

Abstract
The USFDA-approved immunosuppressive drug rapamycin (Rapa), despite its potency, is limited by poor bioavailability and a narrow therapeutic index. In this study, we sought to improve bioavailability of Rapa with subcutaneous (SC) administration and to test its therapeutic feasibility and practicality in a murine model of Sjögren’s syndrome (SS), a systemic autoimmune disease with no approved therapies. To improve its therapeutic index, we formulated Rapa with a carrier termed FAF, a fusion of the human cytosolic FK506-binding protein 12 (FKBP12) and an elastin-like polypeptide (ELP). The resulting 97 kDa FAF (i) has minimal burst release, (ii) is “humanized”, (iii) is biodegradable, (iv) solubilizes two Rapa per FAF, and (v) avoids organic solvents or amphiphilic carriers. Demonstrating high stability, FAF remained soluble and monodisperse with a hydrodynamic radius of 8 nm at physiological temperature. A complete pharmacokinetic (PK) analysis of FAF revealed that the bioavailability of SC FAF was 60%, with significantly higher blood concentration during the elimination phase compared to IV FAF. The plasma concentration of Rapa delivered by FAF was 8-fold higher with a significantly increased plasma-to-whole blood ratio relative to free Rapa, 24 h after injection. To evaluate therapeutic effects, FAF–Rapa was administered SC every other day for 2 weeks to male non-obese diabetic (NOD) mice, which develop an SS-like autoimmune-mediated lacrimal gland (LG) inflammation and other characteristic features of SS. Both FAF–Rapa and free Rapa exhibited immunomodulatory effects by significantly suppressing lymphocytic infiltration, gene expression of IFN-γ, MHC II, type I collagen and IL-12a, and cathepsin S (CTSS) activity in LG compared to controls. Serum chemistry and histopathological analyses in major organs revealed no apparent toxicity of FAF–Rapa. Given its improved PK and equipotent therapeutic efficacy compared to free Rapa, FAF–Rapa is of further interest for systemic treatments for autoimmune diseases like SS.
Keywords: elastin-like polypeptides, FK506-binding protein, rapamycin, dacryoadenitis, lacrimal gland, Sjögren’s syndrome, cathepsin S, non-obese diabetic mouse
Graphical Abstract
INTRODUCTION
Rapamycin (Rapa), also known as sirolimus (an oral formulation of Rapa), is a macrolide first isolated from Streptomyces hygroscopicus.1 In the human body, Rapa binds to cytosolic FK506-binding proteins (FKBPs), where the complex targets and inhibits mammalian Target Of Rapamycin (mTOR) to alter interleukin-2 (IL-2)-mediated signal transduction.2 This inhibition of IL-2 signaling can cause cell cycle arrest in the G1/S phase of T-lymphocytes, thereby suppressing their activation and proliferation.3–5 As a result of its cytostatic and immunosuppressive properties, Rapa is approved by the USFDA to suppress kidney transplant rejection (Wyeth Pharmaceuticals Inc., approved in 1999)6 and to treat lymphangioleiomyomatosis, a rare progressive lung disease (Pfizer Inc., approved in 2015).7 Despite its potency, its use is associated with severe adverse events such as pulmonary- and nephrotoxicity8,9 and metabolic complications.10 Moreover, Rapa’s narrow therapeutic index, low water solubility (2.6 μg/mL),11 low oral bioavailability (<14%),12 and extremely low plasma-to-whole blood ratio13 often necessitates therapeutic drug monitoring.
Efforts to improve the therapeutic index of Rapa include development of more water-soluble analogues (or Rapalogs) such as everolimus, temsirolimus, and ridaforolimus or formulation strategies to control its release. The ability to overcome dose-limiting factors may expand its use for new indications including autoimmune diseases such as rheumatic diseases, sclerosis, or Sjögren’s syndrome (SS).14,15 From our hypothesis that reformulated Rapa for SC administration will improve poor oral bioavailability and exhibit therapeutic efficacy that is comparable to or better than unformulated Rapa, the research described here focuses on how a polypeptide fusion carrier, Berunda (two-headed) polypeptide, can improve pharmacokinetic parameters and affect the therapeutic potential of Rapa toward autoimmune LG inflammation (dacryoadenitis) in a murine model of SS.
To facilitate the absorption of Rapa without rapid burst release, genes encoding FKBP12 (12 kDa) were genetically fused to the N- and C-terminus of the 73 kDa ELP named A192 (amino acid sequence: (Val–Pro–Gly–Ala–Gly)192) to create a soluble, biocompatible, and biodegradable 97 kDa fusion Rapa carrier named FAF16 (Figure 1). FKBP12 is a member of the endogenous cytosolic immunophilin family of proteins that presents the highest affinity for Rapa (EC50 = 3.8 nM) and the strongest inhibition (IC50 = 0.7 nM) toward mTOR.17 The strong binding between FKBP12 and Rapa enables site-specific, affinity-mediated encapsulation of Rapa to FAF (FAF–Rapa), which brings Rapa’s approximate solubility up to that of FAF. ELPs are recombinant polypeptides comprised of pentameric repeats, (Val–Pro–Gly–Xaa–Gly)n. Hydrophilicity of the guest residue (Xaa), number of repeats (n), ELP concentration in the solution, and ionic strength can be modulated individually or simultaneously to control the reversible phase transition properties of the given ELP.18 For this particular study, 73 kDa ELP that contains alanine as a guest residue is chosen to retain high drug and carrier solubility at physiological temperatures and avoid burst renal clearance. ELP-mediated phase separation was optimized to purify the construct.
Figure 1.

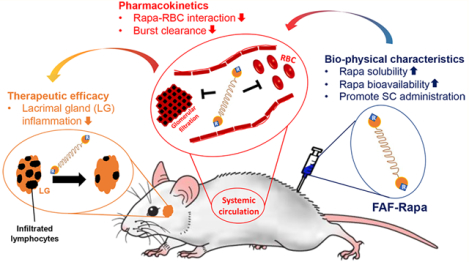
Berunda polypeptides are humanized fusions of the FKBP12 protein that promote solvent-free, burst-free subcutaneous (SC) administration of Rapa to a murine model of autoimmune dacryoadenitis. Genes encoding the FK506-binding protein (12 kDa) were fused to each end of an ELP called A192 (73 kDa) to create a biheaded, biocompatible, and biodegradable drug carrier known as FAF. FAF was expressed via bacterial fermentation, purified at high yield by ELP-mediated purification, and used to solubilize Rapa. To explore the immunosuppressive properties of this formulation, FAF–Rapa was evaluated after SC injection to 14 week old male non-obese diabetic (NOD) mice every other day for 2 weeks. Male NOD mice of this age have developed autoimmune inflammation of the lacrimal gland (LG), also known as dacryoadenitis, leading to reduced tear production and dry eyes. At the termination of the study (Day 16), LG, tears, tissues, and serum were collected and analyzed using histology, gene expression, serum biochemistry, and the activity of a tear and tissue biomarker for SS known as cathepsin S.
SS is a systemic and chronic autoimmune disease with no approved therapies that affects about 4 000 000 people in the US.19 Lymphocytic infiltration and inflammation in the lacrimal gland (LG) and salivary gland (SG) cause severe dry eye and dry mouth, respectively. Mild-to-moderate dry eye symptoms can be treated with ophthalmic solutions such as Restasis (cyclosporine A)20 or Xiidra (lifitegrast);21 however, these topical formulations are not often efficacious, because they target local ocular surface inflammation and not the source of dryness, the inflammation of the LG. These topical formulations also have poor patient compliance due to burning and itching on the ocular surface.22 Progression of SS not only damages visual and oral health but also affects kidney, lung, liver, and thyroid, which necessitates management at the systemic level.23 While several reports from others24–26 and by us27,28 showed subconjunctival, intra-LG, topical, or IV delivery of reformulated Rapa exhibits therapeutic efficacy toward dry eye symptoms, this manuscript characterizes SC delivery of Rapa through the FAF carrier to diversify options for this particular route of administration. While nothing would be easier than topical administration, systemic delivery is likely more efficacious at treating symptoms of SS that occur throughout the body. For our purpose, male non-obese diabetic (NOD) mice were selected as a well-established model of autoimmune dacryoadenitis in SS.29–33 Male NOD mice spontaneously develop autoimmune dacryoadenitis (or autoimmune inflammation of the LG) by 8–12 weeks of age and are used as models of the ocular manifestations of SS.34 In contrast, female NOD mice spontaneously develop autoimmune sialoadenitis by 12–16 weeks of age and are used as models of the oral manifestations of SS.35
This study suggests that FAF is a pharmacokinetically suitable SC Rapa carrier. FAF–Rapa was also therapeutically efficacious in that it significantly suppressed LG lymphocytic infiltration, inflammatory and fibrotic gene expression, and cathepsin S (CTSS) activity (a biomarker of autoimmune disease) in the LG. Collectively, these findings show the potential of FAF–Rapa as a SC-injectable biotherapeutic for SS.
EXPERIMENTAL SECTION
Synthesis, Expression, and Purification of FAF.
The pET-25b(+) vector was originally purchased from Novagen (#69753) and further modified for ELP fusion cloning.36 FAF was biosynthesized as previously described.16 Briefly, the gene encoding the human FKBP1237 was cloned to the N- and C-terminus of ELP A192 (comprised of 192 repeats with Xaa = Ala). The cloned construct was sequenced, transformed into, and expressed in BLR(DE3) competent E. coli (#69053, Novagen, Madison, WI), fermented in terrific broth media for 16–18 h at 37 °C without IPTG induction. After bacterial cell lysis (S-4000 Ultrasonic Disintegrator Sonicator Liquid Processor, Misonix, Inc. NY; Amplitude 9, 18 repeats of 10 s on + 20 s off cycle) and clarification of cell debris by centrifugation at 16 100 rcf for 10 min at 4 °C in a Beckman J2–21 Centrifuge, the supernatant was subjected to ELP-mediated phase separation in 2 M sodium chloride at 37 °C. Coacervates were pelleted at 5000 rcf for 10 min at 37 °C using a Sorvall RC-3C Plus Centrifuge immediately after the phase separation was observed (hot-spin). At the end of the hot-spin, soluble impurities (supernatant) were removed, and FAF coacervates (pellet) were resolubilized in ice-cold PBS. Thoroughly resolubilized FAF was centrifuged at 16 100 rcf for 10 min at 4 °C in an Eppendorf 5415R Centrifuge (cold-spin). At the end of the cold-spin, insoluble impurities (pellet) were again removed by transferring the supernatant to a clean tube. Cycles of hot-spin followed by cold-spin were repeated three times to achieve the necessary purity (Figure 2A).
Figure 2.

High molecular weight FAF–Rapa has the purity, size, and concentration–temperature phase behavior necessary for stability at body temperature. (A) Identity, purity, and fluorescence of FAF, FAF–Rapa, and rhodamine-labeled FAF–Rapa (Rho–FAF–Rapa) were analyzed by Coomassie blue staining and fluorescence imaging of SDS–PAGE. (B) Dynamic light scattering shows that all three formulations (Table 1) remain monodisperse at 37 °C (10 μM, PBS). (C) Optical density of FAF was monitored as a function of temperature at 350 nm to confirm solubility in PBS at physiological temperatures (shaded area). The Tt of FAF at 25 μM was 57.0 °C. (D) Using optical density, the phase transition temperature was plotted vs. concentration as a phase diagram, below which FAF remains soluble (n = 3, Mean ± SD). The shaded area indicates physiological temperatures. Dotted lines show a 95% confidence interval (CI) of the mean.
Biophysical Characterization of FAF.
The purity of FAF was analyzed using SDS–PAGE. The molar extinction coefficient (ε) of FAF was calculated at 20 615 M−1·cm−1.38 Serial dilutions of FAF in Edelhoch buffer were prepared, measured, and averaged to acquire the best estimate of FAF concentration in PBS using eq 1.38,39
| (1) |
The hydrodynamic radius (Rh) of FAF at 20 and 37 °C was determined using dynamic light scattering (DLS). Triplicates of sterile filtered (200 nm pore, #PN 4612, Pall Corp., NY) FAF, FAF–Rapa, or rhodamine-labeled FAF–Rapa (50 μL of 10 μM) in PBS were loaded onto a 384-well plate followed by layering with 15 μL of mineral oil to prevent evaporation, and the whole plate was centrifuged for 1 min at 1000 rcf to remove any remaining air bubbles. Samples were measured using a Wyatt Dynapro plate reader and analyzed by built-in software DYNAMICS V7 (Wyatt Tech. Co., CA). Rh was measured first at 20 °C, and then, the temperature was immediately increased to 37 °C, where the second measurement was made. For the optical density profile, absorbance at 350 nm was measured in a DU800 UV–vis spectrophotometer (Beckman Coulter, CA) under a temperature gradient of 0.5 °C/min. The transition temperature at each concentration of FAF was defined as the temperature at which the maximum first derivative was achieved within each optical density profile with respect to the temperature. The transition temperature from each concentration was used to plot the phase diagram and fit with eq 2.
| (2) |
Encapsulation of Rapa with FAF.
A two-phase solvent evaporation method was employed to bind Rapa to FAF. An aqueous phase (PBS) containing 400 μM FAF was mixed with an equal volume of organic phase (90% hexane/10% EtOH) containing 630 μM Rapa (molar ratio = 1:1.58). Under constant stirring at 29 °C for 20 min to evaporate organic solvents (Laborota 4011 digital rotary evaporator, Heidolph Instruments GmbH & Co. KG, Schwabach, Germany), Rapa was gradually displaced into the aqueous phase, where it was solubilized through binding to FAF. The resulting solution was then centrifuged (16 100 rcf, 10 min, 4 °C), filtered (200 nm pore, #PN 4612, Pall Corp., NY), and dialyzed against PBS (>4 h, 4 °C) to remove any excess or insoluble Rapa and residual solvent. To determine the encapsulation efficiency, 30 μM FAF in MeOH was injected into a C-18 reverse phase (RP)-HPLC column (150 × 4.6 mm, particle size = 5 μm, YMC CO. Ltd., Japan) and eluted in a H2O/MeOH gradient from 50 to 100% over 15 min. The chromatogram was monitored at 280 nm at a flow rate of 0.75 mL/min. Linear standard curves were created between the logarithm of the area under the curve (AUC) vs logarithm of either FAF concentrations (12.5, 25, 50, 75, 100 μM) or Rapa concentrations (6.25, 12.5, 25, 50, 75, 100 μM). FAF and Rapa concentrations in the sample were calculated based on their respective calibrated standard curve. The molar ratio of Rapa to FAF was 1.76:1 (encapsulation ratio of Rapa to FKBP = 0.88:1). The loading of Rapa to FAF was 1.64% (w/w).
Pharmacokinetic Study of FAF.
Procedures and analysis were adopted and modified from methods previously described by MacKay et al.40 FAF–Rapa in PBS was incubated with NHS–rhodamine (#46406, Thermo Fisher, IL) for 1.5 h at room temperature under constant rotation and excess dye was removed using a desalting column (#87772, Thermo Fisher, IL). The labeling efficiency was measured as 100% using eq 3 (FAF to rhodamine = 1:1), where the εrhodamine is 80 000 (M−1 cm−1) at 555 nm, and the concentration of CFAF was measured before labeling to be 200 μM. Fluorophore labeling did not alter the Rapa encapsulation ratio, which was confirmed by RP-HPLC.
| (3) |
To measure the pharmacokinetics of the carrier, rhodamine-labeled Rho–FAF–Rapa (1.0 mg of Rapa/kg of BW) was injected either intravenously (IV, n = 4) via tail vein or SC (n = 5) at the right flank of the animal. A blood sample (20 μL) was collected from the tail vein at 5 min (3 min for IV) and 30 min as well as 1, 2, 4, 8, 12, 24, 36, 48, and 72 h after injection and immediately diluted into heparinized PBS (1000 U/mL, #H4784, Sigma, MO). The blood was centrifuged (16 100 rcf, 10 min, 4 °C), and plasma was stored at −20 °C for further analysis. Collected plasma samples were loaded onto a 384-well plate (#784076, Greiner Bio One International GmbH, NC), and fluorescence intensity was determined (Synergy H1 Hybrid Multi-Mode Reader, BioTek Instruments, Inc., VT; Ex: 542 nm/Em: 585 nm). A linear standard curve was created between the logarithm of the background-subtracted fluorescence vs. logarithm of the FAF concentrations (13.7, 41.2, 123.5, 370.4, 1111.1, 3333.3, 10 000.0 nM). FAF concentration was then calculated based on fluorescence intensity.
Both noncompartmental and compartmental methods were applied to analyze the PK profiles of FAF after IV or SC administration. Noncompartmental analysis was primarily based on the determination of the area under the plasma concentration curve (AUC) and the area under the moment curve (AUMC), which were first estimated by the trapezoidal method. The mean residence time (MRT) of both IV and SC and the SC mean absorption time (MAT) were calculated as follows
| (4) |
| (5) |
Using these estimates of AUC, the plasma clearance (CL) of both IV and SC and the SC bioavailability (F) were estimated as follows
| (6) |
| (7) |
Similarly, kelimination and T1/2,elimination were estimated in each individual mouse by fitting the last three time points to the following equation.
| (8) |
| (9) |
The volume of distribution of both IV and SC was estimated as follows
| (10) |
To obtain pharmacokinetic parameters for the compartmental analysis, the data set of each individual mouse was fit to either a one-compartment (IV) or three-compartment (SC) pharmacokinetic model using SAAM II (University of Washington, WA). It was not possible to account for the late peak times observed using a one-compartment model of absorption from the injection site; however, the data was well-fit by two-step absorption from the SC injection site to an intermediate interstitial compartment and then to the plasma compartment. To estimate the magnitude of the absorption parameters in the SC data set, it was necessary to assume that the kinetic rate constant kinjection site→interstitial fluid is equal to kinterstitial fluids→systemic circulation, which is denoted as kabsorption (kabs). The dose and the plasma concentrations were fit to these models to determine the volume of distribution (Vd), elimination rate constant (kelimination), and kabs. On the basis of these fit parameters, the maximum plasma concentration (Cmax), clearance (CL), and elimination half-life (T1/2,elimination) were estimated for individual mice. Average values for Vd and kelimination from the IV data set were adopted to perform fits for the SC data sets. Equations used for compartmental analyses are as follows
| (11) |
| (12) |
| (13) |
| (14) |
To optimize administration frequency through modeling, average plasma concentration from five mice administered with SC Rho–FAF–Rapa were solved with fixed values of Vd, kabsorption, kelimination, and kdegradation, as reported in Table 2 using SAAM II.
Table 2.
Pharmacokinetic Parameters of IV- or SC-Delivered FAF–Rapa Analyzed Using Compartmental Analysis and Noncompartmental Analysis (Mean ± SD)
| route of administration | ||||
|---|---|---|---|---|
| parameter (unit) | IV | SC | IV | SC |
| preferred model | one compartment | three compartments | noncompartment | |
| AUC (μM·hr) | 141 ± 16 | 92 ± 19 | 207 ± 33 | 109 ± 14 |
| AUMC (μM·hr2) | - | - | 2207 ± 344 | 2214 ± 328 |
| F (%) | 100 | 65.5 ± 13.2 | 100 | 52.7 ± 6.7 |
| CL (mL/h) | 0.15 ± 0.01 | 0.15a | 0.10 ± 0.01 | 0.09 ± 0.01 |
| CL/F (mL/h) | 0.15 ± 0.01 | 0.23 ± 0.04 | 0.10 ± 0.01 | 0.18 ± 0.03 |
| Vd (mL) | 1.46 ± 0.2 | 1.46a | 0.88 ± 0.1 | 0.85 ± 0.1 |
| Cmax (μM) | 13.4 ± 2.4 | 3.3 ± 0.1 | 11.7 ± 2.9 | 4.4 ± 0.7 |
| Tmax (hr) | 0.0 | 12.4 ± 0.7 | 0.0 | 12.0 |
| MRT (hr) | - | - | 10.7 ± 0.3 | 20.3 ± 1.1 |
| MAT (hr) | - | - | - | 9.6 ± 1.1 |
| Tl/2,absorption (hr) | - | 4.2 ± 0.4 | - | - |
| Tl/2,elimination (hr) | 6.9 ± 0.5 | 6.9a | 6.2 ± 0.4 | 6.4 ± 0.7 |
| kabsorption (hr−1) | - | 0.16b | - | - |
| kelimination (hr−1) | 0.10 ± 0.01 | 0.10a | 0.11 ± 0.01 | 0.11 ± 0.01 |
| kdegradation (hr−1) | - | 0.09 ± 0.05 | - | - |
Compartmental values from IV analysis were adopted to estimate other SC parameters.
To fit the observed time to peak concentration, the assumption was required that kabsorption = kinjection site→interstitial = kinterstitial fluid→systemic circulation.
Plasma and Whole Blood Levels of Rapa.
To measure the relative plasma and whole blood levels of Rapa, 1.0 mg of Rapa/kg of BW of rhodamine-labeled FAF–Rapa or free Rapa was injected either IV (n = 5 and n = 4, respectively) via tail vein or SC (n = 4, each) at the right flank of the animal. Blood was collected using a heparinized syringe at 24 h after injection via cardiac puncture. The collected blood was split into a “whole blood sample” and a “plasma sample”. For whole blood, three cycles of freeze–thaw using −80 °C were applied to lyse RBCs, to release RBC-bound Rapa, and stored at −20 °C for further LC–MS analysis. For plasma samples, samples were centrifuged (16,100 rcf, 10 min, 4 °C) and the supernatant (plasma) was stored at −20 °C for further LC–MS analysis. For LC–MS analysis, 25 μL of internal standard (1000 ng/mL Prograf (Tacrolimus), Astellas Pharma US, Inc., IL) was added to a 50 μL plasma sample followed by 350 μL of acetonitrile. The entire mixture was vortexed and then centrifuged (16 100 rcf, 5 min, 4 °C). A 40 μL aliquot of supernatant was transferred to HPLC microvials, and 30 μL was injected into the LC–MS. The same method was used to analyze the whole blood samples. For the standard curve, 50 μL serial dilutions of free Rapa or FAF–Rapa were added to a mixture of 25 μL of internal standard (1000 ng/mL of Tacrolimus), 50 μL of blank mouse plasma, and 300 μL of acetonitrile to generate 16, 80, 400, 2000, 10 000 ng/mL Rapa standard solutions. Samples were admixed thoroughly and centrifuged (16 100 rcf, 5 min, 4 °C). A 40 μL aliquot of supernatant was transferred to HPLC microvials, and 30 μL was injected into the LC–MS. Fluorescence intensity measured from the same plasma sample (Synergy H1 Hybrid Multi-Mode Reader, BioTek Instruments, Inc., VT; Ex: 542 nm/Em: 585 nm) was used to calculate the Rapa to FAF ratio in vivo.
Therapeutic Study in Male Non-Obese Diabetic (NOD) Mice, a Model of SS.
Methods described here are adopted and adjusted from the methods used by Shah et al.28 The male NOD mice were bred in-house from breeding pairs purchased from Taconic (strain: NOD/MrkTac, Taconic Biosciences, Inc., NY). Analysis and treatments were initiated when animals were 13–15 weeks of age, when SS-like autoimmune dacryoadenitis is established. A total of 60 mice were randomly assigned to four different groups receiving either vehicle, free Rapa, carrier (FAF only), or FAF–Rapa SC at a dose of 1.0 mg of Rapa/kg at the right flank of the animal, every other day for 2 weeks (seven injections). Treatments were formulated as follows. For the vehicle, 1 mL of polysorbate 80 (EM8.22187.0500, EMD Millipore, MA) was mixed with 9 mL of endotoxin-free water to generate 10% polysorbate 80. A 1 mL aliquot of polyethylene glycol (PEG) 400 (#202398, Sigma, MO) was also mixed with 9 mL of endotoxin-free water to generate 10% PEG 400. From each dilution, 2.5 mL was mixed together (10% polysorbate 80 + 10% PEG 400, 5 mL) and further diluted with 20 mL of endotoxin-free water to generate a 2% polysorbate 80 + PEG 400 solution. For free Rapa, 100 mg of rapamycin (R-5000, LC Laboratories Inc., MA) was dissolved in 100% EtOH to make 50 mg/mL stock. A 100 μL aliquot of 50 mg/mL stock was mixed with 4.9 mL of polysorbate 80 + PEG 400 mixture (1:1 ratio) to make 1.0 mg/mL rapamycin stock. A 5 mL aliquot of this stock was further diluted with 20 mL of endotoxin-free water to generate a 0.2 mg/mL Rapa stock in 2% polysorbate 80 + PEG 400 solution. For FAF Rapa, based on the molar ratio of Rapa to FAF (1.76:1), a dose of 1.0 mg of Rapa/kg of BW was delivered (i.e, 1.64 mg of FAF–Rapa was injected to a 30 g mouse to deliver 1.0 mg of Rapa/kg of BW). For carrier (FAF only), the same amount of FAF without Rapa was injected to serve as a control. Injection volumes varied from 100–160 μL per injection based on the weight of individual mouse. On the basis of a power calculation using the variance within the LG infiltration in the NOD mice, the therapeutic study was designed as n = 15 per treatment group. All animal use was in full compliance with policies approved by the University of Southern California Institutional Animal Care and Use Committee (IACUC) and the ARVO statement for the Use of Animals in Ophthalmic and Vision Research.
Quantitative Real-Time PCR.
To analyze expression of genes of interest after experimental treatments, total RNA was isolated from half of the LG using the RNeasy plus Universal Mini Kit (#73404, Qiagen, Germany). The LG was cut into half immediately after collection and stored in an ice-chilled bead-prefilled tube (#Z763780, Sigma, MO) supplemented with 900 μL of lysis reagent (provided with the kit). Tissues were homogenized (BeadBlaster 24 High-Throughput Benchtop Homogenizer, Benchmark Scientific, Inc., NJ; speed: 7, cycle: 2, time: 1 min, intermission: 30 s), and clarified lysates were further processed in accord with the manufacturer’s protocol. Complementary DNA was prepared from 4 μg of RNA using the quantitative real-time PCR (qRT-PCR), which was performed with the TaqMan gene expression assays on the QuantStudio 12K Flex Real-Time PCR System for 384-well block (#4471134, Applied Biosystems, CA) using the following probes: TNF-α (Mm00443258_m1), IFN-γ (Mm01168134_m1), Akt3 (Mm00442194_m1), MHCII (Mm00439216_m1), CTSS (Mm01255859_m1), Col1A1 (Mm00801666_g1), and IL-12a (Mm00434165_m1). The GAPDH (Mm99999915_g1) was used as a control gene. Each reaction (10 μL) consisted of 0.5 μL of cDNA from the reverse transcription reaction, 4 μL of nuclease-free water, 0.5 μL of assay primer, and 5 μL of TaqMan Universal PCR Master Mix (#4304437, Applied Biosystems, CA). Each sample was run in triplicate. The thermal profile consisted of preheating the samples at 105 °C for 10 min followed by 40 repeats of 95 °C for 15 s + 60 °C for 1 min. The relative expression levels were calculated using the built-in comparative CT method (ΔΔCT method) in the default ABI software.
Stimulated Tear Collection and Cathepsin S Activity Analysis in Tears and LG Lysates.
Stimulated tear collection was performed under full anesthesia (intraperitoneal (I.P.) injection of 100 mg/kg ketamine + 10 mg/kg xylazine) as a terminal procedure. A small bilateral incision on the axis between the outer junction of the eyelid and the ear was made to expose the LG on both sides. Then, 3 μL of 50 μM carbachol (#L06674, Alfa Aesar, MA) was applied directly onto the LG to stimulate tear secretion, followed by tear collection from both eyes by placing 2 μL microcapillary tubes (#1-000-0020, Drummond Sci. Co., PA) at the tear meniscus in the medial canthus for 5 min. The stimulation was performed for a total of three times. Collected tears were recorded as a function of the length in the capillary tube in millimeters and then converted to microliters (1 mm = 0.625 μL). After tear collection, the LG was cut into half and placed in an ice-chilled bead-prefilled tube (D1032–10, Benchmark Scientific, NJ) supplemented with 300 μL of lysis buffer (provided with the kit). Tissues were homogenized (BeadBlaster 24 High-Throughput Benchtop Homogenizer, Benchmark Scientific, Inc., NJ; speed: 7, cycle: 2, time: 1 min, intermission: 30 s). The homogenate was centrifuged at 6000 rcf for 10 min at 4 °C. The supernatant was separated carefully and analyzed immediately. Cathepsin S (CTSS) activity analysis was measured using the cathepsin S Activity kit (#K144–100, BioVision Inc., CA). Clarified LG lysate (10 μL) was mixed with 40 μL of lysis buffer, and 50 μL of reaction buffer were added on a 96-well plate in duplicates. A 100 μL aliquot of stimulated tear fluid diluted in CTSS reaction buffer was also added on the same 96-well plate in duplicates. CTSS substrate (2 μL) was added to all wells and incubated at 37 °C for 1 h. The amount of resulting fluorescence was measured with a microplate reader (SpectraMax iD3, Molecular Devices, CA). After the experiment, the Bio-Rad protein assay was performed to measure total protein concentration, and the activity was normalized to 50 μg of total protein for both tears and lysates.
Histopathology Analysis and Serum Chemistry.
Lung, spleen, kidney, liver, and one of each pair of LGs from every mouse was collected at the conclusion of the study for histopathology and immediately fixed with 10% neutral buffered formalin (#5701, Thermo Fisher, IL), stored at 4 °C overnight, and then transferred to 70% EtOH. All organs in 70% EtOH were outsourced for paraffin embedding, sectioning, standard H&E staining, and imaging (HistoWiz Inc., NY). For LG, sections from the 25th, 50th, and 75th percentile regions through each gland were selected for quantitation, and the area of the LG occupied by infiltrating lymphocytes was quantified by three blinded reviewers for the best estimation of percentage area of infiltrates per gland as previously described.27,28 The percentage area infiltrated was calculated by measuring the area of the infiltrate divided by the whole area of the gland using ImageJ (v2.0.0, NIH, MD). For lung, spleen, kidney, and liver, two consecutive sections at the 50th percentile regions through each organ were stained and analyzed by a blinded, trained pathologist for any signs of acute toxicity. For serum chemistry, blood collected via cardiac puncture at the conclusion of the study was spun-down (2000 rcf, 10 min, 4 °C) to separate serum and outsourced on the day of collection for standardized analysis (Antech Diagnostics, CA).
Statistics.
All statistical analyses were performed using SPSS v21 (SPSS Inc., IL). A two-way ANOVA served as a primary statistical method to compare the overall effect of the drug and the carrier. If a particular data set showed a significant interaction between Rapa and FAF, then the data was further analyzed either by one-way ANOVA or a Kruskal–Wallis nonparametric test based on Levene’s test. For two-group comparisons, a two-tailed, independent t test was used to compare differences. A p-value less than 0.05 was considered a significant difference.
RESULTS
Biophysical Characterization of FAF, a FKBP-ELP Fusion Protein Polymer That Carries Rapa.
FAF was heterologously overexpressed from a seamlessly cloned synthetic gene in E. coli that encodes the full-length human FKBP12 protein at both the N- and C-terminus of ELP A192 (Table 1). Purification was done via inverse transition cycling,36 a standard nonchromatographic method of ELP fusion protein purification that utilizes ELP-mediated phase separation from cleared bacterial lysates supplemented with 1–2 M NaCl to induce the Hofmeister effect.41 Three rounds of purification yielded ~90 mg/L of FAF with >98% purity, verified by SDS–PAGE (Figure 2A). The precise determination of molecular weight by MALDI-TOF for FAF was reported previously.16 From dynamic light scattering (DLS) analysis, the purified product was consistent with a monomeric and monodisperse population with a hydrodynamic radius of 8 ± 1 nm from 20 to 37 °C, which suggests that FAF remains stable and soluble at physiological temperatures (Figure 2B). The solubility profile and biophysical characteristics of FAF did not change after Rapa encapsulation or fluorescent dye labeling (Table 1). To determine the Tt of FAF, optical density at 350 nm over a range of temperatures was measured (Figure 2C). Fusion of FKBP to ELP did not affect the negative correlation between the Tt and the ELP concentration,18 and the phase diagram was fit by eq 2. This fit yielded a slope, m, of 5.76 (2.23–8.32, 95% CI) °C per 10-fold decrease in concentration and an intercept at 1 μM, b, of 65.7 °C (60.34–69.89, 95% CI). This fit allows estimation of Tt over a range of concentrations; furthermore, at no measured plasma concentrations (1–100 μM) in the in vivo therapeutic studies would FAF be expected to phase separate (Figure 2D).
Table 1.
Biophysical Characteristics of FAF, FAF–Rapa, and Rhodamine-Labeled FAF–Rapa
| Rh (nm)c at | ||||
|---|---|---|---|---|
| label | formulation compositiona | MW (kDa)b | 20 °C | 37 °C |
| FAF | FKBP–G(VPGAG)192–FKBP | 96.6 | 8 ± 1 | 8 ± 1 |
| FAF–Rapa | Rapa/FKBP–G(VPGAG)192–FKBP/Rapa | 98.3 | 9 ± 2 | 8 ± 0.3 |
| Rho–FAF–Rapa | rhodamine–[Rapa/FKBP–G(VPGAG)192–FKBP/Rapa] | 98.8 | 9 ± 1 | 7 ± 0.3 |
FKBP amino acid sequence: GVQVETISPGDGRTFPKRGQT CVVHYTGMLEDGKKFDSSRDRNKPFKFMLGKQEVIRGWEEGVAQMSVGQRAKLTISPDYAYGATGHPGIIPPHATLVFDVELLKLE. G(VPGAG)192Y is the amino acid sequence of the elastin-like polypeptide, A192.
Expected molecular weights for FAF–Rapa and Rho–FAF–Rapa were reported based on observed 1:1 molar ratios of Rapa to FKBP and the rhodamine-labeling ratio.
Hydrodynamic radii of each formulation (of 10 μM, PBS) were determined in triplicate by DLS (Mean ± SD).
SC Administration Significantly Improves the Pharmacokinetic Profiles of FAF.
To date, an accurate pharmacokinetic profile for SC FAF has not been reported; therefore, to estimate the bioavailability and PK parameters for FAF, 1.0 mg of Rapa/kg of BW of Rho–FAF–Rapa was injected either IV via tail vein (n = 4) or SC at the right flank (n = 5) of male NOD mice. Fluorescence intensities were analyzed from collected plasma samples at the designated time points and used to plot the plasma FAF concentration–time profile (Figure 3A). The collected data set from each individual mouse was further analyzed using both compartmental and noncompartmental analyses to estimate all relevant pharmacokinetic parameters. For compartmental analysis, a one-compartment model was adopted to interpret IV FAF–Rapa, whereas a three-compartment model was necessary to fit the delayed peak for SC FAF–Rapa (Figure 3B). A simple two-compartment model with only one absorption compartment was unable to fit the observed data. The simplest model that fit SC data was a three-compartment model, which includes an interstitial fluid compartment as an intermediate absorption compartment. This observation is consistent with previous reports of SC-administered biotherapeutics.42 One acceptable interpretation of this model would be initial absorption into interstitial fluid, possibly the lymphatic system, followed by a second absorption process into the systemic circulation. For the best fit of all data sets, it was necessary to assume that both absorption processes were on the same order of magnitude. Had either absorption step been much slower than the other, it would have become rate-limiting, and the simpler two-compartment model would fit the data.43 On the basis of this, we assumed that the kinetic rate constant kinjection site→interstitial is equal to kinterstitial fluid→systemic circulation, of which both are designated as kabsorption (kabs, Figure 3B, Table 2). To allow for loss of Rho–FAF–Rapa prior to appearance in the plasma, a kinetic constant of removal from the interstitial fluid is denoted as kdegradation. IV FAF–Rapa showed a monoexponential elimination after 8 h of a plateau; however, this phenomenon was not captured by the compartmental model parameter estimates. Thus, parameter estimates from both compartmental analysis and noncompartmental analysis are reported (Table 2). It should be noted that the statistically significantly higher plasma concentration of SC FAF–Rapa compared to IV FAF–Rapa during the elimination phase (36~72 h, Figure 3A) suggests that FAF may be a more effective carrier for sustained release when given SC versus IV.
Figure 3.

Pharmacokinetic analysis reveals that SC administration of FAF–Rapa improves pharmacokinetic properties of Rapa. (A–D) 1.0 mg of Rapa/kg of BW of Rho–FAF–Rapa was injected either IV (n = 4) or SC (n = 5) to male NOD mice. (A) Data for the first 10 h are shown in the inset. SC administration yielded significantly higher Rho–FAF concentrations at 36, 48, and 72 h (Mean ± SD). A Student’s t test was used to compare groups. (B) Data were well-fit by either a one-compartment (IV) or three-compartment (SC) pharmacokinetic model as indicated. kabs = kabsorption. (C) On the basis of these parameters, pharmacokinetic modeling was performed to explore several dosing options prior to initiating a therapeutic study. (D–F) Male NOD mice were injected with 1.0 mg of Rapa/kg of BW as free Rapa IV (n = 4), free Rapa SC (n = 4), FAF–Rapa IV (n = 5), or FAF–Rapa SC (n = 4). Plasma and whole blood samples were collected via cardiac puncture after 24 h. (D) For each sample, Rapa concentration analyzed by LC–MS was compared with its fluorescence intensity analyzed by a plate reader to measure the Rapa to FAF ratio (min, mean, and max are depicted). A Student’s t test was used to compare groups, which revealed that at 24 h, SC administration retained nearly the starting ~2:1 ratio of Rapa:FAF, while FAF–Rapa-administered IV had lost about half of the bound drug. (E) Rapa concentration from each sample was analyzed by LC–MS (Mean ± SD). A Student’s t test and one-way ANOVA were used to compare groups. (F) For each sample, Rapa concentration in the plasma was compared to that of the whole blood (WB) (min, mean, and max are depicted). A two-way ANOVA was used to compare groups, which revealed that FAF reduces accumulation of Rapa in blood cells compared to the free drug.
Using acquired compartmental parameters, the administration frequency of FAF–Rapa was explored for a 2 week study (Figure 3C). On the basis of modeling, the plasma concentration of Rapa reached its peak concentration of 3.47 μM at 12 h, becoming 10-fold lower at 45.6 h and 100-fold lower at 69.6 h after administration. Therefore, a 48 h interval between doses prevents either progressive accumulation of FAF–Rapa in the body (24 h interval) or much more complete clearance of FAF–Rapa before the next dose (72 h interval).
For SC FAF–Rapa, absorption proceeded during the first 24 h, while the maximum concentration (Cmax) was reached 12 h after injection, followed by a monoexponential decay thereafter (Figure 3A). This behavior can be explained by a mean absorption time of 9.6 h and mean residence time of 20.3 h for SC FAF–Rapa (Table 2). The bioavailability of SC FAF–Rapa was 52.7–65.5% (Table 2), without apparent loss of Rapa during the absorption process (Figure 3D). After 24 h, SC FAF–Rapa maintained its mean loading stoichiometry of 1.77 ± 0.3, which exactly matched the initial molar ratio of 1.76:1, whereas some of the Rapa was dissociated from FAF when delivered IV. This difference may be due to immediate dilution into plasma for IV FAF–Rapa, versus retention in the injection site during the nearly 10 h absorption process for SC FAF–Rapa. Nonetheless, there was undetectable displacement of Rapa from FAF during absorption, which might otherwise have been a limitation of SC administration.
We next explored how FAF affects levels of Rapa in vivo. To do so, 1.0 mg of Rapa/kg of BW of free Rapa or Rho–FAF–Rapa was delivered either IV or SC. Blood was collected and Rapa concentrations in the plasma and in the whole blood were compared by LC–MS analysis, 24 h after injection. FAF delivery resulted in an 8-fold higher plasma Rapa concentration compared to that of the free formulation (Figure 3E). Consistent with the elevated plasma concentrations for Rho–FAF–Rapa beyond 24 h (Figure 3A), SC delivery also increased the Rapa concentration by 1.8-fold compared to IV administration (Figure 3E).
The presence of at least two types of FKBPs in erythrocytes, cytosolic FKBP12 and membrane-bound FKBP13, can fractionate Rapa in systemic circulation by extraction into red blood cells (RBC).44 The interaction between Rapa and RBCs is problematic in that it elicits hematologic toxicities which can cause discontinuation of the treatment.10 We wanted to test whether site-specific encapsulation of Rapa to our carrier, thus masking Rapa’s binding motif to FKBPs, effectively inhibits Rapa–RBC interaction in vivo. We observed a significant improvement in the plasma-to-whole blood ratio of Rapa when delivered by FAF. The plasma-to-whole blood ratio of free Rapa was 0.9:1 for IV and 1:1 for SC, but this ratio increased to 1.4:1 for IV (1.5-fold increase) and 1.5:1 for SC (1.5-fold increase) when delivered by FAF (Figure 3F). These elevated ratios are consistent with the retention of Rapa in the plasma by FAF even 1 day after administration of the formulation.
Rapa Significantly Suppresses Lymphocytic Infiltration in the LG.
To evaluate the effect of SC FAF–Rapa and free Rapa on the characteristic autoimmune dacryoadenitis associated with SS, one of each pair of LGs collected from control and treated mice at the conclusion of the study were sectioned and histopathologically quantified to compare the extent of lymphocytic infiltration (Figure 4A). Relative to healthy LG, an inflamed LG shows evidence of lymphocytic infiltration throughout the whole organ (Figure 4B, outlined in blue). Both FAF–Rapa (9 ± 5.6%) and free Rapa (7 ± 3.7%) significantly suppressed lymphocytic infiltration in the LG compared to FAF alone (15 ± 8.4%) or vehicle (21 ± 10.3%, percent area covered, mean ± SD) control (Figure 4C). The ability of FAF–Rapa to suppress lymphocytic infiltration did not differ from free Rapa.
Figure 4.

Rapa reduces lymphocytic infiltration in the LG of male NOD mice. (A) One of each pair of LGs from each mouse in the cohort was collected at the conclusion of the study. The 25th, 50th, and 75th percentile sections from each LG were quantified by three blinded observers to determine the average percentage area of infiltrate per gland (n = 15). (B) Inflamed LGs show areas of purple nuclear staining, which indicate foci of infiltrating lymphocytes (outlined in blue). Lymphocytic infiltration was reduced by FAF–Rapa (middle panel). The scale bar represents 200 μm. (C) The percentage area of infiltration was calculated using ImageJ (Mean ± SD).
SC Rapa Modulates Gene Expression of Proteins Involved in Inflammation, Antigen Presentation, and Fibrosis.
The health of the LG is not only governed by lymphocytic infiltration but also fibrosis, autophagy, and levels of cytokines.45 Elevations in gene expression of major histocompatibility complex II (MHC II) in various autoimmune and inflammatory diseases is well-known,46 a factor that may drive lymphocytic infiltration in the LG.47,48 Moreover, major proinflammatory cytokines, such as IL-12a, IFN-γ, and TNF-α, are significantly elevated in the inflamed LG.49–51 The downregulation of genes encoding these proteins was seen in our previous gene expression profiling in the LG upon mTOR inhibition by IV Rapa treatment.27 We analyzed the expression profiles of genes responsible for autoimmune activation and inflammation to assess changes in immune and inflammatory environments with treatment with free Rapa or FAF–Rapa. Indeed, with both treatments, a decrease in lymphocytic infiltration in the LG was linked with decreased gene expression levels of MHC II, IL-12a, and IFN-γ (Figure 5A–C). Rapa caused no difference in the gene expression level of TNF-α (Figure 5D).
Figure 5.

Gene expression profile of proteins involved in inflammation, antigen presentation, and autophagy in LG of male NOD mice treated with subcutaneous Rapa. One of each pair of LGs from mice in the treatment cohorts was collected at the conclusion of the study for mRNA extraction. Extracted mRNAs were reverse transcribed to cDNA and further analyzed by quantitative real-time PCR. Gene expression levels were normalized to vehicle (mean ± SD, n = 9). Two-way ANOVA was used to compare effects of drug and carrier. On the basis of a significant interaction between Rapa and FAF for Col1A1 (p = 0.025), one-way ANOVA and post hoc comparisons revealed significant differences between vehicle vs FAF–Rapa (p = 0.003); free Rapa vs FAF (p = 0.007); and FAF vs FAF–Rapa (p < 0.001).
One of the hallmarks of the LG in SS patients is degradation of extracellular matrix and deposition of collagen, leading to LG fibrosis.52 An antifibrotic effect of Rapa on renal,53 skin,54 cardiac,55 pulmonary,56 and hepatic57 fibrosis models has been reported, with a common feature manifested as a decrease in type I collagen expression, and its secretion to and deposition at the extracellular space. To see whether free Rapa and FAF–Rapa suppresses LG fibrosis, we analyzed the gene expression level of type I collagen, α1 (Col1A1) in the LG. Free Rapa-and FAF–Rapa-delivered SC significantly lowered gene expression of Col1A1 (p < 0.001, Figure 5E), which suggests Rapa may protect the LG from fibrosis. This is consistent with our previous findings from topical instillation of Rapa as an ophthalmic emulsion or IV administration via micellar FKBP-ELP fusion protein (unpublished data). Although more studies are required, we propose that Rapa may act through inhibition of mTOR, which decreases type 1 collagen expression and reduces LG fibrosis; furthermore, this may proceed via mTOR’s kinase activity toward LARP6 as recently reported by Zhang and Stefanovic.58
Gene expression changes in Akt3 were collected as a surrogate marker for autophagy. The role of autophagy in LG inflammation is controversial, as some researchers show autophagy is highly activated or at least dysregulated in the inflamed LG, thus worsening the disease,59 whereas others report that activation of autophagy during the disease progression can aid the gland in processing accumulated cell debris, thus promoting its health.60 Our previous gene expression profiling showed downregulation of Akt3 in the LG upon IV Rapa treatment,27 associated with induction of autophagy. This change was reconfirmed in a recent topical Rapa study over a 12 week period;28 however, no significant gene expression change was observed in the LG in this 2 week treatment (Figure 5F).
We did not observe an enhanced effect of FAF–Rapa compared to free Rapa on proinflammatory gene expression profiles, suggesting that FAF–Rapa is similar in effect to free Rapa.
Rapa Reduces Cathepsin S (CTSS) Activity in the LG.
Among several candidates proposed as SS tear biomarkers, cathepsin S (CTSS) activity has emerged.61,62 An endolysosomal protease, CTSS is a member of the cysteine cathepsin protease family, with dysregulated activity linked to various diseases including cancer and autoimmune disorders.63 When it is dysregulated, CTSS may be secreted extracellularly and digest various molecules that maintain homeostasis. Its elevated activity has been confirmed in clinical studies showing that increased tear CTSS activity distinguishes SS patients from patients with non-autoimmune dry eye disease, other autoimmune diseases, and healthy controls.61,62 In the NOD mice, CTSS gene expression and activity are increased in LG lysates as well as in tears30,64 and its reduction was correlated with the therapeutic activity of Rapa.27,28 To see if FAF–Rapa affected LG CTSS, activity in LG lysates from male NOD mice treated with FAF–Rapa was compared with that of controls. Results show that SC FAF–Rapa successfully reduced CTSS activity in the LG (Figure 6A) with no significant effect on CTSS gene expression in the LG and CTSS activity in tears (Figure 6B,C). The ability for FAF–Rapa to reduce CTSS activity was not statistically significantly different from that of the free Rapa. The lack of effect on tear CTSS levels may be because the SS-like disease was not yet fully developed within the cohort of mice enrolled to this study. The degree of autoimmune dacryoadenitis in different cohorts of NOD mice can be variable. We observed lower LG infiltration by lymphocytes in this cohort (~20%, Figure 4) compared to some previous studies in NOD mice (35–45%).27,28 Since disease may not have been as developed, CTSS activity might not have been elevated enough in tears to reveal a response to Rapa. Another reason might be the bioavailability of SC FAF–Rapa. Compared to 100% bioavailability for IV FAF–Rapa, 53–66% bioavailability for SC FAF–Rapa may be sufficient only to reduce CTSS activity in the LG. Dosing and administration frequency optimization may be able to improve therapeutic efficacy of SC FAF–Rapa to further decrease tear CTSS at the ocular surface.
Figure 6.

Rapa suppresses proteolytic CTSS activity in the LG but not in the tears. At the conclusion of the 2 week study, LG and tears were collected immediately after euthanasia for CTSS activity and gene expression analysis. (A) A significant decrease in CTSS activity was observed in the LG lysates after SC Rapa treatments (n = 9). (B) No statistical significance was observed for CTSS gene expression level in the LG over a 2 week period (n = 12). (C) No statistical significance was achieved for CTSS activity in tears over a 2 week period (n = 9). Two-way ANOVA was used to compare the effects of the drug and carrier (Mean ± SD).
Histopathological Evaluation Reveals FAF–Rapa as a Biocompatible System for SC Delivery of Rapa.
At the conclusion of the study, major organs and blood samples were collected for histopathological and toxicological evaluation upon FAF–Rapa treatment. Organs subjected to H&E staining did not show any evidence of systemic toxicity (Figure 7 and Supplemental Table 1), and organ weights remained in the normal range across all treatment groups (Table 3). Rapa may be favored over calcineurin inhibitors (cyclosporin A or tacrolimus) because of its manageable renal toxicity65,66 and thus is approved for use in renal allograft recipients; however, concerns regarding nephrotoxicity of Rapa remain.67 On the other hand, Rapa damages the liver in humans because of an abundance of FKBP51 in liver tissue, which is 12:1 in molar ratio to FKBP12;68 thus, Rapa is not indicated for liver transplantation. Its hepatotoxicity has been verified in mouse models.69,70 To observe if FAF–Rapa induced any systemic toxicity during our therapeutic study, serum alanine aminotransferase (ALT), alkaline phosphatase (ALP) (indicators of liver damage), blood urea nitrogen (BUN), and creatinine levels (indicators of kidney damage) were analyzed.71,72 As compared in Table 3, there was no apparent nephro or hepatotoxicities upon FAF–Rapa treatment in this study. Free Rapa- and FAF–Rapa-treated groups showed a trend to an increase ALT, which was statistically insignificant. It should be noted that 1 out of 15 mice treated with FAF–Rapa showed an unusually high level of ALT (824 IU/L), which led to an overall higher value for the FAF–Rapa group; however, the ALP, BUN, creatinine, and BUN/creatinine ratio of this particular mouse remained normal. One parameter that was significantly different for the FAF–Rapa-treated group compared to the other three groups was a body weight change. Only FAF–Rapa-treated mice experienced a body weight loss over a 2 week period. This may be due to a significantly increased blood concentration of FAF–Rapa compared to free Rapa (Figure 3E).
Figure 7.

Histopathology of mouse organs reveals no systemic toxicity of FAF–Rapa at a therapeutic dose. At the conclusion of the study, organs and bloods from mice were sampled upon euthanasia. Organs were fixed, paraffin-embedded, sectioned, and stained with H&E. Kidney (n = 3), spleen (n = 3), lung (n = 3), and liver (n = 5) were analyzed by a blinded, trained pathologist. Images of organs from one representative mouse from each group were selected by a pathologist and shown. Black bar represents 100 μm.
Table 3.
Toxicity Assessment of FAF–Rapa via Serum Chemistry and Organ/Body Weight Measurements (Mean ± SD)
| category | parameters | vehicle | free Rapa | carrier (FAF) | FAF-Rapa |
|---|---|---|---|---|---|
| serum chemistry (n = 13) | ALT (IU/L)a | 34.1 ± 20.8 | 82.3 ± 97.4 | 49.0 ± 43.2 | 158.3 ± 225.6 |
| ALP (IU/L) | 43.1 ± 5.7 | 45.0 ± 9.5 | 34.7 ± 9.3 | 48.0 ± 9.6 | |
| BUN (mg/dL) | 23.6 ± 2.2 | 22.1 ± 2.6 | 23.3 ± 3.0 | 22.5 ± 2.1 | |
| creatinine (mg/dL) | 0.2 ± 0.00 | 0.2 ± 0.00 | 0.2 ± 0.00 | 0.2 ± 0.04 | |
| BUN/creatinine ratio | 118.2 ± 11.2 | 110.4 ± 12.8 | 116.5 ± 14.9 | 106.8 ± 16.6 | |
| organ weights (n = 15) | lung (% BW) | 0.60 ± 0.07 | 0.63 ± 0.06 | 0.57 ± 0.07 | 0.63 ± 0.09 |
| liver (% BW) | 4.7 ± 0.5 | 4.9 ± 0.4 | 4.5 ± 0.4 | 5.0 ± 0.5 | |
| spleen (% BW) | 0.29 ± 0.05 | 0.28 ± 0.09 | 0.33 ± 0.06 | 0.28 ± 0.09 | |
| kidney (% BW) | 1.6 ± 0.1 | 1.6 ± 0.1 | 1.5 ± 0.2 | 1.6 ± 0.1 | |
| body weight | change (% BW)b | 1.5 ± 2.9 | 1.7 ± 2.7 | 3.3 ± 2.3 | −1.8 ± 3.2 |
Two-way ANOVA showed a statistically significantly different ALT level from Rapa treatment (Vehicle + FAF vs free Rapa + FAF–Rapa, p = 0.03) but no significant interaction between Rapa and FAF (p = 0.4). No statistical significance was achieved among groups using the Kruskal–Wallis nonparametric test.
Kruskal–Wallis nonparametric test was performed for body weight change (n = 15) based on a statistical significance achieved by Rapa treatment (Vehicle + FAF vs free Rapa + FAF–Rapa, p = 0.001) and significant interaction between Rapa and FAF (p < 0.001) using two-way ANOVA. From Kruskal–Wallis nonparametric test, the body weight change of FAF–Rapa-treated group was statistically significantly different compared to vehicle (p = 0.012), free Rapa (p = 0.006), and carrier (p < 0.001).
Termination of Rapa Treatment Resolves Hyperglycemia.
One side effect associated with Rapa or Rapalogs (temsirolimus, everolimus, and ridaforolimus), also seen with other mTOR inhibitors, is metabolic complications, such as hyperglycemia, hypercholesterolemia, and hypertriglyceridemia.10 It has been reported that about 70% of patients who received Rapalogs experienced all-grade metabolic disorders of any kind, of which 25% are all-grade hyperglycemia. Within this 25%, only 7% of patients are diagnosed with grade 3–4 (250–500 mg/dL or higher), while the majority experienced mild-to-moderate hyperglycemia.73 To distinguish the elevated glucose level as a result of spontaneous development of diabetes in male NOD mice74 from Rapa treatment, blood glucose levels were monitored before and after the study. FAF–Rapa-treated mice experienced elevated blood glucose, even compared to free Rapa-treated mice (Figure 8A). Among 15 mice treated with FAF–Rapa, 6 mice were identified as hyperglycemic (>250 mg/dL). The mean difference in blood glucose before and after treatment with FAF–Rapa (n = 15) was +150 ± 99 mg/dL, whereas this value was +49 ± 62 mg/dL for free Rapa-treated mice (n = 15) (Mean ± SD). Mice treated with vehicle (+19 ± 85 mg/dL, n = 15) or carrier (+12 ± 36 mg/dL, n =15) did not show significant differences in blood glucose before and after the treatment. While the FAF–Rapa-treated population had a higher incidence of body weight loss and blood glucose, within individual subjects, there was no correlation between the percent body weight change and the severity of hyperglycemia (Figure 8B and Table 3).
Figure 8.

FAF–Rapa induces temporary hyperglycemia in NOD mice that resolves after termination of the treatment. (A) At the conclusion of the study as described in Figure 1, blood glucose levels of individual mice were measured. The dotted line shows 250 mg/dL, which is a criterion for hyperglycemia. A Kruskal–Wallis nonparametric test was performed based on a statistical significance achieved by Rapa treatment (Vehicle + FAF vs free Rapa + FAF–Rapa, p < 0.001) and significant interaction between Rapa and FAF (p = 0.004) using two-way ANOVA (Mean ± SD, n = 15). Results of the Kruskal–Wallis nonparametric test are presented. (B) Each mouse treated with FAF–Rapa was further analyzed to correlate percent body weight change to blood glucose change, before and after the treatment. Mice with final blood glucose less than 200 mg/dL (circle), between 200 and 250 mg/dL (triangle), and above 250 mg/dL (square) were plotted. (C,D) In two additional studies, FAF–Rapa was administered as described in Figure 1 (shaded area) to either (C) male NOD mice (n = 5) or (D) male Balb/C mice (n = 5), and the blood glucose levels of individual mice (M1~M5) were monitored for 2 weeks after termination of the treatment.
In the clinic, hyperglycemia can be resolved by a combination of dietary control, oral medication, and insulin, as recommended by the American Diabetes Association (www.diabetes.org).73,75 To confirm that FAF–Rapa-induced hyperglycemia was reversible, we replicated the treatment procedure described in Figure 1 in age-matched male NOD mice and healthy Balb/C mice and monitored their blood glucose changes during and after the treatment (Figure 8C,D). The glucose levels in male NOD mice upon FAF–Rapa treatment showed a heterogeneous characteristic of that seen in the human population. One (M1) mouse with a high baseline blood glucose progressed to levels consistent with diabetes, which may be representative of a subset of human populations with baseline-elevated blood glucose, which have a higher incidence of hyperglycemia and diabetes with this drug.76 Two (M2, M4) out of five mice were unaffected by FAF–Rapa. Two mice (M2, M4) with elevated blood glucose upon treatment showed levels restored to normal immediately after FAF–Rapa was removed. The lag time of 4–5 days until restoration to the baseline is consistent with the pharmacokinetic behavior of SC FAF–Rapa presented in Figure 3A Unlike the NOD mice, BALB/c mice did not experience hyperglycemia under the same treatment schedule. This suggests that the observed elevated glucose level is not a result of spontaneous development of diabetes in male NOD mice and the metabolic complications induced by SC FAF–Rapa are not permanent and can be managed with conventional methods. While the NOD mouse is also a model of T-cell-mediated type I diabetes, it remains unclear how FAF–Rapa-mediated hyperglycemia would progress in otherwise healthy humans.
DISCUSSION
The subcutaneous (SC) space has long been favored for drug delivery, whereby the injection site behaves as a drug depot. In comparison to intravenous (IV) administration, SC delivery is fast and can be performed easily at home.77 For example, a recent study compared IV to SC administration of trastuzumab in HER2-positive breast cancer patients. When administered SC, trastuzumab showed equivalent pharmacodynamic, pharmacokinetic, and safety profiles compared to standard IV administration and was highly preferred by patients. Two principal reasons for this preference were the reduced pain during injection (158 out of 236 patients, 67%) and the shorter time for injection (5 min per SC injection vs. 30~90 min for IV infusions). Given that patients commonly receive 17–25 trastuzumab IV infusions per year (every 2–3 weeks), SC injections every 3 weeks were preferred by 91.5% of patients.78 Another study compared time spent by healthcare professionals and costs for SC and IV administration.79 The time spent by healthcare professionals from preparation to administration of SC rituximab was only 0.8 h, compared to 3.8 h for IV administration, while the time that patients spent in treatment rooms was only 1.2 h for SC administration, compared to 4.4 h for IV administration. SC administration decreased the total mean staff cost by 140~160 US dollar equivalents per session. While there are oral formulations of rapamycin, low oral bioavailability creates unpredictable PK profiles and variability in the severity of side effects.8–12 In comparison to drugs with poor oral bioavailability and unfavorable pharmacokinetic profiles, SC administration has been reported to improve therapeutic index and patient compliance.80 While there are no approved SC formulations for rapamycin, this drug has been extensively used in sustained release formulations such as from drug-eluting stents.81
Strategies to treat SS are primarily focused on systemic immune suppression. The majority of clinical trials have targeted B-cells and T-cells, with fewer focused on proinflammatory cytokines, and very few studies have been directed to intracellular pathways.82 In considering the targeting of intracellular pathways, JAK-STAT pathway inhibition (clinicaltrials.gov identifier: ), CTSS inhibition (), and ubiquitin/proteasome inhibition83 have been explored but not yet the mTOR pathway. The study described here, to the best of our knowledge, is the first report targeting mTOR signaling by an SC Rapa formulation to treat autoimmune dacryoadenitis in a murine model of SS. This target, drug, and route of administration has never been tested in the clinical or preclinical setting. On the basis of the data presented, FAF–Rapa is a highly promising SC-injectable therapeutic candidate for SS. As with any other new formulation, extensive preclinical and clinical toxicology studies will be required for further development.
To explore pharmacodynamic advantages of using FAF to deliver SC Rapa, we chose to compare it to free Rapa solubilized using components (PEG and polysorbate 80) found in the oral formulation known as Rapamune. While valuable as a control, solubilization alone during SC administration is unlikely to improve its absorption into circulation, entrapment by red blood cells, or toxicity profile. Instead, we propose that high affinity binding between our FKBP moieties on FAF has the potential to improve SC bioavailability, the plasma-to-whole blood ratio, and the absolute plasma concentration of Rapa. As a drug carrier, such a scenario could be pharmacodynamically advantageous compared to that of a solubilizer-based formulation. While SC FAF–Rapa achieved higher plasma levels than SC free Rapa (Figure 3E), the therapeutic end points related to autoimmune dacryoadenitis explored in this manuscript (Figures 4–6) do not distinguish profound differences between the two formulations. This lack of difference likely stems from the fact that both FAF–Rapa and free Rapa are effective at the relatively high dose evaluated. Even so, the 8-fold increase in the absolute plasma level of SC FAF–Rapa compared to free Rapa strongly suggests the drug remains bound to the carrier in the blood near Cmax. If true, then it remains plausible that FAF–Rapa will remain associated even while interacting with lymphocytes and normal cells of the LG. To support this contention, we recently reported that in vitro cellular uptake of FAF–Rapa occurs through macropinocytosis, which differed significantly from simple diffusion of free Rapa.84 Further studies must now explore if FAF–Rapa exhibits a therapeutic effect at a subtherapeutic dose or frequency of free Rapa. These studies may solve the body weight loss (Table 3) and induction of hyperglycemia (Figure 8) observed during SC FAF–Rapa treatment. It should be noted that our preclinical study presented here collected only Rapa plasma concentrations near the Cmax for FAF, which may be useful for translation to clinical settings. Although the dose in this study (1.0 mg of Rapa/kg) cannot be directly compared to the clinical dose (0.02–0.04 mg of Rapa/kg), 1.0 mg of Rapa/kg in mice achieved a plasma Cmax (0.4 ng of Rapa/mL) that is very similar to plasma Cmax in humans (0.5 ng/mL) reported after oral administration of 2 mg of Rapamune.85
In healthy humans, the plasma-to-whole blood ratio of Rapa is about 1:106.86 This extreme partitioning to human red blood cells differs significantly from that observed in other species, such as rabbits, rats, or mice.87 In male NOD mice, free Rapa gave a plasma-to-whole blood ratio of only 1:1, which was significantly increased to 1.5:1 when formulated as FAF–Rapa (Figure 3F). The observation of 1:1 for the plasma-to-whole blood ratio of Rapa in our murine model is similar to that reported by others.88 It has been proposed that other isoforms of FKBP could play a role in this difference. For example, cytosolic FKBP12 and membrane-bound FKBP13 on RBCs have been proposed to modulate the high plasma-to-whole blood ratio seen in humans.89 The nucleotide sequences encoding the binding pocket for Rapa in human and mouse FKBP12 and FKBP13 are highly conserved, which suggests they may have similar affinities.89 There exists the possibility that differential expression or cellular localization may also play a role; however, we were unable to find data to support or reject this possibility. Another reasonable explanation could be the differential stability of Rapa in whole blood between humans and rodents.87 Rapa’s half-life in whole blood taken from humans is 135 h, while in whole blood taken from rats, it is only 15 h. This suggests that entrapment in human RBCs protects Rapa from degradation better than in the whole blood of rodents. Although the exact mechanism underlying differences in plasma-to-whole blood ratios of Rapa between humans and other species remains unknown, the fact that this study detected a significant 50% increase in plasma-to-whole blood ratio for Rapa (Figure 3F) suggests that FAF may significantly affect hematologic toxicities of Rapa in humans, which commonly result in discontinuation of treatment.10
Current formulations of Rapa (sirolimus) or Rapalogs (everolimus, temsirolimus, ridaforolimus) are limited to oral or IV administration. Despite poor bioavailability and frequent discontinuation due to side effects, free Rapa (sirolimus) and everolimus, 2 mg/day (www.pfizermedicalinformation.com) and 10 mg/day (www.hcp.novartis.com), respectively are administered orally to patients. Temsirolimus and ridaforolimus are slightly more water-soluble and formulated for IV infusion. Temsirolimus is infused as 25 mg/week over 30–60 min (www.pfizermedicalinformation.com). Ridaforolimus is being tested as an IV infusion of 12.5 mg/daily for 5 days every 2 weeks.90,91 Despite the potential opportunities for a SC formulation, none of these Rapalogs are given SC because of the lack of an effective carrier. The results of this manuscript suggest that FAF–Rapa may represent a first-generation SC carrier, which can be further optimized to reduce administration frequency and dose, reduce dose-limiting side effects, and increase patient compliance in the clinic.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the USC Stevens Technology Advancement Grant (TAG), NIH R01EY026635 to J.A.M. and S.H.A., NIH R01GM114839 to J.A.M., NIH RO1 EY011386 to S.H.A., P30 EY029220 to the USC Ophthalmology Center Core Grant for Vision Research, P30 CA014089 to the USC Norris Comprehensive Cancer Center, P30 DK048522 to the Liver Histology Core of the USC Research Center for Liver Diseases, the USC L.K Whittier Foundation, and an unrestricted departmental grant from Research to Prevent Blindness. The authors would like to acknowledge the support of the School of Pharmacy Translational Research Core for these studies. The authors would also like to thank Prof. Driss Zoukhri and Mr. Dillon Hawley (Tufts University) and Mihir Shah, Ph.D. (Allergan plc, Irvine, CA) for discussions and suggestions for this study.
ABBREVIATIONS
- CTSS
cathepsin S
- DLS
dynamic light scattering
- ELPs
elastin-like polypeptides
- FAF
FKBP-A192-FKBP
- FKBP
FK506-binding protein
- ITC
inverse transition cycling
- IV
intravenous
- LG
lacrimal gland
- MHC II
major histocompatibility complex II
- NOD
non-obese diabetic
- Rapa
rapamycin
- RBC
red blood cells
- SS
Sjögren’s syndrome
- SC
subcutanous
- USFDA
United States Food and Drug Administration
- WB
whole blood
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.9b00263.
Pathologist’s evaluation for histology (PDF)
The authors declare the following competing financial interest(s): J.A.M., S.F.H.-A., and S.P. are inventors on patents describing delivery of small molecules using protein polymer fusions related to this work. All other authors declare no competing financial interests.
REFERENCES
- (1).Kojima I; Cheng YR; Mohan V; Demain AL Carbon source nutrition of rapamycin biosynthesis in Streptomyces hygroscopicus. J. Ind. Microbiol 1995, 14 (6), 436–9. [DOI] [PubMed] [Google Scholar]
- (2).Costa MA; Simon DI Molecular basis of restenosis and drug-eluting stents. Circulation 2005, 111 (17), 2257–2273. [DOI] [PubMed] [Google Scholar]
- (3).Thomson AW; Turnquist HR; Raimondi G Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol 2009, 9 (5), 324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bjornsti MA; Houghton PJ The TOR pathway: a target for cancer therapy. Nat. Rev. Cancer 2004, 4 (5), 335–48. [DOI] [PubMed] [Google Scholar]
- (5).Hardinger KL; Koch MJ; Brennan DC Current and future immunosuppressive strategies in renal transplantation. Pharmacotherapy 2004, 24 (9), 1159–76. [DOI] [PubMed] [Google Scholar]
- (6).Tedesco Silva H Jr.; Rosso Felipe C; Medina Pestana JO Reviewing 15 years of experience with sirolimus. Transplant. Res 2015, 4 (S1), 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).McCormack FX; Inoue Y; Moss J; Singer LG; Strange C; Nakata K; Barker AF; Chapman JT; Brantly ML; Stocks JM; Brown KK; Lynch JP 3rd; Goldberg HJ; Young LR; Kinder BW; Downey GP; Sullivan EJ; Colby TV; McKay RT; Cohen MM; Korbee L; Taveira-DaSilva AM; Lee HS; Krischer JP; Trapnell BC Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N. Engl. J. Med 2011, 364 (17), 1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pham PT; Pham PC; Danovitch GM; Ross DJ; Gritsch HA; Kendrick EA; Singer J; Shah T; Wilkinson AH Sirolimus-associated pulmonary toxicity. Transplantation 2004, 77 (8), 1215–20. [DOI] [PubMed] [Google Scholar]
- (9).Marti HP; Frey FJ Nephrotoxicity of rapamycin: an emerging problem in clinical medicine. Nephrol., Dial. Transplant 2005, 20 (1), 13–5. [DOI] [PubMed] [Google Scholar]
- (10).Pallet N; Legendre C Adverse events associated with mTOR inhibitors. Expert Opin. Drug Saf 2013, 12 (2), 177–86. [DOI] [PubMed] [Google Scholar]
- (11).Simamora P; Alvarez JM; Yalkowsky SH Solubilization of rapamycin. Int. J. Pharm 2001, 213 (1–2), 25–9. [DOI] [PubMed] [Google Scholar]
- (12).Stenton SB; Partovi N; Ensom MH Sirolimus: the evidence for clinical pharmacokinetic monitoring. Clin. Pharmacokinet 2005, 44 (8), 769–86. [DOI] [PubMed] [Google Scholar]
- (13).Zimmerman JJ; Lasseter KC; Lim HK; Harper D; Dilzer SC; Parker V; Matschke K Pharmacokinetics of sirolimus (rapamycin) in subjects with mild to moderate hepatic impairment. J. Clin. Pharmacol 2005, 45 (12), 1368–72. [DOI] [PubMed] [Google Scholar]
- (14).Mayer DF; Kushwaha SS Transplant immunosuppressant agents and their role in autoimmune rheumatic diseases. Curr. Opin. Rheumatol 2003, 15 (3), 219–25. [DOI] [PubMed] [Google Scholar]
- (15).Neuhaus O; Kieseier BC; Hartung HP Immunosuppressive agents in multiple sclerosis. Neurotherapeutics 2007, 4 (4), 654–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dhandhukia JP; Li Z; Peddi S; Kakan S; Mehta A; Tyrpak D; Despanie J; MacKay JA Berunda Polypeptides: Multi-Headed Fusion Proteins Promote Subcutaneous Administration of Rapamycin to Breast Cancer In Vivo. Theranostics 2017, 7 (16), 3856–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Marz AM; Fabian AK; Kozany C; Bracher A; Hausch F Large FK506-binding proteins shape the pharmacology of rapamycin. Mol. Cell. Biol 2013, 33 (7), 1357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Despanie J; Dhandhukia JP; Hamm-Alvarez SF; MacKay JA Elastin-like polypeptides: Therapeutic applications for an emerging class of nanomedicines. J. Controlled Release 2016, 240, 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mavragani CP; Moutsopoulos HM Sjogren’s syndrome. Annu. Rev. Pathol.: Mech. Dis 2014, 9, 273–85. [DOI] [PubMed] [Google Scholar]
- (20).Dastjerdi MH; Hamrah P; Dana R High-frequency topical cyclosporine 0.05% in the treatment of severe dry eye refractory to twice-daily regimen. Cornea 2009, 28 (10), 1091–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tong AY; Passi SF; Gupta PK Clinical Outcomes of Lifitegrast 5% Ophthalmic Solution in the Treatment of Dry Eye Disease. Eye Contact Lens 2019. [DOI] [PubMed] [Google Scholar]
- (22).de Paiva CS; Pflugfelder SC Rationale for anti-inflammatory therapy in dry eye syndrome. Arq Bras Oftalmol 2008, 71 (6), 89–95. [DOI] [PubMed] [Google Scholar]
- (23).Mitsias DI; Kapsogeorgou EK; Moutsopoulos HM Sjogren’s syndrome: why autoimmune epithelitis? Oral Dis 2006, 12 (6), 523–32. [DOI] [PubMed] [Google Scholar]
- (24).Linares-Alba MA; Gomez-Guajardo MB; Fonzar JF; Brooks DE; Garcia-Sanchez GA; Bernad-Bernad MJ Preformulation Studies of a Liposomal Formulation Containing Sirolimus for the Treatment of Dry Eye Disease. J. Ocul. Pharmacol. Ther 2016, 32 (1), 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ratay ML; Balmert SC; Acharya AP; Greene AC; Meyyappan T; Little SR TRI Microspheres prevent key signs of dry eye disease in a murine, inflammatory model. Sci. Rep 2017, 7 (1), 17527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang S; Wang M; Liu Y; Hu D; Gu L; Fei X; Zhang J Effect of Rapamycin Microspheres in Sjogren Syndrome Dry Eye: Preparation and Outcomes. Ocul. Immunol. Inflammation 2018, 1–8. [DOI] [PubMed] [Google Scholar]
- (27).Shah M; Edman MC; Janga SR; Shi P; Dhandhukia J; Liu S; Louie SG; Rodgers K; Mackay JA; Hamm-Alvarez SF A rapamycin-binding protein polymer nanoparticle shows potent therapeutic activity in suppressing autoimmune dacryoadenitis in a mouse model of Sjogren’s syndrome. J. Controlled Release 2013, 171 (3), 269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shah M; Edman MC; Reddy Janga S; Yarber F; Meng Z; Klinngam W; Bushman J; Ma T; Liu S; Louie S; Mehta A; Ding C; MacKay JA; Hamm-Alvarez SF Rapamycin Eye Drops Suppress Lacrimal Gland Inflammation In a Murine Model of Sjogren’s Syndrome. Invest. Ophthalmol. Visual Sci 2017, 58 (1), 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Takahashi M; Ishimaru N; Yanagi K; Haneji N; Saito I; Hayashi Y High incidence of autoimmune dacryoadenitis in male non-obese diabetic (NOD) mice depending on sex steroid. Clin. Exp. Immunol 1997, 109 (3), 555–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Janga SR; Shah M; Ju Y; Meng Z; Edman MC; Hamm-Alvarez SF Longitudinal analysis of tear cathepsin S activity levels in male non-obese diabetic mice suggests its potential as an early stage biomarker of Sjogren’s Syndrome. Biomarkers 2019, 24 (1), 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ju Y; Janga SR; Klinngam W; MacKay JA; Hawley D; Zoukhri D; Edman MC; Hamm-Alvarez SF NOD and NOR mice exhibit comparable development of lacrimal gland secretory dysfunction but NOD mice have more severe autoimmune dacryoadenitis. Exp. Eye Res 2018, 176, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lodde BM; Mineshiba F; Kok MR; Wang J; Zheng C; Schmidt M; Cotrim AP; Kriete M; Tak PP; Baum BJ NOD mouse model for Sjogren’s syndrome: lack of longitudinal stability. Oral Dis 2006, 12 (6), 566–72. [DOI] [PubMed] [Google Scholar]
- (33).Wang CS; Maruyama CL; Easley JT; Trump BG; Baker OJ AT-RvD1 Promotes Resolution of Inflammation in NOD/ShiLtJ mice. Sci. Rep 2017, 7, 45525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Schenke-Layland K; Xie J; Angelis E; Starcher B; Wu K; Riemann I; MacLellan WR; Hamm-Alvarez SF Increased degradation of extracellular matrix structures of lacrimal glands implicated in the pathogenesis of Sjogren’s syndrome. Matrix Biol. 2008, 27 (1), 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yamano S; Atkinson JC; Baum BJ; Fox PC Salivary gland cytokine expression in NOD and normal BALB/c mice. Clin. Immunol 1999, 92 (3), 265–75. [DOI] [PubMed] [Google Scholar]
- (36).Janib SM; Pastuszka M; Aluri S; Folchman-Wagner Z; Hsueh PY; Shi P; Lin YA; Cui H; Mackay JA A quantitative recipe for engineering protein polymer nanoparticles. Polym. Chem 2014, 5 (5), 1614–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Standaert RF; Galat A; Verdine GL; Schreiber SL Molecular cloning and overexpression of the human FK506-binding protein FKBP. Nature 1990, 346 (6285), 671–4. [DOI] [PubMed] [Google Scholar]
- (38).Pace CN; Vajdos F; Fee L; Grimsley G; Gray T How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4 (11), 2411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Edelhoch H Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 1967, 6 (7), 1948–54. [DOI] [PubMed] [Google Scholar]
- (40).MacKay JA; Chen M; McDaniel JR; Liu W; Simnick AJ; Chilkoti A Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat. Mater 2009, 8 (12), 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cho Y; Zhang Y; Christensen T; Sagle LB; Chilkoti A; Cremer PS Effects of Hofmeister anions on the phase transition temperature of elastin-like polypeptides. J. Phys. Chem. B 2008, 112 (44), 13765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kagan L Pharmacokinetic modeling of the subcutaneous absorption of therapeutic proteins. Drug Metab. Dispos 2014, 42 (11), 1890–905. [DOI] [PubMed] [Google Scholar]
- (43).Guo H; Lee C; Shah M; Janga SR; Edman MC; Klinngam W; Hamm-Alvarez SF; Andrew Mackay J A novel elastin-like polypeptide drug carrier for cyclosporine A improves tear flow in a mouse model of Sjogren’s syndrome. J. Controlled Release 2018, 292, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Biagiotti S; Paoletti MF; Fraternale A; Rossi L; Magnani M Drug delivery by red blood cells. IUBMB Life 2011, 63 (8), 621–31. [DOI] [PubMed] [Google Scholar]
- (45).Zoukhri D Mechanisms involved in injury and repair of the murine lacrimal gland: role of programmed cell death and mesenchymal stem cells. Ocul Surf. 2010, 8 (2), 60–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jones EY; Fugger L; Strominger JL; Siebold C MHC class II proteins and disease: a structural perspective. Nat. Rev. Immunol 2006, 6 (4), 271–82. [DOI] [PubMed] [Google Scholar]
- (47).Mircheff AK; Gierow JP; Lee LM; Lambert RW; Akashi RH; Hofman FM Class II antigen expression by lacrimal epithelial cells. An updated working hypothesis for antigen presentation by epithelial cells. Invest Ophthalmol Vis Sci 1991, 32 (8), 2302–2310. [PubMed] [Google Scholar]
- (48).Guo Z; Azzarolo AM; Schechter JE; Warren DW; Wood RL; Mircheff AK; Kaslow HR Lacrimal gland epithelial cells stimulate proliferation in autologous lymphocyte preparations. Exp. Eye Res 2000, 71 (1), 11–22. [DOI] [PubMed] [Google Scholar]
- (49).Meng Z; Klinngam W; Edman MC; Hamm-Alvarez SF Interferon-gamma treatment in vitro elicits some of the changes in cathepsin S and antigen presentation characteristic of lacrimal glands and corneas from the NOD mouse model of Sjogren’s Syndrome. PLoS One 2017, 12 (9), No. e0184781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ji YW; Byun YJ; Choi W; Jeong E; Kim JS; Noh H; Kim ES; Song YJ; Park SK; Lee HK Neutralization of ocular surface TNF-alpha reduces ocular surface and lacrimal gland inflammation induced by in vivo dry eye. Invest. Ophthalmol. Visual Sci 2013, 54 (12), 7557–66. [DOI] [PubMed] [Google Scholar]
- (51).Vosters JL; Landek-Salgado MA; Yin H; Swaim WD; Kimura H; Tak PP; Caturegli P; Chiorini JA Interleukin-12 induces salivary gland dysfunction in transgenic mice, providing a new model of Sjogren’s syndrome. Arthritis Rheum. 2009, 60 (12), 3633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Sato EA; Matsumoto Y; Dogru M; Kaido M; Wakamatsu T; Ibrahim OM; Obata H; Tsubota K Lacrimal gland in Sjogren’s syndrome. Ophthalmology 2010, 117 (5), 1055–1055. [DOI] [PubMed] [Google Scholar]
- (53).Wu MJ; Wen MC; Chiu YT; Chiou YY; Shu KH; Tang MJ Rapamycin attenuates unilateral ureteral obstruction-induced renal fibrosis. Kidney Int. 2006, 69 (11), 2029–36. [DOI] [PubMed] [Google Scholar]
- (54).Yoshizaki A; Yanaba K; Yoshizaki A; Iwata Y; Komura K; Ogawa F; Takenaka M; Shimizu K; Asano Y; Hasegawa M; Fujimoto M; Sato S Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum. 2010, 62 (8), 2476–87. [DOI] [PubMed] [Google Scholar]
- (55).Yu SY; Liu L; Li P; Li J Rapamycin inhibits the mTOR/p70S6K pathway and attenuates cardiac fibrosis in adriamycin-induced dilated cardiomyopathy. Thorac. Cardiovasc. Surg 2013, 61 (3), 223–8. [DOI] [PubMed] [Google Scholar]
- (56).Korfhagen TR; Le Cras TD; Davidson CR; Schmidt SM; Ikegami M; Whitsett JA; Hardie WD Rapamycin prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol 2009, 41 (5), 562–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bridle KR; Popa C; Morgan ML; Sobbe AL; Clouston AD; Fletcher LM; Crawford DH Rapamycin inhibits hepatic fibrosis in rats by attenuating multiple profibrogenic pathways. Liver Transpl 2009, 15 (10), 1315–24. [DOI] [PubMed] [Google Scholar]
- (58).Zhang Y; Stefanovic B mTORC1 phosphorylates LARP6 to stimulate type I collagen expression. Sci. Rep 2017, 7, 41173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Byun YS; Lee HJ; Shin S; Chung SH Elevation of autophagy markers in Sjogren syndrome dry eye. Sci. Rep 2017, 7 (1), 17280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Corum DG; Tsichlis PN; Muise-Helmericks RC AKT3 controls mitochondrial biogenesis and autophagy via regulation of the major nuclear export protein CRM-1. FASEB J. 2014, 28 (1), 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Hamm-Alvarez SF; Janga SR; Edman MC; Madrigal S; Shah M; Frousiakis SE; Renduchintala K; Zhu J; Bricel S; Silka K; Bach D; Heur M; Christianakis S; Arkfeld DG; Irvine J; Mack WJ; Stohl W Tear cathepsin S as a candidate biomarker for Sjogren’s syndrome. Arthritis Rheumatol. 2014, 66 (7), 1872–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Edman MC; Janga SR; Meng Z; Bechtold M; Chen AF; Kim C; Naman L; Sarma A; Teekappanavar N; Kim AY; Madrigal S; Singh S; Ortiz E; Christianakis S; Arkfeld DG; Mack WJ; Heur M; Stohl W; Hamm-Alvarez SF Increased Cathepsin S activity associated with decreased protease inhibitory capacity contributes to altered tear proteins in Sjogren’s Syndrome patients. Sci. Rep 2018, 8 (1), 11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Wilkinson RD; Williams R; Scott CJ; Burden RE Cathepsin S: therapeutic, diagnostic, and prognostic potential. Biol. Chem 2015, 396 (8), 867–82. [DOI] [PubMed] [Google Scholar]
- (64).Li X; Wu K; Edman M; Schenke-Layland K; MacVeigh-Aloni M; Janga SR; Schulz B; Hamm-Alvarez SF Increased expression of cathepsins and obesity-induced proinflammatory cytokines in lacrimal glands of male NOD mouse. Invest. Ophthalmol. Visual Sci 2010, 51 (10), 5019–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kahan BD Two-year results of multicenter phase III trials on the effect of the addition of sirolimus to cyclosporine-based immunosuppressive regimens in renal transplantation. Transplant. Proc 2003, 35 (3 Suppl), S37–S51. [DOI] [PubMed] [Google Scholar]
- (66).Asante-Korang A; Carapellucci J; Krasnopero D; Doyle A; Brown B; Amankwah E Conversion from calcineurin inhibitors to mTOR inhibitors as primary immunosuppressive drugs in pediatric heart transplantation. Clin. Transplant 2017, 31 (10), No. e13054, ARTN. [DOI] [PubMed] [Google Scholar]
- (67).Marti HP; Frey FJ Nephrotoxicity of rapamycin: an emerging problem in clinical medicine. Nephrol., Dial., Transplant 2005, 20 (1), 13–15. [DOI] [PubMed] [Google Scholar]
- (68).Baughman G; Wiederrecht GJ; Chang F; Martin MM; Bourgeois S Tissue distribution and abundance of human FKBP51, and FK506-binding protein that can mediate calcineurin inhibition. Biochem. Biophys. Res. Commun 1997, 232 (2), 437–43. [DOI] [PubMed] [Google Scholar]
- (69).Massoud O; Wiesner RH The use of Sirolimus should be restricted in liver transplantation. J. Hepatol 2012, 56 (1), 288–290. [DOI] [PubMed] [Google Scholar]
- (70).Umemura A; Park EJ; Taniguchi K; Lee JH; Shalapour S; Valasek MA; Aghajan M; Nakagawa H; Seki E; Hall MN; Karin M Liver Damage, Inflammation, and Enhanced Tumorigenesis after Persistent mTORC1 Inhibition. Cell Metab. 2014, 20 (1), 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Lala V; Minter DA Liver Function Tests Stat Pearls, Treasure Island (FL), 2019. [Google Scholar]
- (72).Kim SY; Moon A Drug-induced nephrotoxicity and its biomarkers. Biomol. Ther 2012, 20 (3), 268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Su DW; Mita M; Mita AC The Clinical Pharmacology and Toxicity Profile of Rapalogs. In mTOR Inhibition for Cancer Therapy: Past, Present and Future 2016, 161–189. [Google Scholar]
- (74).Leiter EH; Prochazka M; Coleman DL The non-obese diabetic (NOD) mouse. Am. J. Pathol 1987, 128 (2), 380–383. [PMC free article] [PubMed] [Google Scholar]
- (75).Busaidy NL; Farooki A; Dowlati A; Perentesis JP; Dancey JE; Doyle LA; Brell JM; Siu LL Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J. Clin. Oncol 2012, 30 (23), 2919–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Johnston O; Rose CL; Webster AC; Gill JS Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J. Am. Soc. Nephrol 2008, 19 (7), 1411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Turner MR; Balu-Iyer SV Challenges and Opportunities for the Subcutaneous Delivery of Therapeutic Proteins. J. Pharm. Sci 2018, 107 (5), 1247–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Pivot X; Gligorov J; Muller V; Barrett-Lee P; Verma S; Knoop A; Curigliano G; Semiglazov V; Lopez-Vivanco G; Jenkins V; Scotto N; Osborne S; Fallowfield L Preference for subcutaneous or intravenous administration of trastuzumab in patients with HER2-positive early breast cancer (PrefHer): an open-label randomised study. Lancet Oncol. 2013, 14 (10), 962–970. [DOI] [PubMed] [Google Scholar]
- (79).Rule S; Collins GP; Samanta K Subcutaneous vs intravenous rituximab in patients with non-Hodgkin lymphoma: a time and motion study in the United Kingdom. J. Med. Econ 2014, 17 (7), 459–68. [DOI] [PubMed] [Google Scholar]
- (80).Jin JF; Zhu LL; Chen M; Xu HM; Wang HF; Feng XQ; Zhu XP; Zhou Q The optimal choice of medication administration route regarding intravenous, intramuscular, and subcutaneous injection. Patient Prefer Adherence 2015, 9, 923–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Moses JW; Leon MB; Popma JJ; Fitzgerald PJ; Holmes DR; O’Shaughnessy C; Caputo RP; Kereiakes DJ; Williams DO; Teirstein PS; Jaeger JL; Kuntz RE Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N. Engl. J. Med 2003, 349 (14), 1315–1323. [DOI] [PubMed] [Google Scholar]
- (82).Brito-Zeron P; Retamozo S; Gheitasi H; Ramos-Casals M Treating the Underlying Pathophysiology of Primary Sjogren Syndrome: Recent Advances and Future Prospects. Drugs 2016, 76 (17), 1601–1623. [DOI] [PubMed] [Google Scholar]
- (83).Jakez-Ocampo J; Atisha-Fregoso Y; Llorente L Refractory primary Sjogren syndrome successfully treated with bortezomib. J. Clin Rheumatol 2015, 21 (1), 31–2. [DOI] [PubMed] [Google Scholar]
- (84).Peddi S; Pan X; MacKay JA Intracellular Delivery of Rapamycin From FKBP Elastin-Like Polypeptides Is Consistent With Macropinocytosis. Front. Pharmacol 2018, 9, 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Brattstrom C; Sawe J; Jansson B; Lonnebo A; Nordin J; Zimmerman JJ; Burke JT; Groth CG Pharmacokinetics and safety of single oral doses of sirolimus (rapamycin) in healthy male volunteers. Ther. Drug Monit 2000, 22 (5), 537–44. [DOI] [PubMed] [Google Scholar]
- (86).Zimmerman JJ; Patat A; Parks V; Moirand R; Matschke K Pharmacokinetics of sirolimus (rapamycin) in subjects with severe hepatic impairment. J. Clin. Pharmacol 2008, 48 (3), 285–92. [DOI] [PubMed] [Google Scholar]
- (87).Ferron GM; Jusko WJ Species differences in sirolimus stability in humans, rabbits, and rats. Drug Metab. Dispos 1998, 26 (1), 83–84. [PubMed] [Google Scholar]
- (88).Yanez JA; Forrest ML; Ohgami Y; Kwon GS; Davies NM Pharmacometrics and delivery of novel nanoformulated PEG-b-poly(epsilon-caprolactone) micelles of rapamycin. Cancer Chemother. Pharmacol 2007, 61 (1), 133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Hendrickson BA; Zhang W; Craig RJ; Yong-Jiu J; Bierer BE; Burakoff S; DiLella AG Structural organization of the genes encoding human and murine FK506-binding protein (FKBP) 13 and comparison to FKBP1. Gene 1993, 134 (2), 271–275. [DOI] [PubMed] [Google Scholar]
- (90).Rizzieri DA; Feldman E; Dipersio JF; Gabrail N; Stock W; Strair R; Rivera VM; Albitar M; Bedrosian CL; Giles FJ A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin. Cancer Res 2008, 14 (9), 2756–62. [DOI] [PubMed] [Google Scholar]
- (91).Mita MM; Mita AC; Chu QS; Rowinsky EK; Fetterly GJ; Goldston M; Patnaik A; Mathews L; Ricart AD; Mays T; Knowles H; Rivera VM; Kreisberg J; Bedrosian CL; Tolcher AW Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J. Clin. Oncol 2008, 26 (3), 361–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.