

Abstract
DAG-lactones afford a synthetically accessible, high affinity platform for probing structure activity relationships at the C1 regulatory domain of protein kinase C (PKC). Given the central role of PKC isoforms in cellular signaling, along with their differential biological activities, a critical objective is the design of isoform selective ligands. Here, we report the synthesis of a series of DAG-lactones varying in their side chains, with a particular focus on linoleic acid derivatives. We evaluated their selectivity for PKC epsilon versus PKC alpha both under standard lipid conditions (100% phosphatidylserine, PS) as well as in the presence of a nuclear membrane mimetic lipid mixture (NML). We find that selectivity for PKC epsilon versus PKC alpha tended to be enhanced in the presence of the nuclear membrane mimetic lipid mixture and, for our lead compound, report a selectivity of 32-fold.
Keywords: Protein kinase C, Diacylglycerol lactone, PKC-ε
1. Introduction
Protein kinase Cs (PKCs) represent a family of serine/threonine kinases which are central signaling elements downstream of the numerous cellular receptors that are coupled to phospholipase C activation [1]. PKCs display complex regulation, with membrane phospholipids and diacylglycerol being critical regulators and elevated Ca2+ additionally being important for the classical PKCs. PKC isoforms divide into three groups; classical PKCs (cPKCs: PKCα, PKCβI, PKCβII and PKCγ), novel PKCs (nPKCs: PKCδ, PKCε, PKCη and PKCθ) and atypical PKCs (aPKCs: PKCξ, PKCι/λ), distinguished by their Ca2+ dependency and cofactors for activation [2,3]. Additionally, individual isozymes vary in their patterns of tissue distribution, subcellular localization, substrate specificity, and biological function [4,5]. For example, different isoforms may be growth promoting or growth inhibitory, with stimulation of one isoform being functionally antagonistic of another [6]. Although the high level of conservation of the regulatory domains of PKC isoforms poses a challenge for the development of isoform selective ligands, the complexity of their regulation also provides promising opportunities [7].
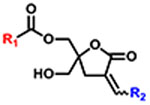
Among the PKC isozymes, particular attention has focused on protein kinase C epsilon (PKC-ε), one of the calcium-independent but phorbol ester/diacylglycerol dependent novel PKC isoforms. Structurally, it contains an N-terminal regulatory domain and C-terminal kinase domain composed on four conserved (C1–C4) and five variable (V1–V5) regions. The regulatory domain contains a pseudosubstrate domain which inhibits PKC activity before activation, a tandem repeat of C1 domain zinc finger structures that bind diacylglycerol (DAG) and phorbol ester, and a C2-like domain which contributes to membrane interaction [8,9]. Additionally, PKC-ε possesses a unique actin-binding site [10] between the first and second cysteine rich C1-domains which promotes association with actin filaments in intact cells. Due to its specific tissue distribution and localization pattern, it has been reported that PKC-ε has critical roles in both the cardiovascular and central nervous system [11] such as cardioprotection [12], neurite outgrowth [13], synapse formation, neurotransmitter release, synaptic loss prevention [14], and sensitization to pain through TRPV1 [15] (see Fig. 1).
Fig. 1.

The structure of DAG-lactone.
As an initial effort to design and develop PKC-ε selective ligands, our attention was drawn to reports that saturated and unsaturated fatty acids may influence PKC-ε activity. Recently, Nishizaki and coworkers described that a linoleic acid derivative with cyclopropane rings replacing the cis-double bonds, viz. 8-[2-(2-pentyl-cyclopropylmethyl)-cyclopropyl]-octanoic acid (DCP-LA, Fig. 2. Compound 5) acted as a selective and direct activator of PKC-ε [16]. In biological studies, DCP-LA effectively prevented both deposition of amyloid plaques and loss of synaptic connections [17].
Fig. 2.

Linoleic acid derivatives: unsaturated, branched, and saturated alkyl chains.
Previously, we have demonstrated that diacylglycerol lactones (DAG-lactones), which are rigidified structures derived from the endogenous PKC ligand DAG, function as DAG analogs to potently bind to the regulatory C1 domain of PKC and cause PKC activation (Fig. 1) [18]. For this binding, both the acyl (R1) and α-alkylidene (R2) positions are critical elements in controlling biological activity and PKC isozyme selectivity [19]. Modeling reveals that there are two different modes of binding, involving interaction between the C1 domain and either the sn-1 carbonyl or the sn-2 carbonyl. The alkyl chain which is not involved in hydrogen bonding to the binding cleft of the C1 domain contributes to the binding energy through its interactions with the C1 domain surface and the lipid bilayer.
In this study, we have evaluated whether the incorporation of linoleic and DCP-LA derivatives into the side chains of DAG-lactones would provide combined structures with enhanced selectivity for PKC-ε. The newly designed linoleic acid derivatives, incorporating cyclo-saturated (2, 5), linear-unsaturated (10, 12), linear-saturated (11), and branched alkyl chains (13–15), were alternatively attached to the carbonyl group for acyl branching (R1) or connected to the lactone via a methylene group for α-alkylidene branching (R2). Novel features of the study include both the specific class of DAG-lactones themselves as well as their analysis, directed at comparing selectivities for the novel PKC-ε isoform relative to that for the classical PKC-α isoform.
2. Result and discussion
2.1. Chemistry
As illustrated in Fig. 2, three branched groups as well as linoleic acid and five different linoleic acid derivatives in which the cis-double bonds were replaced by other groups were introduced as R1 and R2 side chains. Commercially available linoleic acid (1), stearic acid (11), octadec-9-ynoic acid (12), and the branched compounds pivaloyl chloride (13) and isovaleraldehyde (15) were directly used for alkylation. Among the branched-substituents, 3-isobutyl-5-methylhexanoyl chloride (14) [20] and 3-isobutyl-5-methylhexanal (16) [21] were prepared according to previously published methods.
Starting from linoleic acid 1, cyclo-saturated acid derivatives were synthesized. Di-epoxy compound (DEP-LA, 2) was achieved by the conventional mCPBA epoxidation (Scheme 1). Simmons-smith cyclopropanation [22] of methyl (9Z, 12Z)-octadeca-9,12-dienoate (3) followed by hydrolysis of the ester gave the dicyclopropane compound (DCP-LA, 5). The diyne core of compound 10 was assembled by a Cadiot–Chodkiewicz coupling [23] between bromoalkyne 7 and a suitably acid functionalized terminal alkyne 9 (Scheme 2). As illustrated in Scheme 3, the aldehydes 20–22 were generated from the alcohols 17–19, which were derived from the corresponding esters (3, 4) and acid (11), by treatment with the Dess-Martin periodinane.
Scheme 1.

Synthesis of cyclo-saturated alkyl acids. Reagents and conditions: (a) mCPBA, CH2Cl2, 0 °C; (b) SOCl2, CH3OH, r.t.; (c) Et2Zn,CH2I2,CH2Cl2, −5 °C; (d) 1 N LiOH, dioxane, 60 °C.
Scheme 2.

Synthesis of the unsaturated-diyne derivative. Reagents and conditions: (a) NBS, AgNO3, Acetone, r.t.; (b) CrO3, H2SO4, H2O, Acetone, −5 °C; (c) CuCl, 30% n-BuNH2, NH2OHHCl, Et2O, r.t.
Scheme 3.

Synthesis of aldehydes for sn-2 alkylation. Reagents and conditions: (a) LAH, Et2O, reflux; (b) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C; (c) DMP, CH2Cl2, 0 °C.
Depending on the specific substituents, different protecting groups were selected (Table 1). Specially, the TBDPS group was used for the synthesis of DEP-LA (90, 91) due to its sensitivity to the deprotection conditions with BCl3 and CAN.
Table 1:
The synthesized DAG-lactones.
 |
|||||
|---|---|---|---|---|---|
| Geo | R1 | R2 | Protection |
||
| PG1 | PG2 | ||||
| 86 | Z | CH3(CH2)4HC=CHCH2HC=CH(CH2)7 | CH2(i-Pr) | Bn | PMP |
| 87 | E | CH3(CH2)4HC=CHCH2HC=CH(CH2)7 | CH2(i-Pr) | Bn | PMP |
| 88 | Z | CH2(i-Pr) | Bn | PMP | |
| 89 | E | CH2(i-Pr) | Bn | PMP | |
| 90 | Z | CH2(i-Pr) | TBDPS | TBDPS | |
| 91 | E | CH2(i-Pr) | TBDPS | TBDPS | |
| 92 | Z | CH3(CH2)16 | CH2(i-Pr) | PMP | Bn |
| 93 | E | CH3(CH2)16 | CH2(i-Pr) | PMP | Bn |
| 94 | Z | [CH2(i-Pr)]2CHCH2 | CH2(i-Pr) | Bn | PMP |
| 95 | E | [CH2(i-Pr)]2CHCH2 | CH2(i-Pr) | Bn | PMP |
| 96 | Z | CH3(CH2)7C≡C(CH2)7 | CH2(i-Pr) | PMP | Bn |
| 97 | E | CH3(CH2)7C≡C(CH2)7 | CH2(i-Pr) | PMP | Bn |
| 98 | Z | CH3(CH2)4C≡C – C≡C(CH2)7 | CH2(i-Pr) | Bn | PMP |
| 99 | E | CH3(CH2)4C≡C – C≡C(CH2)7 | CH2(i-Pr) | Bn | PMP |
| 100 | Z | (CH3)3C | CH3(CH2)4HC═CHCH2HC═CH(CH2)7 | Bn | PMP |
| 101 | E | (CH3)3C | CH3(CH2)4HC═CHCH2HC═CH(CH2)7 | Bn | PMP |
| 102 | Z | (CH3)3C | Bn | PMP | |
| 103 | E | (CH3)3C | Bn | PMP | |
| 104 | E | (CH3)3C | CH2CH[CH2(i-Pr)]2 | Bn | PMP |
| 105 | Z | (CH3)3C | CH3(CH2)16 | Bn | PMP |
| 106 | E | (CH3)3C | CH3(CH2)16 | Bn | PMP |
The synthesis of DAG-lactone analogues started from the racemic lactone having p-methoxyphenyl (PMP) and benzyl (Bn) (23) or tert-butyldiphenylsilyl (TBDPS) (24) as protecting groups (Scheme 4). Different aldehydes (RCHO, 20–22) were reacted with the DAG-lactone to form aldol intermediates and β-hydroxy-lactones were eliminated by aldol condensation to give the corresponding olefins (25–45). After separation of geometric E/Z-isomers by silica column chromatography, parallel deprotection of 25–43 with BCl3 or ceric ammonium nitrate (CAN) afforded 46–64 and di-deprotections of TBDPS protected compounds 44–45 with TBAF afforded 65–66. The compounds were individually converted to the corresponding DAG-lactones having different R1-alkyl side chains by conventional coupling methods (90–91). Target compounds 86–89, 92–106 were obtained from additional deprotection of 67–85 using BCl3 or CAN.
Scheme 4.

General scheme for alkylation. Reagents and conditions: (a) i) LiHMDS, THF, R2CHO, −78 °C. ii) MsCl, CH2Cl2, DBU; (b) BCl3, CH2Cl2, −78 °C or CAN, CH3CN/H2O; (c) TBAF, THF; (d) EDC, DMAP, CH2Cl2, r.t.; (e) TEA, DMAP, CH2Cl2, r.t.; (f) BCl3, CH2Cl2, −78 °C or CAN, CH3CN/H2O.
2.2. Biological activity
Diacylglycerol lactones 86–106 were prepared and investigated. For the biological studies, we selected the classical PKC isoform PKC-α as the standard for comparison, since it has been used as the standard PKC isoform against which all the DAG-lactones have routinely been characterized in our previous studies. The interaction of the target DAG-lactones with PKC-α and PKC-ε was assessed, as before, by measurement of the ability of the ligands to compete for binding of [20-3H] phorbol 12,13-dibutylate (PDBU) to recombinant PKC [24]. The IC50 values were determined by least squares fit of the data points to the theoretical competition curve, and the Ki values for inhibition of binding were calculated from the corresponding IC50 values (Tables 2 and 3).
Table 2:
Binding affinities for PKCα and PKCε in 100% phosphatidylserine (PS).
| Geo | logP |
Ki (nM)a |
Ratio α/ε | Geo | logP |
Ki (nM)a |
Ratio α/ε | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PKCα | PKCε | PKCα | PKCε | ||||||||
| PDBU Kd (nM) | 0.28 | 0.22 | 1.3 | 96 | Z | 8.01 | 12.6 | 7.12 | 1.8 | ||
| 86 | Z | 8.29 | 7.05 | 12.2 | 0.6 | 97 | E | 8.01 | 8.7 | 4.79 | 1.8 |
| 87 | E | 8.29 | 4.33 | 2.14 | 2.0 | 98 | Z | 7.82 | 9.8 | 13.4 | 0.7 |
| 88 | Z | 9.19 | 15.4 | 9.80 | 1.6 | 99 | E | 7.82 | 8.31 | 5.24 | 1.6 |
| 89 | E | 9.19 | 10.20 | 5.5 | 1.9 | 100 | Z | 8.25 | 7.20 | 5.06 | 1.4 |
| 90 | Z | 5.69 | 16.5 | 16.3 | 1.0 | 101 | E | 8.25 | 8.26 | 4.1 | 2.0 |
| 91 | E | 5.69 | 13.2 | 7.46 | 1.8 | 102 | Z | 9.15 | 33.1 | 15.2 | 2.2 |
| 92 | Z | 8.72 | 43.9 | 19.3 | 2.3 | 103 | E | 9.15 | 26.7 | 8.30 | 3.2 |
| 93 | E | 8.72 | 30.8 | 11.4 | 2.7 | 104 | E | 5.03 | 8.3 | 1.03 | 8.1 |
| 94 | Z | 5.06 | 4.74 | 2.37 | 2.0 | 105 | Z | 8.68 | 31.6 | 11.5 | 2.8 |
| 95 | E | 5.06 | 2.78 | 1.21 | 2.3 | 106 | E | 8.68 | 24.5 | 5.9 | 4.2 |
Values represent the mean of at least three independent experiments.
Table 3:
Binding affinities for PKCα and PKCε at nuclear membrane.
| Geo | logP |
Ki (nM)a |
Ratio α/ε | Geo | logP |
Ki (nM)a |
Ratio α/ε | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PKCα | PKCε | PKCα | PKCε | ||||||||
| PDBU Kd (nM) | 1.44 | 0.59 | 2.44 | 96 | Z | 8.01 | 38.7 | 34.6 | 1.1 | ||
| 86 | Z | 8.29 | 26.2 | 19.5 | 1.3 | 97 | E | 8.01 | 43.8 | 9.1 | 4.8 |
| 87 | E | 8.29 | 24.7 | 13.1 | 1.9 | 98 | Z | 7.82 | 44.8 | 40.3 | 1.1 |
| 88 | Z | 9.19 | 44 | 30.7 | 1.4 | 99 | E | 7.82 | 60.8 | 12.8 | 4.8 |
| 89 | E | 9.19 | 60 | 19.84 | 3.0 | 100 | Z | 8.25 | 31.1 | 9.6 | 3.2 |
| 90 | Z | 5.69 | 103 | 63 | 1.6 | 101 | E | 8.25 | 25.7 | 5.2 | 4.9 |
| 91 | E | 5.69 | 116 | 16.9 | 6.9 | 102 | Z | 9.15 | 73 | 58.8 | 1.2 |
| 92 | Z | 8.72 | 106 | 34 | 3.1 | 103 | E | 9.15 | 110.3 | 41 | 2.7 |
| 93 | E | 8.72 | 99 | 47 | 3.1 | 104 | E | 5.03 | 46 | 1.43 | 32.2 |
| 94 | Z | 5.06 | 57 | 15.6 | 3.6 | 105 | Z | 8.68 | 84.1 | 45.1 | 1.9 |
| 95 | E | 5.06 | 15.4 | 3.0 | 5.1 | 106 | E | 8.68 | 117 | 34.2 | 3.4 |
Values represent the mean of at least three independent experiments.
Our initial objective was to examine the possible selectivity of the DAG-lactones for PKC-ε under standard lipid conditions, viz. 100 μg/ml phosphatidylserine. We determined the binding affinities for PKC-α and PKC-ε and we calculated the Ki ratios for PKC-α/PKC-ε of all compounds (Tables 2–4). Evaluation of the elements contributing to the relative structure activity relations focused on geometry (E or Z), position of substitution (sn-1 or sn-2), and lipophilicities of the compounds.
Table 4:
Affinity ratios of PKCα to PKCε at plasma membrane and nuclear membrane.
| Z-isomer |
Ki ratio PKCα/PKCε |
E-isomer |
Ki ratio PKCα/PKCε |
||||
|---|---|---|---|---|---|---|---|
| PS | NML | PS | NML | ||||
| 86 | sn-1 | 0.6 | 1.3 | 87 | sn-1 | 2.0 | 1.9 |
| 88 | sn-1 | 1.6 | 1.4 | 89 | sn-1 | 1.9 | 3.0 |
| 100 | sn-2 | 1.4 | 3.2 | 101 | sn-2 | 2.0 | 4.9 |
| 102 | sn-2 | 2.2 | 1.2 | 103 | sn-2 | 3.2 | 2.7 |
| 104 | sn-2 | 8.1 | 32.2 | ||||
| 90 | sn-1 | 1.0 | 1.6 | 91 | sn-1 | 1.8 | 6.9 |
| 92 | sn-1 | 2.3 | 3.1 | 93 | sn-1 | 2.7 | 2.1 |
| 94 | sn-1 | 2.0 | 3.6 | 95 | sn-1 | 2.3 | 5.1 |
| 96 | sn-1 | 1.8 | 1.1 | 97 | sn-1 | 1.8 | 4.8 |
| 98 | sn-1 | 0.7 | 1.1 | 99 | sn-1 | 1.6 | 4.8 |
| 105 | sn-2 | 2.8 | 1.9 | 106 | sn-2 | 4.2 | 3.4 |
The binding affinities of ligands for different PKC isozymes may show different dependence on the lipid composition of the membranes with which the PKC is associated. We therefore evaluated the ligand selectivity not only under our standard assay conditions, 100 μg/ml of phosphatidylserine, which provides a highly anionic surface, but also using 100 mg/ml of a lipid mixture [1-palmutoyl-2-oleoyl-sn-glycero-3-phosphocholine(POPC)/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine(POPE)/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS)/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoinositol(POPI)/Cholesterol (61:21:4:7:6)], a much less anionic lipid composition approximating that of the nuclear membrane.
Table 2 displays binding affinities of DAG-lactones under standard (100% phosphatidylserine) conditions. Most of the compounds showed modestly stronger binding affinity for PKC-ε than for PKC-α. The compounds with the strongest PKC-ε binding affinities were characterized as having an E-geometry with a sn-1 carbonyl substituted with a branched alkyl chain or an unsaturated alkyl chain (Table 2, compounds 95 and 104). The best, albeit limited PKC-ε selectivity was seen for the sn-2 carbonyl substituted with a saturated alkyl chain (cyclo-103, branched 104, linear 105 and 106).
The PKC-ε selectivity of the compounds was further investigated for the nuclear membrane mimetic assay system (Table 3). The changes resulting from lipid composition were substantial, particularly for compound 104. The difference in lipid composition led to a reduction in overall binding affinity and caused a different selectivity pattern to emerge. Compounds 91, 97, 99, and 104 showed over a 3-fold enhanced selectivity (Table 3). In particular, compound 104 retained its affinity to PKC-ε and showed the highest selectivity (32-fold) under the nuclear membrane mimetic assay conditions (Table 3). The E-isomers having an sn-1 carbonyl substituted with an unsaturated alkyl chain showed good binding affinities and selectivity in this system. A partial correlation with lipophilicity was observed among the compounds having over 5-fold selectivity.
Another important observation was the clear disparities in selectivity shown by the two geometrical isomers (Table 4), with the E-isomer being favored particularly in the nuclear membrane mimetic assay conditions.
3. Conclusion
In conclusion, different types of alkyl chains derived from linoleic acid were introduced as substituents for the DAG-lactone and their effects on selectivity for PKC-ε relative to PKC-α was explored. Because cells contain a variety of membranes with different compositions, we also assessed the influence of membrane lipid contribution on the measured isozyme selectivity. Most of the compounds showed at least modest PKC-ε selectivity with a number approaching 5-fold in the presence of the nuclear lipid mixture. The most dramatic example of selectivity, 32-fold, was not with any of the linoleic derivative substituted DAG-lactones, however, suggesting that the incorporation of the linoleic acid motif into the DAG-lactone did not confer the hoped for selectivity.
Structure activity relationships were assessed for selected compounds showing the greater extents of selectivity for PKC-ε. When 100% phosphatidylserine was used as the lipid, the E-conformation was preferred for compounds in which the sn-2 carbonyl was substituted with a saturated alkyl chain (branched > linear > cyclo). In contrast, in the presence of the nuclear membrane mimetic lipids, sn-1 substituted compounds with an E-conformation showed PKC-ε selectivity regardless of whether the alkyl chains were saturated or unsaturated.
The most selective compound under both lipid conditions was (E)-(2-(hydroxymethyl)-4-(3-isobutyl-5-methylhexylidene)-5-oxotetrahydrofuran-2-yl) methyl pivalate 104, which has the sn-2 carbonyl substituted with a saturated-branched alkyl chain with an E-conformation. Compound 104 showed a dramatic increase of selectivity (8-fold in the presence of phosphatidylserine, 32-fold in the presence of the nuclear membrane mimetic lipid) in the nuclear membrane conditions, reflecting that its binding affinity for PKC-α was significantly weaker under those conditions while its affinity for PKC-ε was largely retained. In contrast, the other compounds showed reduced binding affinities both for PKC-α and PKC-ε. This result suggests that the lipid composition of the nuclear membrane enhanced the binding affinity to PKC-ε for 104, reflecting enhanced interactions between the DAG-lactone side chain and the protein–membrane interface. The diverse affinity patterns of compounds from this work provide guidance for future approaches. In particular, the high selectivity of 104 suggests that further exploration of this class of DAG-lactones may be productive for obtaining PKC-ε specific compounds and understanding of factors driving subcellular localization of PKC isoforms. Naturally, validation of the selectivity of the optimal PKC-ε selective ligand will ultimately require characterization relative to the complete range of targets for DAG and phorbol esters, e.g. PKC family members, RasGRP, the chimaerins, and include measures of activity in intact cells.
4. Experimental
4.1. Chemistry
All chemical reagents were commercially available. Silica gel column chromatography was performed on silica gel 60, 230–400 mesh, Merck. Nuclear magnetic resonance (1H NMR) spectra were recorded on a JEOL JNM-LA 300 and a Bruker Avance 400 MHz FT-NMR spectrometer. Chemical shifts are reported in ppm units with Me4Si as a reference standard. Mass spectra were recorded on a VG Trio-2 GC-MS and a 6460 Triple Quad LC/MS. Elemental analyses were performed with an EA 1110 Automatic Elemental Analyzer, CE Instruments.
4.1.1. Preparation of alkyl side chains
4.1.1.1. 8-(3-((3-Pentyloxiran-2-yl)methyl)oxiran-2-yl)octanoic acid (2).
A solution of linoleic acid 1 (500 mg, 1.783 mmol) and meta-chloroperoxybenzoic acid (1.8 g, 10.698 mmol) in 10 ml of CH2Cl2 was reacted at 0 °C for 3 h. The excess reagent was decomposed by addition of aqueous Na2CO3 and stirring of the mixture for 1 h, after which the product was isolated by CH2Cl2 extraction. The organic layer was washed with saturated aqueous sodium bicarbonate and saturated brine, dried with MgSO4, and evaporated. The crude product was purified by silica gel flash column chromatography (n-hexane/EtOAc, 4:1) and 2 (469 mg, 82%) was obtained as a white solid.
4.1.1.2. Methyl (9Z,12Z)-octadeca-9,12-dienoate (3).
Thionyl chloride (0.5 ml, 6.239 mmol) was added dropwise to commercially available linoleic acid 1 (700 mg, 2.495 mmol) dissolved in MeOH at 0 °C. The reaction was stirred at room temperature and monitored by TLC. Upon completion, H2O was added and the reaction mixture was concentrated in vacuo. The resulting solution was extracted with ethyl acetate, dried over MgSO4, and concentrated in vacuo. The crude product was purified by silica gel flash column chromatography (n-hexane/EtOAc, 10:1) and 3 (712 mg, 96%) was obtained as a brown oil.
4.1.1.3. Methyl 8-(2-((2-pentylcyclopropyl)methyl)cyclopropyl)octanoate (5).
To a solution of linoleic acid methyl ester 3 (1 g, 3.395 mmol) in CH2Cl2, a 1.1 M n-hexane solution of diethyl zinc (7 ml, 40.751 mmol) was added under a N2 atmosphere with cooling with an ice-water bath (−5 °C to 0 °C) and was stirred for 1 h. Diiodomethane (7 ml, 81.502 equiv.) was then added and the mixture was stirred at ambient temperature overnight. After removal of solvent by evaporation, the product was extracted by ethyl acetate (EA) and purified by chromatography on silica gel to give methyl 8-(2-((2-pentylcyclopropyl)methyl)cyclopropyl)octanoate 4 (969 mg, 88%) as an oil. A mixture of this ester 4, 1 N LiOH (6.6 ml), and dioxane was stirred at 60 °C overnight. The product was purified by acid-base workup with 1 N HCl and 1 N NaOH and 5 (740 mg, 80%) was obtained as an oil.
4.1.1.4. Octadeca-9,11-diynoic acid (10).
Copper (I) chloride (8.8 mg, 0.089 mmol) was added to a 30% w/v n-BuNH2 aqueous solution (10 ml) at room temperature, which resulted in the formation of a blue solution. A few crystals of hydroxylamine hydrochloride was added to discharge the blue color. Dec-9-ynoic acid 9 (300 mg, 1.78 mmol) was then added neat, and the mixture was stirred at 0 °C for 10 min by which time the formation of a bright yellow precipitate (probably the alkynyl cuprate) was observed. Bromoalkyne 6 (404 mg, 2.14 mmol) dissolved in diethyl ether was then added to the cooled reaction mixture and the cooling bath was removed after 10–15 min. Occasional addition of a small amount of hydroxylamine hydrochloride was necessary to keep the color of the reaction light yellow. The reaction mixture was stirred at room temperature for 2 h during which more crystals of hydroxylamine hydrochloride were added whenever the reaction mixture started to turn blue or light green. The reaction was then quenched and adjusted to pH ~2 by adding 1 N HCl and the organic compounds were extracted with ethyl acetate 3 times. The combined layer was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel flash column chromatography (n-hexane/EtOAc, 4:1) and 10 (423 mg, 86%) was obtained as a yellow oil.
4.1.1.4.1. 1-Bromohept-1-yne (7).
The terminal alkyne 6 (1 ml, 6.350 mmol) was dissolved in acetone. N-bromosuccinimide (1.4 g, 7.866 mmol) and AgNO3 (108 mg, 0.635 mmol) were added to the resulting solution in this order, and the mixture was stirred at room temperature for 3 h, until complete consumption of the starting material according to TLC. It was then poured into iced water. The aqueous layer was extracted with pentane (3 times), and the combined organic extracts were dried over MgSO4, filtered, and the solvent removed by evaporation under reduced pressure. The residue was purified by silica gel flash column chromatography (n-hexane/EtOAc, 10:1) to yield bromo-alkyne 7 (1.07 g, 89%) was obtained as a colorless oil.
4.1.1.4.2. Dec-9-ynoic acid (9).
To a solution of 9-decyn-1-ol 8 (5 ml, 28.20 mmol) in acetone at −5 °C was slowly added chromic acid solution prepared from CrO3 (4.23 g, 42.30 mmol), water (4 ml), and conc. H2SO4 (3 ml) at 0 ° C. The mixture was then stirred for 2 h and left overnight at room temperature, after which it was extracted with diethyl ether, washed with water and brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel flash column chromatography (n-hexane/EtOAc, 7:1) to yield dec-9-ynoic acid 9 (3.8 g, 81%) as clear oil.
4.1.2. Procedure for aldehyde preparation
4.1.2.1. Alcohol preparation (17–19).
Starting material (2, 5, 8) was dissolved in ether. Lithium aluminum hydride (2 equiv.) was added at 0 °C and it was refluxed during 30 min H2O was added dropwise at 0 °C; the solution was extracted with ether and the combined extracts were dried over MgSO4 and concentrated in vacuo. Finally, purification by silica gel flash column chromatography (n-hexane/EtOAc, 10:1) by column chromatography yielded the alcohols (17–19).
4.1.2.2. Aldehyde preparation (20–22)
4.1.2.2.1. Method A.
A solution of oxalyl chloride (1.5 equiv.) in distilled CH2Cl2 was cooled to −78 °C, and DMSO (3 equiv.) was carefully added under a N2 atmosphere. After stirring for 15 min, a solution of alcohol (17, 18) in CH2Cl2 was added followed by Et3N (6 equiv.). The cooling bath was removed, and the reaction mixture was allowed to warm to room temperature and stirred for 2.5 h. The solvent was removed under reduced pressure, and the residue was extracted with ethyl acetate. The extract was washed with saturated aqueous Na2CO3 solution and brine, and was then dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, the crude product was purified by silica gel flash column chromatography (n-hexane/EtOAc, 10:1) and the aldehyde (20, 21) was obtained as 75–80%.
4.1.2.2.2. Method B.
To a solution of octadecan-1-ol 19 (560 mg, 1 equiv.) in CH2Cl2 was added Dess-Martin periodinane (1.3 g, 1.5 equiv.) at room temperature and the resulting solution was stirred for 2 h at room temperature. The reaction mixture was quenched with a saturated solution of Na2SO3 and NaHCO3. The reaction mixture was passed through a pad of Celite, the organic compound was extracted three times with CH2Cl2. The combined extracts were washed with water, dried over anhydrous MgSO4, and filtrated. The filtrate was concentrated under vacuum, and purified by silica gel flash column chromatography (n-hexane/EtOAc, 10:1) by column chromatography to afford the stearaldehyde 22 (456 mg, 82%).
4.1.3. General procedure for alkylation
A stirred solution of lactone 23 (or 24) (1 equiv.) in anhydrous THF (5 ml/mmol) was cooled to −78 °C under nitrogen and treated dropwise with LiHMDS (3 equiv., 2 M solution in THF). The mixture was stirred at −78 °C for 30–60 min and lithium enolate formation was detected by TLC (n-hexane/EtOAc, 4:1). A solution of the corresponding aldehyde (1.4–4 equiv.) in anhydrous THF (1 ml/mmol) was added dropwise to the enolate at the same temperature and stirring at −78 °C was continued for 1.5–2 h. The reaction was warmed to room temperature and quenched by the slow addition of a saturated aqueous solution of ammonium chloride. The aqueous layer was extracted three times with Et2O, and the combined organic extracts were washed three times with water, twice with brine, dried over MgSO4, and filtered. The filtrate was concentrated under vacuum to afford the crude alkylation reaction product, which was partially purified by silica gel flash column chromatography after eluting with the appropriate solvent. The obtained mixture of β-hydroxy-lactone diastereomers were typically used directly in the following step without further purification.
4.1.4. General procedure for mesylation–olefination
A solution of the alkylation product in CH2Cl2 at 0 °C was treated with methanesulfonyl chloride (2 equiv.) and triethylamine (4 equiv.). The mixture was stirred at 0 °C for 30 min and then for 2 h at room temperature. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 5 equiv.) was added at 0 °C, and the resulting solution was stirred overnight at ambient temperature. The reaction mixture was concentrated to a brown syrup and the residue was extracted with EtOAc followed by 1 N HCl. The combined organic extracts were washed three times with water, twice with brine, dried over MgSO4, and filtered. The combined organics were purified by silica gel flash column chromatography after eluting with the appropriate solvent.
4.1.5. General procedure for deprotection
4.1.5.1. PMP-deprotection.
A stirred solution of the olefination product (1 equiv.) in acetonitrile/water (4/1) was treated with CAN (3 equiv.) while under nitrogen at 0 °C, and the reaction was monitored by TLC. After 30 min, the reaction was quenched by addition of aqueous NaHCO3 solution and the resulting solution was extracted with CH2Cl2, dried over MgSO4, and concentrated in vacuo. It was purified by silica gel flash column chromatography (n-hexane/EtOAc, 2:1) and the deprotected compound was obtained.
4.1.5.2. Benzyl deprotection.
BCl3 (3 equiv.) was added slowly to a stirred solution of the olefination product (1 equiv.) in CH2Cl2 at −78 °C. The reaction was monitored by TLC and quenched by addition of MeOH at 0 °C. The reaction mixture was concentration in vacuo and purification by silica gel flash column chromatography (n-hexane/EtOAc, 2:1) gave the desired deprotected compounds.
4.1.5.3. TBDPS deprotection.
TBAF (2 equiv.) was added slowly to a stirred solution of the olefination product (1 equiv.) in THF at 0 °C and stirring continued as the temperature was allowed to rise to room temperature. The reaction was monitored by TLC and the solution was concentrated in vacuo upon completion without further work-up. Purification by silica gel flash column chromatography (n-hexane/EtOAc, 1:1) gave the desired de-protected diol-lactone.
4.1.6. General procedure for acylation
A solution of mono-deprotected compound (1 equiv.) in CH2Cl2 was treated with Et3N (3 equiv.) and dimethylaminopyridine (DMAP, 2.5 equiv.) and reacted with the corresponding acid chloride (RCOCl, 1.2–1.5 equiv.). The reaction was stirred at room temperature and monitored by TLC. Upon completion, the reaction was concentrated in vacuo and purified by silica gel flash column chromatography give the desired compound.
4.1.6.1. Monoacylation (di-epoxy compounds, DEP-LA).
A solution of 65 (or 66) (1 equiv.) in CH2Cl2 was treated with EDC (1.1 equiv.) and dimethylaminopyridine (DMAP, 0.3 equiv.) and reacted with 8-(3-((3-pentyloxiran-2-yl)methyl)oxiran-2-yl)octanoic acid (1.5 equiv.). The reaction was stirred at room temperature and monitored by TLC. Upon completion, the reaction was terminated by adding H2O. The organic layer was extracted with CH2Cl2, dried over MgSO4, concentrated in vacuo and purified by silica gel flash column chromatography (n-hexane/EtOAc, 4:1) to give the mono-substituted desired compound 90–91.
4.1.7. ((Z)-2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl-(9Z,12Z)-octadeca-9,12-dienoate (86)
Yield 63%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.24 (t, J = 7.76 Hz, 1H, >C═CHCH2CH(CH3)2), 5.39–5.30 (m, 4H, –CH═CHCH2CH═CH–), 4.26 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 4.14 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 3.58–3.69 (dq, J = 12.00, 7.20, 6.56 Hz, 2H, HOCH2–), 2.88 (AB d, J = 16.44 Hz, 1H, H-3a), 2.75 (t, J = 6.24 Hz, 2H, –CH═CHCH2CH═CH–), 2.72 (AB d, J = 16.44 Hz, 1H, H-3b), 2.60 (t, J = 7.44 Hz, 2H, >C═CHCH2CH(CH3)2), 2.31 (t, J = 7.56 Hz, 2H, Alkyl-CH2(O)COCH2), 2.04 (q, J = 6.88 Hz, 4H, –CH2CH═CHCH2CH═CHCH2–), 1.96 (m, 1H, HOCH2–), 1.71 (septet, J = 6.74 Hz, 1H, >C═CHCH2CH(CH3)2), 1.59 (m, 2H, Alkyl-CH2CH2(O)COCH2), 1.28 (m, 14H, CH3(CH2)3CH2CH═CHCH2CH═CHCH2(CH2)4CH2–), 0.92 (d, J = 6.68 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3(CH2)3CH2CH═CH–); MS (FAB) m/z 477 (M+H).
4.1.8. ((E)-2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl (9Z, 12Z)-octadeca-9,12-dienoate (87)
Yield 76%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.78 (t, J = 7.76 Hz, 1H, >C═CHCH2CH(CH3)2), 5.39–5.30 (m, 4H, –CH═CHCH2CH═CH–), 4.26 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 4.14 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 3.58–3.69 (dq, J = 12.00, 7.20, 6.56 Hz, 2H, HOCH2–), 2.73–2.81 (m, 3H, H-3a, –CH=CHCH2CH═CH–), 2.62 (AB d, J = 17.04 Hz, 1H, H-3b), 2.31 (t, J = 7.56 Hz, 2H, Alkyl-CH2(O)COCH2), 2.04 (q, J = 6.88 Hz, 4H, –CH2CH═CHCH2CH═CHCH2–), 1.96 (m, 1H, HOCH2–), 1.71 (septet, J = 6.74 Hz, 1H, >C═CHCH2CH(CH3)2), 1.59 (m, 2H, Alkyl-CH2CH2(O)COCH2), 1.28 (m, 14H, CH3(CH2)3CH2CH═CHCH2CH–CHCH2(CH2)4CH2–), 0.92 (d, J = 6.68 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3(CH2)3CH2CH═CH–)–); MS (FAB) m/z 477 (M+H).
4.1.9. (Z)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl 8-(2-((2-pentylcyclopropyl)methyl)cyclopropyl)octanoate (88)
Yield 71%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.24 (t, J = 7.76 Hz, 1H, >C═CHCH2CH(CH3)2), 4.26 (AB d, J = 11.8 Hz, 1H, Alkyl-(O) COCHH), 4.14 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 3.58–3.69 (dq, J = 12.1, 7.20, 6.56 Hz, 2H, HOCH2), 2.88 (AB d, J = 16.52 Hz, 1H, H-3a), 2.69 (AB q, J = 16.44 Hz, 1H, H-3b), 2.60 (t, J = 7.44 Hz, 2H, >C═CHCH2CH(CH3)2), 2.31 (t, J = 7.60 Hz, 2H, Alkyl-CH2(O)COCH2), 2.04 (t, J = 4.00 Hz, 1H, HOCH2), 1.69 (septet, J = 6.72 Hz, 1H, >C═CHCH2CH(CH3)2), 1.59 (m, 2H, Alkyl-CH2CH2(O)COCH2), 1.37 (bs, 6H, Alkyl), 1.28 (bs, 12H, Alkyl), 1.10 (m, 2H, Alkyl), 0.92 (d, 6H, J = 6.60 Hz, >C═CHCH2CH(CH3)2), 0.87 (t, J = 6.44 Hz, 3H, CH3-Alkyl-(O)COCH2), 0.75 (m, 2H, –CHCH2CH–), 0.66 (m, 2H, –CHCH2CH–), 0.57 (m, 2H, –CHCH2CH–), −0.30 (m, 2H, –CHCH2CH–); MS (FAB) m/z 505 (M+H).
4.1.10. (E)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl 8-(2-((2-pentylcyclopropyl) methyl)cyclopropyl)octanoate (89)
Yield 73%, Oil; 1H NMR(CDCl3, 400 MHz) δ 6.78 (t, J = 7.76 Hz, 1H, >C═CHCH2CH(CH3)2), 4.26 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 4.14 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 3.58–3.69 (dq, J = 12.1, 7.20, 6.56 Hz, 2H, HOCH2), 2.88 (AB d, J = 16.52 Hz, 1H, H-3a), 2.69 (AB q, J = 16.44 Hz, 1H, H-3b), 2.31 (t, J = 7.60 Hz, 2H, Alkyl-CH2(O)COCH2), 2.04 (m, 3H, HOCH2, >C═CHCH2CH(CH3)2)), 1.69 (septet, J = 6.72 Hz,1H, >C═CHCH2CH(CH3)2), 1.59 (m, 2H, Alkyl-CH2CH2(O) COCH2), 1.37 (bs, 6H, Alkyl), 1.28 (bs, 12H, Alkyl), 1.10 (m, 2H, Alkyl), 0.92 (d, 6H, J = 6.60 Hz, >C═CHCH2CH(CH3)2), 0.87 (t, J = 6.44 Hz, 3H, CH3-Alkyl-(O)COCH2), 0.75 (m, 2H, –CHCH2CH–), 0.66 (m, 2H, –CHCH2CH–), 0.57 (m, 2H, –CHCH2CH–), −0.30 (m, 2H, –CHCH2CH–); MS (FAB) m/z 505 (M+H).
4.1.11. (Z)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl 8-(3-((3-pentyloxiran-2-yl)methyl)oxiran-2-yl)octanoate (90)
Yield 35%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.24 (t, J = 6.24 Hz, 1H, >C═CHCH2CH(CH3)2), 4.26 (AB d, J = 9.56 Hz, 1H, Alkyl-(O)COCHH), 4.14 (AB d, J = 9.56 Hz, 1H, Alkyl-(O)COCHH), 3.59–3.68 (m, 2H, HOCH2–), 3.09 (bs, 2H, –CH(O)CH-), 2.95 (bs, 2H, –CH(O)CH–), 2.78 (AB d, J = 13.00 Hz, 1H, H-3a), 2.68 (AB d, J = 13.00 Hz, 1H, H-3b), 2.60 (t, J = 5.72 Hz, 2H, >C═CHCH2CH(CH3)2), 2.31 (t, J = 5.96 Hz, 2H, Alkyl-CH2(O)COCH2–), 1.81 (m, 1H, >C═CHCH2CH(CH3)2), 1.70 (m, 3H, –CH(O)CHCH2CH(O)CH–), 1.31–1.40 (m, 20H, Alkyl), 0.92 (d, J = 5.28 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3-Alkyl-); MS (FAB) m/z 509 (M+H).
4.1.12. (E)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl 8-(3-((3-pentyloxiran-2-yl)methyl)oxiran-2-yl)octanoate (91)
Yield 32%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.24 (m, 1H, >C═CHCH2CH(CH3)2), 4.26 (AB d, J = 9.52 Hz, 1H, Alkyl-(O)COCHH), 4.14 (AB d, J = 9.52 Hz, 1H, Alkyl-(O)COCHH), 3.60–3.71 (dq, J = 9.66, 5.64, 5.24 Hz, 2H, HOCH2–), 3.09 (bs, 2H, –CH(O)CH–), 2.95 (bs, 2H, –CH(O)CH–), 2.78 (AB d, J = 14.60 Hz, 1H, H-3a), 2.62 (AB d, J = 14.60 Hz, 1H, H-3b), 2.31 (t, J = 6.00 Hz, 2H, Alkyl-CH2(O)COCH2–), 2.11 (m, 1H, HOCH2–), 2.06 (m, 2H, >C═CHCH2CH(CH3)2), 1.81 (m, 1H, >C═CHCH2CH(CH3)2), 1.70 (m, 3H, –CH(O)CHCH2CH(O)CH–), 1.44–1.49 (m, 8H, Alkyl), 1.22–1.32 (m, 12H, Alkyl), 0.92 (d, J = 5.28 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3-Alkyl-); MS (FAB) m/z 509 (M+H).
4.1.13. (Z)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl stearate (92)
Yield 60%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.24 (t, J = 7.72 Hz, 1H, >C═CHCH2CH(CH3)2), 4.26 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 4.14 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 3.64 (m, 2H, HOCH2–), 2.90 (AB d, J = 16.34 Hz, 1H, H-3a), 2.69 (AB d, J = 16.34 Hz, 1H, H-3b), 2.60 (t, J = 7.48 Hz, 2H, >C═CHCH2CH(CH3)2), 2.30 (t, J = 7.44 Hz, 2H, Alkyl-CH2(O)COCH2–), 2.04 (m, 1H, HOCH2–), 1.71 (m, 1H, >C═CHCH2CH(CH3)2), 1.58 (m, 2H), 1.23 (m, 28H, Alkyl), 0.92 (d, J = 6.64 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3-Alkyl-); MS (FAB) m/z 481 (M+H).
4.1.14. (E)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl stearate (93)
Yield 60%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.78 (m, 1H, >C–CHCH2CH(CH3)2), 4.26 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 4.15 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 3.66 (q, J = 11.04 Hz, 2H, HOCH2–), 2.79 (AB d, J = 16.00 Hz, 1H, H-3a), 2.63 (AB d, J = 16.00 Hz, 1H, H-3b), 2.30 (t, J = 7.64 Hz, 2H, Alkyl-CH2(O)COCH2–), 2.04 (m, 3H, HOCH2–, >C═CHCH2CH(CH3)2)), 1.80 (septet, J = 6.74 Hz, 1H, >C═CHCH2CH(CH3)2), 1.23 (m, 32H, Alkyl), 0.92 (d, J = 6.64 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3-Alkyl-); MS (FAB) m/z 481 (M+H).
4.1.15. (Z)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl 3-isobutyl-5-methylhexanoate (94)
Yield 59%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.26 (tt, J = 7.28, 2.3 Hz, 1H, >C═CHCH2CH(CH3)2), 4.20 (AB q, J = 11.9 Hz, 2H, Alkyl-(O)COCH2), 3.66 (AB q, J = 12.2 Hz, 2H, HOCH2–), 2.90 (AB d, J = 16.4 Hz, 1H, H-3a), 2.72 (AB d, J = 16.4 Hz, 1H, H-3b), 2.62 (m, 2H, >C═CHCH2CH(CH3)2), 2.24 (m, 2H, ((CH3)2CHCH2)2CHCH2(O)COCH2–), 1.80 (m, 1H, >C═CHCH2CH(CH3)2), 1.68–1.78 (m, 1H, ((CH3)2CHCH2)2CHCH2(O)COCH2–), 1.58–1.62 (m, 2H, ((CH3)2CHCH2)2CHCH2(O)COCH2–), 1.3 (m, 4H, ((CH3)2CHCH2)2CHCH2(O)COCH2–), 0.96 (d, J = 6.70 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (m, 12H, ((CH3)2CHCH2)2CHCH2(O)COCH2–); MS (FAb) m/z 383 (M+H).
4.1.16. (E)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl 3-isobutyl-5-methylhexanoate (95)
Yield 62%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.80 (tt, J = 7.70, 2.3 Hz, 1H, >C═CHCH2CH(CH3)2), 4.31 (AB d, J = 3.12 Hz, 1H, Alkyl-(O)COCHH), 4.17 (AB d, J = 2.76 Hz, 1H, Alkyl-(O)COCHH), 3.68 (m, 2H, HOCH2-), 2.83 (AB d, J = 17.01 Hz, 1H, H-3a), 2.65 (AB d, J = 17.01 Hz, 1H, H-3b), 2.22–2.24 (m, 2H, ((CH3)2CHCH2)2CHCH2(O)COCH2–), 2.04 (m, 3H, HOCH2–, >C═CHCH2CH(CH3)2), 1.82 (m, 1H, >C═CHCH2CH(CH3)2), 1.61 (m, 1H, ((CH3)2CHCH2)2CHCH2(O)COCH2–), 1.58 (m, 2H, ((CH3)2CHCH2)2CHCH2(O)COCH2–), 1.03 (m, 4H, ((CH3)2CHCH2)2CHCH2(O)COCH2–), 0.96 (d, J = 6.70 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (m, 12H, ((CH3)2CHCH2)2CHCH2(O) COCH2–); MS (FAB) m/z 383 (M+H).
4.1.17. (Z)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl octadec-9-ynoate (96)
Yield 80%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.24 (m, 1H, >C═CHCH2CH(CH3)2), 4.26 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 4.14 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 3.61 (q, J = 12.2 Hz, 2H, HOCH2–), 2.88 (AB d, J = 17.32, 2.2 Hz, 1H, H-3a), 2.63 (AB d, J = 17.32, 2.2 Hz, 1H, H-3b), 2.60 (tt, J = 7.24, 2.0 Hz, 2H, >C═CHCH2CH(CH3)2), 2.30 (t, J = 7.52 Hz, 2H, Alkyl-CH2(O)COCH2–), 2.12 (t, J = 6.24 Hz, 4H, –CH2C≡CCH2–), 1.71 (septet, J = 6.72 Hz, 1H, >C═CHCH2CH(CH3)2), 1.59 (m, 7H), 1.44 (m, 5H), 1.25 (m, 11H), 0.92 (d, J = 6.68 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3-Alkyl); MS (FAB) m/z 477 (M+H).
4.1.18. (E)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl octadec-9-ynoate (97)
Yield 77%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.74 (m, 1H, >C–CHCH2CH(CH3)2), 4.26 (AB d, J = 11.88 Hz, 1H, Alkyl-(O)COCHH), 4.17 (AB d, J = 11.88 Hz, 1H, Alkyl-(O)COCHH), 3.66 (m, 2H, HOCH2–), 2.81 (AB d, J = 17.19 Hz, 1H, H-3a), 2.64 (AB d, J = 17.19 Hz, 1H, H-3b), 2.32 (t, J = 7.68 Hz, 2H, Alkyl-CH2(O)COCH2–), 2.13 (t, J = 6.24 Hz, 4H, –CH2C≡CCH2–), 2.07 (m, 2H, >C═CHCH2CH(CH3)2), 1.71 (septet, J = 6.72 Hz, 1H, >C═CHCH2CH(CH3)2), 1.60 (m, 7H), 1.27–1.47 (m, 21H), 0.95 (d, J = 6.60 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3-Alkyl); MS (FAB) m/z 477 (M+H).
4.1.19. (Z)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl octadeca-9,11-diynoate (98)
Yield 74%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.25 (tt, J = 7.82, 7.78 Hz, 1H, >C═CHCH2CH(CH3)2), 4.25 (AB d, J = 12.00 Hz, 1H, Alkyl-(O)COCHH), 4.14 (AB d, J = 12.00 Hz, 1H, Alkyl-(O)COCHH), 3.65 (dq, J = 7.2, 6.52 Hz, 2H, HOCH2–), 2.88 (AB d, J = 16.52 Hz, 1H, H-3a), 2.70 (AB d, J = 16.52 Hz, 1H, H-3b), 2.60 (tt, J = 6.84, 2.04, 2.00 Hz, 2H, >C═CHCH2CH(CH3)2), 2.30 (t, J = 7.48 Hz, 2H, Alkyl-CH2(O)COCH2–), 2.22 (t, J = 6.80 Hz, 4H, CH3(CH2)4CH2–C≡C–C≡C–CH2-), 2.06 (m, 1H, HOCH2–), 1.71 (septet, J = 6.72 Hz, 1H, >C═CHCH2CH(CH3)2), 1.59 (m, 2H, Alkyl), 1.49 (m, 4H), 1.33–1.39 (m, 3H), 1.22–1.29 (m, 3H, Alkyl), 0.93 (d, J = 6.68 Hz, 6H, >C–CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3-Alkyl); MS (FAB) m/z 473 (M+H).
4.1.20. (E)-(2-(Hydroxymethyl)-4-(3-methylbutylidene)-5-oxotetrahydrofuran-2-yl)methyl octadeca-9,11-diynoate (99)
Yield 74%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.78 (tt, J = 7.80, 7.68 Hz, 1H, >C═CHCH2CH(CH3)2), 4.26 (AB d, J = 11.84 Hz, 1H, Alkyl-(O)COCHH), 4.15 (AB d, J = 11.84 Hz, 1H, Alkyl-(O)COCHH), 3.66 (dq, J = 7.00, 6.44 Hz, 2H, HOCH2–), 2.79 (AB d, J = 16.81 Hz, 1H, H-3a), 2.62 (AB d, J = 16.81 Hz, 1H, H-3b), 2.30 (t, J = 7.34 Hz, 2H, Alkyl-CH2(O)COCH2–), 2.22 (t, J = 6.92 Hz, 4H, CH3(CH2)4CH2–C≡C–C≡C–CH2–), 2.06 (m, 3H, HOCH2–, >C═CHCH2CH(CH3)2), 1.80 (septet, J = 6.64 Hz, 1H, >C═CHCH2CH(CH3)2), 1.49 (m, 4H, Alkyl), 1.33–1.39 (m, 3H), 1.22–1.29 (m, 9H, Alkyl), 0.93 (d, J = 6.64 Hz, 6H, >C═CHCH2CH(CH3)2), 0.86 (t, J = 6.08 Hz, 3H, CH3-Alkyl); MS (FAB) m/z 473 (M+H).
4.1.21. ((Z)-2-(Hydroxymethyl)-4-((9Z,12Z)-octadeca-9,12-dien-1-ylidene)-5 oxotetrahydrofuran-2-yl)methyl pivalate (100)
Yield 54%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.22 (t, J = 7.76 Hz, 1H, >C═CH(CH2)7–), 5.32 (m, 4H, –CH═CHCH2CH═CHCH2–), 4.28 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 4.12 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 3.58–3.69 (dq, J = 12.1, 7.04, 6.64 Hz, 2H, HOCH2), 2.80 (AB q, J = 14.64 Hz, 1H, H-3a), 2.61–2.76 (m, 4H, H-3b, >C═CHCH2–, HOCH2–), 2.02 (m, 4H, –CH2CH═CHCH2CH═CHCH2–), 1.72 (m, 2H, –CH2CH═CHCH2CH═CHCH2–), 1.28 (bs, 16H, >C═CH(CH2)5CH2–, –(CH2)3CH3), 1.17 (s, 9H, (CH3)3(O) COCH2), 0.85 (m, 3H, Alkyl-CH3); MS (FAB) m/z 477 (M+H).
4.1.22. ((E)-2-(Hydroxymethyl)-4-((9Z,12Z)-octadeca-9,12-dien-1-ylidene)-5 oxotetrahydrofuran-2-yl)methyl pivalate (101)
Yield 50%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.75 (m, 1H, >C═CH(CH2)7–), 5.32 (m, 4H, –CH═CHCH2CH═CHCH2–), 4.28 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 4.12 (AB d, J = 11.8 Hz, 1H, Alkyl-(O)COCHH), 3.58–3.69 (dq, J = 12.1, 7.04, 6.64 Hz, 2H, HOCH2–), 2.80 (AB d, J = 17.08 Hz,1H, H-3a), 2.63 (AB d, J = 17.08 Hz,1H, H-3b), 2.14 (m, 2H, >C═CHCH2–), 2.02 (m, 4H, –CH2CH═CHCH2CH═CHCH2-), 1.72 (m, 2H, –CH2CH═CHCH2CH═CHCH2–), 1.28 (bs, 16H, >C═CH(CH2)5CH2–, –(CH2)3CH3), 1.17 (s, 9H, (CH3)3(O)COCH2), 0.85 (m, 3H, Alkyl-CH3); MS (FAB) m/z 477 (M+H).
4.1.23. (Z)-(2-(Hydroxymethyl)-5-oxo-4-(8-(2-((2-pentylcyclopropyl)methyl)cyclopropyl)octylidene)tetrahydrofuran-2-yl)methylpivalate (102)
Yield 55%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.22 (t, J = 7.36 Hz, 1H, >C═CH(CH2)7–), 4.25 (AB d, J = 12.00 Hz, 1H, (CH3)3(O)COCHH–), 4.11 (AB d, J = 12.00 Hz, 1H, (CH3)3(O)COCHH–), 3.63 (dq, J = 18.96, 7.24, 6.60 Hz, 2H, HO–CH2), 2.88 (AB d, J = 15.54 Hz, 1H, H-3a), 2.67 (m, 3H, H-3b, >C═CHCH2(CH2)6–), 2.04 (t, J = 7.2 Hz, 1H, HO–CH2–), 1.11–1.46 (m, 30H, Alkyl), 0.82 (m, 3H, –Alkyl-CH3); MS (FAB) m/z 505 (M+H).
4.1.24. (E)-(2-(Hydroxymethyl)-5-oxo-4-(8-(2-((2-pentylcyclopropyl)methyl)cyclopropyl)octylidene)tetrahydrofuran-2-yl)methylpivalate (103)
Yield 60%, Oil; 1H NMR(CDCl3, 400 MHz) δ 6.75 (t, J = 7.2 Hz,1H, >C═CH(CH2)7–), 4.28 (AB d, J = 12.00 Hz, 1H, (CH3)3(O)COCHH–), 4.13 (AB d, J = 12.00 Hz, 1H, (CH3)3(O)COCHH–), 3.59–3.72 (dq, J = 14.52, 6.96, 6.48 Hz, 2H, HO–CH2–), 2.80 (AB d, J = 17.24 Hz, 1H, H-3a), 2.64 (AB-d, J = 17.24 Hz, 1H, H-3b), 2.15 (m, 2H, >C═CHCH2(CH2)6–), 2.05 (t, J = 6.84 Hz, HO–CH2), 1.10–1.52 (m, 22H, Alkyl), 1.05 (s, 9H, (CH3)3(O)COCH2–), 0.87 (m, 3H, –Alkyl-CH3), 0.64–0.69 (m, 2H), 0.56–0.61 (m, 1H); MS (FAB) m/z 505 (M+H).
4.1.25. (E)-(2-(Hydroxymethyl)-4-(3-isobutyl-5-methylhexylidene)-5-oxotetrahydrofuran-2-yl)methyl pivalate (104)
Yield 59%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.78 (m, 1H, >C═CHCH2CH(CH2(CH3)2)2), 4.29 (AB d, J = 11.8 Hz, 1H, (CH3)3(O)COCHH–), 4.11 (AB d, J = 11.8 Hz,1H, (CH3)3(O)COCHH–), 3.59–3.72 (dq, J = 13.2, 7.16, 6.6 Hz, 2H, HO–CH2), 2.79 (AB d, J = 17.1 Hz, H-3a), 2.62 (AB d, J = 17.1 Hz, 1H, H-3b), 2.11 (t, J = 6.04 Hz, 2H, >C═CHCH2CH(CH2(CH3)2)2), 2.03 (t, J = 6.88 Hz, 1H, HO–CH2), 1.69 (pentet, J = 6.56 Hz, 1H,>C═CHCH2CH(CH2(CH3)2)2), 1.06 (septet, J = 6.72 Hz, 2H, >C═CHCH2CH(CH2CH(CH3)2)2), 1.17 (s, 9H, (CH3)3(O)COCH2–), 1.04–1.10 (m, 4H, >C═CHCH2CH(CH2CH(CH3)2)2), 0.85 (d, J = 6.44 Hz, 12H, >C═CHCH2CH(CH2CH(CH3)2)2); MS (FAB) m/z 383 (M+H).
4.1.26. (Z)-(2-(Hydroxymethyl)-4-octadecylidene-5-oxotetrahydrofuran-2-yl)methyl pivalate (105)
Yield 68%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.22 (tt, 1H, >C═CH(CH2)16CH3), 4.25 (AB d, J = 11.8 Hz, 1H, (CH3)3(O)COCHH), 4.11 (AB d, J = 11.8 Hz, 1H, (CH3)3(O)COCHH), 3.63 (dq, J = 6.76, 6.08 Hz, 2H, HOCH2), 2.87 (AB q, J = 16.68 Hz, 1H, H-3a), 2.66–2.71 (m, 3H, H-3b,>C═CHCH2–), 2.09 (t, 1H, HOCH2–), 1.23 (s, 30H, >C═CHCH2(CH2)15CH3), 1.17 (s, 9H, (CH3) 3(O)COCH2), 0.85 (t, J = 7.04 Hz, 3H, Alkyl-CH3); MS (FAB) m/z 481 (M+H).
4.1.27. (E)-(2-(Hydroxymethyl)-4-octadecylidene-5-oxotetrahydrofuran-2-yl)methyl pivalate (106)
Yield 62%, Oil; 1H NMR (CDCl3, 400 MHz) δ 6.75 (tt, J = 3.12, 3.08, 2.96 Hz, 1H, >C═CH(CH2)16CH3), 4.27 (AB d, J = 11.8 Hz, 1H, (CH3)3(O)COCHH), 4.12 (AB d, J = 11.8 Hz, 1H, (CH3)3(O)COCHH), 3.66 (dq, J = 7.04, 6.6 Hz, 2H, HOCH2), 2.80 (AB q, J = 16.88 Hz, 1H, H-3a), 2.63 (AB d, J = 16.88 Hz, 1H, H-3b), 2.14 (m, 3H, HOCH2–, >C═CHCH2(CH2)15CH3), 1.23 (s, 30H, >C═CH(CH2)15CH3), 1.17 (s, 9H, (CH3)3(O)COCH2), 0.85 (t, J = 7.04 Hz, 3H, Alkyl-CH3); MS (FAB) m/z 481 (M+H).
Acknowledgments
This research was supported by research grants from the National Research Foundation of Korea (R11-2007-107-02001-0) and by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, NCI (Project Z1A BC 005270).
References
- [1].Nacro K, Bienfait B, Lee J, Han K-C, Kang J-H, Benzaria S, Lewin NE, Bhattacharyya DK, Blumberge PM, Marquez VE, Conformationally constrained analogues of diacylglycerol (DAG). 16. How much structural complexity is necessary for recognition and high binding affinity to protein kinase C? J. Med. Chem 43 (2000) 921–944. [DOI] [PubMed] [Google Scholar]
- [2].Dries DR, Gallegos LL, Newton AC, A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production, J. Biol. Chem 282 (2007) 826–830. [DOI] [PubMed] [Google Scholar]
- [3].Kiley SC, Jaken S, Whelan R, Parker PJ, Intracellular targeting of protein kinase C isozymes: functional implications, Biochem. Soc. Trans 23 (1995) 601–605. [DOI] [PubMed] [Google Scholar]
- [4].Duan D, Sigano DM, Kelly JA, Lai CC, Lewin NE, Kedei N, Peach ML, Lee J, Abeyweera TP, Rotenberg SA, Kim H, Kim YH, Kazzouli SE, Chung J-U, Young HA, Young MR, Baker A, Colburn NH, Haimovitz-Friedman A, Truman JP, Parrish DA, Deschamps JR, Perry NA, Surawski RJ, Blumberg PM, Marquez VE, Conformationally constrained analogues of diacylglycerol. 29. Cells sort diacylglycerol-lactone chemical zip codes to produce diverse and selective biological activities, J. Med. Chem 51 (2008) 5198–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nishizuka Y, Protein kinase C and lipid signaling for sustained cellular responses, FASEB J. 9 (1995) 484–496. [PubMed] [Google Scholar]
- [6].Griner EM, Kazanietz MG, Protein kinase C and other diacylglycerol effectors in Cancer, Nat. Rev. Cancer 7 (2007) 281–294. [DOI] [PubMed] [Google Scholar]
- [7].Blumberg PM, Kedei N, Lewin NE, Yang D, Czifra G, Pu Y, Peach ML, Marquez VE, Wealth of opportunity – the C1 domain as a target for drug development, Curr. Drug Targets 9 (2008) 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jaken S, Protein kinase C isozymes and substrates, Curr. Opin. Cell Biol 8 (1996) 168–173. [DOI] [PubMed] [Google Scholar]
- [9].Farah CA, Sossin WS, The role of C2 domains in PKC signaling, Adv. Exp. Med. Biol 740 (2012) 663–683. [DOI] [PubMed] [Google Scholar]
- [10].Akita Y, Protein kinase c-ε (PKC-ε): Its unique structure and function, J. Biochem 132 (2002) 847–852. [DOI] [PubMed] [Google Scholar]
- [11].Prekeris R, Mayhew MW, Cooper JB, Terrian DM, Identification and localization of an actin-binding motif that is unique to the epsilon isoform of protein kinase C and participates in the regulation of synaptic function, J. Cell Biol 132 (1996) 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Inagaki K, Churchill E, Mochly-Rosen D, Epsilon protein kinase C as a potential therapeutic target for the ischemic heart, Cardiovasc. Res 70 (2006) 222–230. [DOI] [PubMed] [Google Scholar]
- [13].Zeidman R, Troller U, Raghunath A, Pahlman S, Larsson C, Protein kinase C ε actin-binding site is important for neurite outgrowth during neuronal differentiation, Mol. Biol. Cell 13 (2002) 12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shirai Y, Adachi N, Saito N, Protein kinase C ε: function in neurons, FEBS J. 275 (2008) 3988–3994. [DOI] [PubMed] [Google Scholar]
- [15].Srinivasan R, Wolfe D, Goss J, Watkins S, de Groat WC, Sculptoreanu A, Glorioso JC, Protein kinase C epsilon contributes to basal and sensitizing responses of TRPV1 to capsaicin in rat dorsal root ganglion neurons, Eur. J. Neurosci 28 (2008) 1241–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kanno T, Yamamoto H, Yaguchi T, Hi R, Mukasa T, Fujikawa H, Nagata T, Yamamoto S, Tanaka A, Nishizaki T, The linoleic acid derivative DCP-LA selectively activates PKC-ε, possibly binding to the phosphatidylserine binding site, J. Lipid Res 47 (2006) 1146–1156. [DOI] [PubMed] [Google Scholar]
- [17].Hongpaisan J, Sun M-K, Alkon DL, PKC ε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer’s disease transgenic mice, J. Neurosci 31 (2011) 630–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Marquez VE, Blumberg PM, Synthetic diacylglycerols (DAG) and DAG-lactones as activators of protein kinase C (PK-C), Acc. Chem. Res 36 (2003) 434–443. [DOI] [PubMed] [Google Scholar]
- [19].Malolanarashimhan K, Kedei N, Sigano DM, Kelley JA, Lai CC, Lewin NE, Surawski RJ, Pavlyukovets VA, Garfield SH, Wincovitch S, Blumberg PM, Marquez VE, Conformationally constrained analogues of diacylglycerol (DAG). 27. Modulation of membrane translocation of protein kinase C (PKC) isozymes γ and δ by diacylglycerol lactones (DAG-Lactones) containing rigid-rod acyl groups, J. Med. Chem 50 (2007) 962–978. [DOI] [PubMed] [Google Scholar]
- [20].Denmark SE, Harmata MA, White KS, Studies on the addition of allyl oxides to sulfonylallenes. Preparation of highly substituted allyl vinyl ethers for carbanionic claisen rearrangements, J. Org. Chem 52 (1987) 4031–4042. [Google Scholar]
- [21].Kang JH, Peach ML, Pu Y, Lewin NE, Nicklaus MC, Blumberg PM, Marquez VE, Conformationally constrained analogues of diacylglycerol (DAG). 25. exploration of the sn-1 and sn-2 carbonyl functionality reveals the essential role of the sn-1 carbonyl at the lipid interface in the binding of DAG-lactones to protein kinase C, J. Med. Chem 48 (2005) 5738–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tanaka A, Nishizaki T, The newly synthesized linoleic acid derivative FR236924 induces a long-lasting facilitation of hippocampal neurotransmission by targeting nicotinic acetylcholine receptors, Bioorg. Med. Lett 13 (2003) 1037–1040. [DOI] [PubMed] [Google Scholar]
- [23].Shun ALKS, Tykwinski RR, Synthesis of naturally occurring polyynes, Angew. Chem. Int. Ed 45 (2006) 1034–1057. [DOI] [PubMed] [Google Scholar]
- [24].Lewin NE, Blumberg PM, [3H]Phorbol 12,13-dubutyrated binding assay for protein kinase C and related proteins, Methods Mol. Biol 233 (2003) 129–156. [DOI] [PubMed] [Google Scholar]