat# 115922; RRID: AB_2303663 |
| anti-CD107a (1D4B) | BioLegend | Cat# 121626; RRID: AB_2572055 |
| anti-IFNγ (XMG1.2) | BioLegend | Cat# 505810; RRID: AB_315404 |
| anti-TNFα (MP6-XT22) | BioLegend | Cat# 506324; RRID: AB_2256076 |
| anti-IL-2 (JES6-5H4) | BioLegend | Cat# 503808; RRID: AB_315302 |
| anti-Granzyme B (GB11) | BD Biosciences | Cat# 562462; RRID: AB_2737618 |
| anti-p-ERK (T202/Y204; 20A) | BD Biosciences | Cat# 562644; RRID: AB_2737698 |
| anti-p-AKT (S473; M89-61) | BD Biosciences | Cat# 561670; RRID: AB_10896328 |
| anti-CD3 (HIT3a) | BD Biosciences | Cat# 740073; RRID: AB_2687538 |
| anti-CD4 (RPA-T4) | BD Biosciences | Cat# 555347; RRID: AB_395752 |
| anti-CD8 (RPA-T8) | BD Biosciences | Cat# 555366; RRID: AB_395769 |
| anti-CCR7 (G043H7) | BioLegend | Cat# 353212; RRID: AB_10916390 |
| anti-CD45RA (HI100) | BioLegend | Cat# 304134; RRID: AB_2563814 |
| anti-SIRT1 (D60E1) | Cell Signaling Technology | Cat# 3931; RRID: AB_1642293 |
| anti-DUSP6 (EPR129Y) | Abcam | Cat# ab76310; RRID: AB_1523517 |
| anti-b-actin (13E5) | Cell Signaling Technology | Cat# 4970; RRID: AB_2223172 |
| anti-Rabbit IgG HRP | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| Bacterial and Virus Strains | ||
| LCMV Armstrong | Laboratory of Rafi Ahmed | Ahmed et al., 1984 |
| Listeria-GP61-80 (Lm-gp61) | Laboratory of Rafi Ahmed | Williams et al., 2008 |
| Biological Samples | ||
| Peripheral blood from Individuals previously recovered from WNV infection | University of Arizona College of Medicine | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fixable Viability Dye | Thermo Fisher Scientific | Cat# 65-0866-14 |
| PE Streptavidin | BioLegend | Cat# 405204 |
| IAb LCMV GP 66–77 (DIYKGVYQKSV) tetramer | NIH tetramer core facility | N/A |
| Db LCMV GP 33–41 (KAVYNFATM) tetramer | NIH tetramer core facility | N/A |
| Db LCMV GP 276–286 (SGVENPGGYCL) tetramer | NIH tetramer core facility | N/A |
| Db LCMV NP 396–404 (FQPQNGQFI) tetramer | NIH tetramer core facility | N/A |
| HLA-A*02 WNV Env 430–438 (SVGGVFTSV) tetramer | NIH tetramer core facility | N/A |
| DRB1*01:01 WNV NS1 205–220 (RLNDTWKLERAVLGEVK) tetramer | NIH tetramer core facility | N/A |
| DRB1*04:01 WNV NS1 205–220 (RLNDTWKLERAVLGEVK) tetramer | NIH tetramer core facility | N/A |
| LCMV GP61–80 peptide (GLKGPDIYKGVYQFKSVEFD) | Anaspec | Cat# AS-64851 |
| LCMV GP33–41 peptide (KAVYNFATM) | Anaspec | Cat# AS-61296 |
| LCMV GP276–286 peptide (SGVENPGGYCL) | Anaspec | Cat# AS-62539 |
| LCMV NP396–404 peptide (FQPQNGQFI) | Anaspec | Cat# AS-61700 |
| BD GolgiStop | BD Biosciences | Cat# 554724 |
| Critical Commercial Assays | ||
| Naive CD4+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat# 130-104-453 |
| CD8a+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat# 130-104-075 |
| Pan T Cell Isolation Kit II, mouse | Miltenyi Biotec | Cat# 130-095-130 |
| Anti-Biotin MicroBeads | Miltenyi Biotec | Cat# 130-090-485 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat# 554714 |
| BD Cytofix Fixation Buffer | BD Biosciences | Cat# 554655 |
| BD Phosflow Perm Buffer III | BD Biosciences | Cat# 558050 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| miRNeasy Micro Kit | QIAGEN | Cat# 217084 |
| miRCURY LNA RT Kit | QIAGEN | Cat# 339340 |
| TaqMan Universal Master Mix II | Thermo Fisher Scientific | Cat# 4440047 |
| TaqMan MicroRNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat# 4366596 |
| TaqMan MicroRNA Assays, miR-181a | Thermo Fisher Scientific | Assay ID 000480 |
| TaqMan MicroRNA Assays, snoRNA202 | Thermo Fisher Scientific | Assay ID 001232 |
| SMARTer® RACE 5’/3’ Kit | Takara Bio USA, Inc. | Cat# 634859 |
| MiSeq Reagent Kit v3 | Illumina | Cat# MS-102-3003 |
| Deposited Data | ||
| TRB sequencing data | This paper | SRA: PRJNA558782 |
| Experimental Models: Cell Lines | ||
| BHK cells | ATCC | ATCC CCL-10 |
| Vero cells | ATCC | ATCC CCL-81 |
| Plat-E cells | Cell Biolabs, Inc. | Cat# RV-101 |
| Experimental Models: Organisms/Strains | ||
| C57BL6/J mice | Jackson Laboratory | Stock No: 000664 |
| distal Lck-Cre mice | Jackson Laboratory | Stock No: 012837 |
| Rosa26-YFP mice | Jackson Laboratory | Stock No: 006148 |
| miR-181ab1fl/fl mice | Laboratory of CZ Chen | Fragoso et al., 2012 |
| SMARTA TCR transgenic mice | Laboratory of Rafi Ahmed | Williams et al., 2008 |
| Oligonucleotides | ||
| hsa-miR-181a-5p miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00206081 |
| U6 snRNA(hsa, mmu) miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00203907 |
| SNORD48(hsa) miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00203903 |
| TRB amplicon; forward primer 5’-GGCATTCCTGC TGAACC GCTCTTCCGATCTNNNNACTAGGTAAAGCAGTGGTATCAA CGCAGAGT-3’ | This paper | N/A |
| TRB amplicon; reverse primer 5’-ACACTCTTTCCCTACAC GACGCTCTTCCGATCTNNNNxxxxxxxxTGGCTCAAACAA GGAGACCT-3’ | This paper | N/A |
| Dusp6 shRNA sequence; 5’-TGCTGTTGACAGTGAGCGAACA CATCGAATCTGCCATTAATAGTGAAGCCACAGATGTATTAA TGGCAGATTCGATGTGTGTGCCTACTGCCTCGGA-3’ | This paper | N/A |
| miR30 common; forward primer 5’-CAGAAGGCTCGAGAAGG TATATTGCTGTTGACAGTGAGCG-3’ | This paper | N/A |
| miR30 common; reverse primer 5’-CTAAAGTAGCCCCTTGA ATTCCGAGGCAGTAGGCA-3’ | This paper | N/A |
| Recombinant DNA | ||
| LMPd-shCtrl vector | Laboratory of YS Choi | Choi and Crotty, 2015 |
| LMPd-shDusp6 vector | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | TreeStar | RRID:SCR_008520 |
| Prism | GraphPad Software | RRID:SCR_002798 |
| IMGT | Lefranc and Lefranc, 2001 | http://www.imgt.org/ |
| MiXCR | Bolotin et al., 2015 | https://mixcr.readthedocs.io/en/master/ |
Highlights.
miR-181a deficiency in naive T cells is a hallmark of human and murine T cell aging
miR-181a deficiency in T cells from young mice resembles aged T cell responses
miR-181a deficiency in T cells impairs expansion, viral clearance, and recall response
miR-181a deficiency impairs generation of liver-residing memory T cells
ACKNOWLEDGMENTS
We thank Dr. C.Z. Chen (Stanford University) for generously providing miR-181ab1fl/fl mice and Dr. R. Ahmed (Emory University) and Dr. S. Hale (University of Utah) for providing SMARTA mice and LCMV-Armstrong and Lm-gp61 constructs. We thank Y.S. Choi (Seoul National University, South Korea) for LMPd-Ametrine vector and the NIH tetramer core facility (Atlanta, GA) for providing tetramers. This work was supported by the National Institutes of Health (NIH) (R01 AI108891, R01 AG045779, U19 AI057266, R01 AI129191 to J.J.G.; R01 AR042527, R01 HL117913, R01 AI108906, and P01 HL129941 to C.M.W.; and N01 AI00017 to J.N.-Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.044.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agius E, Lacy KE, Vukmanovic-Stejic M, Jagger AL, Papageorgiou AP, Hall S, Reed JR, Curnow SJ, Fuentes-Duculan J, Buckley CD, et al. (2009). Decreased TNF-alpha synthesis by macrophages restricts cutaneous immunosurveillance by memory CD4+ T cells during aging. J. Exp. Med 206, 1929–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed R, Salmi A, Butler LD, Chiller JM, and Oldstone MB (1984). Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med 160, 521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, McGuire DJ, Cohen KW, et al. (2017). Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender BS, Johnson MP, and Small PA (1991). Influenza in senescent mice: impaired cytotoxic T-lymphocyte activity is correlated with prolonged infection. Immunology 72, 514–519. [PMC free article] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381. [DOI] [PubMed] [Google Scholar]
- Brien JD, Uhrlaub JL, Hirsch A, Wiley CA, and Nikolich-Žugich J (2009). Key role of T cell defects in age-related vulnerability to West Nile virus. J. Exp. Med 206, 2735–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, and Crotty S (2015). Retroviral vector expression in TCR transgenic CD4+ T cells. Methods Mol. Biol 1291, 49–61. [DOI] [PubMed] [Google Scholar]
- Cramer JP, Dubischar K, Eder S, Burchard GD, Jelinek T, Jilma B, Kollaritsch H, Reisinger E, and Westritschnig K (2016). Immunogenicity and safety of the inactivated Japanese encephalitis vaccine IXIAROâ in elderly subjects: open-label, uncontrolled, multi-center, phase 4 study. Vaccine 34, 4579–4585. [DOI] [PubMed] [Google Scholar]
- Czesnikiewicz-Guzik M, Lee WW, Cui D, Hiruma Y, Lamar DL, Yang ZZ, Ouslander JG, Weyand CM, and Goronzy JJ (2008). T cell subset-specific susceptibility to aging. Clin. Immunol 127, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Acremont V, Herzog C, and Genton B (2006). Immunogenicity and safety of a virosomal hepatitis A vaccine (Epaxal) in the elderly. J. Travel Med. 13, 78–83. [DOI] [PubMed] [Google Scholar]
- Daniels MA, and Teixeiro E (2015). TCR signaling in T cell memory. Front. Immunol 6, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert PJ, Jiang S, Xie J, Li QJ, and Davis MM (2009). An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nat. Immunol 10, 1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoso R, Mao T, Wang S, Schaffert S, Gong X, Yue S, Luong R, Min H, Yashiro-Ohtani Y, Davis M, et al. (2012). Modulating the strength and threshold of NOTCH oncogenic signals by mir-181a-1/b-1. PLoS Genet. 8, e1002855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goronzy JJ, and Weyand CM (2013). Understanding immunosenescence to improve responses to vaccines. Nat. Immunol 14, 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goronzy JJ, and Weyand CM (2017). Successful and maladaptive T cell aging. Immunity 46, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goronzy JJ, and Weyand CM (2019). Mechanisms underlying T cell ageing. Nat. Rev. Immunol 19, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson CE, Cavanagh MM, Jin J, Weyand CM, and Goronzy JJ (2019). Functional pathways regulated by microRNA networks in CD8 T-cell aging. Aging Cell 18, e12879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J, Williams A, Goff LA, Staron M, Licona-Limón P, Kaech SM, Nakayama M, Rinn JL, and Flavell RA (2013). The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity 38, 984–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herati RS, Reuter MA, Dolfi DV, Mansfield KD, Aung H, Badwan OZ, Kurupati RK, Kannan S, Ertl H, Schmader KE, et al. (2014). Circulating CXCR5+PD-1+ response predicts influenza vaccine antibody responses in young adults but not elderly adults. J. Immunol 193, 3528–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James EA, Gates TJ, LaFond RE, Yamamoto S, Ni C, Mai D, Gersuk VH, O’Brien K, Nguyen QA, Zeitner B, et al. (2016). Neuroinvasive West Nile infection elicits elevated and atypically polarized T cell responses that promote a pathogenic outcome. PLoS Pathog. 12, e1005375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelley-Gibbs DM, Brown DM, Dibble JP, Haynes L, Eaton SM, and Swain SL (2005). Unexpected prolonged presentation of influenza antigens promotes CD4 T cell memory generation. J. Exp. Med 202, 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jílková E, Vejvalková P, Stiborová I, Skorkovský J, and Král V (2009). Serological response to tick-borne encephalitis (TBE) vaccination in the elderly–results from an observational study. Expert Opin. Biol. Ther 9, 797–803. [DOI] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, and Ahmed R (2001). Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naı¨ve cells. Nat. Immunol 2, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapasi ZF, Murali-Krishna K, McRae ML, and Ahmed R (2002). Defective generation but normal maintenance of memory T cells in old mice. Eur. J. Immunol 32, 1567–1573. [DOI] [PubMed] [Google Scholar]
- Kim C, Fang F, Weyand CM, and Goronzy JJ (2017). The life cycle of a T cell after vaccination—where does immune ageing strike? Clin. Exp. Immunol 187, 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Hu B, Jadhav RR, Jin J, Zhang H, Cavanagh MM, Akondy RS, Ahmed R, Weyand CM, and Goronzy JJ (2018). Activation of miR-21-regulated pathways in immune aging selects against signatures characteristic of memory T cells. Cell Rep. 25, 2148–2162.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, Cook SL, Chen J, Bahroos N, Sievert E, Corse E, Chong A, and Sciammas R (2017). The IRF4 gene regulatory module functions as a read-write integrator to dynamically coordinate T helper cell fate. Immunity 47, 481–497.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Connors TJ, and Farber DL (2018). Human T cell development, localization, and function throughout life. Immunity 48, 202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley JM, Risi G, Caldwell M, Gilderman L, Berwald B, Fogarty C, Poling T, Riff D, Baron M, Frenette L, et al. (2011). Dose-sparing H5N1 A/Indonesia/05/2005 pre-pandemic influenza vaccine in adults and elderly adults: a phase III, placebo-controlled, randomized study. J. Infect. Dis 203, 1729–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M-P, and Lefranc G.r. (2001). The T Cell Receptor FactsBook (Academic Press; ). [Google Scholar]
- Lelic A, Verschoor CP, Ventresca M, Parsons R, Evelegh C, Bowdish D, Betts MR, Loeb MB, and Bramson JL (2012). The polyfunctionality of human memory CD8+ T cells elicited by acute and chronic virus infections is not influenced by age. PLoS Pathog. 8, e1003076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, et al. (2007). miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell 129, 147–161. [DOI] [PubMed] [Google Scholar]
- Li G, Yu M, Lee WW, Tsang M, Krishnan E, Weyand CM, and Goronzy JJ (2012). Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat. Med 18, 1518–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey NP, Staples JE, Lehman JA, and Fischer M; Centers for Disease Control and Prevention (CDC) (2010). Surveillance for human West Nile virus disease—United States, 1999–2008. MMWR Surveill. Summ 59, 1–17. [PubMed] [Google Scholar]
- McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM, and Swain SL (2014). Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat. Commun 5, 5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, Buenrostro JD, Weyand CM, Greenleaf WJ, and Goronzy JJ (2017). Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol 2, eaag0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolich-Žugich J (2018). The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol 19, 10–19. [DOI] [PubMed] [Google Scholar]
- Nikolich-Žugich J, Li G, Uhrlaub JL, Renkema KR, and Smithey MJ (2012). Age-related changes in CD8 T cell homeostasis and immunity to infection. Semin. Immunol 24, 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obst R, van Santen HM, Mathis D, and Benoist C (2005). Antigen persistence is required throughout the expansion phase of a CD4+ T cell response.J. Exp. Med 201, 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallett LJ, Davies J, Colbeck EJ, Robertson F, Hansi N, Easom NJW, Burton AR, Stegmann KA, Schurich A, Swadling L, et al. (2017). IL-2high tissue-resident T cells in the human liver: sentinels for hepatotropic infection.J. Exp. Med 214, 1567–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pera A, Campos C, Corona A, Sanchez-Correa B, Tarazona R, Larbi A, and Solana R (2014). CMV latent infection improves CD8+ T response to SEB due to expansion of polyfunctional CD57+ cells in young individuals. PLoS ONE 9, e88538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza P, McMurtrey CP, Lelic A, Cook RL, Hess R, Yablonsky E, Borowski L, Loeb MB, Bramson JL, Hildebrand WH, and Rinaldo CR (2010). Surface phenotype and functionality of WNV specific T cells differ with age and disease severity. PLoS ONE 5, e15343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, Olshen RA, Weyand CM, Boyd SD, and Goronzy JJ (2014). Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. USA 111, 13139–13144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roukens AH, Soonawala D, Joosten SA, de Visser AW, Jiang X, Dirksen K, de Gruijter M, van Dissel JT, Bredenbeek PJ, and Visser LG (2011). Elderly subjects have a delayed antibody response and prolonged viraemia following yellow fever vaccination: a prospective controlled cohort study. PLoS ONE 6, e27753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, and Ahmed R (2008). Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med 205, 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffert SA, Loh C, Wang S, Arnold CP, Axtell RC, Newell EW, Nolan G, Ansel KM, Davis MM, Steinman L, and Chen CZ (2015). mir-181a-1/b-1 modulates tolerance through opposing activities in selection and peripheral T cell function. J. Immunol 195, 1470–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz AR, Mälzer JN, Domingo C, Jürchott K, Grützkau A, Babel N, Nienen M, Jelinek T, Niedrig M, and Thiel A (2015). Low thymic activity and dendritic cell numbers are associated with the immune response to primary viral infection in elderly humans. J. Immunol 195, 4699–4711. [DOI] [PubMed] [Google Scholar]
- Smithey MJ, Renkema KR, Rudd BD, and Nikolich-Žugich J (2011).Increased apoptosis, curtailed expansion and incomplete differentiation of CD8+ T cells combine to decrease clearance of L. monocytogenes in old mice. Eur. J. Immunol 41, 1352–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithey MJ, Venturi V, Davenport MP, Buntzman AS, Vincent BG, Frelinger JA, and Nikolich-Žugich J (2018). Lifelong CMV infection improves immune defense in old mice by broadening the mobilized TCR repertoire against third-party infection. Proc. Natl. Acad. Sci. USA 115, E6817–E6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snook JP, Kim C, and Williams MA (2018). TCR signal strength controls the differentiation of CD4+ effector and memory T cells. Sci. Immunol 3, eaas9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JJ, Lee CY, Ibrahim S, Watts P, Shlomchik M, Weigert M, and Litwin S (1997). A Shannon entropy analysis of immunoglobulin and T cell receptor. Mol. Immunol 34, 1067–1082. [DOI] [PubMed] [Google Scholar]
- Targonski PV, Jacobson RM, and Poland GA (2007). Immunosenescence: role and measurement in influenza vaccine response among the elderly. Vaccine 25, 3066–3069. [DOI] [PubMed] [Google Scholar]
- Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, and Fukuda K (2003). Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289, 179–186. [DOI] [PubMed] [Google Scholar]
- Van Epps P, Banks R, Aung H, Betts MR, and Canaday DH (2014). Age-related differences in polyfunctional T cell responses. Immun. Ageing 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukmanovic-Stejic M, Rustin MH, Nikolich-Žugich J, and Akbar AN (2011). Immune responses in the skin in old age. Curr. Opin. Immunol 23, 525–531. [DOI] [PubMed] [Google Scholar]
- Weinberger B, Haks MC, de Paus RA, Ottenhoff THM, Bauer T, and Grubeck-Loebenstein B (2018). Impaired immune response to primary but not to booster vaccination against hepatitis B in older adults. Front. Immunol 9, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, and Bevan MJ (2004). Shortening the infectious period does not alter expansion of CD8 T cells but diminishes their capacity to differentiate into memory cells. J. Immunol 173, 6694–6702. [DOI] [PubMed] [Google Scholar]
- Williams MA, Ravkov EV, and Bevan MJ (2008). Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity 28, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Li G, Kim C, Hu B, Jadhav RR, Weyand CM, and Goronzy JJ (2018). Regulation of miR-181a expression in T cell aging. Nat. Commun 9, 3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DJ, Wang Q, Wei J, Baimukanova G, Buchholz F, Stewart AF, Mao X, and Killeen N (2005). Selective expression of the Cre recombinase in late-stage thymocytes using the distal promoter of the Lck gene. J. Immunol 174, 6725–6731. [DOI] [PubMed] [Google Scholar]
- Zhou B, Li C, Qi W, Zhang Y, Zhang F, Wu JX, Hu YN, Wu DM, Liu Y, Yan TT, et al. (2012). Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia 55, 2032–2043. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Li GY, Ren JP, Wang L, Zhao J, Ning SB, Zhang Y, Lian JQ, Huang CX, Jia ZS, et al. (2016). Protection of CD4+ T cells from hepatitis C virus infection-associated senescence via DNp63-miR-181a-Sirt1 pathway. J. Leukoc. Biol 100, 1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for TRB sequencing data reported in this paper is SRA: PRJNA558782.