

Abstract
As an initial step in designing a simplified bryostatin hybrid molecule, three bryostatin analogues bearing a diacylglycerol lactone-based C-ring, which possessed the requisite pharmacophores for binding to protein kinase C (PKC) together with a modified bryostatin-like A- and B-ring region, were synthesized and evaluated. Merle 46 and Merle 47 exhibited binding affinity to PKC alpha with Ki values of 7000 ± 990 and 4940 ± 470 nM, respectively. Reinstallation of the trans-olefin and gem-dimethyl group present in bryostatin 1 in Merle 48 resulted in improved binding affinity, 363 ± 42 nM. While Merle 46 and 47 were only marginally active biologically, Merle 48 showed sufficient activity on the U937 cells to confirm that it was PMA-like for growth and attachment, as predicted by the substitution pattern of its A-and B-rings.
Graphical Abstract
The bryostatins are a class of 20 natural products that consist of a 20-membered macrolactone bearing three embedded pyran rings and were originally isolated by Pettit from the marine bryozoan Bugula neritina (Figure 1).1 Bryostatin 1 was shown to be a potent antitumor agent in vivo and has been evaluated in over 80 clinical trials to date.2–4 Bryostatin 1 was shown to exhibit synergy in combination with a number of known chemotherapeutic agents, but results as a single agent in humans were unsatisfactory. Bryostatin 1 has demonstrated the ability to activate latent HIV in vitro, potentially allowing for complete destruction of the virus when used in combination with the anti-retroviral therapy HAART.5 Additionally, bryostatin 1 has been investigated as a treatment for stroke and was shown to reduce the severity of the effects of stroke in rats.6 Bryostatin 1 is also in clinical trials for the treatment of Alzheimer’s disease.7
Figure 1.

Bryostatin family of natural products.
Bryostatin 1 binds with single-digit nanomolar affinity to the C1 domains of protein kinase C (PKC) and several related families of signaling proteins, such as RasGRP. These proteins mediate central cellular signaling pathways and play a role in numerous disease processes, including cancer and neurological conditions.8,9 The bryostatins are ultrapotent mimetics of diacylglycerols (DAGs), which represent the endogenous ligands for the C1 domains of PKC.10 Many PKC activators (such as the phorbol esters, of which phorbol 12-myristate 13-acetate (PMA) is the prototype) are potent tumor promotors.11 Bryostatin 1, in contrast, is not tumor-promoting and has been shown to both block and actually reverse the effects of phorbol esters in a dose-dependent manner.12,13 It is this unique combination of high binding affinity for PKC coupled with the functional ability to block many PKC-mediated responses which sets the bryostatins apart from the other C1 domain targeted PKC ligands.
A goal of functional synthesis is to preserve the desired functional activities of a compound while eliminating irrelevant synthetic complexity. The bryostatins incorporate two critical functional activities. First, they bind to the C1 domain of PKC. Second, they act as functional antagonists of many of the PKC-mediated responses to the phorbol esters. The critical structural element conferring on bryostatin the C1 binding activity is the lower (C-ring) region. In contrast, appropriately functionalized A- and B-rings are necessary for the ability of bryostatin to act as an antagonist rather than as an analogue of the phorbol esters.14–16 Importantly, neristatin 1 showed that the functionalized A- and B-rings were not only necessary but sufficient to confer bryostatin-like biological activity, provided that the compound could still bind to PKC; the C-region of neristatin 1 is distinct from that of bryostatin but is still capable of binding.17 These results suggested that it might be possible to develop simplified bryostatin analogues retaining the unique biological functionality of bryostatin by coupling a bryostatin-like A- and B-region to a bottom half of convenience that was capable of conferring C1 binding activity, provided that region maintained whatever interactions were necessary between the two halves. Among such templates with appropriate binding activity, the DAG-lactones represent a combination of structural simplicity and potential high affinity.18
The current study represents an initial exploration of the feasibility of this strategy. Previous work conducted by our group has resulted in a library of bryostatin analogues bearing simplified AB-ring top halves.19–21 All of our previous work has relied upon the synthesis and utilization of a fully functionalized C-ring through a 17–19 step route.22
Several models have been reported for the binding of PKC ligands to the C1 domain.23 The binding hypothesis proposed by Kang and co-workers appears to be consistent with the experimental results reported to date (Figure 2).20 All binding models have relied upon a three-point pharmacophore hypothesis; any simplified C-ring would have to contain suitable surrogate functionality in the proper orientation for binding. Our current understanding of bryostatin binding is that the C1 ester, the C26 alcohol, and C21 enoate carbonyl are largely responsible for PKC binding.20 The C3 hydroxyl and C19 hemiketal are also very important as these are involved, along with the A and B-ring pyran oxygens, in forming an intramolecular H-bonding network, providing rigidity to the macrocycle. The C9 hydroxyl group present in the natural product is not required for high affinity but presumably may help anchor the top half of the molecule to the protein surface.20
Figure 2.

Bryostatin binding hypothesis. H-bonding to PKC utilizes the residues circled.
The Marquez diacylglycerol lactones are potent PKC ligands that can still retain low nanomolar affinity for PKC when constrained into macrocyclic rings that are similar in size to the bryostatins.24,25 The Marquez lactones bear functionality similar to that of the binding motif of bryostatin 1. The most potent DAG lactones possess a Z-enoate with a bulky end substituent, a free γ-hydroxymethyl, and a γ-hydroxymethyl esterified to a bulky carboxylic acid.26 We envisioned that a DAG lactone subunit could perhaps function as a suitable replacement for the complex C-ring (primary binding subunit) of bryostatin 1. Depending on ring size and associated lipophilicity, macrocyclic DAG lactones have been described displaying affinities ranging from 6.1 nM to 1.8 μM (Figure 3). At least for the nonmacrocyclic DAG lactones, log P values of 4–6 appear to be optimal for PKC binding.27
Figure 3.

Marquez lactones. Binding affinities to PKC alpha are indicated. Log P values were calculated by ChemDraw.
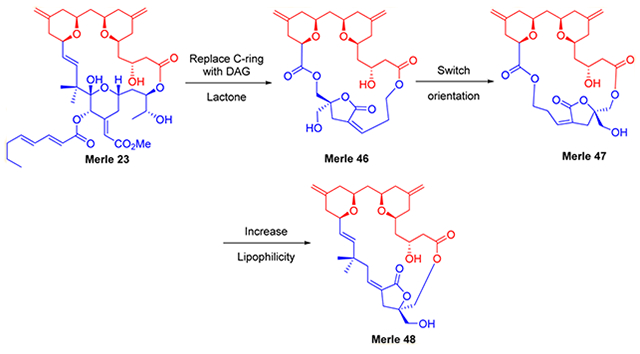
Replacement of the natural C-ring of bryostatin 1 in our most studied analogue, Merle 23, with a Marquez DAG lactone was envisioned to be a suitable platform for testing this idea. Marquez and co-workers demonstrated that the optimum configuration for PKC binding of DAG lactones is opposite from that of the endogenous DAGs due to the constrained nature of the DAG lactone.25 The optimum attachment points for the DAG lactone to the simplified AB-ring system were unknown, thus two analogues, Merles 46 and 47 were initially targeted to determine the optimum orientation. Suitable linker groups for the enoate and ester were chosen to retain the optimum ring size of the macrocyclic Marquez lactones, although this, of course, would not simultaneously provide a match for the lipophilicity of bryostatin-like compounds.15
RESULTS AND DISCUSSION
The retrosynthesis of the first two analogues involved the disconnection of the C1 ester and the C16 ester, giving AB carboxylic acid 1 and enoate 2. The bispyran 1 could be approached by pyran annulation as in previous reports from our group, while the enoate 2 could be accessed by an aldol condensation (Scheme 1).
Scheme 1.

Retrosynthesis of Merles 46 and 47
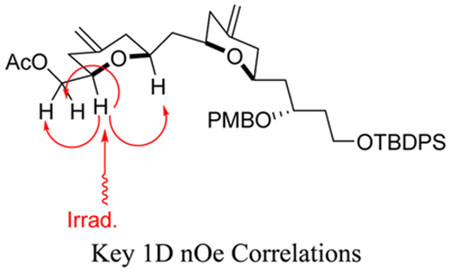
The AB acid 1 was accessed by an optimized route from known β-hydroxyallylsilane 3.28 Pyran annulation29 of known aldehyde 430 with β-hydroxyallylsilane 3 produced bispyran 5 in moderate yield. Deprotection of the C16 TBS group in the presence of the C1 TBDPS was accomplished with ZrCl4 in 2-propanol, producing alcohol 6 in good yield.31 An acetate derivative 6b of alcohol 6 was also prepared and used to confirm the stereochemistry of the new pyran ring via 1D NOESY NMR spectroscopy. Parikh–Doering32 oxidation of alcohol 6, followed by Pinnick33 oxidation, produced carboxylic acid 1 in excellent yield (Scheme 2).
Scheme 2.

Synthesis of Carboxylic Acid 1 and Relevant NOESY Correlations of Acetate 6b
Enoate 2 was envisioned to be synthesized via modification of Marquez’s optimized DAG lactone route.34 Known epoxy alcohol 7 was accessed by the route reported by Marquez and co-workers.34 BOM protection of epoxy alcohol 7 gave epoxide 8 in moderate yield. Epoxide 8 was treated with lithium acetylide ethylenediamine complex,34 producing homopropargylic alcohol 9. Lindlar hydrogenation34 produced olefin 10 in excellent yield. Hydroboration–oxidation with 9-BBN35 gave diol 11 in moderate yield. Parikh–Doering and subsequent Pinnick oxidations gave a mixture of the corresponding γ-hydroxy acid and lactone 12. This mixture was reacted under Shiina conditions36 to give the desired lactone 12 in acceptable yield (Scheme 3).
Scheme 3.

Initial Synthesis of Lactone 12
Even though this route could be used to produce gram quantities of lactone 12, a shorter route was devised using diethyl malonate as a 2-carbon synthon. Ring opening of epoxide 8 with the anion of diethyl malonate, followed by subsequent intramolecular cyclization,37 produced intermediate lactone 13 as an uncharacterized mixture of diastereomers. Hydrolysis with NaOH and subsequent decarboxylation mediated by quinoline38 at 80 °C produced lactone 12 in excellent yield, spectroscopically identical with the lactone prepared by the previously described route; double resonance 1H NMR and COSY spectroscopy were used to confirm the identity of the product. This route afforded the desired lactone in four steps from epoxide 8 as compared to seven steps using the modified Marquez route (Scheme 4).
Scheme 4.

Optimized Synthesis of Lactone 12
Synthesis of enoate 2 from 12 began with the formation of the lithium enolate and aldol reaction with the known aldehyde 14.39 This was followed by mesylation of the hydroxyl group and elimination with DBU to give enoate 2 as a 1.2:1 mixture of Z- and E-enoates in moderate yield.34 All attempts at Z-selective Horner–Wadsworth–Emmons (HWE) olefinations failed to deliver usable amounts of the desired product. After separation of the desired Z-enoate 2, the trityl group was selectively removed in the presence of the BOM and homoallylic TBS by exposure to a 1:1 mixture of TFA and TFAA at 0 °C, yielding alcohol 15.40 Synthesis of the requisite alcohol for Merle 47 involved the removal of the homoallylic TBS of 2 using TBAF buffered with NH4F,41 providing primary alcohol 16 in good yield (Scheme 5).
Scheme 5.

Synthesis of Enoate 2 and Deprotections Producing Alcohols 15 and 16
Union of carboxylic acid 1 and alcohol 15 was accomplished by the Keck–Boden-modified Steglich esterification,42 providing tricyclic ester 17 in good yield. Deprotection of the C1 TBDPS in the presence of the C24 TBS under conditions43 previously used for other analogues to give alcohol 18 proved unselective (Scheme 6).
Scheme 6.

C1 Deprotection Attempt
Selective removal of the C26 TBS of 17 was accomplished using Corey conditions, 3:1:1 AcOH/THF/H2O 45 °C,44 to give alcohol 19 in acceptable yield. Alcohol 19 was protected as the ethoxyethyl ether. Removal of the C1 TBDPS of acetal 20 provided C1 alcohol 21 in moderate yield. Parikh-Doering and Pinnick oxidations of the C1 alcohol 21 to the corresponding carboxylic acid 22 proceeded in good overall yield. Removal of the ethoxyethyl group was accomplished with PPTS in MeOH,45 affording seco-acid 23 in moderate yield. Yamaguchi macrolactonization of seco-acid 23 provided macrolactone 24.46 Deprotection of the C3 PMB of 24 was accomplished with DDQ and finally, the C24 BOM group was removed using Lipshutz conditions, LiBF4 in MeCN/H2O47 at 80 °C, to give Merle 46 (Scheme 7).
Scheme 7.

Completion of Merle 46
Synthesis of the reversed analogue Merle 47 commenced with esterification of carboxylic acid 1 with C-ring alcohol 16. In this case, the Keck–Boden-modified Steglich esterification was found to be low yielding, while a modified Yamaguchi esterification48 was found to provide tricyclic ester 25 in somewhat better yields (Scheme 8).
Scheme 8.

Esterification Optimization
Removal of the C1 TBDPS group of 25 was accomplished by reaction with HF·pyridine, providing alcohol 26 in good yield. Parikh–Doering and subsequent Pinnick oxidations provided the corresponding carboxylic acid 27 in good overall yield. Deprotection of the trityl group with formic acid in ether49 followed by Yamaguchi macrolactonization provided macrolactone 28 in moderate yield. Finally, deprotection of the PMB was effected with DDQ and the BOM group was removed with LiBF4 in MeCN/H2O in low yield, producing analogue Merle 47. The low yield may be attributable to elimination of the δ ester, resulting in ring opening in the final deprotection step (Scheme 9).
Scheme 9.

Completion of Merle 47
Merle 46 was found to have a binding affinity of 7000 ± 990 nM for PKCα, which is 4 orders of magnitude less potent than the model compound Merle 23 (0.70 nM with PKCα). This low potency suggests some caution in interpretation of biological effects since the compound may also be having some influence on membrane properties. Evaluation in Toledo leukemia cells, a cell line in which bryostatin 1 and phorbol esters both inhibit proliferation, confirms the large loss of potency for Merle 46 (Figure 4). In the U937 leukemia cells, Merle 46, like PMA, inhibited cell proliferation, albeit only at very high concentrations (80 μM). Unlike PMA, Merle 46 failed to induce significant attachment of the U937 cells. This latter result did not indicate that Merle 46 was behaving like bryostatin 1, however, in that the combined treatment of Merle 46 and PMA was similar to that of PMA alone. The most likely conclusion is that Merle 46 is PMA-like but of marginal potency in the U937 cells. We cannot exclude that it is even less potent in these cells and that the growth inhibition observed was the result of toxicity.
Figure 4.

Biological evaluation of Merle 46.
Merle 47 inhibited PDBu binding to PKCα with a binding affinity of 4940 ± 470 nM (mean ± SE), a value approximately similar to that of Merle 46. Biological activity of Merle 47 on the U937 cells was not detected, and only marginal activity was found on the Toledo cells (data not shown), possibly due to its low potency together with the instability of the ester linkage to the δ-carbon of the enoate under biological conditions. The low potency of these two compounds can be attributed at least partially to their reduced lipophilicity. Their log P value, −0.23, is much below the optimal range for DAG lactones of log P of 4–6.27 One contributor to the low log P is the ester linkage to the B-ring and concomitant loss of lipophilic functionality in that region of the structure, relative to the natural bryostatins and previous analogues containing the natural C-ring and a simplified AB-ring system. A potential additional contributor to the loss of affinity might be the disruption of the internal hydrogen-bonding network present in bryostatin and previous analogues due to deletion of the C20 hemiketal. This hydrogen-bonding network present in the natural product and previous analogues is thought to preorganize and rigidify the structures in conformations that facilitate their binding to PKC.
All of the members of the bryostatin class of natural products contain an E-olefin and a bulky gem-dimethyl group on the carbon chain which links the C-ring to the AB-ring system on the left-hand side of the molecule. Reintroduction of this functionality into a DAG lactone bryostatin analogue should increase the lipophilicity and the binding affinity of the resulting ligand for PKC. Such an analogue, Merle 48, could be disconnected via macrolactonization and pyran annulation into the known hydroxyallylsilane 3 and aldehyde 29. The C-ring aldehyde 29 could be derived from aldehyde 30 and lactone 31. Aldehyde 30 could be elaborated from ester 32 that was previously used for the synthesis of the natural bryostatin C-ring. Lactone 31 could be produced from previously prepared lactone 12 (Scheme 10).50
Scheme 10.

Retrosynthesis of Merle 48
A new route to ester 32 was developed based upon an aldol dehydration sequence starting from methyl isobutyrate. Formation of the lithium enolate of methyl isobutyrate by treatment with LDA at −78 °C and subsequent reaction with aldehyde 14 gave hydroxy ester 33 in excellent yield. Mesylation and elimination with DBU at 115 °C gave ester 32 (Scheme 11).
Scheme 11.

Synthesis of Ester 32
Reduction of ester 32 with LiAlH4, followed by Parikh–Doering oxidation of the resulting alcohol, provided aldehyde 34 in excellent yield. Wittig olefination of aldehyde 34 produced olefin 35 in good yield. Hydroboration/oxidation of olefin 35 with 9-BBN gave alcohol 36. Parikh–Doering oxidation of 36 gave aldehyde 30 in moderate yield. The trityl group of lactone 12 was removed with TFA/TFAA, followed by reprotection of the resulting alcohol 37 as the TIPS ether to give 31. Aldol condensation of the lithium enolate of lactone 31 with aldehyde 30, followed by mesylation–elimination, afforded enoate 38 as an inseparable mixture of isomers (Scheme 12).
Scheme 12.

Initial Approach to Enoate 38
In order to circumvent this selectivity issue, a Z-selective HWE olefination was investigated to construct the Z-enoate. The lithium enolate of lactone 31 was reacted with diethylchlorophosphate at −78 °C in the presence of HMPA; in situ treatment of the O-phosphonate with an excess of LDA provided the C-phosphonate, which was not further characterized.51 Treatment with KHMDS in the presence of 18-crown-6 at −78 °C, addition of aldehyde 30, and rapid warming to rt afforded the kinetic Z-enoate 38 over two steps with 5:1 Z/E-selectivity.52 Subsequent deprotection of the TBS group was effected using Corey conditions of 3:1:1 AcOH/THF/H2O, 45 °C, producing alcohol 39 in good yield. Finally, Parikh–Doering oxidation of alcohol 39 gave the requisite aldehyde 29 (Scheme 13).
Scheme 13.

Selective HWE Olefination
Pyran annulation of β-hydroxyallylsilane 3 with aldehyde 29 was conducted with TMSOTf in Et2O at −78 °C, producing bispyran 40 in good overall yield and as a single diastereomer via 1H NMR. One-dimensional ROESY NMR spectroscopy confirmed the desired stereochemistry of the B-ring pyran (Scheme 14).
Scheme 14.

Synthesis of Bispyran 40 and Relevant 1D ROESY Correlations
Deprotection of the TBDPS with HF·pyridine gave alcohol 41, which was processed to afford carboxylic acid 42 via sequential Parikh–Doering and Pinnick oxidations. Removal of the TIPS group was accomplished by stirring carboxylic acid 42 with HF·pyridine at rt for 2 days. The resulting seco-acid was cyclized under Yamaguchi conditions to give macrolactone 43 in low yield over two steps. Removal of the PMB group was accomplished with DDQ, and finally, the BOM group was removed with LiBF4 in MeCN/H2O at 80 °C, providing analogue Merle 48 (Scheme 15).
Scheme 15.

Completion of Merle 48
Merle 48 was found to have a binding affinity of 363 ± 42 nM (mean ± SE) toward PKCα, which was a 13-fold increase compared to that with Merle 47. This higher binding affinity of Merle 48 as compared to that of Merles 46 and 47 is consistent with its higher lipophilicity (log P = 1.94), although this log P is still appreciably lower than optimal for the DAG lactones. Merle 48 was PMA-like in the U937 cells. It inhibited proliferation and induced attachment, which is a pattern of response similar to that of Merle 23. Like PMA, the inhibition of proliferation and the induction of attachment by Merle 48 were both inhibited by bryostatin 1. Like both PMA and bryostatin 1, Merle 48 inhibited the growth of Toledo cells (Figure 5). Its greater binding affinity relative to that of Merle 46 was reflected in its greater potency for the inhibition of the Toledo cell growth. The finding that Merle 48 was PMA-like is consistent with the relatively unfunctionalized nature of the A- and B-rings, which correspond to those of Merle 23, which is PMA-like.14
Figure 5.

Biological evaluation of Merle 48.
Molecular modeling studies of Merles 46, 47, and 48 indicated that the intramolecular hydrogen-bonding network present in bryostatin 1 and Merle 23 was lost due to the absence of the hemiketal hydroxyl group and the conformational preferences of the molecule differed dramatically as a result (Figure 6). In the energy-minimized conformation, the A- and B-rings are nearly perpendicular to each other which has not been observed in previous modeling studies of the bryostatins.20 Since the A- and B-rings largely dictate the unique biological activity of the bryostatins, this conformational change raises the concern that, even if properly substituted, the A- and B-rings linked to a C-region binding template that does not preserve the conformation may not function as a bryostatin mimetic.
Figure 6.

Overlay of energy-minimized conformations of Merle 46 and bryostatin 1.
An energy-minimized conformation of Merle 46 was docked into the C1b domain of PKCδ (Figure 7). This model indicates that the binding domain of the analogue has two functionalities that interact with the C1b domain instead of the three that are seen in bryostatin and the phorbol esters. The diacylglycerol lactone portion of Merle 46 was bound to Thr 242, Leu 251, and Glu 253 as previously seen for bryostatin 1, but the interaction of the macrolactone C25 oxygen with Gln 257 seen in previous analogues is absent.20 The conformation of Merle 46 precludes interaction of the C25 oxygen, which may lessen the binding affinity as compared to bryostatin 1.
Figure 7.

Merle 46 docked to the C1b domain of PKC.
CONCLUSION
In summary, we have synthesized several bryostatin analogues containing a dramatically simplified C-ring binding domain. Replacement of the natural C-ring of bryostatin for a diacylglycerol lactone using an ester linker to the B-ring produced analogues with low affinity for PKC. Deletion of this ester linkage and reintroduction of the natural olefin and gem-dimethyl group modulated the lipophilicity of the resulting analogue and improved its affinity toward PKC. Modeling studies indicated that the overall conformation of the resulting analogues was altered due to deletion of the intramolecular hydrogen-bonding network present in the natural product. Docking studies with PKC suggest that these analogues bind using just two functionalities instead of three functionalities, as seen in many highly potent PKC ligands. While the first two compounds possessed only marginal biological activity, the derivative incorporating the natural olefin and gem-dimethyl group was shown to be PMA-like, not bryostatin-like, in its activity on U937 cells. This result is consistent with the simplified structure of its A- and B-rings, which corresponds to those of the PMA-like derivative Merle 23. The results from this work provide guidance for the design and synthesis of compounds embodying a similar strategy but with enhanced binding affinities and with more highly functionalized A- and B-rings that may confer bryostatin-like activity.
EXPERIMENTAL SECTION
General Considerations.
Diisopropylamine, diisopropylethylamine, pyridine, triethylamine, EtOAc, and CH2Cl2 were distilled from CaH2. Reagent-grade DMF, DMSO, and acetone were purchased, stored over 4 Å molecular sieves, and used without further purification. Et2O, THF, and toluene were distilled from Na under an atmosphere of N2. MeOH was distilled from dry Mg turnings. The titer of n-BuLi was determined by the method of Baclawski and Kofron.53 All other reagents were used without further purification. Yields were calculated for material judged homogeneous by thin layer chromatography and nuclear magnetic resonance (NMR). Thin layer chromatography was performed on Merck Kieselgel 60 Å F254 plates or Silicycle 60 Å F254 eluting with the solvent indicated, visualized by a 254 nm UV lamp, and stained with an ethanolic solution of 12-molybdophosphoric acid, a solution of p-anisaldehyde in ethanol acidified with sulfuric acid, an aqueous potassium permanganate solution, or a solution of ceric ammonium molybdate, acidified with sulfuric acid. Flash column chromatography was performed with silica gel 40–63 μm or silica gel 60–200 μm slurry packed with hexanes in glass columns. Glassware for reactions was oven-dried at 125 °C and cooled under a dry atmosphere prior to use. Liquid reagents and solvents were introduced by oven-dried syringes through septum-sealed flasks under a nitrogen atmosphere. Nuclear magnetic resonance spectra were acquired at 300 and 500 MHz for 1H and 75 and 125 MHz for 13C. Chemical shifts for proton nuclear magnetic resonance (1H NMR) spectra are reported in parts per million relative to the signal of residual CDCl3 at 7.27 ppm or (CH3)4Si at 0.00 ppm. Chemicals shifts for carbon nuclear magnetic resonance (13C NMR and DEPT) spectra are reported in parts per million relative to the centerline of the CDCl3 triplet at 77.23 ppm. Chemical shifts of the unprotonated carbons (“C”) for DEPT spectra were obtained by comparison with the 13C NMR spectrum. The abbreviations s, d, dd, ddd, dddd, ddddd dt, quint, t, and m stand for the resonance multiplicity singlet, doublet, doublet of doublets, doublet of doublet of doublets, doublet of doublet of doublet of doublets, doublet of doublet of doublet of doublet of doublets, doublet of triplets, quintet, triplet, and multiplet, respectively. Optical rotations (Na D line) were obtained using a microcell with a 1 dm path length. Specific rotations [α] (deg·mL/g·dm) are based on the equation α = (100·α)/(l·c) and are reported as unitless numbers, where the concentration c is in g/l00 mL and the path length l is in decimeters. Mass spectrometry was performed on a time-of-flight (TOF) high-resolution mass spectrometer. Compounds were named using ChemBioDraw 14.0.0. Biological evaluation of the bryostatin analogues was conducted according to previously published procedures.54
Bispyran (5).
To a 25 mL flask fitted with a stir bar were added silane 3 (262 mg, 0.367 mmol, 1.0 equiv) and aldehyde 4 (319 mg, 1.83 mmol, 5.0 equiv). The flask was flushed with N2, and Et2O (10.5 mL, 0.035 M) was added. After being cooled to −78 °C, a solution of TMSOTf in Et2O (610 μL of 0.9 M, 0.55 mmol, 1.5 equiv) was added dropwise to the stirring solution. After 3 h, the reaction mixture was quenched by dropwise addition of i-Pr2NEt (1.0 mL). After 5 min, saturated aqueous NaHCO3 solution (5.0 mL) was added and the mixture was stirred until it reached rt. The mixture was extracted with EtOAc (3 × 10 mL), and the organic phase was washed with brine (25 mL) and dried over Na2SO4, then filtered and concentrated. The crude product was purified using a 3 × 10 cm silica gel column, eluting with 4% EtOAc/hexanes, collecting 10 mL fractions. Fractions 4–14 were concentrated to yield the product (272 mg, 93%) as a colorless oil: Rf = 0.44 (10% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 7.71–7.68 (m, 4H), 7.46–7.38 (m, 6H), 7.20 (d, J = 9.0 Hz, 2H), 6.86 (d, J = 8.0 Hz, 2H), 4.73 (dd, J = 8.0, 1.5 Hz, 2H), 4.69 (d, J = 1.5 Hz, 1H), 4.60 (d, J = 1.5 Hz, 1H), 4.43 (ABq, J = 11 Hz, Δν = 30.4 Hz, 2H), 3.98–3.93 (m, 1H), 3.85–3.80 (m, 1H), 3.81 (s, 3H), 3.78–3.75 (m, 1H), 3.71 (dd, J = 10, 5.0 Hz, 1H), 3.59–3.48 (m, 4H), 3.40–3.36 (m, 1H), 2.28 (d, J = 13 Hz, 3H), 2.18 (d, J = 13.5 Hz, 1H), 2.03–1.90 (m, 5H), 1.86–1.73 (m, 2H), 1.70–1.58 (m, 3H), 1.08 (s, 9H), 0.91 (s, 9H), 0.07 (d, J = 2.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 159.3, 145.0, 144.4, 135.83, 135.81, 134.1, 134.0 131.2, 129.8, 129.6, 127.9, 127.8, 114.0, 109.0, 108.7, 79.0, 75.1, 75.0, 74.9, 72.7, 72.2, 66.8, 60.6, 55.5, 43.0, 42.6, 41.5, 41.0, 40.9, 38.1, 37.7, 27.1, 26.2, 19.4, 18.6, −5.0; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 27.1, 26.2, −5.0; CH δ 109.0, 108.7, 72.2, 66.8, 60.6, 43.0, 42.6, 41.5, 41.0, 40.9, 38.1, 37.7; CH δ 135.83, 135.81, 129.8, 129.6, 127.9, 127.8, 114.0, 79.0, 75.1, 75.0, 74.9, 72.7; C δ 159.3, 145.0, 144.4, 134.1, 134.0, 131.2, 19.4, 18.6; HRMS (ESI) calcd 821.4609 for C48H70O6NaSi2 (M + Na), found 821.4614.
Pyran Alcohol (6).
To a stirring solution of 5 (220 mg, 0.275 mmol, 1.0 equiv) in i-PrOH (6.9 mL, 0.04 M) contained in a 15 mL flask under N2 was added ZrCl4 (32 mg, 0.14 mmol, 0.5 equiv). The reaction mixture was stirred at rt for 60 h. The reaction mixture was quenched with saturated aqueous NaHCO3 solution (25 mL) and extracted with EtOAc (3 × 25 mL), then washed with brine (25 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 3 × 10 cm silica gel column, eluting with a gradient of 10–30% EtOAc/hexanes, collecting 10 mL fractions. Fractions 16–28 were concentrated to yield the product alcohol (132 mg, 70%) as a colorless oil: Rf = 0.58 (25% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 7.70–7.76 (m, 4H), 7.45–7.37 (m, 6H), 7.19 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 4.73 (dd, J = 11, 2.0 Hz, 2H), 4.69 (s, 1H), 4.47 (s, 1H), 4.42 (ABq, J = 10.5 Hz, Δν = 41.7 Hz, 2H), 3.96–3.82 (m, 1H), 3.84–3.80 (m, 1H), 3.81 (s, 3H), 3.77–3.73 (m, 1H), 3.65–3.63 (m, 1H), 3.60–3.49 (m, 3H), 3.48–3.39 (m, 2H), 2.27 (t, J = 13 Hz, 2H), 2.17 (d, J = 13 Hz, 1H), 2.08 (q, J = 12 Hz, 3H), 2.02–1.90 (m, 4H), 1.86–1.73 (m, 2H), 1.69–1.57 (m, 3H), 1.07 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 159.3, 144.8, 143.7, 135.83, 135.81 134.1, 134.0 131.2, 129.8, 129.6, 127.9, 127.8 114.0, 109.5, 108.8, 78.7, 75.2, 75.0, 74.9, 72.6, 72.0, 66.2, 60.6, 55.5, 42.9, 42.5, 41.4, 41.1, 40.8, 37.9, 36.5, 27.1, 19.4; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 27.1; CH2 δ 109.5, 108.8, 72.0, 66.2, 60.6, 42.9, 42.5, 41.4, 41.1, 40.8, 37.9, 36.5; CH δ 135.8, 129.8, 129.6, 127.8, 114.0, 78.7, 75.2, 75.0, 74.9, 72.6; C δ 159.3, 144.8, 143.7, 134.1, 131.2, 19.4; IR 3456, 3072, 2935, 2857, 1652, 1613, 1514, 1471, 1428, 1362, 1303, 1248, 1174, 1106, 1036, 891, 823, 738, 703 cm−1; HRMS (ESI) calcd 707.3744 for C42H56O6NaSi (M + Na), found 707.3755.
Acetate Ester (6b).
To a stirring solution of alcohol (6) (36 mg, 0.053 mmol, 1.0 equiv) in pyridine (1.75 mL, 0.03 M) contained in a 5 mL flask under N2 was added Ac2O (150 μL, 1.6 mmol, 30 equiv). The reaction mixture was stirred overnight at rt and quenched with saturated aqueous NaHCO3 solution (25 mL). The mixture was extracted with EtOAc (3 × 10 mL), dried over Na2SO4, filtered, and concentrated. The crude product was purified using a 2 × 10 cm silica gel column, eluting with 15% EtOAc/hexanes, collecting 5 mL fractions. Fractions 6–12 were concentrated to yield the product (36 mg, 92%) as a colorless oil: Rf = 0.76 (50% EtOAc/hexanes); [α]D20 = +3.2 (c = 0.76, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.70–7.67 (m, 4H), 7.45–7.37 (m, 6H), 7.19 (d, J = 9.0 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 4.72 (d, J = 10 Hz, 2H), 4.69 (s, 1H), 4.59 (s, 1H), 4.42 (ABq, J = 10.5 Hz, Δν = 37.8 Hz, 2H), 4.11 (dd, J = 12, 6.5 Hz, 1H), 4.06 (dd, J = 12, 4.0 Hz, 1H), 3.94–3.92 (m, 1H), 3.83–3.80 (m, 1H), 3.80 (s, 3H), 3.77–3.74 (m, 1H), 3.57–3.50 (m, 3H), 3.47–3.46 (m, 1H), 2.27 (t, J = 15.5 Hz, 2H), 2.17 (d, J = 12.5 Hz, 2H), 2.06 (s, 3H), 2.02–1.91 (m, 5H), 1.84–1.74 (m, 2H), 1.67–1.58 (m, 3H), 1.06 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 171.2, 159.4, 145.0, 143.4, 135.9, 134.1, 131.2, 129.8, 129.6, 127.9, 114.0, 109.8, 108.7, 76.1, 75.3, 75.1, 74.9, 72.7, 72.2, 67.1, 60.6, 55.5, 42.8, 42.5, 41.4, 41.0, 40.4, 38.0, 37.0, 27.2, 21.1, 19.4; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 27.2, 21.1; CH2 δ 109.8, 108.7, 72.2, 67.1, 60.6, 42.8, 42.5, 41.4, 41.0, 40.4, 38.0, 37.0; CH δ 135.9, 129.8, 129.6, 127.9, 114.0, 76.1, 75.3, 75.1, 74.9, 72.7; C δ 171.2, 159.4, 145.0, 143.4, 134.1, 131.2, 19.4; IR (neat) 3071, 2939, 2858, 1740, 1684, 1653, 1558, 1515, 1472, 1428, 1362, 1248, 1111, 1040, 823, 703 cm−1; HRMS (ESI) calcd 749.3850 for C44H58O7NaSi (M + Na), found 749.3856.
Carboxylic Acid (1).
A solution of alcohol (6) (131 mg, 0.19 mmol, 1.0 equiv) was prepared in CH2Cl2 (1.9 mL, 0.1 M) in a 10 mL flask under N2. After being cooled to 0 °C, i-Pr2NEt (230 μL, 1.33 mmol, 7.0 equiv) was added, followed by DMSO (140 μL, 1.9 mmol, 10 equiv). After 10 min, SO3·Pyr (121 mg, 0.76 mmol, 4.0 equiv) was added in 4 equal portions over 20 min to the stirring solution. The reaction mixture was stirred for 1 h at 0 °C and then quenched with saturated aqueous NaHCO3 solution (25 mL), extracted with EtOAc (3 × 10 mL), washed with brine (25 mL), dried over Na2SO4, filtered, and concentrated. The crude product was purified using a 3 × 10 cm silica gel column, collecting 10 mL fractions. The product containing fractions were concentrated to yield the aldehyde (127 mg, 97%) which was used immediately for the subsequent oxidation step.
A solution of the previously prepared aldehyde (127 mg, 0.190 mmol, 1.0 equiv) in t-BuOH (2.65 mL) was prepared in a 10 mL flask, and 2-methyl-2-butene (2.65 mL) was added, along with an aqueous solution of KH2PO4 (1.35 M. 930 μL, 6.0 equiv) to the stirring solution. The reaction mixture was cooled to −10 °C, and powdered 80% NaClO2 (106 mg, 5.0 equiv) was added. The reaction mixture was stirred overnight, allowing the cooling bath to expire. The mixture was then quenched with pH 4.0 acetate buffer (0.1 M, 15 mL). After being extracted with EtOAc (3 × 10 mL), the organic phase was washed with brine (25 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 3 × 10 cm silica gel column, eluting with a 60:15:1 toluene/EtOAc/AcOH mixture, collecting 5 mL fractions. Fractions 6–16 were concentrated to yield the carboxylic acid product (119 mg, 92%) as a colorless oil: Rf = 0.21 (60:15:1 toluene/EtOAc/AcOH); 1H NMR (500 MHz, CDCl3) δ 7.71–7.68 (m, 4H), 7.46–7.38 (m, 6H), 7.19 (dd, J = 8.5, 2.0 Hz, 2H), 6.86 (dd, J = 8.5, 2.0 Hz, 2H), 4.77 (d, J = 21 Hz, 2H), 4.74 (s, 1H), 4.66 (s, 1H), 4.38 (ABq, J = 10.5 Hz, Δν = 35.6 Hz, 2H), 3.943.88 (m, 2H), 3.85–3.79 (m, 1H), 3.81 (s, 3H), 3.78–3.73 (m, 1H), 3.70–3.65 (m, 1H), 3.56–3.52 (m, 1H), 3.46–3.41 (m, 1H), 2.62 (d, J = 13 Hz, 1H), 2.37–2.31 (m, 1H), 2.26–2.17 (m, 3H), 2.08–1.91 (m, 5H), 1.88–1.81 (m, 1H), 1.80–1.73 (m, 1H), 1.72–1.60 (m, 3H), 1.07 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 173.2, 159.3, 144.4, 141.4, 135.8, 133.99, 133.97, 131.1, 129.8, 129.6, 127.87, 127.85, 114.0, 111.3, 109.1, 76.1, 76.1, 75.1, 74.6, 72.7, 71.9, 60.6, 55.5, 42.6, 42.4, 41.3, 41.1, 39.8, 37.8, 37.2, 27.1, 19.4; HRMS (ESI) calcd 721.3537 for C42H54O7NaSi (M + Na), found 721.3553.
Epoxide (8).
A solution of known epoxy alcohol 722 (2.01 g, 5.80 mmol, 1.0 equiv) was prepared in CH2Cl2 (58 mL, 0.1 M) in a 100 mL flask equipped with a magnetic stir bar. i-Pr2NEt (2.5 mL, 14.5 mmol, 2.5 equiv) was added, followed by BOM-Cl (1.80 mL, 13.3 mmol, 2.3 equiv) to the stirring solution. After being stirred overnight, the mixture was quenched with saturated aqueous NaHCO3 solution (60 mL) and extracted with CH2Cl2 (3 × 25 mL). The combined organic phases were washed with brine (60 mL), dried over MgSO4, filtered, and concentrated. The product was purified using a 5 × 20 cm silica gel column using 10% EtOAc/hexanes, collecting 30 mL fractions. Fractions 18–32 were combined and concentrated, yielding an oil that crystallized upon chilling overnight in a −20 °C freezer (2.0 g, 75%): Rf = 0.47 (25% EtOAc/hexanes); [α]D20 = −8.04 (c = 3.65, CHCl3); mp: 75–86 °C; 1H NMR (300 MHz, CDCl3) δ 7.47–7.41 (m, 6H), 7.36–7.19 (m, 14H), 4.71 (s, 2H), 4.51 (s, 2H), 3.83 (ABq, J = 11.4 Hz, Δν = 9.75 Hz, 2H), 3.29 (q, J = 10.5 Hz, 2H), 2.78 (dd, J = 15, 4.8 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 143.9, 137.8, 128.9, 128.6, 128.2, 128.1, 127.9, 127.3, 94.9, 86.9, 69.6, 68.0, 64.1, 58.1, 49.4; DEPT (125 MHz, CDCl3) CH3 δ CH δ 94.9, 69.6, 68.0, 64.1, 49.4; CH δ 128.9, 128.6, 128.2, 128.1, 127.9, 127.3; C δ 143.9, 137.8, 86.9, 58.1; IR (neat) 3060, 3031, 2932, 2880, 1597, 1491, 1449, 1213, 1166, 1104, 1073, 1048, 900, 747, 703, 631 cm−1; HRMS (ESI) calcd 489.2042 for C31H30O4Na (M + Na), found 489.2047.
Homopropargylic Alcohol (9).
A solution of epoxide 8 (2.7 g, 5.8 mmol, 1.0 equiv) was prepared in DMSO (14 mL) in a 50 mL flask under N2. Lithium acetylide ethylenediamine complex (1.34 g, 14.5 mmol, 2.5 equiv) was weighed out in a drybox and transferred to the stirring solution of the epoxide. After 2 h, the solution was cooled in an ice bath and slowly quenched by the addition of saturated aqueous NH4Cl solution (25 mL). The solution was then extracted with CH2Cl2 (3 × 25 mL). The organic phase was then dried over MgSO4, filtered, and concentrated. The product was dissolved in the minimum amount of CHCl3 and purified using a 5 × 20 cm silica gel column, eluting with 10% EtOAc/hexanes, collecting 30 mL fractions. Fractions 10–38 were concentrated yielding the product (2.5 g, 88%) as a colorless oil: Rf = 0.26, (25% EtOAc/hexane); [α]D20 = −5.55 (c = 12.2, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.47–7.41 (m, 6H), 7.34–7.20 (m, 14H), 4.72 (s, 2H), 4.53 (s, 2H), 3.73 (ABq, J = 10.2 Hz, Δν = 8.8 Hz, 2H), 3.24 (s, 2H), 2.74 (s, 1H), 2.60 (d, J = 2.4 Hz, 2H), 1.95 (t, J = 2.4 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 143.8, 137.7, 128.9, 128.6, 128.1, 128.0, 128.0, 127.3, 95.3, 86.9, 80.0, 73.4, 71.3, 71.3, 69.7, 65.4, 25.7; DEPT (125 MHz, CDCl3) CH3 δ CH2 δ 95.4, 71.4, 69.8, 65.5, 25.8; CH δ 129.0, 128.7, 128.2, 128.1, 128.0, 127.4; C δ 143.8, 137.7, 86.9, 80.0, 73.4, 71.3; IR (neat) 3556, 3454, 3295, 3060, 3031, 2936, 2881, 2119, 1597, 1492, 1449, 1047, 991, 903, 767, 746, 702, 633 cm−1; HRMS (ESI) calcd 515.2198 for C33H32O4Na (m + Na), found 515.2202.
Homoallylic Alcohol (10).
A solution of homopropargylic alcohol 9 (1.99 g, 4.0 mmol, 1.0 equiv) was prepared in 50% EtOAc/hexanes (35 mL) in a 100 mL three-neck flask. The center neck of the flask was fitted with a valve connected to a balloon filled with H2. The other two necks were closed with rubber septa. Lindlar’s catalyst containing 5% Pd (0.90 g) and quinoline (0.84 mL, 7.3 mmol, 1.8 equiv) were then added, and the flask was flushed with H2. The reaction was complete by TLC after 2 h of stirring. The mixture was filtered through a 3 × 2 cm bed of Celite and concentrated. The crude product was purified using a 3 × 20 cm silica gel column, eluting with 10% EtOAc/hexanes, collecting 10 mL fractions. Fractions 12–30 were concentrated, yielding the product (1.91 g, 96%) as a colorless oil: Rf = 0.33 (25% EtOAc/hexanes); [α]D20 = −2.5 (c = 0.96, CDCl3); 1H NMR (300 MHz, CDCl3) δ 7.46–7.41 (m, 6H), 7.36–7.19 (m, 14H), 5.72 (dddd, J = 14.7, 9.9, 7.2, 7.2 Hz, 1H), 5.08–5.02 (m, 1H), 5.02–4.95 (m, 1H), 4.72 (s, 2H), 4.53 (s, 2H), 3.65 (ABq, J = 9.9 Hz, Δν = 33.7 Hz, 2H), 3.12 (s, 2H), 2.58 (s, 1H), 2.38 (d, J = 7.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 143.9, 137.7, 133.2, 128.9, 128.7, 128.1, 128.0, 127.3, 118.7, 95.4, 86.8, 73.6, 72.2, 69.7, 65.7, 39.6; DEPT (125 MHz, CDCl3) CH3 δ CH2 δ 118.7, 95.4, 72.2. 69.7, 65.7, 39.6; CH δ 133.2, 128.9, 128.7, 128.1, 128.0, 127.3; C δ 143.9, 137.7, 86.8, 73.6; IR (neat) 3563, 3456, 3062, 3030, 2935, 2880, 1641, 1597, 1492, 1449, 1046, 701 cm−1; HRMS (ESI) calcd 517.2355 for C33H34O4Na (M + Na), found 517.2350.
Diol (11).
A solution of homoallylic alcohol 10 (1.91 g, 3.86 mmol, 1.0 equiv) was prepared in THF (19 mL) in a 100 mL flask under N2 with stirring. A solution of 9-BBN (0.5 M, 19.0 mL, 11.6 mmol, 3.0 equiv) was added, and the mixture was stirred for 15 min. The mixture was then placed in an ultrasonic bath for 1 h, after which time no further conversion to product was noticeable via TLC. The mixture was chilled in an ice bath, and aqueous NaOH (2.0 M, 10 mL) was carefully added. Aqueous H2O2 (30%, 5.0 mL) was then carefully added in small portions to prevent loss of the reaction mixture due to the exothermic nature of the oxidation of the organoborane. After being stirred for 1 h at rt, the mixture was extracted with EtOAc (3 × 30 mL) and then washed with brine (50 mL). The organic phase was then dried over MgSO4, filtered, and concentrated. The product was then purified using a 5 × 20 cm silica gel column, eluting with a 10–35% gradient of EtOAc/hexanes, collecting 10 mL fractions. Fractions 96–120 were combined and concentrated to yield the product (1.61 g, 81%) as a viscous oil: Rf = 0.28 (25% EtOAc/hexanes); [α]D20 = −2.2 (c = 2.1, CDCl3); 1H NMR (300 MHz, CDCl3) δ 7.44–7.41 (m, 6H), 7.32–7.21 (m, 14H), 4.72 (s, 2H), 4.53 (s, 2H) 3.69 (ABq, J = 9.9 Hz, Δν = 46.5 Hz, 2H), 3.69 (t, J = 6.0 Hz, 2H), 3.14 (dd, J = 12.9, 8.5 Hz, 2H), 2.96 (s, 1H), 2.19 (s, 1H), 1.70–1.61 (m, 2H), 1.53–1.45 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 143.9, 137.7, 128.9, 128.7, 128.1, 128.0, 128.0, 127.3, 95.5, 86.8, 73.6, 72.4, 69.8, 65.4, 63.4, 31.5, 26.2; DEPT (125 MHz, CDCl3) CH3 δ CH2 δ 95.5, 72.4, 69.8, 65.4, 63.4, 31.5, 26.2; CH δ 129.0, 128.7, 128.1, 128.1, 128.0, 127.3; C δ 143.9, 137.7, 86.8, 73.6; IR (neat) 3387, 3086, 3060, 2934, 2878, 1597, 1491, 1449, 1049, 746, 701 cm−1; HRMS (ESI) calcd 535.2498 for C33H36O5Na (M + Na), found 535.2450.
Lactone (12).
A solution of diol 11 (1.32 g 2.58 mmol, 1.0 equiv) was prepared in CH2Cl2 (26 mL) in a 100 mL flask fitted with a magnetic stir bar and septum under N2. After being chilled to 0 °C, i-Pr2NEt (3.20 mL, 18.1 mmol, 7.0 equiv) and DMSO (1.80 mL, 25.8 mmol, 10.0 equiv) were added to the stirred solution. SO3·Pyr (1.65 g, 10.3 mmol, 4.0 equiv) was added in 4 portions over 20 min. After 1 h, the reaction mixture was quenched by the addition of saturated aqueous NaHCO3 solution (50 mL) and extracted with CH2Cl2 (3 × 25 mL). The combined organic phases were washed with brine (50 mL), dried over MgSO4, filtered, and concentrated to yield a yellow colored oil that was used in the next step without purification.
A solution of the crude aldehyde and hemiacetal from the previous step was prepared in t-BuOH (36 mL) in a 250 mL flask, 2-methyl-2-butene (36 mL) was added, along with an aqueous solution of KH2PO4 (1.25 M, 12.4 mL). After being cooled to −10 °C, NaClO2 (80%, 1.45 g, 16.1 mmol, 5.0 equiv) was added in 4 portions over 20 min. The reaction mixture was then allowed to warm to rt overnight. The reaction mixture was then extracted with Et2O (3 × 25 mL), and the combined organic phases were washed with brine (50 mL). The organic phase was then dried over MgSO4, filtered, and concentrated to yield the crude carboxylic acid as a viscous yellow oil. The product was purified using a 2.5 × 15 cm silica gel column, eluting with 3% MeOH/CH2Cl2, collecting 10 mL fractions. Fractions 22–48 contained a mixture of the carboxylic acid and the desired lactone and were concentrated to yield an oil which was used in the subsequent step without characterization (1.08 g, 80%, two steps).
The crude product from the previous step (0.20 g, 0.38 mmol, 1.0 equiv) was dissolved in toluene (58 mL, 0.006 M) in a 100 mL flask. DMAP (0.27 g, 2.2 mmol, 6.1 equiv) was added, followed by the slow addition of benzoyl chloride (60 μL, 0.5 mmol, 1.3 equiv). The reaction mixture was then quenched with saturated aqueous NaHCO3 solution (10 mL) after being stirred overnight. After the organic phase was separated, the aqueous phase was extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine (10 mL) and dried over MgSO4. The mixture was filtered and the solvent removed. The product was purified using a 2.5 × 20 cm silica gel column, eluting with a gradient from 10–75% EtOAc/hexanes, collecting 5 mL fractions. Fractions 28–38 were concentrated to yield the product (0.15 g, 79%) as a viscous oil: Rf = 0.65 (50% EtOAc/hexanes); [α]D20 = +6.6 (c = 4.6, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.44–7.33 (m, 6H), 7.36–7.24 (m, 14H), 4.73 (s, 2H), 4.55 (s, 2H), 3.71 (ABq, J = 10.0 Hz, Δν = 59.6 Hz, 2H), 3.24 (ABq, J = 10.0 Hz, Δν = 62.2 Hz, 2H), 2.64 (t, J = 9.0 Hz, 2H), 2.15–2.00 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 177.3, 143.6, 137.7, 128.9, 128.7, 128.19, 128.17, 128.0, 127.4, 95.1, 87.3, 86.7, 70.9, 69.9, 66.4, 29.4, 26.8; DEPT (125 MHz, CDCl3 CH3 δ CH2 δ 95.0, 70.8, 69.9, 66.4, 29.4, 26.8; CH δ 128.9, 128.7, 128.2, 128.18, 128.0, 127.5; C δ 177.3, 143.6, 137.7, 87.3, 86.7; IR (neat) 3060, 3031, 2938, 2879, 1779, 1597, 1492, 1450, 1414, 1381, 1048, 747, 704, 634 cm−1; HRMS (ESI) calcd 531.2147 for C33H32O5Na (M + Na), found 531.2148.
Diester (13).
To a 500 mL flask containing EtOH (190 mL, 0.1 M) and a stir bar under N2 was slowly added 60% NaH (6.84 g, 171 mmol, 9.0 equiv) at 0 °C. After 20 min, the ice bath was removed and diethyl malonate (29 mL, 190 mmol, 10.0 equiv) was added. After being stirred for 30 min at rt, a solution of epoxide 8 (8.90 g, 19.0 mmol, 1.0 equiv) in THF (30 mL) was added via cannula; THF (20 mL) was used to complete the transfer. After being stirred for 8 days at rt, the reaction was quenched with saturated aqueous NH4Cl solution (250 mL) and diluted with H2O (200 mL). The solution was then extracted with 50% EtOAc/hexanes (3 × 100 mL), washed with brine (2 × 100 mL), dried over Na2SO4, filtered, and concentrated. The crude product was then purified on a 5 × 20 cm silica gel column, eluting with 15% EtOAc/hexanes, collecting 30 mL fractions. Fractions 18–82 contained the product as well as diethyl malonate. The product was heated under high vacuum 0.05 mmHg to remove the remaining diethyl malonate, yielding the product as a mixture of two diastereomers (9.87 g, 89%) as a light yellow oil that was used without characterization.
A solution of diester 13 (3.0 g, 5.2 mmol, 1.0 equiv) was prepared in EtOH (52 mL, 0.1 M) in a 100 mL flask. To the stirred solution was added aqueous NaOH (1.0 M, 12.9 mL, 12.9 mmol, 2.5 equiv). After being stirred for 24 h, the solution was concentrated and then quenched with a solution of AcOH (2.0 mL) in brine (50 mL). The solution was then extracted with EtOAc (3 × 25 mL), washed with brine (50 mL), dried over Na2SO4, filtered, concentrated, and evaporated with toluene to remove any remaining AcOH. The crude carboxylic acid was used as is (2.82 g, 99%).
To a stirring solution of the previously produced carboxylic acid (2.82 g, 5.1 mmol, 1.0 equiv) in toluene (102 mL, 0.05 M) in a 250 mL flask was added quinoline (730 μL, 6.1 mmol, 1.2 equiv). A reflux condenser was attached, and the mixture was flushed with N2 and heated at 80 °C overnight. After being cooled to rt, the solution was washed with aqueous HCl (1.0 M, 2 × 50 mL), saturated aqueous NaHCO3 solution (100 mL), brine (100 mL), and dried over Na2SO4. After filtration and concentration, the product was purified using a 3 × 15 cm silica gel column, eluting with 25% EtOAc/hexanes, collecting 10 mL fractions. Fractions 8–24 were combined and concentrated to yield the product (2.33 g, 90%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.46–7.39 (m, 6H), 7.36–7.22 (m, 14H), 4.73 (s, 2H), 4.54 (s, 2H), 3.71 (ABq, J = 10.0 Hz, Δν = 59.4 Hz, 2H), 3.25 (ABq, J = 10.0 Hz, Δν = 62.2 Hz, 2H), 2.64 (t, J = 8.5 Hz, 2H), 2.13–1.99 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 177.3, 143.5, 137.7, 128.8, 128.7, 128.18, 128.15, 128.0, 127.4, 95.0, 87.3, 86.7, 70.9, 69.9, 66.4, 29.4, 26.8.
Enoate (2).
A 1.0 M solution of LDA in THF was prepared by adding i-Pr2NH, (0.77 mL, 5.5 mmol, 1.1 equiv) to a 5.0 mL volumetric flask under N2. The solution was chilled to −78 °C, and n-BuLi (2.5 M, 2.0 mL, 5.0 mmol, 1.0 equiv) was then added dropwise. After 5 min, the mixture was transferred to a 0 °C bath and THF added to make 5 mL of solution. THF (2.0 mL) was added to a 15 mL flask under N2, which was chilled to −78 °C, and the previously prepared solution of LDA (1.0 M, 0.52 mL, 0.52 mmol, 1.1 equiv) was added. A solution of lactone 12 (238 mg, 0.47 mmol, 1.0 equiv) in THF (1.0 mL) was then added via cannula to the LDA-containing solution; the transfer was completed with THF (1.7 mL). After being stirred for 45 min at −78 °C, a solution of known aldehyde 14 (133 mg, 0.71 mmol, 1.5 equiv) in THF (1.0 mL) was added. THF (1.0 mL) was used to complete the transfer of the aldehyde-containing solution. The reaction mixture was stirred for 2 h, allowing the −78 °C bath to expire. The reaction mixture was then quenched with saturated aqueous NH4Cl solution (25 mL), extracted with EtOAc (3 × 15 mL), and washed with brine (50 mL). After being dried over Na2SO4, the solution was filtered and concentrated. The product was purified using a 2 × 10 cm silica gel column, eluting with 25% EtOAc/hexanes, collecting 5 mL fractions. Fractions 6–12 contained the product, which was used as is without further characterization.
The aldol product (326 mg) was taken up in CH2Cl2 (4.7 mL, 0.1 M) contained in a 15 mL flask under N2. The stirring solution was cooled to 0 °C. Et3N (200 μL, 1.40 mmol, 3.0 equiv) was added to the solution, followed by MsCl (68 μL, 0.75 mmol, 1.6 equiv). After being stirred for 10 min, DBU (210 μL, 1.40 mmol, 3.0 equiv) was added dropwise. After 15 min, all starting material was consumed via TLC; the reaction mixture was then quenched by the addition of a solution of saturated aqueous NaHCO3 (50 mL). After extraction with CH2Cl2 (3 × 15 mL), the organic phase was washed with brine (50 mL) and dried over Na2SO4. The solution was filtered and concentrated, and the resulting oil was purified using a 2 × 15 cm silica gel column, eluting with 10% EtOAc/hexanes, collecting 5.0 mL fractions. Fractions 12–22 contained the desired Z-isomer (150 mg, 47%) as a colorless oil. Fractions 23–38 contained the E-isomer (112 mg, 35%): Rf = 0.64 (25% EtOAc/hexanes); [α]D20 = −1.73 (c = 1.62, CHCl3); 1H NMR (500 MHz, CDCl3) 7.48–7.46 (m, 6H), 7.36–7.32 (m, 14H), 6.31 (dddd, J = 9.0, 4.0, 1.5, 1.5 Hz, 1H), 4.70 (s, 2H), 4.53 (s, 2H) 3.75–3.67 (m, 4H), 3.25 (ABq, J = 10 Hz, Δν = 61.2 Hz, 2H), 3.06–3.01 (m, 1H), 2.97–2.93 (m, 1H), 2.81 (d, J = 16.5 Hz, 1H), 2.72 (d, J = 18.5 Hz, 1H), 0.91 (s, 9H), 0.08 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 169.2, 143.6, 140.4, 137.7, 128.8, 128.8, 128.6, 128.1, 127.9, 127.3, 126.3, 94.9, 87.1, 83.0, 70.2, 69.7, 65.9, 62.3, 34.1, 31.4, 26.1, 18.6, −5.1; DEPT (125 MHz, CDCl3) CH3 δ 26.1, −5.1; CH2 δ 94.9, 70.2, 69.7, 65.9, 62.3, 34.1, 31.4; CH δ 140.4, 128.8, 128.6, 128.1, 127.9, 127.3, 126.3 C δ 169.2, 143.6, 137.7, 128.8, 87.1, 83.0, 18.6; IR (neat) 3058, 2953, 2928, 2857, 1758, 1491, 1449, 1363, 1256, 1201, 1178, 1095, 1047, 992, 941, 836, 777, 746, 700, 668, 632 cm−1; HRMS (ESI) calcd 701.3280 for C42H50O6SiNa (M + Na), found 701.3284.
Enoate Alcohol (15).
To a stirring solution of enoate 2 (80 mg, 0.12 mmol, 1.0 equiv) in CH2Cl2 (2.4 mL, 0.05 M) at 0 °C was added a 50:50 TFA/TFAA solution in CH2Cl2 (0.5 M, 720 μL, 0.36 mmol, 3.0 equiv). The reaction mixture turned bright yellow and was stirred at 0 °C for 45 min. The reaction mixture was quenched with Et3N (0.34 mL, 2.4 mmol, 20 equiv). After 5 min, the mixture was evaporated with methanol (3 × 25 mL), followed by toluene (10 mL). The product was purified using a 2 × 13 cm silica gel column, eluting with 25% EtOAc/hexanes, collecting 5 mL fractions. Fractions 6–24 were concentrated to yield the product (45.5 mg, 69%) as a colorless oil: Rf = 0.14 (25% EtOAc/hexanes); [α]D20 = −8.1 (c = 0.61, MeOH); 1H NMR (500 MHz, CDCl3) 7.37–7.27 (m, 5H), 6.35 (dddd, J = 9.5, 4.5, 2.0, 2.0 Hz, 1H), 4.77 (s, 2H), 4.60 (s, 2H), 3.75–3.64 (m, 6H), 2.93 (dd, J = 13.5, 6.0 Hz, 2H), 2.86–2.76 (m, 2H), 2.25 (s, 1 H), 0.90 (s, 9H), 0.06 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 169.2, 141.8, 137.7, 128.7, 128.1, 128.0, 125.7, 95.2, 83.5, 70.0, 69.7, 65.4, 62.3, 33.2, 31.4, 26.1, 18.5, 5.1; DEPT (125 MHz, CDCl3) CH3 δ 26.1, −5.1; CH2 δ 95.2, 70.0, 69.7, 65.4, 62.3, 33.2, 31.4; CH δ 141.8, 128.7, 128.1, 128.0; C δ 169.2, 137.7, 125.7, 83.5, 18.5; IR (neat) 3443, 3033, 2954, 2930, 2885, 2858, 1755, 1672, 1575, 1498, 1472, 1463, 1381, 1364, 1257, 1200, 1177, 1101, 1046, 939, 837, 778 cm−1; HRMS (ESI) calcd 459.2179 for C23H36O6SiNa (M + Na), found 459.2181.
Enoate Alcohol (16).
To a solution of enoate 2 (150 mg, 0.22 mmol, 1.0 equiv) in THF (4.40 mL, 0.05 M) contained in a 15 mL polyethylene centrifuge tube was added NH4F (41 mg, 1.1 mmol, 5.0 equiv). TBAF (1.0 M, 1.11 mL, 1.11 mmol, 5.0 equiv) was added. After being stirred for 1.5 h, the reaction mixture was quenched with brine (20 mL) and extracted with EtOAc (3 × 10 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. The product was purified using a 2 × 10 cm silica gel column, eluting with 35% EtOAc/hexanes, collecting 5 mL fractions. Fractions 18–44 were concentrated to yield the product as a colorless oil (118 mg, 95%): Rf = 0.40 (50% EtOAc/hexanes); [α]D20 = +0.48 (c = 1.67, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.44–7.43 (m, 6H), 7.39–7.24 (m, 14H), 6.28 (dddd, J = 7.5, 4.5, 2.0, 2.0 Hz, 1H), 4.72 (s, 2H), 4.53 (s, 2H), 3.83–3.74 (m, 2H), 3.70 (q, J = 10 Hz, 2H), 3.34 (d, J = 9.5 Hz, 1H), 3.17 (d, J = 11.5 Hz, 1H), 3.10–3.04 (m, 1H), 3.00–2.94 (m, 1H), 2.83 (dd, J = 14, 2.0 Hz, 1H), 2.74 (dd, J = 16.5, 2.0 Hz, 1H), 1.78 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 169.9, 143.6, 139.4, 137.7, 128.8, 128.6, 128.1, 128.1, 128.0, 127.7, 127.4, 95.0, 87.2, 83.5, 70.2, 69.8, 65.9, 62.1, 34.2, 31.2; DEPT (125 MHz, CDCl3) CH3 δ CH2 δ 95.0, 70.2, 69.8, 65.9, 62.1, 34.2, 31.2; CH δ 139.4, 128.8, 128.6, 128.1, 128.0, 127.4; C δ 169.9, 143.6, 137.7, 127.7, 87.2, 83.5; IR (neat) 3472, 3087, 2928, 2876, 1754, 1672, 1491, 1449, 1370, 1214, 1172, 1078, 1045, 992, 944, 900, 747, 700 cm−1; HRMS (ESI) calcd 587.2410 for C36H36O6Na (M + Na), found 587.2420.
Pyran Ester (17).
A stirring solution of carboxylic acid 1 (91 mg, 0.13 mmol, 1.0 equiv) and alcohol 15 (68 mg, 0.16 mmol, 1.2 equiv) was prepared in CH2Cl2 (1.3 mL, 0.1 M) in a 5 mL flask under N2. After being cooled to 0 °C, EDCI·HCl (100 mg, 0.52 mmol, 4.0 equiv), DMAP (48 mg, 0.39 mmol, 3.0 equiv), and DMAP·HCl (41 mg, 0.26 mmol, 2.0 equiv) were then added. After being warmed to rt, the reaction mixture was stirred for 6 h. The reaction mixture was then quenched with saturated aqueous NaHCO3 solution (25 mL), extracted with CH2Cl2 (3 × 15 mL), washed with brine (25 mL), and dried over Na2SO4. After filtration and concentration, the product was purified using a 3 × 10 cm silica gel column, eluting with a gradient consisting of 100 mL of 10% EtOAc/hexanes, 300 mL of 25% EtOAc/hexanes, collecting 10 mL fractions. Fractions 36–64 were concentrated to yield the product (135 mg, 93%) as a colorless oil: Rf = 0.44 (25% EtOAc/hexanes); [α]D20 = +7.5 (c = 0.85, PhCH3); 1H NMR (500 MHz, CDCl3) δ 7.70–7.68 (m, 4H), 7.45–7.33 (m, 11H), 7.18 (d, J = 9.0 Hz, 2H), 6.86 (d, J = 9.0 Hz, 2H), 6.36 (t, J = 7.5 Hz, 1H), 4.76 (s, 2H), 4.73 (d, J = 4.0 Hz, 2H), 4.71 (s, 1H), 4.62(s, 1H), 4.60 (s, 2H), 4.40 (ABq, J = 10.5 Hz, Δν = 42.7 Hz, 2H), 4.28 (ABq, J = 12 Hz, Δν = 64.9 Hz, 2H), 3.92 (dd, J = 12, 2.5 Hz, 2H), 3.83–3.79 (m, 1H), 3.80 (s, 3H), 3.77–3.71 (m, 3H), 3.67 (dd, J = 15, 10 Hz, 2H), 3.62–3.50 (m, 2H), 3.50–3.42 (m, 1H), 3.06–2.97 (m, 1H), 2.92–2.84 (m, 1H), 2.85 (d, J = 16.5 Hz, 1H), 2.75 (d, J = 16.5 Hz, 1H), 2.44 (d, J = 13.5 Hz, 1H), 2.30–2.23 (m, 3H), 2.18 (t, J = 12 Hz, 1H), 2.09–1.90 (m, 4H), 1.86–1.72 (m, 2H), 1.71–1.59 (m, 3H), 1.06 (s, 9H), 0.90 (s, 9H), 0.07 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 170.3, 168.5, 159.3, 144.8, 142.04, 142.01, 137.7, 135.8, 134.1, 134.0, 131.2, 129.8, 129.5, 128.7, 128.08, 128.06, 127.87, 127.85, 125.0, 114.0, 110.8, 108.8, 95.2, 81.2, 76.6, 75.6, 75.0, 74.6, 72.8, 72.0, 70.1, 69.6, 66.0, 62.2, 60.6, 55.5, 42.6, 42.5, 41.4, 41.1, 39.9, 37.9, 37.6, 33.8, 31.4, 27.2, 26.1, 19.4, 18.5, −5.1; DEPT (125 MHz, CDCl3) CH3 δ 55.3, 27.2, 26.1, −5.1; CH2 δ 110.8, 108.8, 95.2, 72.0, 70.1, 69.6, 66.0, 62.2, 60.6, 42.6, 42.5, 41.4, 41.1, 39.9, 37.9, 37.6, 33.8, 31.4; CH δ 170.3, 142.1, 135.8, 129.8, 129.5, 128.7, 128.08, 128.06, 127.87, 127.85, 114.0, 76.6, 75.6, 75.0, 74.6, 72.8; C δ 168.5, 159.3, 144.8, 142.04, 142.01, 137.7, 134.1, 134.0, 131.2, 125.0, 81.2, 19.4, 18.5; IR (neat) 2933, 2857, 1762, 1653, 1612, 1514, 1472, 1429, 1362, 1249, 1173, 1106, 1048, 893, 836, 777, 737, 702 cm−1; HRMS (ESI) calcd for C65H88O12Si2Na (M + Na), 1139.5712 found 1139.5618.
C24 Alcohol (19).
A solution of 17 (175 mg, 0.157 mmol, 1.0 equiv) was prepared in 3:1:1 AcOH/THF/H2O (15.7 mL, 0.01 M) in a 25 mL flask. The stirring solution was heated at 45 °C for 1.5 h, after which the reaction was complete. The reaction mixture was diluted with brine (100 mL) and extracted with EtOAc (3 × 25 mL). After being dried over Na2SO4 and filtered, the solution was concentrated. The product was purified using a 2 × 15 cm silica gel column, eluting with a gradient of 25–50% EtOAc/hexanes, collecting 5 mL fractions. Fractions 35–72 were concentrated to yield the product (117 mg, 74%) as a colorless oil: Rf = 0.28 (50% EtOAc/hexanes); [α]D20 = +9.7 (c = 0.57, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.70–7.67 (m, 4H), 7.45–7.30 (m, 11H), 7.18 (d, J = 9.0 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.33 (dddd, J = 12, 7.5, 2.0, 2.0 Hz, 1H), 4.77 (s, 2H), 4.74–4.72 (m, 3H), 4.61 (s, 1H), 4.57 (s, 2H), 4.40 (ABq, J = 10.5 Hz, Δν = 45.6 Hz, 2H), 4.30 (ABq, J = 12 Hz, Δν = 42 Hz, 2H), 3.93 (dd, J = 11.5, 2.0 Hz, 1H), 3.94–3.88 (m, 1H), 3.84–3.80 (m, 1H), 3.80 (s, 3H), 3.77–3.72 (m, 3H), 3.68 (s, 2H), 3.61–3.56 (m, 1H), 3.56–3.50 (m, 1H), 3.49–3.43 (m, 1H), 2.96 (dd, J = 13.5, 6.5 Hz, 2H), 2.82 (ABq, J = 19 Hz, Δν = 33.9 Hz, 2H), 2.43 (d, J = 13.5 Hz, 1H), 2.31–2.23 (m, 3H), 2.17 (d, J = 13.5 Hz, 1H), 2.09–2.04 (m, 2H), 2.02–1.90 (m, 3H), 1.84–1.74 (m, 2H), 1.69–1.61 (m, 3H), 1.06 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 170.3, 169.0, 159.3, 144.7, 142.0, 140.9, 137.6, 135.8, 134.0, 133.96, 131.1, 129.8, 129.5, 128.7, 128.1, 127.87, 127.85, 126.3, 114.0, 110.8, 108.9, 95.2, 81.6, 76.6, 75.6, 75.1, 74.6, 72.8, 72.1, 70.1, 69.6, 66.1, 61.9, 60.6, 55.5, 42.6, 42.4, 41.3, 41.1, 39.8, 37.9, 37.5, 33.9, 31.1, 27.1; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 27.1; CH2 δ 110.8, 108.9, 95.2, 72.1, 70.1, 69.6, 66.1, 61.9, 60.6, 42.6, 42.4, 41.3, 39.8, 37.9, 37.5, 33.9, 31.1, 27.1, 19.4; CH δ 140.8, 135.8, 129.8, 129.5, 128.7, 128.1, 127.9, 114.0, 76.6, 75.6, 75.1, 74.6, 72.8; C δ 170.3, 169.0, 159.3, 144.7, 142.0, 137.6, 134.0, 131.1, 126.3, 81.6, 70.1, 19.4; IR (neat) 3496, 3070, 2939, 2888, 2857, 1760, 1671, 1654, 1612, 1514, 1472, 1428, 1363, 1248, 1173, 1111, 1048, 895, 823, 739, 703 cm−1; HRMS (ESI) calcd 1025.4847 for C59H74O12NaSi (M + Na), found 1025.4835.
Ethoxyethyl acetal (20).
A solution of alcohol 19 (38.2 mg, 0.038 mmol, 1.0 equiv) in a 4:1 mixture of CH2Cl2/ethyl vinyl ether (3.80 mL, 0.01 M) was prepared in a 10 mL flask under N2. After being cooled to 0 °C, PPTS (1.0 mg, 0.004 mmol, 0.1 equiv) was added, and the mixture was then stirred at 0 °C for 1 h. The reaction mixture was warmed to rt and stirred for 1 h. The reaction mixture was quenched with brine (10 mL), extracted with EtOAc (3 × 10 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 1 × 10 cm silica gel column, eluting with a gradient of 100 mL of 15% EtOAc, followed by 100 mL of 25% EtOAc/hexanes, collecting 5 mL fractions. Fractions 18–30 were concentrated to yield the product (32 mg, 78%) as a colorless oil: Rf = 0.77 (50% EtOAc/hexanes); [α]D20 = +6.5 (c = 1.6, CHCl3); 1H NMR (500 MHz, CDCl3) 7.70–7.67 (m, 4H), 7.45–7.28 (m, 11H), 7.18 (d, J = 8.5 Hz, 2H) 6.86 (d, J = 8.5 Hz, 2H), 6.34 (t, J = 7.0 Hz, 1H), 4.76 (s, 2H), 4.75–4.68 (m, 4H), 4.61 (s, 1H), 4.59 (s, 2H), 4.40 (ABq, J = 10.5 Hz, Δν = 43.8 Hz, 2H), 4.28 (ABq, J = 12.5 Hz, Δν = 64.5 Hz, 2H), 3.94–3.88 (m, 2H), 3.84–3.81 (m, 1H), 3.80 (s, 3H), 3.77–3.72 (m, 1H), 3.70–3.64 (m, 4H), 3.63–3.51 (m, 3H), 3.50–3.44 (m, 2H), 3.09–3.02 (m, 1H), 2.98–2.91 (m, 1H), 2.85 (d, J = 16.5 Hz, 1H), 2.75 (d, J = 16.5 Hz, 1H), 2.43 (d, J = 13 Hz, 1H), 2.30–2.15 (m, 4H), 2.06 (ddd, J = 14, 8.5, 6.5 Hz, 1H), 2.02–1.94 (m, 2H), 1.91 (t, J = 12.5 Hz, 1H), 1.80 (ddddd, J = 13, 13, 13, 6.5, 6.5 Hz, 2H), 1.73–1.63 (m, 3H), 1.31 (d, J = 5.0 Hz, 3H), 1.21 (t, J = 6.5 Hz, 3H), 1.06 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 170.3, 168.5, 159.3, 144.8, 142.0, 141.7, 137.6, 135.8, 134.04, 134.01, 131.1, 129.8, 129.5, 128.7, 128.1, 127.86, 127.84, 125.3, 114.0, 110.8, 108.8, 99.9, 99.8, 95.2, 81.3, 76.6, 75.6, 75.0, 74.6, 72.8, 72.0, 70.1, 69.5, 66.0, 64.02, 63.99, 61.3, 60.6, 55.5, 42.6, 42.5, 41.3, 41.0, 39.9, 37.9, 37.6, 33.7, 28.6, 27.2, 20.1, 19.4, 15.5; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 27.1, 20.1, 15.5; CH2 δ 110.8, 108.9, 95.1, 72.1, 70.1, 69.5, 66.0, 64.0, 61.3, 60.6, 42.6, 42.5, 41.4, 41.0, 39.9, 37.9, 37.6, 33.7, 28.6; CH δ 141.7, 135.8, 129.8, 129.5, 128.7, 128.1, 127.9, 114.0, 99.8, 76.6, 75.6, 75.0, 74.6, 72.8; C δ 170.3, 168.5, 159.3, 144.8, 142.0, 137.6, 134.1, 131.1, 125.3, 81.3, 19.4; IR (neat) 2939, 2887, 1761, 1656, 1612, 1513, 1428, 1378, 1248, 1172, 1110, 1049, 894, 823, 738, 703 cm−1.
C1 Alcohol (21).
A solution of 20 (90.3 mg, 0.084 mmol, 1.0 equiv) was prepared in DMF (1.70 mL, 0.05 M) in a 5 mL flask. A solution of AcOH in DMF (1.0 M, 84 μL, 1.0 equiv) and TBAF in THF (1.0 M, 84 μL, 1.0 equiv) were mixed together and added to the reaction mixture. The mixture was stirred at rt overnight, quenched by the addition of a solution of saturated aqueous NaHCO3 (5.0 mL), and diluted with brine (10 mL). After extraction with EtOAc (3 × 10 mL), the solution was dried over Na2SO4, filtered, and concentrated. The crude product was then purified using a 1 × 10 cm silica gel column, eluting with a gradient of 25–60% EtOAc/hexanes, collecting 5 mL fractions. Fractions 50–90 were concentrated to yield the product (70.1 mg, 100%) as a colorless oil: Rf = 0.15 (50% EtOAc/hexanes); [α]D20 = +9.5 (c = 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) 7.36–7.29 (m, 5H), 7.26 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 6.34 (t, J = 7.0 Hz, 1H), 4.77 (s, 2H), 4.77 (s, 1H), 4.72 (s, 2H), 4.70 (q, J = 5.5 Hz, 1H), 4.67 (s, 1H), 4.59 (s, 2H), 4.47 (ABq, J = 10.5 Hz, Δν = 26.5 Hz, 2H), 4.28 (ABq, J = 11.5 Hz, Δν = 42.0 Hz, 2H), 3.95 (dd, J = 12.5, 2.5 Hz, 1H), 3.90–3.85 (m, 1H), 3.83–3.81 (m, 1H), 3.80 (s, 3H), 3.75–3.62 (m, 6H), 3.56–3.53 (m, 2H), 3.50–3.44 (m, 3H), 3.08–3.02 (m, 1H), 3.00–2.94 (m, 1H,), 2.89–2.71 (m, 2H), 2.45 (d, J = 13 Hz, 1H), 2.25 (m, 2H), 2.18 (d, J = 13.5 Hz, 2H), 2.07–2.01 (m, 1H), 1.99–1.94 (m, 3H), 1.92–1.89 (m, 1H), 1.80–1.70 (m, 2H), 1.68–1.63 (m, 2H), 1.31 (d, J = 5.5 Hz, 3H), 1.21 (t, J = 6.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 170.3, 168.5, 159.6, 144.4, 142.1, 141.7, 137.6, 130.6, 129.7, 128.7, 128.1, 125.3, 114.2, 110.8, 109.1, 99.86, 99.84, 95.2, 81.3, 76.7, 75.8, 75.4, 75.3, 74.8, 72.0, 70.1, 69.5, 66.1, 64.0, 61.3, 60.4, 55.5, 42.5, 41.7, 41.4, 40.9, 39.9, 37.6, 36.8, 33.8, 31.2, 28.6, 20.1, 15.5; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 20.1, 15.5; CH2 δ 110.8, 109.1, 95.2, 72.0, 70.1, 69.5, 66.1, 64.0, 61.3, 60.4, 42.5, 41.7, 41.4, 40.9, 39.9, 37.6, 36.8, 33.8, 31.2, 28.6; CH δ 141.7, 129.7, 128.7, 128.1, 114.2, 99.9, 76.7, 75.8, 75.4, 75.3, 74.8; C δ 170.3, 168.5, 159.6, 144.4, 142.1, 137.6, 130.6, 125.3, 81.3; IR (neat) 3478, 2940, 2886, 1760, 1653, 1612, 1514, 1441, 1378, 1249, 1173, 1108, 1048, 898, 822, 747, 700 cm−1; HRMS (ESI) calcd 859.4245 for C47H64O13Na (M + Na), found 859.4240.
C1 Carboxylic Acid (22).
To a stirring solution of 21 (17.1 mg, 0.02 mmol, 1.0 equiv) in CH2Cl2 (200 μL, 0.1 M) in a 5 mL vial was added i-Pr2NEt (20 μL, 0.13 mmol, 7.0 equiv), followed by DMSO (10 μL, 0.19 mmol, 10.0 equiv). The solution was chilled to 0 °C, and SO3·Pyr (12 mg, 0.08 mmol, 4.0 equiv) was added in one portion. After being stirred for 1.5 h at 0 °C, the reaction mixture was quenched with saturated aqueous NaHCO3 solution (2.0 mL). After extraction with EtOAc (3 × 5 mL), the solution was dried over Na2SO4, filtered, and concentrated. The crude aldehyde was purified on a plug of silica gel in a Pasteur pipet, eluting with 25% EtOAc/hexanes. The crude aldehyde was used as is without characterization.
The aldehyde (0.02 mmol, 1.0 equiv) was taken up in t-BuOH (270 μL) in a 5 mL vial. To a stirring solution of aldehyde, 2-methyl-2-butene (270 μL) was added, followed by aqueous KH2PO4 solution (1.25 M, 90 μL, 0.11 mmol, 6.0 equiv). The rapidly stirred mixture was chilled to −10 °C in a MeOH/ice bath. To the cold solution was added 80% NaClO2 (11 mg, 0.1 mmol, 5.0 equiv) in one portion. After being stirred for 1 h at −10 °C, the reaction was quenched with aqueous pH 4.0 acetate buffer (0.1 M, 5.0 mL). After being extracted with EtOAc (4 × 5.0 mL), the solution was dried over Na2SO4, filtered, and concentrated. The product was purified using a 0.5 × 5 cm Pasteur pipet silica gel column, eluting with 80/20/1 toluene/EtOAc/AcOH, collecting 1.0 mL fractions. Fractions 4–14 were concentrated to yield the product (14 mg, 82%) as a colorless oil: Rf = 0.37 (80/20/1 PhCH3/EtOAc/AcOH); [α]D20 = +14 (c = 0.45, CHCl3); 1H NMR (500 MHz, CDCl3) 7.38–7.28 (m, 5H), 7.25 (d, J = 8.5 Hz, 2H), 6.88 (d, J = 9.0 Hz, 2H), 6.34 (ddd, J = 5.0, 5.0, 2.0 Hz, 1H), 4.77 (s, 2H), 4.76–4.69 (m, 5H), 4.60 (s, 2H), 4.49 (ABq, J = 11 Hz, Δν = 60 Hz, 2H), 4.32 (dd, J = 13, 4.5 Hz, 1H), 4.28 (dd, J = 15, 3.5 Hz, 1H), 4.09–4.07 (m, 1H), 3.94 (dd, J = 11.5, 2.0 Hz, 1H), 3.80 (s, 3H), 3.72–3.62 (m, 4H), 3.58–3.54 (m, 4H), 3.43–3.37 (m, 1H), 3.10–2.92 (m, 2H), 2.81 (ABq, J = 16.5 Hz, Δν = 40.8 Hz, 2H), 2.63 (m, 2H), 2.45 (d, J = 12.5 Hz, 1H), 2.27–2.19 (m, 3H), 2.15 (d, J = 12.5 Hz, 1H), 2.08–2.02 (m, 1H), 1.96 (m, 3H), 1.79 (dd, J = 14, 8.0 Hz, 1H), 1.70 (ddd, J = 10, 5.0, 5.0 Hz, 1H), 1.65 (ddd, J = 11, 5.5, 5.0 Hz, 1H), 1.32 (t, J = 5.5 Hz, 3H), 1.21 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.8, 170.8, 168.6, 159.5, 144.3, 141.8, 141.3, 137.6, 130.4, 129.6, 128.7, 128.1, 125.5, 114.1, 111.0, 109.1, 99.8, 95.2, 81.4, 76.6, 75.9, 75.1, 75.0, 73.3, 72.0, 70.1, 69.6, 66.5, 64.0, 61.3, 55.5, 42.7, 42.0, 41.2, 41.0, 40.1, 40.0, 37.5, 33.9, 28.5, 20.0, 15.5; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 20.0, 15.5; CH2 δ 111.0, 109.1, 95.2, 72.0, 70.1, 69.6, 66.5, 64.0, 61.3, 42.7, 42.0, 41.2, 40.1, 40.0, 37.5, 33.9, 28.5; CH δ 141.3, 129.6, 128.7, 128.1, 114.1, 99.8, 76.6, 75.9, 75.1, 75.0, 73.3, 41.0; C δ 173.8, 170.8, 168.6, 159.5, 144.3, 141.8, 137.6, 130.4, 125.5, 81.4; IR (neat) 2938, 1761, 1684, 1653, 1514, 1379, 1249, 1173, 1108, 1047, 899, 823, 740, 699 cm−1; HRMS (ESI) calcd 873.4037 for C47H62O14Na (M + Na), found 873.4047.
seco-Acid (23).
To a stirring solution of carboxylic acid 22 (46 mg, 0.05 mmol, 1.0 equiv) in MeOH (5.4 mL, 0.01 M) in a 10 mL flask under N2 was added PPTS (6.4 mg, 0.03 mmol, 0.47 equiv). After being stirred overnight, the reaction mixture was diluted with brine (25 mL) and extracted with EtOAc (3 × 10 mL). After being dried over Na2SO4, the solution was filtered and concentrated. The crude product was purified using a 1 × 10 cm silica gel column, eluting with 50 mL of 50% EtOAc/hexanes and then with 50/40/10 hexanes/EtOAc/MeOH, collecting 5 mL fractions. Fractions 12–42 were concentrated to yield the seco-acid (39.2 mg, 93%) as a colorless oil: Rf = 0.21 (50/40/10 Hexanes/EtOAc/MeOH); [α]D20 = +12.1 (c = 0.860, MeOH); 1H NMR (500 MHz, CDCl3) 7.38–7.29 (m, 5H), 7.25 (d, J = 8.0 Hz, 2H), 6.88 (d, J = 9.0 Hz, 2H), 6.34 (t, J = 7.5 Hz, 1H), 4.78 (m, 3H), 4.75–4.72 (m, 2H), 4.68 (s, 1H), 4.61 (s, 2H), 4.49 (ABq, J = 10.5 Hz, Δν = 52.2 Hz, 2H), 4.32 (ABq, J = 12 Hz, Δν = 27.9 Hz, 2H), 4.11–4.08 (m, 1H), 3.95 (dd, J = 12, 2.5 Hz, 1H), 3.80 (s, 3H), 3.82–3.74 (m, 2H), 3.68 (dd, J = 11, 1.0 Hz, 2H), 3.58–3.52 (m, 1H), 3.47 (t, J = 10.5 Hz, 1H), 3.42–3.35 (m, 1H), 3.10 (dddd, J = 13.5, 7.0, 7.0, 7.0 Hz, 1H), 3.00–2.98 (m, 3H), 2.63 (d, J = 6.0 Hz, 2H), 2.45 (d, J = 13 Hz, 1H), 2.29–2.20 (m, 3H), 2.16 (d, J = 13 Hz, 1H), 2.08–2.02 (m, 2H), 2.02–1.90 (m 3H), 1.79 (dd, J = 13, 8.0 Hz, 1H), 1.72–1.62 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 170.7, 169.3, 159.5, 144.2, 141.9, 140.4, 137.6, 130.4, 129.6, 129.3, 128.7, 128.4, 128.1, 125.5, 114.1, 111.0, 109.2, 95.2, 81.8, 76.7, 75.9, 75.3, 74.9, 73.6, 72.1, 70.2, 69.8, 66.7, 61.9, 55.5, 42.8, 42.0, 41.2, 41.1, 40.3, 39.9, 37.6, 34.0, 30.9; DEPT (125 MHz, CDCl3) CH3 δ 55.5; CH2 δ 111.0, 109.2, 95.2, 72.1, 70.2, 69.8, 66.7, 61.9, 42.8, 42.0, 41.2, 41.1, 40.3, 39.9, 37.6, 34.0, 30.9; CH δ 140.4, 129.6, 128.7, 128.4, 128.1, 114.1, 76.7, 75.9, 75.3, 74.9, 73.6; C δ 170.7, 169.3, 159.5, 144.2, 141.9, 137.6, 130.4, 129.3, 125.5, 81.8; IR (neat) 3467, 3072, 3032, 2936, 2891, 1757, 1673, 1654, 1613, 1586, 1514, 1498, 1455, 1439, 1421, 1399, 1374, 1302, 1249, 1174, 1108, 1081, 1045, 898, 848, 822 cm−1; HRMS (ESI) calcd 801.3462 for C43H54O13Na (M + Na), found 801.3462.
Macrolactone (24).
To a 100 mL flask under N2 was added toluene (14.5 mL), followed by 2,4,6-trichlorobenzoyl chloride (230 μL of 0.1 M, 0.023 mmol, 1.0 equiv), Et3N (460 μL of 0.1 M, 0.046 mmol, 2.0 equiv), and DMAP (23 μL of 0.1 M, 0.002 mmol, 0.1 equiv). All of these solutions were prepared in toluene. A solution of seco-acid 23 (17.8 mg, 0.02 mmol, 1.0 equiv) in toluene (14.5 mL) was added via syringe pump at 0.75 mL/h to the stirring solution at 40 °C. The addition was complete after 20 h. The transfer was completed by washing the syringe with toluene (1.0 mL) and adding this to the reaction mixture via syringe pump at 0.75 mL/h. After being stirred at 40 °C for 3 h, the reaction mixture was cooled to rt and quenched with aqueous NaHCO3 solution (50 mL). The mixture was extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 1 × 10 cm silica gel column, eluting with 25% EtOAc/hexanes, collecting 5 mL fractions. Fractions 17–37 were concentrated to the yield the product (9.3 mg, 54%) as a colorless oil: Rf = 0.56 (50% EtOAc/hexanes); [α]D20 = +27 (c = 0.69, CHCl3); 1H NMR (500 MHz, CDCl3) 7.39–7.31 (m, 5H), 7.22 (d, J = 9.0 Hz, 2H), 6.88 (d, J = 8.5 Hz, 2H), 6.28 (t, J = 7.0 Hz, 1H), 4.81 (s, 1H), 4.80 (s, 2H), 4.75 (s, 1H), 4.69 (s, 2H), 4.62 (s, 2H), 4.41 (s, 2H), 4.27 (ddd, J = 15, 8.0, 3.5 Hz, 1H), 4.16–4.09 (m, 3H), 3.81 (s, 3H), 3.74 (dd, J = 12, 2.5 Hz, 1H), 3.71(s, 2H), 3.60 (t, J = 11 Hz, 1H), 3.43 (t, J = 11 Hz, 1H), 3.27 (t, J = 11 Hz, 1H), 3.18–3.10 (m, 1H), 2.97–2.88 (m, 1H), 2.83 (dd, J = 17, 2.5 Hz, 1H), 2.76 (dd, J = 15.5, 2.5 Hz, 1H), 2.72 (d, J = 17 Hz, 1H), 2.56 (dd, J = 15.5, 10.5 Hz, 1H), 2.46 (d, J = 13 Hz, 1H), 2.35 (d, J = 15 Hz, 1H), 2.27 (t, J = 12.5 Hz, 1H), 2.20 (t, J = 11.5 Hz, 2H), 2.06–2.00 (m, 1H), 1.97–1.92 (m, 3H), 1.80 (m, 1H), 1.75–1.69 (m, 1H), 1.66–1.61 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 171.5, 170.0, 168.8, 159.5, 143.9, 141.6, 138.1, 137.6, 130.6, 129.5, 128.8, 128.2, 127.3, 114.1, 111.3, 109.5, 95.3, 82.3, 77.8, 77.5, 76.6, 75.9, 74.8, 74.4, 73.0, 70.3, 69.9, 67.8, 64.0, 55.5, 43.0, 42.2, 41.7, 41.3, 39.6, 37.6, 34.3, 27.2; DEPT (125 MHz, CDCl3) CH3 δ 55.5; CH2 δ 111.3, 109.5, 95.3, 73.0, 70.3, 69.9, 67.8, 64.0, 43.0, 42.2, 41.7, 41.3, 39.6, 37.6, 34.3, 27.2; CH δ 138.1, 129.5, 128.8, 128.2, 114.1, 77.8, 76.6, 75.9, 74.9, 74.4; C δ 171.5, 170.0, 168.8, 159.5, 143.9, 141.6, 137.6, 130.6, 127.3, 82.3, 77.5; IR (neat) 3073, 2929, 2856, 1819, 1760, 1739, 1692, 1674, 1613, 1581, 1550, 1514, 1454, 1440, 1405, 1369, 1326, 1301, 1249, 1207, 1178, 1083, 1044, 987, 899 cm−1; HRMS (ESI) calcd 783.3356 for C43H52O12Na (M + Na), found 783.3356.
Merle 46.
To a stirring solution of macrolactone 24 (6.9 mg, 0.01 mmol, 1.0 equiv) in CH2Cl2 (0.90 mL, 0.01 M) was added DDQ (21 mg, 0.1 mmol, 10.0 equiv). The reaction mixture was stirred at 0 °C for 2 h and then quenched with saturated aqueous NaHCO3 solution (10 mL). The reaction mixture was extracted with CH2Cl2 (3 × 10 mL), dried over Na2SO4, filtered, and concentrated. The crude product was used without purification.
To the previously described crude alcohol contained in a 5 mL vial was added a solution of LiBF4 in 25:1 MeCN/H2O (0.25 M, 1.62 mL, 0.41 mmol, 45 equiv). After being flushed with N2, the vial was capped and heated at 80 °C overnight with stirring. After being cooled to rt, the solvent was evaporated and the reaction mixture was diluted with brine (10 mL) and extracted with EtOAc (3 × 10 mL). After being dried over Na2SO4, the solution was filtered and concentrated. The product was purified using a 1 × 5 cm silica gel column, eluting with 60% EtOAc/pentane, collecting 1 mL fractions. Fractions 11–56 were concentrated to yield the product (3.4 mg, 73% over two steps) as a colorless oil: Rf = 0.40 (50/40/10 hexanes/EtOAc/MeOH); [α]D20 = +6.2 (c = 0.065, CHCl3); 1H NMR (500 MHz, CDCl3) 6.29 (t, J = 7.5 Hz, 1H), 4.83 (d, J = 18 Hz, 2H), 4.74 (s, 2H), 4.65 (d, J = 12 Hz, 1H), 4.38 (ddd, J = 15, 8.0, 3.5 Hz, 1H), 4.20–4.17 (m, 1H), 4.14 (d, J = 12 Hz, 2H), 3.82 (dd, J = 12.5, 2.5 Hz, 1H), 3.79–3.76 (m, 1H), 3.70 (dd, J = 12, 6.5 Hz, 1H), 3.55–3.49 (m, 1H), 3.46–3.41 (m, 2H), 3.32–3.25 (m, 2H), 2.92 (dd, J = 16.5, 3.0 Hz, 1H), 2.85–2.79 (m, 1H), 2.71 (dd, J = 15.5, 1.5 Hz, 1H), 2.46 (d, J = 13 Hz, 1H), 2.41 (dd, J = 16, 10.5 Hz, 1H), 2.30 (d, J = 14.5 Hz, 1H), 2.24 (d, J = 12.5 Hz, 1H), 2.18 (d, J = 13.5 Hz, 2H), 2.05–1.92 (m, 5H), 1.79–1.66 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 173.6, 173.3, 170.2, 143.8, 141.8, 138.7, 127.7, 111.0, 109.5, 83.8, 76.8, 76.8, 75.2, 67.8, 67.2, 65.1, 64.2, 42.6, 42.4, 41.2, 41.2, 39.7, 37.6, 33.2, 29.9, 27.4; DEPT (125 MHz, CDCl3) CH3 δ CH2 δ 111.0, 109.5, 67.2, 65.1, 64.2, 42.6, 42.4, 41.2, 41.2, 39.7, 37.6, 33.2, 29.9, 27.4; CH δ 138.7, 76.8, 75.2, 67.8; C δ 173.6, 173.3, 170.2, 143.8, 141.8, 127.7, 83.8; IR (neat) 3488, 2924, 1755, 1652, 1408, 1368, 1327, 1262, 1179, 1104, 907, 733 cm−1; HRMS (ESI) calcd 543.2206 for C27H36O10Na (M + Na), found 543.2216.
Pyran Ester (25).
To a stirring solution of carboxylic acid 1 (117 mg, 0.167 mmol, 1.0 equiv) in PhCH3 (1.67 mL, 0.1 M) in a 10 mL flask under N2 was added Et3N (93 μL, 0.67 mmol, 4.0 equiv). After being stirred for 5 min, 2,4,6-trichlorobenzoyl chloride (40 μL, 0.23 mmol, 1.4 equiv) was added. The reaction mixture was stirred for 1 h. A solution of alcohol 16 (165 mg, 0.29 mmol, 1.8 equiv) and DMAP (36 mg, 0.29 mmol, 1.8 equiv) in PhCH3 (500 μL) was added. PhCH3 (2 × 250 μL) was used to complete the transfer. The reaction mixture was stirred for 1 h and then quenched with saturated aqueous NaHCO3 solution (25 mL). The mixture was extracted with EtOAc (3 × 10 mL), washed with brine (15 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 2 × 15 cm silica gel column, eluting with 15% EtOAc/hexanes, collecting 5 mL fractions. Fractions 14–32 were concentrated to yield the product as a colorless oil (127 mg, 61%): Rf = 0.48 (25% EtOAc/hexanes); [α]D20 = +2.48 (c = 1.62); 1H NMR (500 MHz, CDCl3) δ 7.70–7.67 (m, 4H), 7.45–7.37 (m, 12H), 7.35–7.23 (m, 14H), 7.19 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.13 (t, J = 7.0 Hz, 1H), 4.73 (m, 3H), 4.71 (s, 2H), 4.61 (s, 1H), 4.52 (s, 2H), 4.41 (ABq, J = 10.5 Hz, Δν = 41.2 Hz, 2H), 4.25 (ddddd, J = 13.5, 10.5, 10.5, 6.5, 6.5 Hz, 2H), 3.94–3.91 (m, 2H), 3.85–3.81 (m, 1H), 3.80 (s, 3H), 3.77–3.73 (m, 1H), 3.68 (ABq, J = 10.5 Hz, Δν = 22.9 Hz, 2H), 3.63–3.60 (m, 1H), 3.58–3.51 (m, 1H), 3.50–3.45 (m, 1H), 3.32 (d, J = 10 Hz, 1H), 3.17 (d, J = 9.5 Hz, 1H), 3.20–3.14 (m, 1H), 3.13–3.09 (m, 1H), 2.79 (d, J = 16.5 Hz, 1H), 2.71 (d, J = 16.5 Hz, 1H), 2.48 (d, J = 12.5 Hz, 1H), 2.31 (d, J = 13 Hz, 1H), 2.27–2.23 (m, 2H), 2.17 (d, J = 13.5 Hz, 1H), 2.10 (ddd, J = 14.0, 8.0, 5.5 Hz, 1H), 2.01 (t, J = 12 Hz, 2H), 1.93 (t, J = 12 Hz, 1H), 1.84–1.74 (m, 2H), 1.72–1.62 (m, 3H), 1.07 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 170.9, 169.1, 159.3, 144.8, 143.5, 142.3, 137.7, 137.4, 135.8, 134.03, 133.99 131.1, 129.8, 129.5, 128.8, 128.6, 128.1, 127.99, 127.96, 127.87, 127.85, 127.4, 114.0, 110.6, 108.8, 95.0, 87.2, 83.4, 77.0, 75.7, 75.0, 74.6, 72.8, 72.1, 70.2, 69.8, 66.0, 63.9, 60.6, 55.5, 42.6, 42.5, 41.4, 41.1, 39.9, 38.0, 37.7, 34.0, 27.2, 27.2, 19.4; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 27.2; CH2 δ 110.6, 108.8, 95.0, 72.1, 70.2, 69.8, 66.0, 63.9, 60.6, 42.6, 42.5, 41.4, 41.1, 39.9, 38.0, 37.7, 34.0, 27.2; CH δ 137.4, 135.8, 129.8, 129.5, 128.8, 128.6, 128.1, 128.0, 127.9, 127.4, 114.0, 77.0, 75.7, 75.0, 74.6, 72.8; C δ 170.9, 169.1, 159.3, 144.8, 143.5, 142.3, 137.7, 134.0, 131.1, 127.9, 87.2, 83.4, 19.4; IR (neat) 2936, 1758, 1611, 1513, 1449, 1428, 1362, 1248, 1174, 1110, 1046, 898, 823, 745, 703, 632 cm−1; HRMS (ESI) calcd 1267.5943 for C78H88O12NaSi (M + Na), found 1267.5942.
C1 Alcohol (26).
A solution of ester 25 (25 mg, 0.02 mmol, 1.0 equiv) was prepared in THF (200 μL, 0.1 M) in a 2 mL polyethylene vial. To the stirring solution, a 20% HF·pyridine solution in pyridine (500 μL) (25 mL/mmol) was added. After being stirred for 2.5 h, the reaction mixture was quenched with brine (10 mL) and saturated aqueous NaHCO3 solution (5 mL). The mixture was extracted with EtOAc (4 × 5 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 1 × 10 cm silica gel column, eluting with 45% EtOAc/hexanes, collecting 5 mL fractions. Fractions 6–22 were concentrated to yield the product as a colorless oil (16.1 mg, 80%): Rf = 0.37 (50% EtOAc/hexanes); [α]D20 = +2.1 (c = 0.59); 1H NMR (500 MHz, CDCl3) δ 7.42–7.40 (m, 6H), 7.35–7.23 (m, 16H), 6.89 (d, J = 9.0 Hz, 2H), 6.14 (t, J = 7.0 Hz, 1H), 4.77–4.73 (m, 2H), 4.71 (s, 2H), 4.69 (d, J = 16 Hz, 2H), 4.52 (s, 2H), 4.47 (ABq, J = 10.5 Hz, Δν = 19.5 Hz, 2H), 4.30–4.22 (m, 2H), 3.95 (dd, J = 11.5, 2.5 Hz, 1H), 3.91–3.86 (m, 1H), 3.84–3.81 (m, 1H), 3.80 (s, 3H), 3.75–3.71 (m, 1H), 3.68 (q, J = 6.0 Hz, 2H), 3.59–3.55 (m, 1H), 3.51–3.44 (m, 2H), 3.32 (d, J = 10 Hz, 1H), 3.22–3.15 (m, 1H), 3.16 (d, J = 10 Hz, 1H), 3.13–3.08 (m, 1H), 2.80 (dd, J = 16.5, 2.0 Hz, 1H), 2.72 (d, J = 16 Hz, 1H), 2.51 (d, J = 13 Hz, 1H), 2.30–2.23 (m, 3H), 2.19 (d, J = 12.5 Hz, 1H), 2.10–1.90 (m, 5H), 1.81–1.64 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 170.9, 169.1, 159.5, 144.4, 143.6, 142.4, 137.7, 137.4, 130.6, 129.7, 128.8, 128.7, 128.7, 128.1, 128.0, 128.0, 127.4, 114.2, 110.6, 109.1, 95.0, 87.2, 83.4, 77.0, 75.8, 75.5, 75.4, 74.9, 72.0, 70.2, 69.8, 66.0, 63.9, 60.5, 55.5, 42.5, 41.7, 41.4, 41.0, 40.0, 37.7, 36.8, 34.1, 27.2; DEPT (125 MHz, CDCl3) CH3 δ 55.5; CH2 δ 110.6, 109.1, 95.0, 72.0, 70.2, 69.8, 66.0, 63.9, 60.5, 42.5, 41.7, 41.4, 41.0, 40.0, 37.7, 36.8, 34.1, 27.2; CH δ 137.4, 129.7, 128.8, 128.7, 128.1, 128.0, 127.4, 114.2, 77.0, 75.8, 75.5, 75.4, 74.9; C δ 170.9, 169.1, 159.5, 144.4, 143.6, 142.4, 137.7, 130.6, 128.7, 128.0, 87.2, 83.4; IR (neat) 3503, 2936, 2884, 1756, 1684, 1652, 1616, 1516, 1490, 1448, 1362, 1249, 1175, 1078, 1045, 898, 748, 704, 633 cm−1; HRMS (ESI) calcd 1029.4765 for C62H70O12Na (M + Na), found 1029.4774.
Carboxylic Acid (27).
To a stirring solution of alcohol 26 (25 mg, 0.03 mmol, 1.0 equiv) in CH2Cl2 (250 μL, 0.1 M) contained in a 5 mL vial under N2 was added i-Pr2NEt (30 μL, 0.18 mmol, 7.0 equiv), followed by DMSO (20 μL, 0.3 mmol, 10 equiv). After being cooled to 0 °C, SO3·Pyr (16 mg, 0.10 mmol, 4.0 equiv) was added. The reaction mixture was stirred for 2 h at 0 °C and quenched with saturated aqueous NaHCO3 solution (5 mL). After dilution with brine (5 mL), the mixture was extracted with EtOAc (3 × 5 mL), dried over Na2SO4, filtered, and concentrated. After evaporation with PhCH3 (3 × 2 mL), the crude aldehyde was used without purification for the subsequent Pinnick oxidation.
A solution of the previously described aldehyde (0.025 mmol, 1.0 equiv) in t-BuOH (360 μL, 14.3 mL/mmol) and 2-methyl-2-butene (360 μL, 14.3 mL/mmol) was prepared in a 5 mL vial. After the addition of a solution of aqueous KH2PO4 (1.25 M, 120 μL, 0.15 mmol, 6.0 equiv), the reaction mixture was cooled to −10 °C in an ice/MeOH bath. NaClO2 (80%, 14.1 mg, 0.13 mmol, 5.0 equiv) was added in 1 portion to the rapidly stirred mixture. The cooling bath was allowed to expire overnight. The reaction mixture was quenched with aqueous pH 4.0 acetate buffer (0.1 M, 5 mL) and brine (5 mL). After extraction with EtOAc (3 × 5 mL), the organic phase was dried over Na2SO4, filtered, and concentrated. The crude carboxylic acid was purified using a 1 × 10 cm silica gel column, eluting with 80/20/1 PhCH3/EtOAc/AcOH, collecting 5 mL fractions. Fractions 4–12 were concentrated and evaporated with PhCH3 (3 × 5 mL) to remove residual AcOH, yielding the product as a colorless oil (22 mg, 86% over 2 steps): Rf = 0.35 (80/20/1 PhCH3/EtOAc/AcOH); [α]D20 = +6.33 (c = 1.08); 1H NMR (500 MHz, CDCl3) δ 7.42–7.41 (m, 6H), 7.33–7.29 (m, 10H), 7.27–7.23 (m, 6H), 6.88 (d, J = 9.0 Hz, 2H), 6.14 (t, J = 7.0 Hz, 1H), 4.78 (s, 1H), 4.74–4.69 (m, 5H), 4.55 (d, J = 11 Hz, 1H), 4.52 (s, 2H), 4.45 (d, J = 10.5 Hz, 1H), 4.30–4.23 (m, 2H), 4.10 (dddd, J = 11.5, 7.5, 5.5, 5.5 Hz, 1H), 3.98 (dd, J = 11.5, 2.5 Hz, 1H), 3.79 (s, 3H), 3.69 (q, J = 11 Hz, 2H), 3.54–3.42 (m, 3H), 3.32 (d, J = 10 Hz, 1H), 3.24–3.19 (m, 1H), 3.17 (d, J = 9.5 Hz, 1H), 3.11–3.06 (m, 1H), 2.80 (d, J = 16.5 Hz, 1H), 2.72 (d, J = 15.5 Hz, 1H), 2.69–2.61 (m, 2H), 2.51 (d, J = 8.0 Hz, 1H), 2.27–2.22 (m, 3H), 2.15 (q, J = 12.5 Hz, 1H), 2.08 (dd, J = 14.5, 7.0 Hz, 1H), 2.03 (t, J = 12.5 Hz, 1H), 1.96 (t, J = 12 Hz, 2H), 1.82 (ddd, J = 15, 8.5, 2.0 Hz, 1H), 1.73–1.62 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 174.5, 171.3, 169.1, 159.5, 144.3, 143.5, 142.3, 137.6, 137.3, 130.3, 129.7, 128.8, 128.7, 128.4, 128.1, 128.1, 128.0, 127.4, 114.1, 110.7, 109.1, 95.0, 87.2, 83.4, 76.8, 75.7, 75.1, 74.9, 73.2, 72.0, 70.2, 69.8, 65.9, 64.0, 55.5, 42.8, 41.9, 41.2, 41.0, 40.2, 39.9, 37.7, 34.1, 27.2; DEPT (125 MHz, CDCl3) CH3 δ 55.5; CH2 δ 110.7, 109.1, 95.0, 72.0, 70.2, 69.8, 65.9, 64.0, 42.8, 41.9, 41.2, 41.0, 40.2, 39.9, 37.7, 34.1, 27.2; CH δ 137.4, 129.7, 128.8, 128.7, 128.1, 128.0, 127.4, 114.1, 76.8, 75.7, 75.1, 74.9, 73.2 ;C δ 174.5, 171.3, 169.1, 159.5, 144.3, 143.5, 142.3, 137.6, 130.3, 128.4, 128.1, 87.2, 83.4; IR (neat) 3065, 2959, 2894, 1756, 1711, 1653, 1612, 1514, 1449, 1369, 1249, 1176, 1080, 1043, 900, 824, 748, 701 cm−1; HRMS (ESI) calcd 1043.4558 for C62H68O13Na (M + Na), found 1043.4554.
Macrolactone (28).
To a 5 mL vial containing carboxylic acid 27 (22 mg, 0.02 mmol, 1.0 equiv) was added 50:50 Et2O/HCOOH (2.3 mL, 105 mL/mmol). The vial was flushed with N2 and the mixture stirred for 3 h at rt. The solvent was evaporated and the crude product was azeotroped with PhCH3 (3 × 5 mL). The product was purified using a 1 × 10 cm silica gel column, eluting with 50/45/5 hexanes/EtOAc/MeOH, collecting 5 mL fractions. Fractions 4–22 were concentrated, yielding the product as a clear oil which was used without further characterization.
The previously prepared seco-acid was dried overnight under high vacuum in a 5 mL vial. The seco-acid was taken up in THF (820 μL, 0.03 M) and Et3N (20 μL, 0.13 mmol, 6.0 equiv) was added to the stirred solution under N2. After being cooled to 0 °C, a solution of 2,4,6-trichlorobenzoyl chloride in THF (0.1 M, 660 μL, 0.066 mmol, 3.0 equiv) was added dropwise. After being stirred for 5 min at 0 °C, the mixture was then stirred at rt for 3 h. The resulting solution was taken up in PhCH3 (14 mL) in a 25 mL gastight syringe. This solution was then added over 16 h via syringe pump at 0.875 mL/h to a stirring solution of DMAP (54 mg, 0.44 mmol, 20 equiv) in PhCH3 (14 mL) contained in a 50 mL flask under N2 that was heated at 40 °C. This produced a final reactant concentration of 0.0008 M. After the addition was complete, PhCH3 (1.0 mL) was taken up in the syringe and added over 1 h to complete the transfer. After being stirred for 3 h at 40 °C, the reaction mixture was diluted with brine (50 mL), extracted with EtOAc (3 × 10 mL), dried over Na2SO4, filtered, and concentrated. The crude product was purified using a 1 × 10 cm silica gel column, eluting with 50% EtOAc/hexanes, collecting 5 mL fractions. Fractions 2–7 were concentrated to yield the product as a colorless oil (9.2 mg, 55% over 2 steps): Rf = 0.52 (50% EtOAc/hexanes); [α]D20 = +16.1 (c = 0.460); 1H NMR (500 MHz, CDCl3) δ 7.35–7.28 (m, 5H), 7.25 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 8.5 Hz, 2H), 6.17 (t, J = 8.0 Hz, 1H), 4.95 (d, J = 6.0 Hz, 1H), 4.78 (d, J = 8.5 Hz, 1H), 4.78 (s, 1H), 4.72–4.71 (m, 2H), 4.66 (d, J = 11.0 Hz, 1H), 4.64 (s, 1H), 4.62–4.46 (m, 4H), 4.39 (d, J = 11 Hz, 1H), 4.19–4.14 (m, 1H), 4.01 (d, J = 11.5 Hz, 1H), 4.00–3.95 (m, 1H), 3.88 (ddd, J = 11.5, 11.5, 2.0 Hz, 2H), 3.81 (s, 3H), 3.67 (d, J = 10 Hz, 1H), 3.63–3.57 (m, 1H), 3.55–3.49 (m, 1H), 3.38–3.34 (m, 2H), 2.89 (d, J = 15.5 Hz, 1H), 2.81 (dd, J = 15.5, 4.0 Hz, 1H), 2.74–2.62 (m, 1H), 2.59 (d, J = 13 Hz, 1H), 2.55 (d, J = 16 Hz, 1H), 2.45 (dd, J = 14.5, 9.5 Hz, 1H), 2.34 (d, J = 13.5 Hz, 1H), 2.27 (t, J = 12 Hz, 1H), 2.21–2.18 (m, 1H), 2.15 (t, J = 12 Hz, 2H), 1.96 (t, J = 12.5 Hz, 2H), 1.88 (t, J = 12.5 Hz, 1H), 1.70 (m, 2H), 1.63–1.60 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 170.4, 168.8, 159.5, 144.4, 142.4, 138.5, 137.8, 130.4, 129.4, 128.7, 128.1, 128.0, 128.0, 127.9, 114.2, 110.6, 109.1, 94.6, 81.5, 77.6, 76.2, 74.9, 74.7, 73.0, 72.4, 69.9, 66.6, 65.3, 62.6, 55.5, 43.1, 42.4, 41.4, 41.2, 40.8, 40.4, 37.0, 34.7, 26.5; DEPT (125 MHz, CDCl3) CH3 δ 55.5; CH2 δ 110.6, 109.1, 94.6, 72.4, 69.9, 66.6, 65.3, 62.3, 62.5, 43.1, 42.4, 41.4, 41.2, 40.8, 40.4, 37.0, 34.7, 26.5; CH δ 138.5, 129.4, 128.7, 128.0, 114.1, 77.6, 76.2, 74.9, 74.7, 73.0; C δ 170.4, 168.8, 159.5, 144.4, 142.4, 137.8, 130.4, 128.1, 128.0, 127.9, 81.5. IR (neat) 2940, 2886, 1764, 1742, 1612, 1514, 1380, 1249, 1176, 1120, 1047, 892, 821, 740 cm−1: HRMS (ESI) calcd 783.3356 for C43H52NaO12 (M + Na), found 783.3367.
Merle 47.
To a stirring solution of macrolactone 28 (5.3 mg, 0.007 mmol, 1.0 equiv) in CH2Cl2 (700 μL, 0.01 M) contained in a 5 mL vial was added H2O (20 μL). After being cooled to 0 °C, DDQ (16.1 mg, 0.07 mmol, 10 equiv) was added. After being stirred at 0 °C for 2 h, the reaction mixture was quenched with saturated aqueous NaHCO3 solution (25 mL) and extracted with CH2Cl2 (3 × 10 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. After being dried under high vacuum for 1 h, the crude product was used without purification for the subsequent deprotection step.
To the aforementioned product contained in a 5 mL vial, a solution of LiBF4 in 25:1 MeCN/H2O (0.25 M, 1.26 mL, 45 equiv) was added. The vial was flushed with N2, capped, and stirred at 80 °C overnight. After being cooled to rt, the reaction mixture was quenched with brine (10 mL), and extracted with EtOAc (3 × 10 mL). After being dried over Na2SO4, the solution was filtered and concentrated to dryness. The crude product was purified using a 1 × 5 cm silica gel column, eluting with 50% EtOAc/hexanes, collecting 1 mL fractions. Fractions 9–50 contained the desired product. The product was purified a second time by the same procedure to yield the desired macrolactone as an oil (1.3 mg, 36% over 2 steps): Rf = 0.08 (50% EtOAc/hexanes); [α]D20 = −6.2 (c = 0.065); 1H NMR (500 MHz, CDCl3) δ 6.28 (t, J = 7.0, Hz, 1H), 4.61 (dd, J = 10, 1.5 Hz, 2H), 4.57 (s, 2H), 4.50 (d, J = 12 Hz, 1H), 4.45–4.41 (m, 1H), 4.36–4.30 (m, 1H), 4.24 (ddd, J = 10.5, 10.5, 2.5 Hz, 1H), 4.07 (d, J = 12 Hz, 1H), 3.87 (dd, J = 11.5, 2.5 Hz, 1H), 3.72–3.70 (m, 2H), 3.63 (t, J = 10 Hz, 1H), 3.53–3.48 (m, 1H), 3.43 (m, 2H), 2.92 (d, J = 16 Hz, 1H),), 2.90–2.85 (m, 1H), 2.83–2.73 (m, 2H), 2.69 (d, J = 16 Hz, 1H), 2.53 (d, J = 13 Hz, 1H), 2.48 (dd, J = 14, 9.5 Hz, 1H), 2.42 (d, J = 6.0 Hz, 1H), 2.37–2.29 (m, 2H), 2.18 (t, J = 9.0 Hz, 2H), 2.07–1.97 (m, 4H), 1.78–1.73 (m, 1H), 1.70–1.62 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 170.4, 168.8, 143.6, 141.9, 139.2, 127.7, 111.0, 109.6, 83.0, 77.5, 77.0, 76.3, 76.1, 75.5, 65.8, 63.4, 63.2, 43.2, 43.0, 41.9, 41.3, 41.2, 40.6, 37.0, 34.0, 26.9; DEPT (125 MHz, CDCl3) CH3 δ CH2 δ 111.0, 109.6, 77.5, 75.4, 65.8, 63.4, 63.2, 43.2, 41.9, 41.4, 41.2, 40.6, 37.0, 26.9; CH δ 139.2, 77.0, 76.3, 76.1, 75.4, 65.8; C δ 170.4, 168.8, 143.6, 141.9, 127.7, 83.0. IR (neat) 3430, 2943, 1754, 1742, 1581, 1382, 1257, 1194, 1081, 1030, 896, 753 cm−1; HRMS (ESI) calcd for C27H36NaO10 (M + Na) 543.2206, found 543.2207.
Aldol (33).
To a solution of i-Pr2NH (3.0 mL, 21.5 mmol, 1.1 equiv) in THF (15 mL) contained in a 100 mL flask under N2 at −78 °C was added a solution of n-BuLi (2.5 M, 8.6 mL, 21.5 mmol, 1.1 equiv). The solution was stirred at rt for 30 min. After being cooled the solution to −78 °C, a solution of methyl isobutyrate (2.2 mL, 19.5 mmol, 1.0 equiv) in THF (5 mL) was added via cannula. The solution was stirred for 1.5 h at −78 °C, after which a solution of aldehyde 14 (4.4 g, 23.4 mmol, 1.2 equiv) in THF (5 mL) was added via cannula. The reaction mixture was stirred at −78 °C for 2 h. The reaction mixture was then quenched with saturated aqueous NaHCO3 solution (50 mL). The mixture was then extracted with 50% EtOAc/hexanes (3 × 15 mL). The organic phase was washed with brine (2 × 25 mL) and dried over Na2SO4. After filtration, the solvent was evaporated. The crude product was purified using a 4 × 10 cm silica column, eluting with 10% EtOAc/hexanes, collecting 10 mL fractions. Fractions 22–58 were combined and concentrated to yield the product (5.9 g, 94%) as a colorless oil: Rf = 0.54 (25% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 3.94 (ddd, J = 8.0, 6.0, 3.0 Hz, 1H), 3.89 (dd, J = 10, 5.0 Hz, 1H), 3.85–3.81 (m, 1H), 3.70 (s, 3H), 3.43 (d, J = 3.0 Hz, 1H), 1.59 (t, J = 4.5 Hz, 2H), 1.20 (s, 3H), 1.17 (s, 3H), 0.90 (s, 9H), 0.08 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 178.0, 76.3, 62.5, 52.1, 47.3, 33.9, 26.1, 21.5, 20.8, 18.4, −5.3; DEPT (125 MHz, CDCl3) CH3 δ 52.1, 26.1, 21.5, 20.7, −5.3; CH2 δ 62.9, 33.8; CH δ 76.3; C δ 178.0, 47.3, 18.4; IR (neat) 3508, 2954, 2857, 1732, 1471, 1388, 1256, 1090 cm−1; HRMS (ESI) calcd 313.1811 for C14H30O4NaSi (M + Na), found 313.1809.
Ester (32).
To a stirring solution of ester 33 (5.9 g, 20 mmol, 1.0 equiv) in toluene (81 mL, 0.25 M) contained in a 250 mL flask under N2 was added Et3N (5.6 mL, 40 mmol, 2.0 equiv). MsCl (1.9 mL, 24 mmol, 1.2 equiv) was then slowly added. A white precipitate immediately formed; the reaction mixture was then stirred overnight at rt. The reaction mixture was then quenched with saturated aqueous NaHCO3 solution (150 mL). The mixture was then extracted with 50% EtOAc/hexanes (3 × 25 mL). The organic phase was then washed with brine (100 mL) and dried over Na2SO4. After filtration, the solution was concentrated and the crude mesylate used as is.
To a stirring solution of the previously prepared mesylate in DMSO (200 mL, 0.1 M) contained in a 500 mL flask fitted with a reflux condenser under N2 was added DBU (7.5 mL, 50 mmol, 2.5 equiv) The reaction mixture was heated at 115 °C overnight. After being cooled to rt, the reaction mixture was quenched with saturated aqueous NaHCO3 solution (250 mL) and H2O (250 mL). The mixture was extracted with 50% EtOAc/hexanes (3 × 100 mL). The organic phase was washed with brine (200 mL) and dried over Na2SO4. After filtration and concentration, the product was purified using a 4 × 11 cm silica gel column, eluted with 5% EtOAc/hexanes, collecting 10 mL fractions. Fractions 28–44 were combined and concentrated to yield the product (3.5 g, 64%) as a colorless oil, spectroscopically identical to the material previously reported:22 Rf = 0.76 (25% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) 5.84 (dt, J = 15.5, 2.0 Hz, 1H), 5.59 (dt, J = 15.5, 5.0 Hz, 1H), 4.18 (dd, J = 5.0, 1.5, Hz, 2H), 3.67 (s, 3H), 1.31 (s, 6H), 0.91 (s, 9H), 0.07 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 177.1, 134.8, 128.0, 64.0, 52.2, 44.1, 26.2, 25.2, 18.6, −4.9.
Aldehyde (34).
To a stirring solution of ester 32 (3.24 g, 8.7 mmol, 1.0 equiv) in Et2O (43 mL, 0.2 M) contained in a 100 mL flask under N2 at 0 °C was added LiAlH4 (362 mg, 9.6 mmol, 1.1 equiv). After being stirred for 1 h, the reaction mixture was quenched by the slow addition of EtOAc (5 mL), followed by a saturated aqueous solution of potassium sodium tartrate (50 mL). After being stirred for 30 min, the mixture was extracted with 50% EtOAc/hexanes (3 × 25 mL), dried over Na2SO4, filtered, and concentrated. The crude alcohol was used without purification for the subsequent oxidation step.
To a stirring solution of the aforementioned alcohol (2.90 g, 8.7 mmol, 1.0 equiv) in CH2Cl2 (87 mL, 0.1 M) contained in a 250 mL flask under N2 was added DMSO (6.2 mL, 87 mmol, 10.0 equiv) and i-Pr2NEt (10.6 mL, 61 mmol, 7.0 equiv). After being cooled to 0 °C, SO3·Pyr (5.54 g, 35 mmol, 4.0 equiv) was added to the stirring solution. After being stirred for 1 h at 0 °C, the reaction was quenched with saturated aqueous NaHCO3 solution (100 mL). After being stirred at rt for 20 min, the mixture was extracted with CH2Cl2 (3 × 30 mL). The organic phase was washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated. The crude aldehyde was purified using a 4 × 10 cm silica gel column, eluting with 10% EtOAc/hexanes, collecting 10 mL fractions. Product bearing fractions 16–36 were combined and concentrated to yield the aldehyde (2.78 g, 97%, over 2 steps) as a colorless oil: Rf = (25% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 9.38 (s, 1H), 5.64 (s, 2H), 4.20 (t, J = 1.0 Hz, 2H), 1.20 (s, 6H), 0.91 (s, 9H), 0.07 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 202.8, 131.4, 131.3, 63.8, 59.9, 48.4, 26.2, 21.6, 18.6, −5.0; DEPT (125 MHz, CDCl3) CH3 δ 26.2, 21.7, −4.9; CH δ 63.9; CH δ 202.8, 131.5, 131.3; C δ 48.6, 18.7; IR (neat) 2930, 2858, 1730, 1472, 1362, 1255, 1107, 837 cm−1: HRMS (ESI) calcd 265.1600 for C13H26O2SiNa (M + Na), found 265.1608.
Diene (35).
To a 250 mL flask was added KOtBu (1.72 g, 15.3 mmol, 1.8 equiv), followed by methyl triphenylphosphonium bromide (6.1 g, 17 mmol, 2.0 equiv). After being flushed with N2, the mixture was cooled to 0 °C and THF (75 mL) was added. The yellow mixture was stirred at 0 °C for 30 min. A solution of aldehyde 34 (2.06 g, 8.5 mmol, 1.0 equiv) in THF (5 mL) was then added via cannula, using THF (5.0 mL) to complete the transfer. After being stirred at rt for 30 min, the reaction mixture was quenched with saturated aqueous NH4Cl solution (100 mL). The mixture was extracted with 50% EtOAc/hexanes (3 × 25 mL). The organic phase was washed with brine (100 mL), dried over Na2SO4, filtered, and concentrated. The crude olefin was purified using a 2 × 10 cm silica gel column, eluting with 10% EtOAc/hexanes, collecting 5 mL fractions. Fractions 2–14 were combined and concentrated to yield the desired product (1.92 g, 94%) as a colorless oil: Rf = 0.88 (10% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 5.83 (dd, J = 17, 10 Hz, 1H), 5.64 (dt, J = 15.5, 1.5, Hz, 1H), 5.48 (dt, J = 10.5, 5.5 Hz, 1H), 4.94 (ddd, J = 17.5, 10.5, 1.0 Hz, 2H), 4.17 (dd, J = 5.5, 1.5 Hz, 2H), 1.12 (s, 6H), 0.92 (s, 9H), 0.07 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 147.3, 139.3, 126.1, 110.8, 64.4, 59.9, 27.1, 26.2, 18.7, −4.8; DEPT (125 MHz, CDCl3) CH3 δ 27.1, 26.2, −4.8; CH2 δ 110.7, 64.4; CH δ 147.3, 139.3, 126.1; C δ 59.9, 18.7; IR (neat) 3084, 2958, 2929, 2858, 1637, 1472, 1386, 1361, 1255, 1106, 1068, 973, 914, 837, 775, 665 cm−1; HRMS (ESI) calcd 263.1807 for C14H28ONaSi (M + Na), found 263.1805.
Alcohol (36).
To a stirring solution of olefin 35 (1.60 g, 6.65 mmol, 1.0 equiv) in THF (27 mL, 0.25 M) contained in a 100 mL flask was added a solution of 9-BBN in THF (0.5 M, 20 mL, 10.0 mmol, 1.5 equiv). The reaction mixture was then placed in an ultrasonic bath for 1 h. The reaction mixture was quenched with H2O (50 mL), followed by an aqueous solution of NaOH (1 M, 50 mL) and chilled in an ice bath; 30% aqueous H2O2 (20 mL) was then added in small portions. The mixture was stirred for 1 h at 0 °C. The mixture was diluted with brine (50 mL), followed by extraction with 50% EtOAc/hexanes (3 × 25 mL). The organic phase was washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 4 × 15 cm silica gel column, eluting with 5% EtOAc/hexanes until fraction 96 was reached; after this the product was eluted with 25% EtOAc/hexanes, collecting 5 mL fractions. Fractions 110–138 were combined and concentrated to yield the product (1.46 g, 85%) as a colorless oil: Rf = 0.10 (10% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 5.66 (dt, J = 16, 1.5 Hz, 1H), 5.48 (dt, J = 16, 5.5 Hz, 1H), 4.15, (dd, J = 5.0, 1.5 Hz, 2H), 3.65 (t, J = 7.0 Hz, 2H), 1.62 (t, J = 7.5 Hz, 2H), 1.33 (s, 1H), 1.04 (s, 6H), 0.91 (s, 9H), 0.07 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 140.6, 126.0, 64.3, 60.4, 45.7, 35.0, 27.7, 26.2, 18.6, −4.9; DEPT (125 MHz, CDCl3) CH3 δ 27.7, 26.2, −4.9; CH2 δ 64.3, 60.4, 45.6; CH δ 140.6, 125.9; C δ 35.0, 18.6; IR (neat) 3356, 2956, 2857, 1472, 1387, 1362, 1255, 1107, 1060, 1024, 975, 836, 776 cm−1; HRMS (ESI) calcd 281.1913 for C14H30O2NaSi (M + Na), found 281.1915.
Aldehyde (30).
To a 50 mL flask, alcohol 36 (500 mg, 1.93 mmol, 1.0 equiv) was added. After being flushed with N2, CH2Cl2 (19 mL, 0.1 M) was added. The solution was chilled to 0 °C, and i-Pr2NEt (2.4 mL, 14 mmol, 7.0 equiv) was added, followed by DMSO (1.4 mL, 19 mmol, 10.0 equiv). To the solution, SO3–Pyr(1.23 g, 7.7 mmol, 4.0 equiv) was added. The reaction mixture was then stirred at 0 °C for 1 h. The reaction mixture was then quenched with saturated aqueous NaHCO3 solution (50 mL). After being stirred for 15 min, the mixture was extracted with CH2Cl2 (2 × 10 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. The crude product was purified using a 2 × 15 cm silica gel column, eluting with 5% EtOAc/hexanes, collecting 5 mL fractions. Product containing fractions 10–15 were combined and concentrated to yield the product (377 mg, 76%) as a colorless oil: Rf = 0.40 (10% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 9.73 (t, J = 1.8 Hz, 1H), 5.75 (dt, J = 15.5, 1.5 Hz, 1H), 5.53 (dt, J = 15.5, 4.5 Hz, 1H), 4.17 (dd, J = 5.0, 1.5 Hz, 2H), 2.34 (d, J = 3.0 Hz, 2H), 1.16 (s, 6H), 0.91 (s, 9H), 0.07 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 203.5, 138.4, 127.0, 64.0, 55.3, 35.3, 27.9, 26.2, 18.6, −4.9; DEPT (125 MHz, CDCl3) CH3 δ 27.9, 26.2, −4.9; CH2 δ 64.0, 55.2; CH δ 203.5, 138.3, 127.0; C δ 35.3, 18.6; IR (neat) 2958, 2930, 2857, 2731, 1724, 1472, 1388, 1375, 1255, 1108, 1068, 976, 837, 776, 666 cm−1; HRMS (ESI) calcd 279.1756 for C14H28O2NaSi (M + Na), found 279.1760.
Lactone Alcohol (37).
A stirring solution of lactone 12 (750 mg, 1.48, mmol, 1.0 equiv) in CH2Cl2 (30 mL, 0.05 M) contained in a 50 mL flask at 0 °C was prepared under N2. To this solution, a 50:50 TFA/TFAA solution in CH2Cl2 (1.0 M, 4.44 mL, 4.44 mmol, 3.0 equiv) was added dropwise, turning the solution bright yellow in color. After being stirred for 30 min at 0 °C, Et3N (4.1 mL, 30 mmol, 20 equiv) was added and the mixture was stirred for 5 min. The solution was then transferred to a 250 mL flask and evaporated with MeOH (3 × 75 mL). The resulting oil was dissolved in PhCH3 (2.0 mL) and purified using a 3 × 15 cm silica gel column. The column was eluted with 25% EtOAc/hexanes (300 mL), 50% EtOAc/hexanes (200 mL), and 75% EtOAc/hexanes (100 mL), collecting 10 mL fractions. Fractions 46–64 were combined and concentrated to yield the product (342 mg, 87%) as a light yellow oil: Rf = 0.29 (50% EtOAc/hexanes); [α]D20 = +2.18 (c = 6.0); 1H NMR (500 MHz, CHCl3) δ 7.36–7.34 (m, 4H), 7.32–7.30 (m, 1H), 4.77 (dd, J = 9.5, 6.5 Hz, 2H), 4.60 (s, 2H), 3.77 (d, J = 12 Hz, 1H), 3.69 (ABq, J = 10.5 Hz, Δν = 36.3 Hz, 2H), 3.62 (d, J = 14.0 Hz, 1H), 2.71 (s, 1H), 2.64 (ddd, J = 14, 10, 8.0 Hz, 2H), 2.22–2.16 (m, 1H), 2.13–2.07 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 177.6, 137.6, 128.7, 128.1, 128.0, 95.1, 87.6, 70.2, 70.0, 65.4, 29.4, 25.7; DEPT (125 MHz, CDCl3) CH3 δ ; CH δ 95.1, 70.2, 70.0, 65.4, 29.4, 25.7; CH δ 128.7, 128.1, 128.0; C δ 177.6, 137.6, 87.6; IR (neat) 3454, 2940, 1765, 1605, 1461, 1414, 1382, 1208, 1170, 1110, 1043, 946, 736, 699 cm−1; HRMS (ESI) calcd 289.1052 for C14H18O5Na (M + Na), found 289.1052.
TIPS Ether (31).
To a stirring solution of alcohol 37 (933 mg, 3.5 mmol, 1.0 equiv) in CH2Cl2 (35 mL, 0.1 M) contained in a 100 mL flask under N2 was added 2,6-lutidine (1.2 mL, 11 mmol, 3.0 equiv), followed by TIPSOTf (1.4 mL, 5.3 mmol, 1.5 equiv). The reaction mixture was stirred overnight at rt, quenched with saturated aqueous NaHCO3 solution (50 mL), and extracted with CH2Cl2 (3 × 15 mL). The organic phase was washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 3 × 10 cm silica gel column, eluting with 15% EtOAc/hexanes, collecting 10 mL fractions. Fractions 14–30 were combined and concentrated to yield the product (1.28 g, 86%) as a colorless oil: Rf = 0.48 (25% EtOAc/hexanes); [α]D20 = −1.03 (c = 1.95); 1H NMR (500 MHz, CDCl3) δ 7.38–7.29 (m, 5H), 4.78 (dd, J = 10, 6.5 Hz, 2H), 4.61 (s, 2H), 3.81 (ABq, J = 10.5 Hz, Δv = 28 Hz, 2H), 3.71 (dd, J = 17, 10.5 Hz, 2H), 2.68–2.57 (m, 2H), 2.25 (ddd, J = 13.5, 10.5, 7.0 Hz, 1H), 2.14–2.05 (m, 1H), 1.16–1.09 (m, 3H), 1.07 (d, J = 7.0 Hz, 18H); 13C NMR (125 MHz, CDCl3) δ 177.2, 137.8, 128.7, 128.1, 128.0, 95.2, 87.4, 70.6, 69.9, 66.7, 29.5, 26.1, 18.1, 12.5; DEPT (125 MHz, CDCl3) CH3 δ 18.1; CH δ 95.2, 70.6, 69.9, 66.7, 29.5, 26.1; CH δ 128.7, 128.1, 128.0, 12.1; C δ 177.2, 137.8, 87.4; IR (neat) 2943, 2866, 1781, 1463, 1382, 1207, 1171, 1116, 1049, 949, 883, 811, 738, 696 cm−1; HRMS (ESI) calcd 445.2386 for C23H38O5SiNa (M + Na), found 445.2394.
Enoate (38).
To a solution of lactone 31 (150 mg, 0.36 mmol, 1.0 equiv) in THF (3.6 mL, 0.1 M) in a 10 mL flask at −78 °C was added a solution of LDA (1.0 M, 400 μL, 0.4 mmol, 1.1 equiv). After being stirred at −78 °C for 0.5 h, a solution 1 M in HMPA and 1 M in (EtO)2POCl in THF (470 μL, 0.47 mmol, 1.3 equiv) was added to the reaction mixture. After being stirred for 5 min, the reaction mixture was then warmed to rt and stirred for 1 h. After being cooled the reaction mixture to −78 °C, a solution of LDA (1.0 M, 790 μL, 0.79 mmol, 2.2 equiv) was added. The reaction mixture was stirred at −78 °C for 1.5 h. The reaction was then quenched at −78 °C with a solution of AcOH (1.0 mL) in Et2O (5.0 mL). The mixture was diluted with Et2O (50 mL) and filtered through a 4 × 1 cm pad of Celite. The solution was then concentrated to yield the phosphonate as a mixture of diastereomers which were used without further characterization.
To the aforementioned phosphonate (65 mg, 0.12 mmol, 1.0 equiv) contained in a 15 mL flask, 18-crown-6 (159 mg, 0.60 mmol. 5.0 equiv) was added. The flask was flushed with N2 and THF (3.0 mL, 0.04 M) was added. After being cooled to −78 °C, a solution of KHMDS in toluene (0.5 M, 270 μL, 0.13 mmol, 1.1 equiv) was added dropwise to the stirring mixture. After being stirred for 30 min at −78 °C, a solution of aldehyde 33 (37 mg, 0.15 mmol, 1.2 equiv) in THF (500 μL) was added dropwise; the transfer was completed with THF (500 μL). The reaction mixture was promptly placed in a 30 °C water bath and stirred overnight at rt. The reaction mixture was quenched with saturated aqueous NH4Cl solution (10 mL), extracted with EtOAc (3 × 10 mL), washed with brine (25 mL), dried over Na2SO4, filtered, and concentrated. 1H NMR analysis of the crude product indicated a 5:1 ratio of Z- to E-enoates by integration of the enoate proton peaks. The product was purified using a 2 × 10 cm silica gel column, eluting with 5% EtOAc/hexanes, collecting 5 mL fractions. The product containing fractions were combined and concentrated to yield the product (79 mg, 51% over 2 steps) as a colorless oil: Rf = 0.29 (10% EtOAc/hexanes); [α]D20 = −0.69 (c = 1.73, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.37–7.29 (m, 5H), 6.15 (dddd, J = 9.5, 5.5, 2.0, 2.0 Hz, 1H), 5.63 (ddd, J = 15.5, 1.5, 1.5 Hz, 1H), 5.48 (ddd, J = 16, 5.5, 5.5 Hz, 1H), 4.78 (dd, J = 8.5, 6.5 Hz, 2H), 4.60 (s, 2H), 4.17 (dd, J = 5.5, 1.5 Hz, 2H), 3.77 (ABq, J = 10.5 Hz, Δv = 23.8 Hz, 2H), 3.69 (d, J = 1.5 Hz, 2H), 2.90 (dd, J = 17, 2.0 Hz, 1H), 2.81 (q, J = 7.5 Hz, 1H), 2.75–2.67 (m, 2H), 1.15–1.04 (m, 27 H), 0.92 (s, 9H), 0.08 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 169.3, 140.9, 139.8, 137.8, 128.6, 128.1, 128.0, 126.5, 126.4, 95.2, 83.5, 70.0, 69.8, 65.9, 64.4, 39.8, 36.5, 33.5, 27.3, 26.2, 18.6, 18.1, 12.1, −4.8; DEPT (125 MHz, CDCl3) CH3 δ 27.2, 26.2, 18.1, −4.8; CH2 δ 95.2, 70.0, 69.8, 65.9, 64.4, 39.9, 33.5; CH δ 140.9, 139.8, 128.6, 128.1, 128.0, 126.5, 12.1; C δ 169.3, 137.8, 126.4, 83.5, 26.5, 18.6; IR (neat) 2957, 2866, 1761, 1671, 1463, 1367, 1254, 1117, 1049, 882, 837, 777, 684 cm−1; HRMS (ESI) calcd 683.4139 for C37H64O6NaSi2 (M + Na), found 683.4139.
Alcohol (39).
To a 25 mL flask containing enoate 38 (133 mg, 0.20 mmol, 1.0 equiv) was added 3:1:1 AcOH/THF/H2O (10 mL). The flask was stoppered and stirred in an oil bath at 45 °C for 1 h. Upon cooling to rt, the reaction mixture was slowly added to a stirring saturated aqueous NaHCO3 solution (100 mL). The mixture was extracted with EtOAc (3 × 15 mL), washed with saturated aqueous NaHCO3 solution (50 mL) and brine (50 mL). The organic phase was then dried over Na2SO4, filtered, and concentrated. The crude product was purified using a 2 × 10 cm silica gel column, eluting with 25% EtOAc/hexanes, collecting 5 mL fractions. Fractions 10–25 were combined and concentrated to yield the product (91 mg, 83%) as a colorless oil: Rf = 0.15 (25% EtOAc/hexanes); [α]D20 = −5.4 (c = 0.50, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.38–7.29 (m, 5H), 6.14 (dddd, J = 10, 7.5, 5.0, 2.5 Hz, 1H), 5.69 (dd, J = 15.5, 1.5 Hz, 1H), 5.56 (dt, J = 16, 5.5 Hz, 1H), 4.78 (dd, J = 8.5, 6.0 Hz, 2H), 4.60 (s, 2H), 4.12 (dd, J = 6.0, 1.0 Hz, 2H), 3.77 (ABq, J = 10.5 Hz, Δv = 21 Hz, 2H), 3.69 (dd, J = 13, 11 Hz, 2H), 2.91 (dd, J = 16, 2.0 Hz, 1H), 2.85 (dddd, J = 15.5, 8.0, 3.5, 2.0 Hz, 1H), 2.74 (dd, J = 16, 2.0 Hz, 1H), 2.67 (dddd, J = 15, 9.5, 7.5, 2.0 Hz, 1H), 1.12–1.07 (m, 3H), 1.05 (s, 18 H), 1.04 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 169.4, 141.8, 140.5, 137.8, 128.7, 128.1, 128.0, 126.8, 126.1, 95.2, 83.6, 70.0, 69.8, 65.9, 64.1, 39.7, 36.7, 33.5, 27.3, 27.1, 18.1, 12.1; DEPT (125 MHz, CDCl3) CH3 δ 27.3, 27.1, 18.1; CH2 δ 95.2, 70.0, 69.8, 65.9, 64.1, 39.7, 33.5; CH δ 141.8, 140.5, 128.7, 128.1, 128.0, 126.1, 12.1; C δ 169.4, 137.8, 126.1, 83.6, 36.7; IR (neat) 3464, 2943, 2866, 1757, 1684, 1653, 1464, 1383, 1368, 1210, 1170, 1117, 1046, 973, 882, 801, 734, 682 cm−1; HRMS (ESI) calcd 569.3274 for C31H50O6NaSi (M + Na), found 569.3273.
Aldehyde (29).
To a solution of alcohol 39 (91 mg, 0.17 mmol, 1.0 equiv) in CH2Cl2 (3.3 mL, 0.05 M) contained in a 15 mL flask under N2, was added DMSO (120 μL, 1.7 mmol, 10 equiv), followed by i-Pr2NEt (210 μL, 1.2 mmol, 7.0 equiv). After being cooled to 0 °C, SO3·Pyr (108 mg, 0.68 mmol, 4.0 equiv) was added. The reaction mixture was stirred at 0 °C for 1 h and then quenched with saturated aqueous NaHCO3 solution (25 mL). The mixture was extracted with CH2Cl2 (3 × 10 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 2 × 10 cm silica column, eluting with 15% EtOAc/hexanes, collecting 5 mL fractions. Fractions 8–20 were combined and concentrated to yield the product (60 mg, 66%) as a colorless oil: Rf = 0.38 (25% EtOAc/hexanes); [α]D20 = −59.4 (c = 1.75, CHCl3); 1H NMR (500 MHz, CDCl3) δ 9.53 (d, J = 8.0 Hz, 1H), 7.37–7.30 (m, 5H), 6.81 (d, J = 15.5 Hz, 1H), 6.08–6.03 (m, 2H), 4.77 (s, 2H), 4.59 (s, 2H), 3.77 (dd, J = 14, 10.5 Hz, 2H), 3.68 (dd, J = 12.5, 10.5 Hz, 2H), 2.98 (dd, J = 15, 8.0 Hz, 1H), 2.91 (d, J = 16.5 Hz, 1H), 2.81 (dd, J = 15, 7.5 Hz, 1H), 2.75 (d, J = 16 Hz, 1H), 1.16 (s, 6H), 1.12–1.04 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 194.5, 169.2, 166.7, 138.0, 137.8, 130.1, 128.7, 128.3, 128.1, 128.0, 95.2, 83.9, 70.0, 69.9, 66.0, 38.9, 38.5, 33.4, 26.4, 26.3, 18.1, 12.1; DEPT (125 MHz, CDCl3) CH3 δ 26.4, 26.3, 18.1; CH2 δ 95.2, 70.0, 69.9, 66.0, 38.9, 33.4; CH δ 194.5, 166.7, 138.0, 130.1, 128.7, 128.3, 128.1, 12.1; C δ 169.2, 137.8, 128.0, 83.9, 38.2; IR (neat) 2944, 2867, 1757, 1693, 1464, 1369, 1117, 1047, 883, 803, 739 cm−1; HRMS (ESI) calcd 567.3118 for C31H48O6NaSi (M + Na), found 567.3116
Bispyran (40).
To a stirring solution of hydroxyallylsilane 3 (66 mg, 0.092 mmol, 1.0 equiv) and aldehyde 29 (60 mg, 0.11 mmol, 1.2 equiv) in Et2O (2.3 mL, 0.04 M) contained in a 10 mL flask under N2 at −78 °C was added a solution of TMSOTf in Et2O (1.0 M, 120 μL, 0.12 mmol, 1.3 equiv). The reaction mixture was stirred at −78 °C for 3 h and then quenched with i-Pr2NEt (500 μL). After being stirred for 5 min at −78 °C, the solution was added to saturated aqueous NaHCO3 solution (25 mL). The mixture was extracted with EtOAc (3 × 10 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using a 2 × 10 cm silica column, eluting with 10% EtOAc/hexanes, collecting 5 mL fractions. The product containing fractions, 8–22, were combined and concentrated to yield the product (90 mg, 83%) as a colorless oil: Rf = 0.83 (50% EtOAc/hexanes); [α]D20 = +1.56 (c = 3.4, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.71–7.68 (m, 4H), 7.45–7.35 (m, 10H), 7.32–7.29 (m, 1H), 7.20 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.0 Hz, 2H), 6.12 (t, J = 7.5 Hz, 1H), 5.63 (d, J = 16 Hz, 1H), 5.45 (dd, J = 15.5, 5.5 Hz, 1H), 4.78 (s, 2H), 4.73 (d, J = 11.5 Hz, 2H), 4.66 (s, 1H), 4.61 (s, 2H), 4.55 (s, 1H), 4.43 (ABq, J = 10.5 Hz, Δv = 35.1 Hz, 2H), 3.98–3.93 (m, 1H), 3.85–3.83 (m, 1H), 3.80 (s, 3H), 3.78 (ABq, J = 10 Hz, Δv = 20.2 Hz, 2H), 3.76–3.73 (m, 1H), 3.69 (s, 2H), 3.64–3.61 (m, 1H), 3.59–3.55 (m, 1H), 3.51–3.45 (m, 1H), 2.90 (d, J = 16 Hz, 1H), 2.83–2.64 (m, 3H), 2.30 (d, J = 23.5 Hz, 1H), 2.27 (d, J = 24 Hz, 1H), 2.18 (t, J = 15.5 Hz, 2H), 2.07–2.01 (m, 3H), 1.98–1.91 (m, 2H), 1.85–1.73 (m, 2H), 1.70–1.61 (m, 4H), 1.08–1.03 (m, 36H); 13C NMR (125 MHz, CDCl3) δ 169.3, 159.3, 145.0, 144.3, 140.7, 140.6, 137.8, 135.8, 134.1, 134.0, 131.2, 129.8, 129.6, 128.6, 128.1, 128.06, 128.01, 127.85, 127.84 126.8, 114.0, 109.0, 108.7, 95.2, 83.5, 79.2, 75.1, 75.0, 74.8, 72.8, 72.3, 70.0, 69.8, 66.0, 60.6, 55.5, 43.1, 42.6, 41.5, 41.4, 41.2, 40.4, 39.8, 38.0, 36.6, 33.5, 27.2, 27.1, 27.0, 19.4, 18.1, 12.1; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 27.2, 18.1; CH2 δ 109.1, 108.7, 95.2, 72.3, 70.0, 69.8, 66.0, 60.6, 43.1, 42.6, 41.5, 41.4, 41.3, 40.4, 39.8, 38.1, 33.5; CH δ 140.7, 140.6, 135.8, 129.8, 129.6, 128.7, 128.1, 128.0, 128.0, 127.9, 114.0, 79.2, 75.1, 75.0, 74.8, 72.8, 12.1; C δ 169.3, 159.3, 145.0, 144.3, 137.8, 134.1, 134.0, 131.2, 83.5, 36.6, 19.4; IR (neat) 3072, 2941, 1761, 1613, 1514, 1464, 1428, 1362, 1248, 1110, 846, 730, 703 cm−1; HRMS (ESI) calcd 1191.6753 for C71H100O10NaSi2 (M + Na), found 1191.6760.
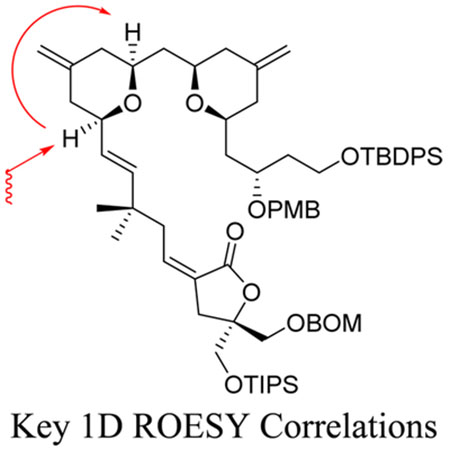
C1 Alcohol (41).
To a stirring solution of ester 40 (59 mg, 0.05 mmol, 1.0 equiv) in THF (1.0 mL, 0.05 M) contained in a 10 mL polyethylene vial at 0 °C was added a solution of 20% HF·Pyr in pyridine (1.25 mL, 25 mL/mmol). The solution was stirred at 0 °C for 5 h, after which it was quenched by slowly pipetting it into stirring, saturated aqueous NaHCO3 solution (30 mL). The mixture was extracted with EtOAc (3 × 10 mL), dried over Na2SO4, filtered, and concentrated. The crude alcohol was purified using a 2 × 10 cm silica gel column, eluting with 30% EtOAc/hexanes, collecting 5 mL fractions. Concentration of fractions 10–36 yielded the product (32 mg, 68%) as a colorless oil: Rf = 0.48 (50% EtOAc/hexanes); [α]D20 = −0.88 (c = 0.57, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.37–7.28 (m, 5H), 7.26 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 6.12 (t, J = 8.0 Hz, 1H), 5.66 (d, J = 16.5 Hz, 1H), 5.45 (dd, J = 15.5, 5.5 Hz, 1H), 4.77 (s, 2H), 4.74 (m, 2H), 4.69 (s, 1H), 4.60 (s, 2H), 4.60 (s, 1H), 4.48 (q, J = 10 Hz, 2H), 4.02–3.97 (m, 1H), 3.85–3.80 (m, 1H), 3.80 (s, 3H), 3.76–3.73 (m, 1H), 3.77 (ABq, J = 10.5 Hz, Δv = 20.9 Hz, 2H), 3.68 (s, 2H), 3.58–3.53 (m, 1H), 3.52–3.43 (m, 2H), 2.90 (d, J = 17 Hz, 1H), 2.80 (dd, J = 16, 8.0 Hz, 1H), 2.73 (d, J = 19 Hz, 1H), 2.69 (dd, J = 15.5, 7.5 Hz, 1H), 2.36 (s, 1H), 2.29–2.18 (m, 4H), 2.06–1.92 (m, 6H), 1.80 (dddd, J = 14.5, 9.0, 8.5, 2.5 Hz, 1H), 1.75–1.62 (m, 4H), 1.10–1.03 (m, 27H); 13C NMR (125 MHz, CDCl3) δ 169.3, 159.6, 144.7, 144.4, 140.7, 140.6, 137.9, 130.7, 129.7, 129.6, 128.7, 128.1, 128.0, 126.7, 114.2, 109.0, 108.9, 95.2, 83.6, 79.4, 77.4, 75.5, 75.3, 75.1, 72.1, 70.1, 69.8, 66.0, 60.5, 55.5, 43.0, 41.8, 41.5, 41.1, 40.5, 39.8, 36.9, 36.6, 33.6, 27.1, 27.1, 18.1, 12.1; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 27.1, 27.0, 18.1; CH2 δ 109.0, 108.9, 95.2, 72.1, 70.0, 69.8, 66.0, 60.5, 43.0, 41.8, 41.5, 41.1, 40.5, 39.8, 36.8, 33.5; CH δ 140.7, 140.6, 129.7, 129.6, 128.6, 128.1, 114.1, 79.4, 77.4, 75.5, 75.2, 75.2, 12.1; C δ 169.3, 159.6, 144.7, 144.4, 137.9, 130.7, 126.7, 83.6, 36.6; IR (neat) 3488, 2942, 1758, 1652, 1612, 1514, 1464, 1366, 1248, 1048, 884, 804, 739, 684 cm−1; HRMS (ESI) calcd 953.5575 for C55H82O10NaSi (M + Na), found 953.5592.
C1 Acid (42).
To a stirring solution of alcohol 41 (37 mg, 0.04 mmol, 1.0 equiv) in CH2Cl2 (800 μL, 0.05 M) contained in a 5 mL vial under N2 was added i-Pr2NEt (70 μL, 0.40 mmol, 10.0 equiv), followed by DMSO (20 μL, 0.28 mmol, 7.0 equiv). The mixture was cooled to 0 °C, and SO3-Pyr (26 mg, 0.16 mmol, 4.0 equiv) was added. After being stirred at 0 °C for 1 h, the reaction mixture was quenched with saturated aqueous NaHCO3 solution (25 mL) and extracted with CH2Cl2 (3 × 10 mL). The organic phase was then dried over Na2SO4, filtered, and concentrated. The product was purified on a 1 × 10 cm silica gel column, eluting with 20% EtOAc/hexanes, collecting 5 mL fractions. Fractions 6–14 were concentrated to yield the intermediate aldehyde as a colorless oil which was used without further characterization (28 mg, 75%).
To a stirring solution of the aforementioned aldehyde (28 mg, 0.030 mmol, 1.0 equiv) in t-BuOH (430 μL, 14.3 mL/mmol) and 2-methyl-2-butene (430 μL, 14.3 mL/mmol) contained in a 5 mL vial was added an aqueous solution of KH2PO4 (1.25 M, 144 μL, 0.18 mmol, 6.0 equiv). The rapidly stirred reaction mixture was cooled to −5 °C, and 80% NaClO2 (17 mg, 0.15 mmol, 5.0 equiv) was added. The reaction mixture was stirred at 0 °C for 1.5 h and then quenched with aqueous pH 4.0 acetate buffer (0.1 M, 25 mL). After extraction with EtOAc (3 × 10 mL), the solution was dried over Na2SO4, filtered, and concentrated. The resulting product was purified using a 2 × 15 cm silica gel column, eluting with 40% EtOAc/hexanes, collecting 5 mL fractions. Fractions 8–28 were combined and concentrated to yield the product as a colorless oil (15 mg, 54%): Rf = 0.48 (20/5/0.3 PhCH3/EtOAc/AcOH); [α]D20 = +9.4 (c = 0.74, CHCl3); 500 MHz 1H NMR (C6D6) δ 7.29 (dd, J = 7.5, 3.0 Hz, 4H), 7.16 (t, J = 7.0 Hz, 2H), 7.07 (t, J = 10 Hz, 1H), 6.81 (d, J = 8.5 Hz, 2H), 6.00 (t, J = 8.0 Hz, 1H), 5.69 (d, J = 16 Hz, 1H), 5.55 (dd, J = 16.5, 6.0 Hz, 1H), 4.74 (s, 2H), 4.65 (d, J = 4.0 Hz, 2H), 4.55 (s, 2H), 4.51 (ABq, J = 11 Hz, Δv = 54.2 Hz, 2H), 4.48 (s, 2H), 3.81 (dd, J = 10, 5.5 Hz, 1H), 3.69–3.62 (m, 2H), 3.67 (d, J = 3.0 Hz, 2H), 3.54–3.47 (m, 2H), 3.51 (d, J = 5.5 Hz, 2H), 3.31 (s, 3H), 3.02 (dd, J = 14.5, 7.5 Hz, 1H), 2.86 (dd, J = 14.5, 7.0 Hz, 1H), 2.69 (dd, J = 16.5, 2.0 Hz, 1H), 2.50–2.42 (m, 3H), 2.18 (t, J = 14 Hz, 2H), 2.12–2.03 (m, 3H), 1.97–1.76 (m, 5H), 1.66–1.54 (m, 2H), 1.04–1.00 (m, 27H); 13C NMR (125 MHz, CDCl3) δ 174.5, 169.3, 159.6, 144.5, 144.3, 140.8, 140.7, 137.8, 130.3, 129.7, 128.7, 128.1, 128.0, 128.0, 126.7, 114.2, 109.1, 109.0, 95.2, 83.6, 79.3, 75.3, 75.2, 75.0, 72.9, 72.4, 70.1, 69.8, 66.0, 55.5, 43.0, 42.0, 41.4, 41.3, 41.0, 40.6, 40.0, 39.8, 36.7, 33.6, 27.1, 27.1, 18.1, 12.1; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 27.1, 27.0, 18.1; CH2 δ 109.1, 109.0, 95.2, 72.4, 70.0, 69.8, 66.0, 43.0, 41.9, 41.4, 41.3, 41.0, 40.6, 40.0, 39.8, 33.5; CH δ 140.7, 140.6, 129.7, 128.6, 128.1, 128.0, 128.0, 114.1, 79.3, 75.2, 75.2, 75.0, 72.9, 12.1; C δ 174.5, 169.3, 159.6, 144.5, 144.3, 137.8, 130.3, 126.7, 83.6, 36.7; IR (neat) 2943, 2867, 1758, 1712, 1653, 1613, 1514, 1464, 1367, 1249, 1172, 1112, 1046, 884, 805, 739 cm−1; HRMS (ESI) calcd 967.5368 for C55H80O11NaSi (M + Na), found 967.5378.
Macrolactone (43).
To a stirring solution of acid 42 (0.037 mmol, 1.0 equiv) in THF (740 μL, 0.05 M) contained in a 10 mL polyethylene vial at rt was added a 20% solution of HF·Pyr in pyridine (925 μL, 25 mmol/mL). The mixture was stirred at rt for 2 days, then quenched by pouring into aqueous pH 4.0 acetate buffer (0.1 M, 25 mL). The mixture was extracted with EtOAc (3 × 10 mL), dried over Na2SO4, filtered, and concentrated. The product was purified on a 1 × 10 cm silica gel column, eluting with 3% MeOH/CH2Cl2, collecting 5 mL fractions. Fractions 4–16 were concentrated to yield the product as a colorless oil which was used without characterization for the subsequent macrolactonization.
To a stirring solution of the aforementioned seco-acid in THF (1.2 mL, 0.03 M) contained in a 10 mL flask under N2 was added a solution of Et3N in THF (1.0 M, 220 μL, 0.22 mmol, 6.0 equiv). The reaction mixture was cooled to 0 °C, and a solution of 2,4,6-trichlorobenzoyl chloride in THF (1.0 M, 110 μL, 0.11 mmol, 3.0 equiv) was added. The reaction mixture was stirred at 0 °C for 5 min and warmed to rt. The reaction mixture was then stirred at rt for 3 h. The resulting solution was taken up into PhCH3 (10 mL) in a 25 mL gastight syringe and added over 16 h via syringe pump to a stirring solution of DMAP (90 mg, 0.74 mmol, 20.0 equiv) in PhCH3 (27 mL) contained in a 100 mL flask under N2 at 40 °C, achieving a final concentration of 0.001 M in substrate. After 16 h, PhCH3 (1.0 mL) was used to complete the transfer of the reactant. The reaction mixture was stirred for an additional h at 40 °C, then cooled to rt, quenched with brine (50 mL), and extracted with EtOAc (3 × 10 mL). After being dried over Na2SO4, the solution was filtered and concentrated. The product was purified using a 1 × 12 cm silica gel column, eluting with 15% EtOAc/hexanes, collecting 5 mL fractions. Fractions 12–24 were combined and concentrated to yield the product as a colorless oil (11 mg, 38%): Rf = 0.25 (25% EtOAc/hexanes); [α]D20 = +22.4 (c = 0.42, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.36–7.28 (m, 5H), 7.24 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 9.0 Hz, 2H), 6.17 (t, J = 8.5 Hz, 1H), 5.59 (d, J = 17 Hz, 1H), 5.40 (dd, J = 16, 4.0 Hz, 1H), 4.90 (d, J = 6.5 Hz, 1H), 4.78 (d, J = 6.5 Hz, 1H), 4.72–4.70 (m, 3H), 4.65 (s, 1H), 4.62 (ABq, J = 12 Hz, Δv = 35.9 Hz, 2H), 4.53 (m, 1H), 4.46 (ABq, J = 11 Hz, Δv = 38.5 Hz, 2H), 4.09 (m, 1H), 4.01 (d, J = 11.5 Hz, 1H), 3.81 (s, 3H), 3.73–3.71 (m, 1H), 3.69 (q, J = 10.5 Hz, 2H), 3.50–3.41 (m, 2H), 3.36–3.32 (m, 1H), 2.81–2.74 (m, 3H), 2.62–2.54 (m, 3H), 2.26 (d, J = 13.5 Hz, 2H), 2.21 (d, J = 13.5 Hz, 1H), 2.15 (d, J = 13.5 Hz, 1H), 2.01–1.89 (m, 5H), 1.79–1.69 (m, 2H), 1.60–1.54(m, 1H), 1.11 (s, 3H), 1.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.7, 168.6, 159.5, 144.6, 144.5, 141.4, 137.8, 137.8, 130.5, 129.5,129.0,128.7,128.0,128.0,125.8,114.1,109.0,108.9,94.9,81.4, 78.5, 77.4, 75.5, 75.4, 73.5, 71.8, 69.9, 65.1, 55.5, 43.7, 42.3, 41.3, 41.3, 41.2, 41.0, 40.3, 37.9, 34.8, 22.6, 14.3; DEPT (125 MHz, CDCl3) CH3 δ 55.5, 14.3; CH2 δ 109.0, 108.9, 94.9, 71.8, 69.9, 65.1, 43.6, 42.3, 41.3, 41.3, 41.2, 41.0, 34.8, 22.5; CH δ 141.4, 137.8, 129.5, 129.0, 128.7, 128.1, 128.0, 114.1, 78.5, 77.5, 75.5, 75.4, 73.5; C δ 170.7, 168.6, 159.5, 144.6, 144.5, 137.8, 130.5, 125.8, 81.4, 37.9; IR (neat) 3057, 2916, 1762, 1744, 1707, 1675, 1632, 1576, 1562, 1496, 1446, 1343, 1220, 1077, 1007, 946, 867, 809, 698 cm−1; HRMS (ESI) calcd 793.3928 for C46H58O10Na (M + Na), found 793.3939.
Merle 48.
To a stirring solution of macrolactone 43 (9.0 mg, 0.012 mmol, 1.0 equiv) in CH2Cl2 (1.2 mL, 0.01 M) contained in a 5 mL vial was added H2O (30 μL). After being cooled to 0 °C, DDQ (27 mg, 0.12 mmol, 10.0 equiv) was added and the reaction mixture was stirred at 0 °C for 1 h. The reaction mixture was quenched with saturated aqueous NaHCO3 solution (10 mL), then extracted with CH2Cl2 (3 × 5 mL), dried over Na2SO4, filtered, and concentrated. The crude alcohol was used without purification for the subsequent reaction.
To the previously prepared alcohol contained in a 5 mL vial, a solution of LiBF4 in 25:1 MeCN/H2O (0.25 M, 2.0 mL, 45.0 equiv) was added. The vial was flushed with N2, capped, and heated at 80 °C overnight. After being cooled to rt, the reaction mixture was concentrated to dryness and taken up in EtOAc (10 mL). The solution was partitioned with brine (25 mL) and subsequently extracted with EtOAc (2 × 10 mL). The combined organic phases were dried over Na2SO4, filtered, and concentrated. The crude product was purified using a 1 × 11 cm silica gel column, eluting with 35% EtOAc/pentane, collecting 5 mL fractions. Fractions 20–48 were combined and concentrated to yield the macrolide as a colorless oil (3.8 mg, 59% over 2 steps): Rf = 0.36 (50% EtOAc/hexanes); [α]D20 = −27 (c = 0.12, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.24 (t, J = 8.5 Hz, 1H), 5.58 (d, J = 16 Hz, 1H), 5.34 (dd, J = 16, 5.5 Hz, 1H), 4.78 (s, 2H), 4.75 (s, 2H), 4.56 (d, J = 11.5 Hz, 1H), 4.34 (m, 1H), 4.00 (d, J = 12 Hz, 1H), 3.70–3.63 (m, 4H), 3.45–3.31 (m, 4H), 2.88 (d, J = 15 Hz, 1H), 2.82 (dd, J = 14, 4.5 Hz, 1H), 2.59 (d, J = 16 Hz, 1H), 2.45 (dd, J = 9.5, 4.0 Hz, 1H), 2.34 (d, J = 13 Hz, 1H), 2.20–2.14 (m, 5H), 2.03–1.95 (m, 4H), 1.90 (t, J = 11.5 Hz, 1H), 1.77 (t, J = 14.5 Hz, 1H), 1.64–1.59 (m, 2H), 1.12 (s, 3H), 1.10 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.1, 169.0, 143.8, 143.6, 141.7, 139.4, 128.3, 126.1, 109.6, 109.4, 82.7, 79.8, 76.1, 75.3, 75.1, 66.0, 65.7, 62.0, 44.6, 43.8, 42.7, 41.8, 41.6, 41.3, 41.3, 40.3, 38.2, 34.6, 30.3, 25.1; DEPT (125 MHz, CDCl3) CH3 δ 30.3, 25.1; CH2 δ 109.6, 109.4, 66.0, 62.0, 44.6, 43.8, 42.7, 41.8, 41.6, 41.3, 41.3, 40.3, 34.6; CH δ 141.7, 139.4, 128.3, 79.8, 76.1, 75.3, 75.1, 65.7; C δ 170.1, 169.0, 143.8, 143.6, 126.1, 82.7, 38.2; IR (neat) 3444, 2940, 1743, 1671, 1653, 1559, 1541, 1437, 1366, 1327, 1281, 1246, 1219, 1161, 1123, 1098, 1072, 1042, 1000, 981, 936, 893, 792 cm−1 HRMS (ESI) calcd 553.2777 for C30H42O8Na (M + Na), found 553.2778.
Molecular Modeling.
Conformational Searching.
The initial structures for Merle 46, Merle 47, and Merle 48 were built based on the crystal structure of bryostatin 155 from the Cambridge Structural Database (reference code BOKKIV) to keep the structure of the (A + B)-rings consistent. All searches were performed using mixed torsional/large-scale low-mode sampling in MacroModel56–58 with the OPLS 2005 force field59 in octanol implicit solvent. During the searches torsions were varied for 10,000 steps with enhanced sampling, but the chiral centers and double bonds were restricted to their crystal conformations. Low mode displacements were between 3 and 18 Å. After each step the resulting structure was energy-minimized to a gradient convergence of 0.05. The minimized structure was then compared to previously stored structures and either kept as a unique conformer or rejected as a duplicate, using a 0.75 Å rmsd cutoff to the heavy atoms in the central macrolide ring structure. A set of low-energy conformers for each structure, with energies with 3 kcal/mol of the global minimum, were passed on to the docking program.
Docking.
The crystal structure of the C1b domain of PKCδ60 was prepared for docking by adding hydrogen atoms and deleting the phorbol-13-acetate ligand. This was saved to a separate file to be used as a template for the similarity constraint (see below). Docking was done using the program GOLD, version 5.2.2,61 which uses a genetic algorithm to optimize the set of interactions between the ligand and the protein. Default settings were used for the genetic algorithm. The binding site was defined as a sphere with a 10.0 Å radius, centered on the Nε atom of residue Gln 257. For each conformer, 20 docking runs were performed, with no early termination, using the GoldScore scoring function with an internal ligand energy offset. Free corners of ligand rings were allowed to flip above or below the plane of their neighboring atoms during docking, and intramolecular hydrogen bonds in the ligand were allowed to form. Torsion angle distributions were from the CSD. A template similarity constraint was added to bias the conformation of docked ligands toward solutions where the acceptor atoms in the ligand were close in space to the acceptor atoms in bound phorbol-13-O-acetate from the crystal structure.
Energy Minimization.
A subset of diverse poses with fitness score >40.0 were further refined by energy minimization in MacroModel,57 using the OPLS_2005 force field and octanol implicit solvent. All atoms in the C1 domain were held fixed, while ligand atoms were free to move, and the complex was minimized using the Polak-Ribiere conjugate gradient scheme to a gradient convergence of 0.05. Hydrogen bonds in the docked poses were preserved using distance constraints.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by the National Institutes of Health through Grant No. GM28961 and was supported in part by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (Project Z1A BC 005270). The project was also funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract HHSN261200800001E.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b01516.
1H and 13C NMR spectra of all new compounds(PDF)
The authors declare no competing financial interest.
DEDICATION
We would like to dedicate this paper to the life and memory of our dear friend and continual source of inspiration, Dr. Merle Haggard, the namesake for the compounds in this series.
REFERENCES
- (1).Pettit GR; Day JF; Hartwell JL; Wood HB Nature 1970, 227, 962. [DOI] [PubMed] [Google Scholar]
- (2).Pettit GR; Herald CL; Hogan F In Anticancer Drug Development; Baguley BC, Kerr DJ, Eds.; Academic Press: San Diego, CA, 2002; pp 203–235. [Google Scholar]
- (3).Hale KJ; Hummersone MG; Manaviazar S; Frigerio M Nat. Prod. Rep 2002, 19, 413–453. [DOI] [PubMed] [Google Scholar]
- (4).Hale KJ; Manaviazar S Chem. - Asian J 2010, 5, 704–754. [DOI] [PubMed] [Google Scholar]
- (5).Mehla R; Bivalkar-Mehla S; Zhang R; Handy I; Albrecht H; Giri S; Nagarkatti P; Nagarkatti M; Chauhan A PLoS One 2010, 5 (6) , e11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sun MK; Hongpaisan J; Nelson TJ; Alkon DL Proc. Natl. Acad. Sci. U. S. A 2008, 105 (36), 13620–13625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Denvir J; Neitch S; Fan J; Niles RM; Boskovic G; Schreurs BG; Primerano DA; Alkon DL J. Alzheimer’s Dis 2015, 46, 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hofmann J Curr. Cancer Drug Targets 2004, 4, 125–146. [DOI] [PubMed] [Google Scholar]
- (9).Battaini F Pharmacol. Res 2001, 44 (5), 353–361. [DOI] [PubMed] [Google Scholar]
- (10).Colon-Gonzalez F; Kazanietz MG Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2006, 1761, 827–837. [DOI] [PubMed] [Google Scholar]
- (11).Hennings H; Boutwell RK Cancer Res. 1970, 30, 312–320. [PubMed] [Google Scholar]
- (12).Hennings H; Blumberg PM; Pettit GR; Herald CL; Shores R; Yuspa SH Carcinogenesis 1987, 8 (9), 1343–1346. [DOI] [PubMed] [Google Scholar]
- (13).Kraft AS; Smith JB; Berkow RL Proc. Natl. Acad. Sci. U. S. A 1986, 83, 1334–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Keck GE; Kraft MB; Truong AP; Li W; Sanchez CC; Kedei N; Lewin NE; Blumberg PM J. Am. Chem. Soc 2008, 130, 6660–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Keck GE; Poudel YB; Welch DS; Kraft MB; Truong AP; Stephens JC; Kedei N; Lewin NE; Blumberg PM Org. Lett 2009, 11 (3), 593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Andrews IP; Ketcham JM; Blumberg PM; Kedei N; Lewin NE; Krische MJ J. Am. Chem. Soc 2014, 136, 13209–13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kedei N; Kraft MB; Keck GE; Herald CL; Melody N; Pettit GR; Blumberg PM J. Nat. Prod 2015, 78, 896–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Marquez VE; Blumberg PM Acc. Chem. Res 2003, 36, 434–443. [DOI] [PubMed] [Google Scholar]
- (19).Keck GE; Li W; Kraft MB; Kedei N; Lewin NE; Blumberg PM Org. Lett. 2009, 11 (11), 2277–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Keck GE; Poudel YB; Rudra A; Stephens JC; Kedei N; Lewin NE; Peach ML; Blumberg PM Angew. Chem., Int. Ed 2010, 49 (27), 4580–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Keck GE; Poudel YB; Rudra A; Stephens JC; Kedei N; Lewin NE; Blumberg PM Bioorg. Med. Chem. Lett 2012, 22 (12), 4084–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Petersen ME Synthesis of structurally simplified bryostatin analogues: Probing the underlying biology and progressing towards more ‘drug like’ compounds; Ph.D. Thesis, University of Utah, Salt Lake City, UT, 2014. [Google Scholar]
- (23).Wender PA; Cribbs CM; Koehler KF; Sharkey NA; Herald CL; Kamano Y; Pettit GR; Blumberg PM Proc. Natl. Acad. Sci. U. S. A 1988, 85, 7197–7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kang JH; Kim SY; Lee J; Marquez VE; Lewin NE; Pearce LV; Blumberg PM J. Med. Chem 2004, 47, 4000–4007. [DOI] [PubMed] [Google Scholar]
- (25).Nacro K; Bienfait B; Lee J; Han KC; Kang JH; Benzaria S; Lewin NE; Bhattacharyya DK; Blumberg PM; Marquez VE J. Med. Chem 2000, 43, 921–944. [DOI] [PubMed] [Google Scholar]
- (26).Lee J; Wang S; Milne GWA; Sharma R; Lewin NE; Blumberg PM; Marquez VE J. Med. Chem 1996, 39, 29–35. [DOI] [PubMed] [Google Scholar]
- (27).Nacro K; Bienfait B; Lee J; Han KC; Kang JH; Benzaria S; Lewin NE; Bhattacharyya DK; Blumberg PM; Marquez VE J. Med. Chem 2000, 43, 921–944. [DOI] [PubMed] [Google Scholar]
- (28).Li W The synthetic and biological study of bryostatin analogues; Ph.D. Thesis, University of Utah, Salt Lake City, UT, 2011. [Google Scholar]
- (29).Keck GE; Covel J; Schiff T; Yu T Org. Lett 2002, 4 (7), 1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Vanier SF; Larouche G; Wurz RP; Charette AB Org. Lett 2010, 12 (4), 672–675. [DOI] [PubMed] [Google Scholar]
- (31).Sharma GVM; Srinivas B; Krishna PR Tetrahedron Lett. 2003, 44, 4689–4691. [Google Scholar]
- (32).Parikh JR; Doering WVE J. Am. Chem. Soc 1967, 89 (21), 5505–5507. [Google Scholar]
- (33).Bal BS; Childers WE; Pinnick HW Tetrahedron 1981, 37 (11), 2091–2096. [Google Scholar]
- (34).Kang JH; Siddiqui MA; Sigano DM; Krajewski K; Lewin NE; Pu Y; Blumberg PM; Lee J; Marquez VE Org. Lett 2004, 6 (14), 2413–2416. [DOI] [PubMed] [Google Scholar]
- (35).Crimmins MT; O’Mahony R Tetrahedron Lett. 1989, 30 (44), 5993–5996. [Google Scholar]
- (36).Shiina I; Kubota M; Oshiumi H; Hashizume M J. Org. Chem 2004, 69, 1822. [DOI] [PubMed] [Google Scholar]
- (37).Marshall JA; Cohen N; Arenson KR J. Org. Chem 1965, 30 (3), 762–766. [DOI] [PubMed] [Google Scholar]
- (38).Solabannavar SB; Helavi VB; Desai UV; Mane RB Tetrahedron Lett. 2002, 43, 4535–4536. [Google Scholar]
- (39).Mortensen MS; Osbourn JM; O’Doherty GA Org. Lett 2007, 9, 3105–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Krainer E; Naider F Tetrahedron Lett. 1993, 34 (11), 1713–1716. [Google Scholar]
- (41).Furstner A; Weintritt H J. Am. Chem. Soc 1998, 120, 2817–2825. [Google Scholar]
- (42).Boden EP; Keck GE J. Org. Chem 1985, 50, 2394–2395. [Google Scholar]
- (43).Higashibayashi S; Shinko K; Ishizu T; Hashimoto K; Shirahama H; Nakata M Synlett 2000, 2000, 1306–1308. [Google Scholar]
- (44).Corey EJ; Venkateswarlu A J. Am. Chem. Soc 1972, 94 (17), 6190–6191. [Google Scholar]
- (45).Tius MA; Fauq AH J. Am. Chem. Soc 1986, 108 (5), 1035–1039. [Google Scholar]
- (46).Inanaga J; Hirata K; Saeki H; Katsuki T; Yamaguchi M Bull. Chem. Soc. Jpn 1979, 52, 1989–1993. [Google Scholar]
- (47).Lipshutz BH; Harvey DF Synth. Commun 1982, 12 (4), 267–277. [Google Scholar]
- (48).Wender PA; Koehler MFT; Wright DL; Irie K Synthesis 1999, 1999, 1401–1406. [Google Scholar]
- (49).Bessodes M; Komiotis D; Antonakis K Tetrahedron Lett. 1986, 27 (5), 579–580. [Google Scholar]
- (50).Cummins TJ The synthesis of the C-ring subunit of bryostatin 1, and the synthesis and biological evaluation of fluorescent bryostatin analogues; Ph.D. Thesis, University of Utah, Salt Lake City, UT, 2015. [Google Scholar]
- (51).Jackson JA; Hammond GB; Wiemer DF J. Org. Chem 1989, 54, 4750–4754. [Google Scholar]
- (52).Yu JS; Wiemer DF J. Org. Chem 2007, 72, 6263–6265. [DOI] [PubMed] [Google Scholar]
- (53).Kofron WG; Baclawski LM J. Org. Chem 1976, 41 (10), 1879–1880. [Google Scholar]
- (54).Andrews IP; Ketcham JM; Blumberg PM; Kedei N; Lewin NE; Peach ML; Krische MJ J. Am. Chem. Soc 2014, 136, 13209–13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Pettit G; Herald C; Doubek D; Herald D; Arnold E; Clardy J J. Am. Chem. Soc 1982, 104 (24), 6846–6848. [Google Scholar]
- (56).MacroModel; Schrödinger, LLC: New York, 2013. [Google Scholar]
- (57).Chang G; Guida WC; Still WC J. Am. Chem. Soc 1989, 111 (12), 4379–4386. [Google Scholar]
- (58).Kolossváry I; Keserü GM J. Comput. Chem 2001, 22 (1), 21–30. [Google Scholar]
- (59).Jorgensen WL; Maxwell DS; Tirado-Rives J J. Am. Chem. Soc 1996, 118 (45), 11225–11236. [Google Scholar]
- (60).Zhang G; Kazanietz MG; Blumberg PM; Hurley JH Cell 1995, 81 (6), 917–924. [DOI] [PubMed] [Google Scholar]
- (61).Jones G; Willett P; Glen RC; Leach AR; Taylor RJ J. Mol. Biol 1997, 267 (3), 727–748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.