

Abstract
Background:
The incidence of cardiovascular disease (CVD) is higher in HIV+ patients than it is in the average population, and combination antiretroviral therapy (cART) is a recognized risk factor for CVD. However, the molecular mechanisms that link cART and CVD are currently unknown. Our study explores the role of the activation of p90RSK, a reactive oxygen species (ROS)-sensitive kinase, in engendering senescent phenotype in macrophages and accelerating atherogenesis in patients undergoing cART.
Methods:
Peripheral whole blood from cART-treated HIV+ individuals and non-treated HIV− individuals was treated with H2O2 (200 μM) for 4 minutes, and p90RSK activity in CD14+ monocytes was measured. Plaque formation in the carotids were also analyzed in these individuals. Macrophage senescence was determined by evaluating their efferocytotic ability, antioxidation-related molecule expression, telomere length, and inflammatory gene expression. The involvement of p90RSK-NRF2 signaling in cART-induced senescence was assessed by p90RSK specific inhibitor (FMK-MEA) or dominant negative p90RSK (DN-p90RSK), and NRF2 activator (NRF2A). Further, the severity of atherosclerosis was determined in myeloid cell-specific wild type and DN-p90RSK transgenic mice.
Results:
Monocytes from HIV+ patients exhibited higher levels of p90RSK activity and were also more sensitive to ROS than monocytes from HIV− individuals. A multiple linear regression analysis involving cART, Reynolds CV risk score, and basal p90RSK activity revealed that cART and basal p90RSK activity were the two significant determinants of plaque formation. Many of the antiretroviral drugs individually activated p90RSK, which simultaneously triggered all components of the macrophage senescent phenotype. cART inhibited antioxidant response element reporter activity via ERK5 S496 phosphorylation. NRF2A reversed the H2O2-induced over-activation of p90RSK in cART-treated macrophages by countering the induction of senescent phenotype. Lastly, the data obtained from our gain- or loss-of-function mice conclusively showed the crucial role of p90RSK in inducing senescent phenotype in macrophages and atherogenesis.
Conclusions:
cART increased monocyte/macrophage sensitivity to ROS in HIV+ individuals by suppressing NRF2-ARE activity via p90RSK-mediated ERK5 S496 phosphorylation, which coordinately elicited senescent phenotypes and pro-inflammatory responses. As such, our report underscores the importance of p90RSK regulation in monocytes/macrophages as a viable biomarker and therapeutic target for preventing CVD, especially in HIV+ patients treated with cART.
Keywords: HIV, atherosclerosis, efferocytosis, antioxidants, telomere length, senescence, p90RSK, cART, ERK5, NRF2, reactive oxygen species
Introduction
Remarkable progress in the treatment of HIV infection has dramatically changed the outlook of this disease from one with no treatment to one of a chronic, manageable disease, at least in developed countries1. Although there has been a steady increase in longevity, the lifespan of patients still falls short of that of the average population because of the premature development of non-AIDS comorbidities such as cardiovascular disease (CVD) and metabolic syndrome2, 3. In addition to the traditional risk factors associated with CVD4, both HIV infection itself and antiretroviral drugs have been proposed to be risk factors5; however, the mechanisms underlying this risk remain unclear.
A potential target of both HIV viral products and antiretroviral drugs is myeloid cells, which are important effectors in atherogenesis. In particular, the use of cART has been recently associated, directly and indirectly, with premature senescence and subsequent CVD6–8. It has been suggested that the following four components of the senescent phenotype would be the key to evoke CVD: 1) telomere (TL) shortening-mediated DNA damage and subsequent p16 and p21 induction, 2) reactive oxygen species (ROS) induction, 3) inflammation, and 4) impairment of efferocytosis9–11. However, how these four events are simultaneously provoked by cART remains largely unknown.
In this study, we expanded on our previous work on CVD that implicated p90RSK12, a unique serine/threonine kinase with two distinct functional kinase domains13, as a key molecule in atherogenesis. p90RSK is a redox-sensitive kinase, and p90RSK-mediated ERK5-S496 phosphorylation is important for endothelial activation (i.e. inflammation) and dysfunction by inhibiting ERK5 and the transcriptional activity of an anti-inflammatory transcriptional factor, KLF (Krüppel-like Factor) 2 14. However, p90RSK may also play a role in efferocytosis, as suggested by the results of our recent study in which macrophage-specific ERK5 activity was found to be increased when these cells were allowed to feed on apoptotic cells or were treated by statins15. Under these conditions, macrophages also enhanced phagocytic activity by up-regulating the expression of efferocytosis-related molecules. In addition, depletion of ERK5 in macrophages increased the expression of inflammation-related genes and atherosclerotic plaque formation and reduced the thickness of the fibrotic cap of advanced plaques. Since p90RSK activation inhibits ERK5 transcriptional activity in endothelial cells14, we hypothesize that macrophage p90RSK activation down-regulates not only inflammation but also macrophage efferocytosis.
Several chemical compounds and drugs are known to directly and indirectly cause mitochondrial oxidative stress and dysfunction and to subsequently induce CVD16. Of note, it has been reported that almost 10% of drugs in the last four decades have been recalled from the clinical market worldwide because of cardiovascular safety concerns, and accumulating evidence points to mitochondrial dysfunction as the causative factor16. cART can induce mitochondrial oxidative stress, and this may be related to the increased incidence of CVD in AIDS patients16, 17. To this end, we determined how p90RSK in monocytes/macrophages plays a major role in mediating the cART-induced atherogenesis that frequently occurs in HIV+ patients. Our studies suggest that the cART regimen activates monocyte/macrophage p90RSK, thereby regulating the genes that are involved in efferocytosis and inflammation. Importantly, peripheral monocytes isolated from cART-treated HIV+ patients exhibited enhanced sensitivity to oxidative stress compared with those from HIV− controls. This heightened sensitivity was triggered by a reduction in nuclear factor erythroid 2-related factor 2 (NRF2)-antioxidant response element (ARE) transcriptional activity, which is known to accelerate the aging process18, 19. In fact, we found that NRF2 activation abolished the induction of the senescent phenotype. Since p90RSK activation in myeloid cells appears to play a crucial role in atherosclerotic plaque formation, we posit that p90RSK activation in peripheral monocytes serves as a viable biomarker and target for the treatment of CVD, especially in individuals undergoing cART for HIV.
Methods
Additional details of the experimental procedures are included in the online supplemental information. The data, analytic methods, and study materials that support the findings of this study are available from the corresponding authors upon reasonable request.
Population studies
The study was approved by the Institutional Review Board of the University of Rochester, and that the subjects gave informed consent.
Statistical analysis (clinical study)
The descriptive characteristics are summarized in Supplementary Table 1. Percentages and frequencies are reported for discrete variables; means and standard deviations are reported for continuous characteristics. Fisher’s exact test was used to compare the proportion of categorical variables such as sex and race between the HIV+ and control groups. Welch’s t-test was used to compare the mean values of continuous variables such as age and the Reynolds risk score between the HIV+ and control groups. For multi-group comparisons, we performed one-way ANOVA followed by Bonferroni post hoc testing. When groups exhibited unequal variances, Welch’s ANOVA was used to perform multiple group comparisons.
All analyses were performed using R 3.3.0 (R Foundation for Statistical Computing, Vienna, Austria), SAS 9.3 (SAS Institute, Cary, NC, USA), and Prism 5.0 (GraphPad Software, La Jolla, CA, USA) software.
Results
Various cART components activate p90RSK in monocytes and macrophages.
First, we determined whether the commonly used cART drugs activate p90RSK in vitro in human peripheral blood monocytes as well as in bone marrow-derived macrophages (BMDMs) obtained from C57BL/6 mice. These cells were used to measure both p90RSK phosphorylation as well as phosphorylation of its downstream targets, ERK5, at the S496 residue (important for inhibiting ERK5 transcriptional activity)14 and at the threonine-glutamic acid-tyrosine residue (TEY) motif (important for ERK5 kinase activity)20. We found that efavirenz (EFV: non-nucleoside reverse transcriptase inhibitor (NNRTI)), alone or in combination with tenofovir (TDF: NRTI), emtricitabine (FTC: NRTI), and maraviroc (MVC, CCR5 antagonist), and TDF/FTC with atazanavir (ATV: protease inhibitor [PI] and ritonavir [RTV: PI]), resulted in significantly increased p90RSK activity, as determined by the phosphorylation of Ser 380 (Fig. 1a, b). This effect of cART on p90RSK activation was completely reversed in the presence of a p90RSK-specific inhibitor, FMK-MEA. Similarly, exposure of BMDMs from C57BL/6 mice to ATV (Supplemental Fig. 1a, b), RTV (Supplemental Fig. 1c, d), MVC (Supplemental Fig. 1e, f), and rilpivirine (RPV: non-NRTI [NNRTI] alone (Supplemental Fig. 1g, h), but not to TDF/FTC backbone alone (Supplemental Fig. 1i), and exposure to DMSO (vehicle; Supplemental Fig. 1a, c, j, and k) also produced equivalent changes in p90RSK activation in a dose- and time-dependent manner.
Figure 1. cART and p90RSK activation in HIV+ patients.

(a) Human monocytes were pre-treated with a p90RSK-specific inhibitor of FMK-MEA (10 μM) or vehicle (DMSO, 0.1%) for 30 min and incubated with EFV (10 μM), TDF/FTC/ATV/RTV (10 μM each), TDF/FTC (10 μM each), or vehicle (DMSO, 0.1%) for 10 min. The cells were collected, and total p90RSK, p90RSK phosphorylation, ERK5 S496 phosphorylation, ERK5 TEY motif phosphorylation, and total ERK5 were detected by Western blotting with the indicated antibodies. Representative images from three independent experiments are shown. (b) Quantification of cART-induced p90RSK activation (S380 phosphorylation; left), ERK5 S496 phosphorylation (middle), and ERK5 activation (TEY motif phosphorylation; right) is shown after normalization by total protein levels. The data represent the mean±SD (n=3). **p<0.01. Blue bar: FMK-MEA pre-treatment, white bar: vehicle (DMSO) control pre-treatment. (c, d) The basal levels (c) and levels after H2O2 stimulation (d) of p90RSK activity in CD14+ peripheral blood monocytes from HIV+ and HIV− patients. p90RSK activity was determined using the ratio between phosphorylated p90RSK and total p90RSK, detected by flow cytometry, as described in the Methods. The p90RSK activity after H2O2 stimulation was significantly higher (p=0.0000363) in the HIV+ group. No significant difference was detected in basal p90RSK activity. (e, f) The basal levels (e) and levels after H2O2 stimulation (f) of p90RSK activity in monocytes from HIV+ patients undergoing treatment with integrase inhibitors, multiple combination treatments, and NNRTI regimens, as described in the Methods. No significant difference was found among these three groups. (g, h) The basal levels (g) and levels after H2O2 stimulation (h) of p90RSK activity by viral load detectable status in the HIV+ group, as described in the Methods. Yes, n=11, No, n=74. The p90RSK activity in both basal (p=0.0012245) and H2O2-stimulated (p=0.0022490) samples was significantly higher in the no virus detectable group than those in the virus detectable group.
Integrase inhibitors constitute an important element of the cART regimen, and it is noteworthy that increasing doses of elvitegravir resulted in the upregulation of p90RSK activity within the indicated time points (Supplemental Fig. 1k, l), while raltegravir failed to exert such an effect on p90RSK (Supplemental Fig. 1j). As we described in the Methods, each drug concentration used in this study was within the therapeutic range of plasma concentration in humans. Taken together, our findings suggest that a large number of cART regimens, either alone or in combination, are capable of activating p90RSK in monocytes and macrophages. Given these results, we used TDF/FTC/ATV/RTV in subsequent experiments unless specified otherwise.
p90RSK in peripheral monocytes is primed for activation by cART in HIV+ individuals.
Next, we determined whether p90RSK activation is also affected by cART in HIV+ patients. As shown in Supplemental Table 1, we enrolled two cohorts in this study: (a) 104 HIV+ patients ≥45 years of age with viral load ≤200 copies/mL (1 on nucleoside reverse transcriptase inhibitors [NRTIs], 23 on non-NRTIs [NNRTIs], 56 on multi-category [for example, NNRTI and integrase inhibitors], and 24 on integrase inhibitor-based cART regimens) on stable cART for at least 1 year and (b) 92 HIV− controls matched for similar cardiovascular risk factors using the Reynolds cardiovascular risk score21. Blood was collected at the time of enrollment from all subjects, and a subset of it (HIV+: n=92; HIV−: n=86) was used to measure levels of total and phosphorylated p90RSK in peripheral CD14+ monocytes by flow cytometry.
Since p90RSK is a redox-sensitive kinase22, the sensitivity of monocytes to ROS as a known inducer of p90RSK activation was also assessed by the ex vivo treatment of monocytes with H2O2 (200 μM) for 4 min. Although the basal levels of phospho-p90RSK remained comparable in all subjects, regardless of their HIV status (p=0.544, Fig. 1c), HIV+ patients receiving cART displayed a sharp increase in the sensitivity of monocytic p90RSK to the exposure of H2O2 (Welch two-sample t-tests, p<0.0001, Fig. 1d), suggesting a progressive mechanism engaged by cART in stimulating p90RSK. The association between HIV status and H2O2-mediated p90RSK activation was also found to be remarkable (p=0.0002) following a controlled multivariate regression analysis of the Reynolds risk score21.
We did not find a significant association with p90RSK activation (both basal and H2O2-treated) among different cART regimens (NNRTIs [n=20], integrase inhibitors [n=22], and multi-category [n=49]). (Fig. 1e, f and Supplemental Table 2). These results suggest that the increased sensitivity of monocyte p90RSK in response to H2O2 was not due to a specific drug in the cART regimen. We also compared the p90RSK sensitivity of monocytes of HIV+ patients with detectable (n=11) and undetectable (n=74) baseline HIV viral loads by Welch t-test. We found that both basal and H2O2-treated p90RSK activation levels overlapped between those with detectable and undetectable viremia, despite the mean being significantly higher in those with undetectable virus (Fig. 1g, h). This is likely a spurious effect of the large sample difference between the two groups. Overall, these data suggest that the p90RSK activation detected in our HIV+ patients was not dependent on the high level of HIV replication. Furthermore, the CD4 counts were not significantly associated with both basal and H2O2-treated p90RSK activation levels (Supplemental Fig. 2a, b).
Next, we assessed whether elevated sensitivity of monocyte-p90RSK to oxidative stress in HIV+ individuals was also associated with plaque formation in carotid vessels. To this end, carotid vascular sonography was performed in study participants; the results showed that 28.3±4.7 % and 35.6±4.7% (mean±SEM.) of HIV+ and HIV− individuals, respectively, exhibited plaques in both carotids. A univariate logistic regression analysis did not reveal any significant difference in plaque formation in both sides of the carotid arteries between these two groups (Supplemental Table 3). A multivariate logistic regression analysis was performed on HIV status, Reynolds score, and basal or H2O2-induced p90RSK activation; while neither HIV status nor basal p90RSK activation was significantly associated with plaque formation, their interaction was associated with a significant increase in the risk of forming plaques on both carotid arteries (p=0.0464, Supplemental Table 4). These results suggest that subjects with both HIV and high basal p90RSK activity in peripheral monocytes are at a significantly higher risk of plaque formation. Last, we studied the relationship between the Reynolds score and p90RSK activation and found no significant correlation with the Reynolds score (Supplemental Fig. 2c, d), suggesting that the activation of p90RSK in human monocytes is independent of conventional CVD risk factors.
cART primes monocytes and macrophages to oxidative stress by decreasing antioxidant expression and driving senescence.
As shown in Fig. 1d, we found that cART appears to prime human monocytes in response to H2O2. To confirm this observation in more amenable cell systems, we isolated bone marrow cells from C57BL/6 mice and cultured them to obtain macrophages. These mouse BMDMs were first treated with cART for 16 h and then with H2O2 (200 μM) for up to 30 min, and p90RSK activation was determined. We found that cART pre-treatment significantly increased p90RSK phosphorylation (Fig. 2a, b).
Figure 2. cART inhibits NRF2-ARE transcriptional activity, shortens TLs, and sensitizes macrophages to oxidative stress.

(a) BMDMs were pre-treated with cART (TDF/FTC/ATV/RTV, 10 μM each) or vehicle for 24 h, and cells were incubated with H2O2 (200 μM) for 0-30 min. p90RSK S380 phosphorylation, ERK5 TEY motif phosphorylation, ERK5-S496 phosphorylation, and ERK5, p90RSK, and tubulin expression were detected by Western blotting with specific antibodies. TNFα stimulation (20 ng/ml for 30 min) was used as the positive control. Representative images from three independent experiments are shown. (b) Quantification of p90RSK activation (S380 phosphorylation; left), ERK5 S496 phosphorylation (middle), and ERK5 TEY motif phosphorylation (right) is shown after normalization by total protein levels. The data are represented as the mean±SD. (n=3). **p<0.01 and *p<0.05. (c) BMDMs were pre-treated with FMK-MEA (10 μM) or vehicle for 1 h, and cells were incubated with cART (TDF/FTC/ATV/RTV, 10 μM each) for 0–24 h. Western blotting was performed using specific antibodies against the proteins indicated on the right. (d) BMDMs were transfected with the ARE luciferase reporter and the constitutively expressing Renilla luciferase vector for 16 h. Cells were pre-treated with FMK-MEA (10 μM; left [red]) for 1 h, NRF2 activator (NRF2A, CAS 1362661 [2 μM]; right [orange]) for 6 h, or vehicle control and then treated with vehicle or cART for 6 h. NRF2-ARE transcriptional activity was measured as described in the Methods. Mean±SD., **p<0.01. (e) BMDMs were pre-treated with FMK-MEA (10 μM; left [red]) for 1 h, NRF2 activator (NRF2A, CAS 1362661 [2 μM]; right [orange]) for 6 h, or vehicle, as in (l), and then treated with vehicle or cART for 18 h. TL lengths were determined by measuring the fluorescent telomeric signal intensity of the fluorescein-conjugated PNA probe, as described in the Methods. Results are represented as the relative TL length (%) (mean±SD). The TL length of human T-cell leukemia cell line (1301) cells was set to 100%. **p<0.01.
To gain insight into the mechanism of this priming effect, we tested the following two potential mechanisms: (a) decreased expression of antioxidants and (b) TL shortening, both of which are known senescent phenotypes, as we discussed in the Introduction. We previously reported that ERK5 transcriptional activity, but not kinase activity, is inhibited by p90RSK activation which induces ERK5 S496 phosphorylation in endothelial cells14, and others have shown that ERK5 can up-regulate the activity of NRF2, which is a transcription factor that is well known to regulate antioxidant expression23, 24. Therefore, we hypothesized that p90RSK activation, induced by cART, inhibits NRF2 activity and anti-oxidant expression via ERK5 S496 phosphorylation.
To test this hypothesis, we first determined whether p90RSK activation by cART inhibits ERK5 transcriptional activity. We co-overexpressed p90RSK and a constitutively active form of MEK5 (CA-MEK5) in BMDMs and found that CA-MEK5-induced ERK5 transactivation was indeed significantly inhibited by p90RSK overexpression (Supplemental Fig. 3a). However, since MEK5 also phosphorylates the ERK5 TEY motif, which can activate both the kinase and transcriptional activities of ERK5, we determined whether cART-activated p90RSK phosphorylates both or just one site. As expected, cART increased p90RSK phosphorylation, but more importantly, we found that only ERK5 S496 was phosphorylated (Fig. 1a, b, 2a, b, and Supplemental Fig. 1). These results are consistent with the human data shown in Fig. 1a and b that the effects of EFV and the combination of TDF/FTC/ATV/RTV and TDF/FTC/MVC on p90RSK activation were completely inhibited by a p90RSK-specific inhibitor, FMK-MEA. We also confirmed that 16 h of cART pre-treatment of BMDMs significantly enhanced H2O2-induced p90RSK activation and ERK5 S496, but not TEY motif, phosphorylation (Fig. 2a, b).
We next tested cART’s effects on ERK5 transcriptional activity in RAW264.7 cells. In line with the observations described above, we found that treatment with EFV alone or exposure to the combination of TDF/FTC/ATV/RTV significantly inhibited ERK5 transcriptional activity, which was reversed by pre-treatment with FMK-MEA (Supplemental Fig. 3b, c). Consistently, such a reduction in ERK5 transcriptional activity by cART was found to be completely abolished in BMDMs that overexpressed ERK5-S496A mutant (ERK5 S496A)14 but not WT ERK5 (ERK5-WT; Supplemental Fig. 3d).
Figure 3. Roles of NRF2 and mitochondrial ROS in the expression of the cART-induced senescent phenotype and p90RSK-ERK5 S496 phosphorylation.

(a) BMDMs were pre-treated with NRF2A (CAS 1362661) or vehicle for 6 h and treated with cART or vehicle for 18 h, as in Fig. 2c. They were then incubated with H2O2 (200 μM) for 10 min. A Western blotting analysis was performed using specific antibodies against the proteins indicated on the right side of the panel. Representative images from three independent experiments are shown. (b, c) Various cART drugs increased p90RSK activity, ERK5 S496 phosphorylation, and ERK5 TEY motif phosphorylation via mitochondrial ROS production. BMDMs from C57Bl/6 mice were treated with MitoTEMPO (20 μM) for 1 h and incubated with various cART drugs, as indicated, for 10 min. A Western blotting analysis was performed using specific antibodies against the proteins indicated in the figure. (c) The graph represents densitometry data of immunoblots from three independent experiments (n=3) in (b), mean±SD. **p<0.01. (d, e) BMDMs from C57Bl/6 mice were incubated with various cART drugs and TNFα with vehicle or MitoTEMPO, as indicated. MitoSox Red (d) and H2DCF-DA (e) were added, and mitochondrial ROS (d) and intracellular ROS (e) levels were detected as described in the Methods. Mean±SD. (n=3), **p<0.01.
We investigated how p90RSK is involved in antioxidant expression. As shown in Fig. 2c and Supplemental Fig. 4a, treatment of BMDMs with cART significantly decreased the expression of the antioxidant molecules such as thioredoxin 1 (Trx1), heme oxygenase 1 (HO1), catalase, and glutathione peroxidase 1 (GPx1). However, this decrease was completely reversed by pre-treatment with FMK-MEA. Since NRF2 regulates the expression of antioxidant proteins, we determined whether cART-induced p90RSK activation negatively regulated NRF2. Although cART did not affect NRF2 expression (including both 60 kDa and 110 kDa isoforms) (Fig. 2c, and Supplemental Fig. 4b), it inhibited ARE reporter luciferase activity (Fig. 2d), and once again, FMK-MEA prevented this inhibition (Fig. 2d, left), along with subsequent antioxidant expression (Fig. 2c, and Supplemental Fig. 4a).
To determine the role of NRF2 in cART-induced reduction of antioxidant molecule expression and TL length, we used a cell-penetrating, NRF2-KEAP1-binding inhibitory peptide, CAS 1362661 (NRF2A), that can specifically activate NRF2 transcriptional activity25. As shown in Fig. 2d (right), the reduction in NRF2-ARE transcriptional activity was completely prevented in BMDMs by pre-treatment with NRF2A. It is well known that senescent cells show pro-inflammatory and pro-oxidative phenotypes26. The G-rich sequence of TL forms quadruplex structures in which oxidized equivalents can be trapped. This structural characteristic of TL protects chromosomes from oxidative damage27. We found that cART significantly decreased TL lengths by activating p90RSK and inhibiting NRF2-ARE activity (Fig. 2e).
We previously reported that macrophage efferocytosis and the expression of the inflammatory phenotype are controlled by ERK515; however, the role of the macrophages p90RSK and NRF2 in efferocytosis and inflammation remains unclear. Therefore, we determined whether the activation of p90RSK and NRF2 by cART plays a role in the expression of Gas6 (an efferocytosis-related molecule) and TNFα (an inflammation-related molecule). As shown in Fig. 2c and Supplemental Fig. 4d, Gas6 expression was inhibited while TNFα expression was increased, both in a p90RSK-dependent manner. We also confirmed that pre-treatment with cART, followed by H2O2 stimulation, inhibited the expression of the antioxidant proteins Trx1 HO1, catalase, and GPx1 and enhanced p90RSK activation and ERK5 S496 phosphorylation (Fig. 3a and Supplemental Fig. 5a). All of these events were profoundly diminished by NRF2A treatment.
cART was found to increase the expression of cell senescence markers (p16 and p21); this increase was inhibited by FMK-MEA (Fig. 2c and Supplemental Fig. 4c) and NRF2A (Fig. 3a and Supplemental Fig. 5b), suggesting that cART-induced p90RSK activation and subsequent regulation of NRF2-ARE play a role in TL dysfunction and an accelerated senescence of macrophages. Taken together, these data suggest that cART increases the sensitivity of monocytes/macrophages to oxidative stress by both down-regulating NRF2 transcriptional activity and accelerating the process of premature aging. Although the role of NRF2 in up-regulating CD36 expression has been implicated in atherosclerotic plaque formation28, such changes in CD36 levels following cART treatment were not detected in our study (Fig. 2c, Supplemental Fig. 4b, Fig. 3a, and Supplemental Fig. 5c). Lastly, we found that cART decreased Gas6 expression and induced TNFα expression, both of which were inhibited by FMK-MEA (Fig. 2c and Supplemental Fig. 4d) and NRF2A (Fig. 3a and Supplemental Fig. 5d); these results suggest that p90RSK activity and NRF2-ARE transactivation contribute to regulating ERK5-mediated efferocytosis and inflammation-related events. Taken together, our study indicates the crucial role of p90RSK-NRF2 signaling in coordinately regulating four components of the senescent phenotype: 1) TL shortening-mediated DNA damage and subsequent p16 and p21 induction, 2) ROS induction, 3) inflammation, and 4) impairment of efferocytosis induced by cART.
Since our study showed that p90RSK is a redox-sensitive kinase22, we investigated the effects of a mitochondrial ROS-targeted antioxidant, MitoTEMPO, on p90RSK activation by cART reagents. MitoTEMPO inhibited the phosphorylation of p90RSK and ERK5 S496 by TNFα (positive control) and cART reagents, except RPV (NNRTI) (Fig. 3b, c). We also measured both intracellular and mitochondrial ROS by using CM-H2DCF-DA and MitoSOX red, respectively. Various cART drugs and TNFα, which up-regulated p90RSK activity, increased both intracellular and mitochondrial ROS, and MitoTEMPO significantly inhibited both mitochondrial and intracellular ROS levels (Fig. 3d, e). These results are consistent with the crucial role of mitochondrial ROS production, induced by most cART regimens, in activating p90RSK and reducing ERK5 transcriptional activity, suggesting that cART targets mitochondria.
Increased p90RSK activity during cART-induced inflammation and efferocytosis.
To further study the role of p90RSK in myeloid cell inflammation and efferocytosis, we used transgenic mice expressing the dominant-negative (i.e. kinase-dead) form of p90RSK (DN-p90RSK) specifically in myeloid cells (DN-p90rsk-MTg). BMDMs were derived from the DN-p90rsk-MTg and corresponding non-transgenic littermate control (NLC) mice. In cells from NLC, but not DN-p90rsk-MTg, mice, cART increased the expression of pro-inflammatory genes (TNFα and iNos) and decreased the expression level of efferocytosis-related genes (Gas6, Anxa1, and Tyro3), both in a p90RSK activation-dependent manner (Fig. 4a–e). Indeed, the cART-induced reduction of Gas6 and induction of TNFα were inhibited by both FMK-MEA and NRF2A (Fig. 2c, Supplemental Fig. 4d, Fig. 3a, and Supplemental Fig. 5d). Furthermore, cART-treated NLC cells exhibited reduced efferocytosis. Cells treated with FMK-MEA and BMDMs expressing DN-p90RSK exhibited significantly improved efferocytosis (Fig. 4f).
Figure 4. cART induces inflammation and inhibits efferocytosis-related gene expression via p90RSK activation and elevates atherosclerosis.
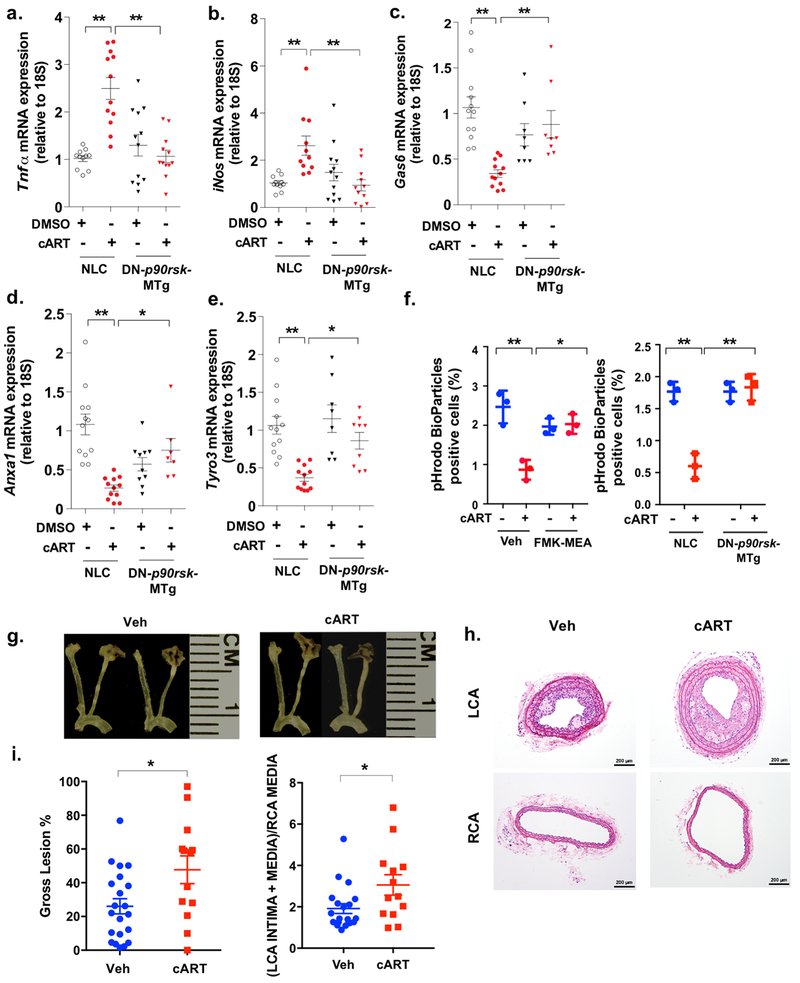
(a-e) BMDMs obtained from NLC and DN-p90rsk-MTg mice were stimulated with cART (red) or vehicle (black) for 16 h. RT-PCR (qRT-PCR) was performed for the five genes indicated, and the levels of expression of each gene were quantified relative to ribosomal 18S RNA. Means±SD. **p<0.01, *p<0.05. (f) BMDMs were pre-treated with FMK-MEA (10 μM) or vehicle for 1 h and treated with cART (red) or vehicle (blue) for 24 h. Cells were then incubated with the IncuCyte pHrodo-labeled apoptosis detection probe, and pHrodo-positive cells were quantified. Mean±SD. (n=3), **p<0.01. (g-i) PCL surgery was performed on the LCA. One week later, mice were treated with cART (TDF/FTC/ATV/RTV) or vehicle for 2 weeks, and the gross plaque size (g, i: left) was determined. Mean±SD. (n=13-20). (h, i: right) The plaque size after PCL in vehicle-treated (veh) and cART-treated Ldlr−/− mice was quantified as described in the Methods. Representative images are shown after H&E staining. Scale bars: 200 μm. (i: right, n=13-20).
cART accelerates plaque formation in Ldlr−/− mice.
Our sonography studies revealed that plaque formation in carotid arteries was correlated with HIV infection (i.e., cART treatment) and increased p90RSK activity. To more closely examine the effect of cART on atherosclerosis, we used the partial carotid artery ligation (PCL) model, which experimentally creates an area of disturbed flow where atherogenic responses are induced in a short period of time29. PCL surgery was performed on Ldlr−/− mice on a high-fat diet and treated with cART or vehicle for two weeks. Three weeks after PCL, mice were euthanized and plaque formation in the left and right carotids evaluated by measuring the gross lesion (%) and the plaque area using histological sections as described in the Methods. cART treatment accelerated plaque formation compared to vehicle treatment (Fig. 4g–i). We did not find any difference in body weight or cholesterol level between cART- and vehicle-treated mice (Supplemental Fig. 6a, b).
Next, we examined whether cART treatment affects the expression of inflammation- and efferocytosis-related molecule expression within the plaque by immunohistochemical analysis. We found that anti-Gas6 staining was significantly lower in the plaques of cART group (Supplemental Fig. 6c, g), and both TNFα (Supplemental Fig. 6d, g) and cleaved caspase 3 expression (Supplemental Fig. 6e, g) levels were increased in the plaques of the cART group compared to vehicle-treated group. Non-immune IgG showed no positive staining (Supplemental Fig. 6f).
Overexpression of p90RSK in myeloid cells accelerates atherosclerosis.
Previously, we reported that overexpression of wild-type p90RSK inhibits ERK5 transcriptional activity via ERK5 S496 phosphorylation in endothelial cells14, and in the present study, we also obtained the same results using macrophages (Supplemental Fig. 3a). We generated mice that specifically overexpressed wild-type p90RSK (WT-p90RSK; LysMCre+/− Tg[WT-p90rskfl]32, referred to as [WT-p90rsk-MTg]) in myeloid cells. WT-p90rsk-MTg mice showed a roughly 2-3-fold increase in WT-p90RSK protein expression and ERK5 S496 phosphorylation (a marker of p90RSK activity) in peritoneal (PE) macrophages compared with their wild-type counterparts, including NLC mice (Fig. 5a). Complete blood cell counts were obtained, which were comparable in NLC and WT-p90rsk-MTg mice (Fig. 5b). The mRNA expression levels of efferocytosis-related genes, as well as an M2-type macrophage marker, Arg1, were significantly lower in cells isolated from WT-p90rsk-MTg mice (Fig. 5c). In contrast, annexin V expression in PE macrophages was not affected (Supplemental Fig. 6h).
Figure 5. Constitutive activation of p90RSK in macrophages down-regulates their expression of efferocytosis-related genes.

(a) p90RSK and ERK5 expression, ERK5 S496 phosphorylation, and tubulin expression were detected in PE macrophages isolated from NLC and WT-p90rsk-MTg mice. (b) Peripheral blood cell counts were performed in NLC and WT-p90rsk-MTg mice. Means±SD. (n=5). (c) Relative mRNA expression of efferocytosis-related genes in NLC and WT-p90rsk-MTg PE macrophages was determined by quantitative RT-PCR. Mean±SD., *p<0.05.
To determine the role of the macrophage p90RSK in the development of atherosclerosis, we bred NLC and WT-p90rsk-MTg mice into the Ldlr−/− background and fed them a high-fat diet for 12 weeks and 16 weeks, respectively. No differences were noted in their body weight or their LDL and HDL cholesterol levels (Supplemental Fig. 7a, b). However, the atherosclerotic lesion area observed in the en face sample of the aorta was significantly larger in WT-p90rsk-MTg/Ldlr−/− mice than in NLC/Ldlr−/− mice (Fig. 6a, c: left, f, g). Larger lesions were also found in histological cross-sections at the aortic valve area in WT-p90rsk-MTg/Ldlr−/− mice than in NLC/Ldlr−/− mice (Fig. 6b, c: right, h, i). Plaque necrosis is the consequence of apoptotic cell accumulation in advanced lesions30. Therefore, we assessed the effect of p90RSK overexpression on apoptotic cell accumulation. The number of TUNEL-positive nuclei was significantly higher in WT-p90rsk-MTg/Ldlr−/− mouse lesions with larger necrotic cores than in NLC/Ldlr−/− mouse lesions (Fig. 6d, e, j). Because annexin V expression in PE macrophages was unaffected by p90RSK overexpression (Supplemental Fig. 6h), it is possible that the apoptotic cell accumulation observed in WT-p90rsk-MTg/Ldlr−/− mouse lesions was not caused by increased apoptosis but by defective clearance of apoptotic cells, or efferocytosis.
Figure 6. Overexpression of p90RSK in myeloid cells accelerates atherosclerosis and necrotic core formation.
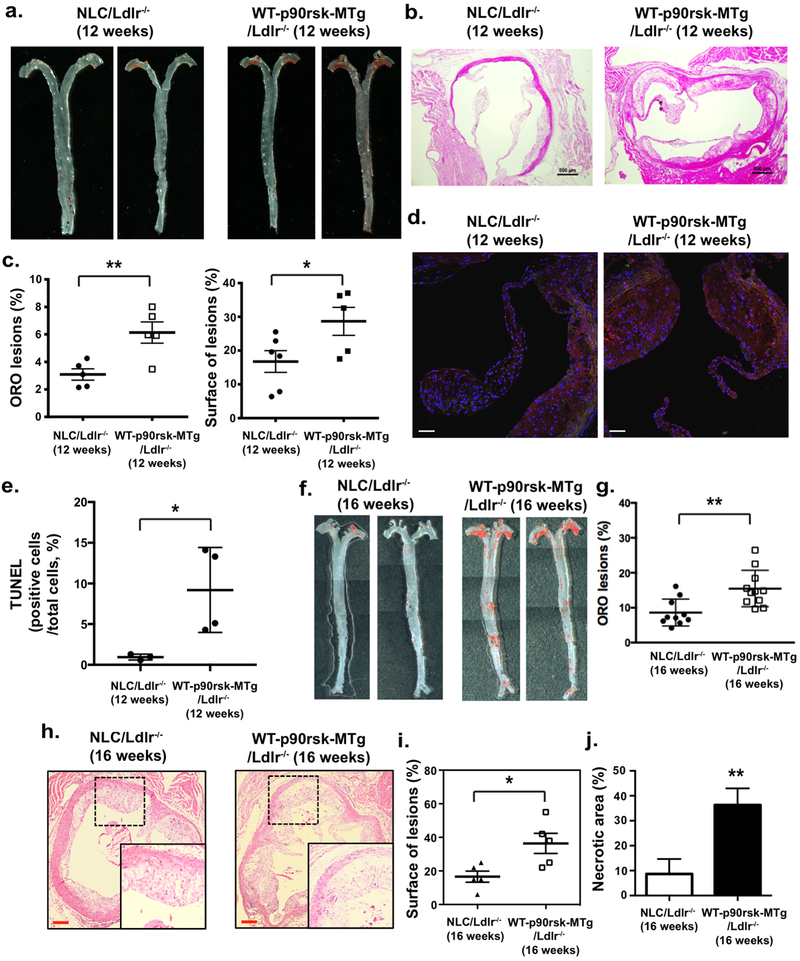
NLC/Ldlr−/− and WT-p90rsk-MTg/Ldlr−/− mice were fed a high-fat diet for 12 weeks (a-e) or 16 weeks (f-j), respectively. WT-p90rsk-MTg/Ldlr−/− mice exhibited increased oil-red O-stained atherosclerotic lesions in the en face whole aorta (a, c: left, f, g) as well as increased H&E-stained sections of the aortic valve region (b, h). Scale bars: 500 μm. Quantified oil-red O-stained lesions (c: left, g) and histologically identified plaque areas (c: right, j) are shown. Mean±SD. (d) Sections of proximal aortas from each group were labeled using TUNEL reagents to detect apoptotic cells and counterstained with DAPI to detect nuclei. Scale bars: 50 μm. (e) The graph shows the percentage of TUNEL-positive cells (TUNEL+ cells/total cells counted) in the lesion area. Over 200 cells were counted for each group. *p<0.05. (j) The area occupied by the necrotic core (acellular lipid core) is shown as the percentage of the total lesion area. Data are expressed as means±SD. n=5 per genotype. **p<0.01.
To further establish the role of myeloid cell-specific p90RSK in atherosclerosis, we generated myeloid cell-specific, dominant-negative p90RSK transgenic (LysM-Cre+/− Tg[DN-p90rskfl]) mice (DN-p90rsk-MTg). To verify the expected dominant negativeness of this model animal, we created BMDMs from DN-p90rsk-MTg and control NLC mice; stimulated them with cART, TNFα, or vehicle for 30 min; and assayed them for ERK5 S496 phosphorylation, a marker for p90RSK activity. We found that the increase in ERK5 S496 phosphorylation due to cART or TNFα treatment was lost in cells isolated from DN-p90rsk-MTg mice (Fig. 7a–d), establishing that macrophages from DN-p90rsk-MTg mice have little to no p90RSK activity. Complete blood cell counts were obtained, which were comparable in NLC and DN animals (Supplemental Fig. 7c). When BMDMs from NLC mice were exposed to cART or TNFα, ARE reporter luciferase activity was blocked. In contrast, this inhibition was not detected in BMDMs from DN-p90rsk-MTg mice (Fig. 7e, f). We also found that cART and TNFα treatment led to reduced protein expression of Gas6, HO-1, and Trx1 and accumulation of TNFα (only by cART), p16, and p21 in cells from NLC mice. These effects, however, were not seen in BMDMs from DN-p90rsk-MTg mice (Fig. 7g, h, and Supplemental Fig. 8). Our results show that cART is not the only primer of macrophage p90RSK. The expression of CD36 (induced by cART) and tubulin remained constant under these experimental conditions (Fig. 7g, Supplemental Fig. 8b, Fig. 7h, and Supplemental Fig. 8f). We also found that both cART and TNFα increased mitochondrial ROS within 10 min after incubation, which was significantly attenuated in BMDMs from DN-p90rsk-MTg mice (Fig. 7i, j). These data lend support to the theory that p90RSK is a key player in regulating efferocytosis, inflammation, antioxidant expression and senescence via mitochondrial ROS production.
Figure 7. cART-induced, CVD-associated phenotypes are reversed in BMDMs obtained from myeloid cells-specific DN-p90rsk-MTg mice.
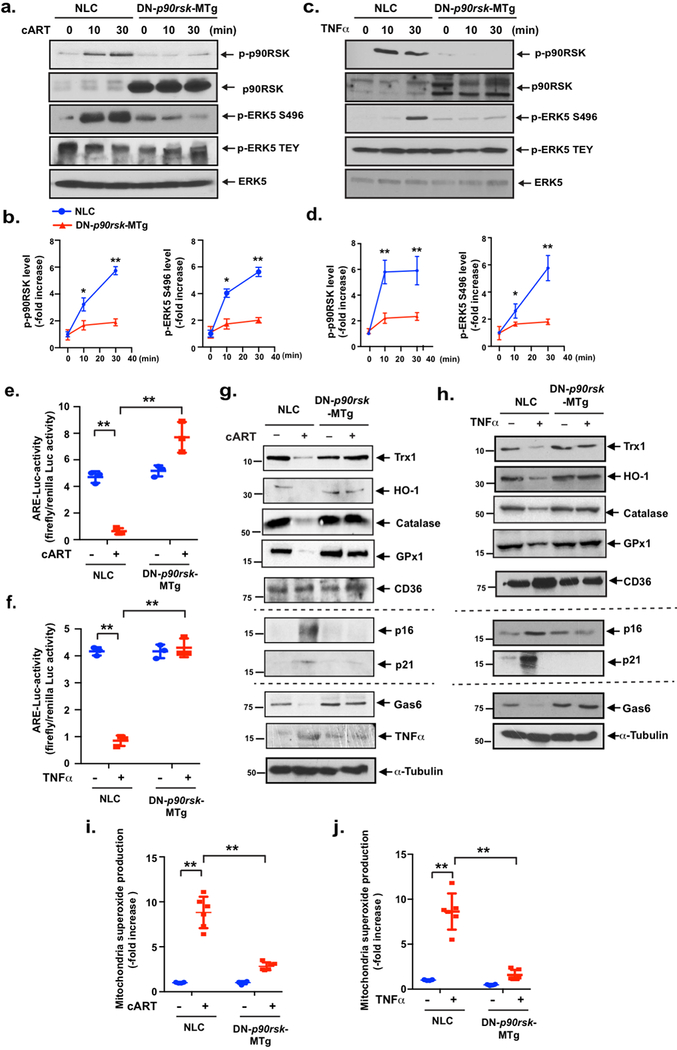
(a-d) BMDMs from NLC and DN-p90rsk-MTg mice were incubated with cART (TDF/FTC/ATV/RTV, 10 μM each) (a) or TNFα (20 ng/ml) (c) for 0–30 min, and p90RSK activation (S380 phosphorylation), ERK5 S496 phosphorylation, ERK5 TEY motif phosphorylation, and total p90RSK and ERK5 expression were detected by Western blotting using specific antibodies, as indicated. (b, d) The graph represents the densitometry data of immunoblots (n=3), similar to those shown in (a) and (c). The data are the means±SD. **p<0.01 and *p<0.05. (e, f) BMDMs from NLC and DN-p90rsk-MTg mice were transfected with the ARE luciferase reporter and the constitutively expressing Renilla luciferase vector for 16 h. Cells were incubated with (e) cART (TDF/FTC/ATV/RTV, 10 μM each) or (f) TNFα (20 ng/ml) for 6 h, and NRF2-ARE transcriptional activity from three independent experiments was measured as described in the Methods. Mean±SD., **p<0.01. (g, h) BMDMs from NLC and DN-p90rsk-MTg mice were incubated with cART (TDF/FTC/ATV/RTV, 10 μM each) or TNFα (20 ng/ml) for 24 h. Western blotting was performed using antibodies against the indicated proteins. (i, j) BMDMs from NLC and DN-p90rsk-MTg mice were incubated with cART (TDF/FTC/ATV/RTV, 10 μM each) (i) or TNFα (20 ng/ml) (j) for 0 (−) or (+) 10 min. and then treated with MitoSOX Red for 10 min, and mitochondrial ROS levels were detected as described in the Methods. Mean±SD. (n=3), **p<0.01.
Next, to determine whether specific activation of p90RSK in macrophages is involved in atherogenesis, we used PCL models of NLC (wild-type C57Bl/6) and DN-p90rsk-MTg mice injected with rAAV8-expressing mouse gain-of-function pro-protein convertase subtilisin/kexin type 9 (PCSK9) mutant of D377Y-PCSK9 (rAAV8-mD377Y-PCSK9)31. Interestingly, the severity of obesity induced by a high-fat diet after this injection was significantly lower in DN-p90rsk-MTg mice than in wild-type mice (Supplemental Fig. 9a). However, there was no notable difference in cholesterol levels among these mice 56 days after rAAV8-mD377Y-PCSK9 injection and 28 days after partial ligation (Supplemental Fig. 9b). Previously, we reported a significant increase in macrophage infiltration into the plaque after PCL in mice with hypercholesteremia, which distinguishes these plaques from the restenosis lesions seen during vascular intimal formation after vascular injury32. When we isolated the carotids, we first examined the ratio of the area of the plaque lesion and total LCA area as the gross lesion (%) as described in the Methods, and found that the gross lesion was much smaller in the DN-p90rsk-MTg mice group than in the NLC group (Fig. 8a, b). Next, we made serial sections through the entire carotid arteries, and the ratio of LCA total area (intima + media) and RCA wall area was calculated as described in the Methods. A significant reduction in LCA plaque size was seen in the DN-p90rsk-MTg mice group compared to that in the NLC group (Fig. 8c, d). Lastly, we determined necrotic core formation as described in the Methods and found a clear necrotic core (non-cellular area) formation in the plaques of the NLC group, but a less developed necrotic core formation in those of the DN-p90rsk-MTg mice group (Fig. 8e).
Figure 8. Atherosclerotic plaque formation is inhibited in myeloid-specific DN-p90rsk-MTg mice.
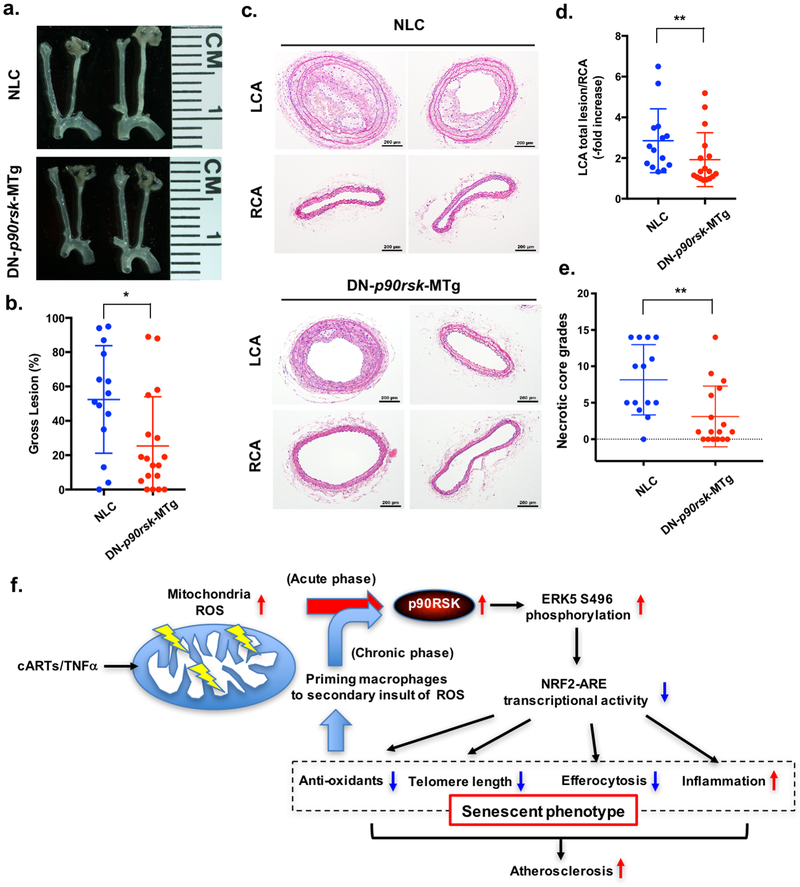
(a, b) Four weeks after the AAV/D377Y-mPCSK9 injection and after being fed a high-fat diet, NLC and DN-p90rsk-MTg mice underwent PCL of the LCA. After 4 weeks, mice were killed and their carotid arteries were harvested. Representative images are shown (a). The gross plaque size (plaque area/total left carotid area) was measured (b). Mean±SD. (n=10-13), *p<0.05. (c, d) H&E-stained cross-sections of the LCA and RCA in NLC and DN-p90rsk-MTg mice (c). The unligated RCA in each mouse was used as the control. Scale bars: 200 μm. The average plaque size was determined by the ratio of the LCA lesion area to RCA media in the cross-sections obtained from 5-10 different positions along the carotid arteries (d), as described in the Methods section and previously32. (e) Necrotic core formation was quantified by a grading system, as described in the Methods. Mean±SD. (n=10-13), **p<0.01. (f) The scheme depicts the mechanisms and roles of p90RSK activation in cART/TNFα-induced atherosclerotic plaque formation. cART and TNFα activate p90RSK by inducing mitochondrial ROS production within 30 min after stimulation (acute phase) and subsequently promote ERK5 S496 phosphorylation and down-regulate NRF2-ARE transcriptional activity without affecting NRF2 and CD36 expression levels. This down-regulation of NRF2-ARE transcriptional activity leads to the up-regulation of 1) inflammation and 2) the expression of senescence-related molecules and the down-regulation of 3) efferocytosis and 4) antioxidant-related molecule expression. After 24 h of cART and TNFα stimulation of macrophages (chronic phase), monocytes and macrophages are sensitized to the secondary insult of ROS; this is completely lost after treatment with NRF2 activator, suggesting that the reduction in NRF2-ARE transcriptional activity, in particular antioxidants and TL shortening, contributes to cART/TNFα-mediated priming of monocytes and macrophages to a secondary insult by ROS.
To determine the effect of cART only, we treated DN-p90rsk-MTg and NLC mice (fed a normal chow diet) with cART twice (20 h and then 4 h before euthanasia) and measured VCAM-1 expression in endothelial cells in the athero-resistant greater curvature area of the aortic arch. We found a significant increase in VCAM-1 expression in the NLC mice, but such an increased VCAM-1 expression was not observed in DN-p90rsk-MTg mice (Supplemental Fig. 9c, d).
Discussion
The use of cART has changed the natural history of HIV infection, allowing patients to live longer. However, this benefit comes at the cost of an increased risk of HIV-associated comorbidities, such as CVD. Indeed, several large retrospective studies have shown an increased incidence of myocardial infarction33–35. HIV-associated CVD is likely the result of multiple interactions, including traditional CVD risk factors, HIV viral proteins, chronic immune activation, and the effects of antiretroviral therapy. Particular attention has been paid to the metabolic abnormalities seen in HIV+ patients on long-term cART. These metabolic changes include insulin resistance and an atherogenic lipid profile that is characterized by elevated low-density lipoprotein cholesterol and triglyceride levels, as well as decreased high-density lipoprotein cholesterol levels36. Some of these metabolic changes have been related to cART-induced mitochondrial dysfunction37. However, how the key events in atherogenesis that lead to CVD are affected by cART in HIV+ individuals is not well known.
In this study, we focused on monocytes because they play a central role in atherogenesis by entering the intima and differentiating to macrophages under pro-inflammatory conditions, thereby actively participating in plaque formation38. We have provided compelling evidence that cART initiates a cycle of p90RSK activation/sensitization via increasing the production of mitochondrial ROS in monocytes/macrophages. p90RSK activation due to elevated oxidative stress causes phosphorylation of ERK5 at S496 residue, in turn decreasing NRF2-ARE transcriptional activity. These signaling pathways sensitize monocytes/macrophages to ROS and accelerate atherosclerosis via inducing the senescent phenotype by reducing monocyte/macrophage efferocytosis activity, reducing anti-oxidant molecules expression and TL lengths, and increasing monocyte/macrophage inflammatory phenotypes (Fig. 8f).
The clinical importance of p90RSK activation in monocytes was confirmed by the observation that monocytes from HIV-infected, cART-treated individuals were more sensitive to oxidative stress (increased levels of p90RSK enzymatic activity) than were those from non-infected, non-treated individuals. Interestingly, the results of multivariate logistic regression analyses indicate that this increased sensitivity of monocytes is not associated with a specific cART regimen or active HIV replication (as measured by detectable viral load) and is not correlated with Reynolds cardiovascular risk score. These findings highlight the role of cART itself broadly (not limited to one specific regimen) in modulating the p90RSK-ERK5 axis, rather than HIV infection, in a manner that is independent of conventional CVD risk factors. Furthermore, basal levels of p90RSK were found to be correlated with plaque formation, as detected by sonography. Similar results were obtained in cART-treated Ldlr−/− mice, wherein the treatment accelerated atherosclerosis and resulted in a larger plaque size.
While we did not detect a significant relationship between plaque formation and HIV status alone, we found a strong link between plaque formation and the combination of basal p90RSK activity and HIV status in a multivariate stepwise regression analysis. No significant relationship between plaque formation and the levels of H2O2-induced p90RSK activation was detected. However, we postulate that basal p90RSK activity in human peripheral monocytes represents the status of monocyte activation after the initial priming of the cells to mitochondrial ROS. Therefore, the basal level of p90RSK activity can demonstrate a stronger relationship to plaque formation than can p90RSK activity after H2O2 treatment. A longitudinal follow-up of the patients is underway that will provide more conclusive results.
A recent report by Beckman et al showed that HIV infection is associated with a 19% increase over the risk of peripheral arterial disease beyond that which can be attributed to traditional CVD risk factors39. However, this increase was found only in individuals with CD4 counts <200 cells/μL. Although their study and ours focus on CVD in HIV+ patients, there are important differences between the two studies, including the clinical endpoint, the cohort composition, and the prospective data acquisition. In our study, the mean CD4 cell count was 754.24/μL, and HIV replication was well controlled. While Beckman et al reported that HIV+ military veterans with time-updated (current) CD4 cell counts >500 cells/μl had no increased risk of peripheral arterial disease39, but we found that the combination of basal p90RSK activation and HIV status was significantly correlated with carotid plaque formation, which was predictive of future cardiovascular events. In addition, the mean age of HIV+ patients was 55.68 years in our study vs 48.0 years in the other study39. Since the HIV prevalence rate among people >50 years old has been steadily increased40, our data on older participants are representative of this population, which is at higher risk for CVD, independently of CD4 cell counts.
In vitro experiments using human and mouse monocytes/macrophages further delineated the mechanisms that underlie cART-mediated sensitization of monocytes/macrophages to ROS via provoking the senescent phenotype. We found that cART decreased the expression of anti-oxidants and TL length via stimulating p90RSK and consequently inhibiting NRF2-ARE transcriptional activity. ERK5 has been shown to up-regulate NRF2 transcriptional activity, and it is plausible that the reduction in ERK5 transcriptional activity induced by p90RSK leads to the inhibition of NRF2-ARE transcriptional activity and subsequent anti-oxidant expression. The protective role of TL against oxidative stress has been well established41, 42, and cART was found to significantly shorten TLs in monocytes/macrophages, thus further sensitizing these cells to oxidative stress. Therefore, p90RSK-NRF2 activation can coordinately regulate various aspects of the senescent phenotype, in that p90RSK activation sensitizes its own activation in response to ROS by inducing the senescent phenotype, which results in reduced anti-oxidant expression and short TL length.
Since p90RSK is activated by mitochondrial ROS22, 43, we investigated the effect of cART on mitochondrial ROS activity and its potential link with p90RSK activation in monocytes. Along similar lines, various studies have investigated the effect of different cART drugs on mitochondrial function34, 35, 44. It is important to emphasize that not only cART but also many other drugs can target mitochondria by accumulating in the matrix or the inner mitochondrial membrane, both of which possess the unique physicochemical characteristics of the mitochondrial matrix17. Therefore, lipophilic compounds of cationic character and weak acids in anionic forms can accumulate in mitochondria and produce ROS and reactive nitrogen species by interfering with the mitochondrial respiratory chain45. In accordance with these reports, our results indicate that cART-induced mitochondrial ROS plays a very crucial role in the subsequent activation of p90RSK in monocytes/macrophages.
To more conclusively establish the key role of p90RSK in atherosclerosis, we used mouse models with both gain- and loss-of-function (myeloid cell-specific overexpression of WT or DN-p90RSK). We did not find any differences in the lipid profile among the different groups of mice examined. This is particularly relevant because integrase inhibitors, which are a recent addition to the groups of cART drugs, are associated with favorable lipid profiles. For this reason, it has been suggested that integrase inhibitors would be preferred drug candidates in patients with elevated cardiovascular risk46, 47. However, we found that elvitegravir, one of the two integrase inhibitors we tested, significantly increased p90RSK activity and subsequent ERK5 S496 phosphorylation in in vitro experiments. In addition, p90RSK activation in macrophages promoted atherosclerosis without affecting the lipid profile in our mouse models. Hence, it is very important to further investigate the effect of integrase inhibitors as the drugs of choice for HIV+ individuals with a high risk for CVD.
In summary, our findings demonstrate that peripheral monocyte p90RSK activity can be a biomarker for cART-induced CVD events. Since the p90RSK-NRF2 signaling pathway in myeloid cells regulates multiple cellular processes, which concurrently induce the senescent phenotype, up-regulate inflammation and oxidative stress response, and down-regulate TL length and efferocytosis, this pathway can be targeted to develop therapeutic strategies for reducing cART-induced atherosclerosis and possibly other causes of CVD, depending on p90RSK activation. One of the limitations of this study was the cross-sectional analysis of the clinical data. Longitudinal data are being collected and will become available in the next few years. However, the data already reported, combined with the in vitro, animal, and human data, strongly suggest that p90RSK activation in myeloid cells is critical in atherogenesis and provides a common pathway for several antiretroviral drugs to contribute to atherosclerosis.
Please refer to Supplemental Figures 10 and 11 for further information on the repeatability of p90RSK activity measurements described in the supplemental methods, and supplementary discussion on Glagov’s effect, respectively. Also please refer to Supplemental Tables 5 and 6 for further information on the real-time PCR described in the supplemental methods, and the supplemental discussion on viremia, plaque, and p90RSK activity in our clinical study, respectively.
Supplementary Material
Clinical Perspective.
What Is New?
p90RSK activation in monocyte/macrophage contributes to the recognized association between combination antiretroviral therapy (cART) and cardiovascular disease (CVD).
cART initiates a cycle of p90RSK activation/sensitization via increasing the production of mitochondrial reactive oxygen species (ROS) in monocytes/macrophages.
p90RSK activation inhibits nuclear factor erythroid 2-related factor 2 (NRF2)-antioxidant response element (ARE) transcriptional activity, which is known to accelerate the aging process.
p90RSK-NRF2 signaling pathway sensitizes monocytes/macrophages to ROS and accelerate atherosclerosis via simultaneously inducing the following four components of the senescent phenotype: 1) telomere shortening and subsequent p16 and p21 induction, 2) ROS induction, 3) inflammation, and 4) impairment of efferocytosis.
What Are the Clinical Implications?
Inhibition of p90RSK activity can be a viable approach to controlling atherosclerosis and reducing CVD in HIV+ patients.
In vitro drug screening using p90RSK activation by incubating monocyte/macrophage could help screening for potential pro-atherogenic drugs, but could also be used to evaluate drugs that could prevent atherogenesis.
H2O2-induced p90RSK activation in CD14+ monocytes obtained from patients may serve as a viable biomarker to predict future CVD events in HIV+ patients.
Novel therapeutic approach aimed at reducing p90RSK activity should be investigated for preventing future CVD events.
Acknowledgements
We thank Jacob Fog Bentzon for his technical advice to use rAAV8-mediated D377Y-mPCSK9; we thank Carolyn J. Giancursio for her technical assistance. We thank to Department of Scientific Publications at University of Texas MD Anderson for proofreading the article. We are thankful to the study coordinators and supporting staff at the University of Rochester for enrolling the study participants and managing data and specimen collection: Emily Cosimano, Mary Adams, Elizabeth Keller, Catherine Bunce, Susanne Heininger, Jill Guary, Valerie Kline, Susan Antenozzi, William Coughlin, Emily Smith, Christopher Foote, and Andrew Moran. We are also grateful for the support received from the infectious Disease Clinics at the University of Rochester (Drs. Peter Mariuz and Michael Keefer) and at Trillium Health (Dr. Michael Mancenido), Rochester, NY. The clinical study would not have been possible without the study participants’ generous contribution of their time. M.V.S., S.K., N.T.L., and K.A.K. performed the experiments, interpreted the data, and wrote the manuscript. K.S.H., Y.F., H.T.V., E.M., T.N.T., Y.W., Y.J.G., Y.T., J.M., N.E.S., and K.J.G. performed the experiments and interpreted the data. J.T. provided FMK-MEA and interpreted the data. N.C., V.D., and M.D. planned the ultrasound studies, supervised the data acquisition, and interpreted the data. C.H. evaluated study participants and contributed to the interpretation of the data. A.T. supervised data management and supported data analyses. W.L. and X.Q. performed the statistical analysis. K.F. performed and interpreted the imaging data. S.B.M., G.S., and J.A. planned and generated the study design, obtained funding, interpreted the data, and wrote the manuscript.
Funding Sources
This work was supported by grants from the National Institutes of Health (NIH) to Drs. Abe, Maggirwar, and Schifitto (HL123346 MPI); Dr. Abe (HL-118462, and HL130193); Dr. Maggirwar (R01 HL128155, R01 NS054578, and R01 NS066801); Dr. Schifitto (R01 AG054328 and R01 MH099921); Dr. Le (HL-134740), and Drs. Singh and Qiu (University of Rochester Center for AIDS Research NIH 5 P30 AI078498-08) and from the American Heart Association to Drs. Fujiwara (11GRNT5850001) and Heo (12SDG11690003).
Footnotes
Disclosure
Dr. Taunton is a cofounder of Principia Biopharma, which has licensed FMK-MEA.
References
- 1.van Sighem AI, Gras LA, Reiss P, Brinkman K, de Wolf F and study Anoc. Life expectancy of recently diagnosed asymptomatic HIV-infected patients approaches that of uninfected individuals. AIDS. 2010;24:1527–1535. [DOI] [PubMed] [Google Scholar]
- 2.Cohen RA, Seider TR and Navia B. HIV effects on age-associated neurocognitive dysfunction: premature cognitive aging or neurodegenerative disease? Alzheimers Res Ther. 2015;7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monsuez JJ, Belin C and Bouchaud O. Microvascular Function in Aging Among Women Living with HIV. Curr HIV/AIDS Rep. 2016;13:392–398. [DOI] [PubMed] [Google Scholar]
- 4.Boccara F Cardiovascular health in an aging HIV population. AIDS. 2017;31 Suppl 2:S157–S163. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan-Lewis E, Aberg JA and Lee M. Atherosclerotic Cardiovascular Disease and Anti-Retroviral Therapy. Curr HIV/AIDS Rep. 2016;13:297–308. [DOI] [PubMed] [Google Scholar]
- 6.Torres RA and Lewis W. Aging and HIV/AIDS: pathogenetic role of therapeutic side effects. Lab Invest. 2014;94:120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith RL, de Boer R, Brul S, Budovskaya Y and van Spek H. Premature and accelerated aging: HIV or HAART? Front Genet. 2012;3:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cruse B, Cysique LA, Markus R and Brew BJ. Cerebrovascular disease in HIV-infected individuals in the era of highly active antiretroviral therapy. J Neurovirol. 2012;18:264–276. [DOI] [PubMed] [Google Scholar]
- 9.Wang JC and Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circulation research. 2012;111:245–259. [DOI] [PubMed] [Google Scholar]
- 10.Li W Phagocyte dysfunction, tissue aging and degeneration. Ageing Res Rev. 2013;12:1005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niccoli T and Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22:R741–752. [DOI] [PubMed] [Google Scholar]
- 12.Itoh S, Ding B, Shishido T, Lerner-Marmarosh N, Wang N, Maekawa N, Berk BC, Takeishi Y, Yan C, Blaxall BC and Abe J. Role of p90 ribosomal S6 kinase-mediated prorenin-converting enzyme in ischemic and diabetic myocardium. Circulation. 2006;113:1787–1798. [DOI] [PubMed] [Google Scholar]
- 13.Frodin M and Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. [DOI] [PubMed] [Google Scholar]
- 14.Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, McClain C, Hurley C, Wang X, Li F, Xu H, Morrell C, Sullivan MA, Cohen MS, Serafimova IM, Taunton J, Fujiwara K and Abe J. A Crucial Role for p90RSK-Mediated Reduction of ERK5 Transcriptional Activity in Endothelial Dysfunction and Atherosclerosis. Circulation. 2013;127:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heo KS, Cushman HJ, Akaike M, Woo CH, Wang X, Qiu X, Fujiwara K and Abe J. ERK5 activation in macrophages promotes efferocytosis and inhibits atherosclerosis. Circulation. 2014;130:180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varga ZV, Ferdinandy P, Liaudet L and Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol. 2015;309:H1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scatena R Mitochondria and drugs. Adv Exp Med Biol. 2012;942:329–346. [DOI] [PubMed] [Google Scholar]
- 18.Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J, Liu GH and Misteli T. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell. 2016;165:1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorbachev AV, Gasparian AV, Gurova KV, Gudkov AV and Fairchild RL. Quinacrine inhibits the epidermal dendritic cell migration initiating T cell-mediated skin inflammation. Eur J Immunol. 2007;37:2257–2267. [DOI] [PubMed] [Google Scholar]
- 20.Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ and Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. Embo J. 1997;16:7054–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook NR, Paynter NP, Eaton CB, Manson JE, Martin LW, Robinson JG, Rossouw JE, Wassertheil-Smoller S and Ridker PM. Comparison of the Framingham and Reynolds Risk scores for global cardiovascular risk prediction in the multiethnic Women’s Health Initiative. Circulation. 2012;125:1748–1756, S1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abe J, Okuda M, Huang Q, Yoshizumi M and Berk BC. Reactive oxygen species activate p90 ribosomal S6 kinase via fyn and ras. J Biol Chem. 2000;275:1739–1748. [DOI] [PubMed] [Google Scholar]
- 23.Hwang AR, Han JH, Lim JH, Kang YJ and Woo CH. Fluvastatin inhibits AGE-induced cell proliferation and migration via an ERK5-dependent Nrf2 pathway in vascular smooth muscle cells. PLoS One. 2017;12:e0178278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim M, Kim S, Lim JH, Lee C, Choi HC and Woo CH. Laminar flow activation of ERK5 protein in vascular endothelium leads to atheroprotective effect via NF-E2-related factor 2 (Nrf2) activation. J Biol Chem. 2012;287:40722–40731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steel R, Cowan J, Payerne E, O’Connell MA and Searcey M. Anti-inflammatory Effect of a Cell-Penetrating Peptide Targeting the Nrf2/Keap1 Interaction. ACS Med Chem Lett. 2012;3:407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Correia-Melo C and Passos JF. Demystifying the role of mitochondria in senescence. Mol Cell Oncol. 2016;3:e1162896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delaney S, Jarem DA, Volle CB and Yennie CJ. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radic Res. 2012;46:420–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, Polotsky VY and Biswal S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS One. 2008;3:e3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ, Visvader JE and Jo H. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood. 2010;116:e66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thorp E and Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;86:1089–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bjorklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG and Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–1689. [DOI] [PubMed] [Google Scholar]
- 32.Heo KS, Le NT, Cushman HJ, Giancursio CJ, Chang E, Woo CH, Sullivan MA, Taunton J, Yeh ET, Fujiwara K and Abe J. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J Clin Invest. 2015;125:1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmberg SD, Moorman AC, Williamson JM, Tong TC, Ward DJ, Wood KC, Greenberg AE, Janssen RS and investigators HIVOS. Protease inhibitors and cardiovascular outcomes in patients with HIV-1. Lancet. 2002;360:1747–1748. [DOI] [PubMed] [Google Scholar]
- 34.Friis-Moller N, Sabin CA, Weber R, d’Arminio Monforte A, El-Sadr WM, Reiss P, Thiebaut R, Morfeldt L, De Wit S, Pradier C, Calvo G, Law MG, Kirk O, Phillips AN, Lundgren JD and Data Collection on Adverse Events of Anti HIVDSG. Combination antiretroviral therapy and the risk of myocardial infarction. N Engl J Med. 2003;349:1993–2003. [DOI] [PubMed] [Google Scholar]
- 35.Bozzette SA, Ake CF, Tam HK, Chang SW and Louis TA. Cardiovascular and cerebrovascular events in patients treated for human immunodeficiency virus infection. N Engl J Med. 2003;348:702–710. [DOI] [PubMed] [Google Scholar]
- 36.Glesby MJ. Coronary heart disease in HIV-infected patients. Curr HIV/AIDS Rep. 2005;2:68–73. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez-Gallego E, Gomez J, Domingo P, Ferrando-Martinez S, Peraire J, Vilades C, Veloso S, Lopez-Dupla M, Beltran-Debon R, Alba V, Vargas M, Castellano AJ, Leal M, Pacheco YM, Ruiz-Mateos E, Gutierrez F, Vidal F, Rull A and Group C-NeS. Circulating metabolomic profile can predict dyslipidemia in HIV patients undergoing antiretroviral therapy. Atherosclerosis. 2018;273:28–36. [DOI] [PubMed] [Google Scholar]
- 38.Tabas I and Lichtman AH. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity. 2017;47:621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beckman JA, Duncan MS, Alcorn CW, So-Armah K, Butt AA, Goetz MB, Tindle HA, Sico JJ, Tracy RP, Justice AC and Freiberg MS. Association of Human Immunodeficiency Virus Infection and Risk of Peripheral Artery Disease. Circulation. 2018;138:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahy M, Autenrieth CS, Stanecki K and Wynd S. Increasing trends in HIV prevalence among people aged 50 years and older: evidence from estimates and survey data. AIDS. 2014;28 Suppl 4:S453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delaney S, Jarem DA, Volle CB and Yennie CJ. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radic Res. 2012;46:420–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Z, Rhee DB, Lu J, Bohr CT, Zhou F, Vallabhaneni H, de Souza-Pinto NC and Liu Y. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 2010;6:e1000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caldiz CI, Garciarena CD, Dulce RA, Novaretto LP, Yeves AM, Ennis IL, Cingolani HE, Chiappe de Cingolani G and Perez NG. Mitochondrial reactive oxygen species activate the slow force response to stretch in feline myocardium. J Physiol. 2007;584:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gardner K, Hall PA, Chinnery PF and Payne BA. HIV treatment and associated mitochondrial pathology: review of 25 years of in vitro, animal, and human studies. Toxicol Pathol. 2014;42:811–822. [DOI] [PubMed] [Google Scholar]
- 45.Kang D and Hamasaki N. Alterations of mitochondrial DNA in common diseases and disease states: aging, neurodegeneration, heart failure, diabetes, and cancer. Curr Med Chem. 2005;12:429–441. [DOI] [PubMed] [Google Scholar]
- 46.Rockstroh JK, Lennox JL, Dejesus E, Saag MS, Lazzarin A, Wan H, Walker ML, Xu X, Zhao J, Teppler H, Dinubile MJ, Rodgers AJ, Nguyen BY, Leavitt R, Sklar P and Investigators S. Long-term treatment with raltegravir or efavirenz combined with tenofovir/emtricitabine for treatment-naive human immunodeficiency virus-1-infected patients: 156-week results from STARTMRK. Clin Infect Dis. 2011;53:807–816. [DOI] [PubMed] [Google Scholar]
- 47.Ofotokun I, Na LH, Landovitz RJ, Ribaudo HJ, McComsey GA, Godfrey C, Aweeka F, Cohn SE, Sagar M, Kuritzkes DR, Brown TT, Patterson KB, Para MF, Leavitt RY, Villasis-Keever A, Baugh BP, Lennox JL, Currier JS and Team ACTGA. Comparison of the metabolic effects of ritonavir-boosted darunavir or atazanavir versus raltegravir, and the impact of ritonavir plasma exposure: ACTG 5257. Clin Infect Dis. 2015;60:1842–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.