

Abstract
GSTpi is a Phase II metabolic enzyme which is originally considered as an important facilitator of cellular detoxification. Here, we found that GSTpi stabilized VE-cadherin in endothelial cell membrane through inhibiting VE-cadherin phosphorylation and VE-cadherin/catenin complex dissociation, and consequently maintained endothelial barrier function. Our findings demonstrated a novel mechanism that GSTpi inhibited VE-cadherin phosphorylation through suppressing the activation of Src/VE-cadherin pathway. Mass spectrometry analysis and molecular docking showed that GSTpi enhanced Src S-glutathionylation at Cys185, Cys245, and Cys400 of Src. More important, we found that GSTpi promoted S-glutathionylation of Src was essential for GSTpi to inhibit Src phosphorylation and activation. Furthermore, in vivo experiments indicated that AAV-GSTpi exerted the protective effect on pulmonary vessel permeability in the animal model of acute lung injury. This study revealed a novel regulatory effect of GSTpi on vascular endothelial barrier function and the importance of S-glutathionylation of Src induced by GSTpi in the activation of Src/VE-cadherin pathway.
Keywords: GSTpi, VE-Cadherin, Src/VE-Cadherin pathway, S-Glutathionylation of Src, Endothelial barrier function
Abbreviations: AAV, adeno-associated virus; AJ, adherens junctions; ECM, endothelial cell medium; ECs, endothelial cells; GSH, glutathione; GSTs, glutathione S-transferases; HPMECs, human pulmonary microvascular endothelial cells; HUVEC, human umbilical vein endothelial cells; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; PMN, polymorphonuclear neutrophils; ROS, reactive oxygen species; Src, proto-oncogene tyrosine-protein kinase; TNF-α, tumor necrosis factor-alpha; TEER, transepithelial electrical resistance; VEGF, vascular endothelial growth factor
Graphical abstract
Highlights
-
•
GSTpi regulates endothelial barrier function in response to pro-inflammatory stress.
-
•
GSTpi inhibits the destabilization of membrane VE-cadherin through suppressing the activation of Src/VE-cadherin pathway.
-
•
GSTpi selectively inhibits Src phosphorylation by S-glutathionylating novel cysteines of Src.
-
•
GSTpi exerts the protective effect on pulmonary vessel permeability in the animal model of acute lung injury.
1. Introduction
The vascular endothelium composed of monolayer endothelial cells (ECs) is on the inner surface of vascular wall. It acts as a semi-selective barrier and the structural integrity of vascular endothelium is essential for the maintenance of vascular homeostasis. Increased vascular endothelial permeability caused by inflammatory stimulation has been established as an early characteristic of vascular dysfunction associated with the pathogenesis of atherosclerosis, ischemia-reperfusion injury, acute respiratory distress syndrome, and septicemia [1,2]. Four main groups of the primary inflammatory mediators including lipids, plasma enzymes, cytokines, and chemokines are involved in vascular endothelial dysfunction. Among the pro-inflammatory cytokines, tumor necrosis factor-alpha (TNF-α) exerts direct effects on vascular endothelial permeability and lead to dysfunction of endothelial barrier [[3], [4], [5], [6]].
Endothelial cells adhere to each other through junctional structures formed by transmembrane adhesive proteins that are responsible for homophilic cell-to-cell adhesion. The junctional structures are essential for controlling the paracellular pathway of vascular endothelium permeability to a considerable extent [7]. The opening of cell-to-cell junctions and/or by rearrangement of their architecture may increase the endothelium permeability. Endothelial cell specific VE-cadherin is the key transmembrane adhesive protein in endothelium adherens junctions [[8], [9], [10], [11]], which dimerizes laterally in-cis and makes head-to-head contacts in-trans, via the most amino-terminal repeats, thus promoting cell-to-cell adhesion. Phosphorylatory modification of VE-cadherin/catenin complex proteins may disorganize this complex, and result in endocytosis of VE-cadherin, thereby increasing the permeability of the endothelium [[12], [13], [14]]. A clear role for VE-cadherin in maintaining permeability was demonstrated by in vivo experiments, in which the injection of anti-VE-cadherin antibodies in mice induced a marked increase in vascular permeability within a few hours [15]. As vascular endothelial cells are directly contact with blood, they are often affected by various stimulators from blood. To maintain barrier function and to prevent intrusion of both endogenous stressors and exogenous pathogens and their quick systemic spread, junctions need to be kept tight and repaired quickly. However, it is unclear how the junctional architecture of endothelial cell is regulated rapidly to maintain the suitable endothelial permeability in response to the various stressors.
Glutathione S-transferase Pi (GSTpi), an important family member of GSTs was originally characterized as a class II detoxification enzyme which catalyzes the nucleophilic attackglutathione (GSH) on electrophilic compounds like by-products of oxidative stress and xenobiotics, thus facilitating their elimination from the cell. In addition to its transferase and detoxification activity, GSTpi also regulates the mitogen-activated protein kinase (MAPK) signaling pathway and other intracellular proteins via its protein-protein binding activity [[16], [17], [18], [19]]. Recent reports show that GSTpi greatly enhances the rate and magnitude of protein S-glutathionylation, and acts as a glutathionylase in S-glutathionylation of redox-sensitive cysteines in proteins [[20], [21], [22], [23]]. Since particularly high levels of GSTpi were found in many kinds of cancers and drug resistant cancer cells, most studies about GSTpi are focus on the relationship between the abnormal GSTpi expression and the occurrence of tumor resistance to chemotherapy drugs [24]. In fact, GSTpi widely distributes in different normal cells and has been reported cytosolic, nuclear and mitochondrial compartment localizations. Depend on the multiple physiological functions such as detoxification, protein-protein binding and protein S-glutathionylation, GSTpi has been found to play some important roles in protecting cells against various stressors and maintaining homeostasis of organs [[25], [26], [27], [28], [29]]. Our previous study demonstrated that overexpression of GSTpi inhibited TRAF2-induced activation of both JNK and p38 [19]. We then found that through inhibiting p38 activation GSTpi prevented the actin polymerization and endothelial permeability increase induced by 6h TNF-α stimulation [28]. We noticed that at early stage of TNF-α stimulation endothelial permeability increased but no significant actin polymerization was observed, and GSTpi inhibited TNF-α-induced the increase of endothelial permeability even if there was no actin polymerization in endothelial cells. Actin polymerization may drive cell retraction and protrusion [30]. Although it is likely that the combination of both cell retraction and junctional changes leads to marked increase in permeability, VE-cadherin−/− endothelial cells present increase in permeability but no cell retraction [31], On the other hand, phosphorylation of VE-cadherin and other junctional proteins and their dissociation from the actin cytoskeleton, may result in increased permeability without actin polymerization and frank appearance of intercellular gaps [32]. These findings suggested that junctional integrity is the key factor for endothelial barrier function maintenance. Thus in the present study, we explored how GSTpi regulated the vascular endothelial permeability in endothelial cells, and analyzed the effect of GSTpi on VE-cadherin phosphorylation and endocytosis in response to early stimulation of proinflammatory factors. Our results demonstrate that GSTpi inhibits the phosphorylation of VE-cadherin through promoting S-glutathionylation of Src, the upstream kinase of VE-cadherin.
2. Materials and methods
2.1. Cell culture and transfection
HUVEC, HPMECs and endothelial cell medium (ECM) supplemented with a low-serum growth supplement were purchased from Sciencell (U.S.A). Cells were maintained at 37 °C in 5% CO2 and 95% ambient air in a humidified cell culture incubator. Only cells from passages 2 to 5 were used in experiments. Cells were used for experiments once they reached >90% confluence. Transient transfection was performed using Lipofectamine® 3000 Reagent according to the manufacturer's instructions. Briefly, transient transfection was performed at a cell density of 60%–80% confluence. Then the plasmid were transfected using Lipofectamine™ 3000 and P3000™ reagent according to the manufacturer's instructions (ratio of plasmid: Lipofectamine™ 3000: P3000™ reagent is 500:1:2, ng: μl: μl, siRNA transfection with no P3000™ reagent). In all cases, the total amount of transfected DNA/siRNA was normalized by addition of empty control plasmids. After 24h transfection, the EC cells were collected and treated for further analysis. The estimated transfection efficiency of EC cells is more than 50%.
2.2. VE-cadherin internalization assay
An mAb directed against the VE-cadherin extracellular domain (BV9) was dialyzed into ECM medium containing 20 mM HEPES and 3% BSA. The dialyzed antibody was incubated with HUVEC cultures at 4 °C for 1 h. Unbound antibody was removed by rinsing cells in ice-cold ECM. Cells were incubated with were treated with TNF-α or vehicle in the presence of 100 μM chloroquine. Chloroquine pre-treatment prevents endosome–lysosome acidification, and thereby allows VE-cadherin internalization but prevents lysosomal degradation [33,34]. After treatment, the cells were rinsed, fixed, and processed for label immunofluorescence.
2.3. Assessment of endothelial permeability In Vitro
Cells were grown on fibronectin-coated Transwell culture plate inserts (polycarbonate membranes with 0.4 μm pore size; EMD Millipore, Thermo Fisher Scientific) in phenol red-free ECM complete medium for 4–5 days to achieve confluence. For measurements, complete medium was replaced with medium containing 0.5% FBS. Each measurement was repeated at least 3–4 times, and there were three parallel samples in each experimental group. For permeability measurements, cells were treated with TNF-α or vehicle, media in the upper compartments were replaced with media containing 1 mg/mL FITC-Dextran 40. Samples from both compartments were taken at different times to determine FITC-Dextran 40 concentrations. The fluorescence intensity of FITC-Dextran 40 was determined with a multi-label counter (Thermo Fisher Scientific, USA).
2.4. Transepithelial electrical resistance (TEER) measurement
TEER was measured with an electric resistance system ERS-2 (Millipore) following the manufacturer's instructions. Briefly, HUVECs were grown on Millicell filter (0.33 cm2 area, 0.4 μm pore diameter, and 6.5 mm diameter) and the culture medium was replaced before TEER measurement. To calculate the actual resistance of the cell monolayer, the mean resistance of filters without cell was subtracted from the monolayer measurement, and the difference between the filter and monolayer areas was corrected. Values measured immediately after treatments were set as 100% to normalize the results.
2.5. Preparation of GSTpi proteins
The full-length cDNA encoding hGSTpi-1 as described previously [16] was amplified by PCR for ligation into pET-28a which contains a hexahistidine N-terminal tag. His6-GSTpi was purified by metal-affinity chromatography by using IDA-Ni2+ affinity column. Contained LPS in purified recombinant proteins were removed by using the Detoxi-Gel™ Endotoxin Removing Gel. Unless other-wise indicated, all purification procedures were carried out at 4 °C [29].
2.6. In Vitro S-Glutathionylation of Src
His-tag-purified Src (1 μg) was incubated with 10 μM H2O2, 250 μM glutathione, 100 ng GSTpi at 37 °C for 30 min. DTT (DTT, 60 mM stock) was then added to the relative tube, Samples were incubated at room temperature (RT) for another 30 min before being mixed with non-reducing loading buffer and boiled for 7 min. The samples were separated by SDS-PAGE under non-reducing conditions. The gels were transferred to nitrocellulose membranes and immunoblotted with anti-S-glutathionylation and anti- Src antibodies.
2.7. Src Kinase Assay
Src Kinase Assay was performed by using the reagents provided with BPS Bioscience following protocols recommended by the manufacturer. For cellular Src kinase assay, Src protein was collected by immunoprecipitation. For in vitro Src kinase assay, Src protein was firstly desalted after the in vitro S-Glutathionylation reaction of Src and then collected by immunoprecipitation. Src kinase was assayed in a reaction (50 μl) containing Kinase assay buffer, 10 μM ATP, and Protein Tyrosine Kinase Substrate (Poly-Glu, Tyr 4:1) and followed for 45 min at 30 °C. After the reaction, add of Kinase-Glo Max reagent to each well, measure luminescence using the microplate reader. The values of all experimental groups minus those of IgG group, and further Src kinase was analyzed by compared the Src protein level.
2.8. Immunofluorescence microscopy
Cells were washed two times with phosphate-buffered saline (PBS) buffer. After washing, cells were fixed with 4% paraformaldehyde for 30 min and then permeabilized with 0.2% Triton X-100 for 20 min. To reduce non-specific binding, cells were blocked in PBS containing 5% bovine serum albumin for 1 h. Then cells were incubated with primary antibodies in blocking buffer overnight. After washing (0.1% Tween-20 in PBS), samples were incubated with relative secondary antibody for 2h in the dark, followed by washing (0.1% Tween-20 in PBS). Finally, cells were incubated with DAPI (4,6-diamidino-2-phenylindole) for 5 min. Cells were washed three times with PBS between incubation steps above and every time 5 min. Slides were mounted and examined under Nikon A1 confocal laser microscope system (Tokyo, Japan). Relative secondary antibodies treatment were set as internal controls to ensure the specificity of primary antibodies.
2.9. AAV preparation
AAV-mCherry, AAV-GSTpi, AAV–NC–shRNA and AAV-GSTpi-shRNA were prepared by Obio Technology Co., Ltd. (Shang hai, China). Viral titer was 2.16 × 1013 vg/mL for AAV-mCherry, 1.06 × 1013 vg/mL for AAV-GSTpi, 2.21 × 1013 vg/mL for AAV–NC–shRNA and 1.32 × 1013 vg/mL for AAV-GSTpi-shRNA.
2.10. Mouse models of Endotoxemia
C57BL/6 mice (8–10 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. The animal study was approved by Medical Laboratory Animal Research Institute of Medical Sciences China [Permit Number: SYXK (Jing) 2014-0004]. All treatments of mice in this study were in strict agreement with guidelines on ARRIVE and recommendations from an NIH sponsored workshop regarding experimental design and reporting standards [35]. All experimental mice were sex- and age-matched. Mice were divided randomly into four groups for GSTpi over-expression and the shRNA knock-down experiment, with each group receiving either adeno-associated virus (AAV)-mCherry, AAV-GSTpi, AAV–NC–shRNA or AAV-GSTpi-shRNA via caudal vein injection. Endotoxemia was induced with injection of lipopolysaccharide (5 mg/kg, IP) (Lv et al., 2018). Relatively equal proportions (ratio 1:1) of male and female mice were used in each experiment. Experimental groups were assigned using a simple randomization procedure by means of drawing lots.
2.11. Evaluation of lung injury parameters
Collection of bronchoalveolar lavage (BAL) fluid was performed using sterile Hanks' balanced saline buffer. The pooled BAL fluid centrifuged at 800 rpm for 5 min at 4 °C and total cells counts were manually measured with a hemocytometer. The BAL protein concentration was determined by BCA™ (bicinchoninic acid) Protein Assay kit (Thermo Scientific, USA). The cells were diluted to a final concentration of 1 × 105 cells per ml. Then, 200 μl cells were resuspended in 30% FBS-PBS buffer and cytospun onto slides at 800 rpm for 5 min with a cytocentrifuge. Slides were stained with Diff-Quick dye and examined by microscopy. The number and percentage of PMNs and macrophages were detected and calculated after counting 300 cells in randomly selected fields. As an additional parameter reflecting increased lung vascular leakiness, Evans blue accumulation in the lung tissue was evaluated. Evans blue-albumin (EBA) at a dose of 30 mg/kg was injected to the caudal vein of mice 40 min before lung collection [36]. Tissues were perfused with cold PBS containing 0.6 mmol/L EDTA, blotted dry, weighed and snap frozen in liquid nitrogen. The tissues were homogenized in 0.5 ml PBS and incubated with 1.0 ml formamide for 18 h at 60 °C followed by centrifuged at 10,000 g for 20 min. The absorption of lung supernatants was determined at 620 nm and corrected for the presence of heme pigments as follows: Corrected A620 = Observed A620 - (1.1649 × A740 + 0.004) (Moitra et al., 2007). The Evans blue index was calculated as the amount of dye in the lung to the weight of lung tissue. Concentrations of TNF-α, IL-1β and IL-6 in BAL samples were measured using ELISA kit available from R&D Systems (Minneapolis, MN) according to manufacturer's instructions.
2.12. Statistical analysis
The experimental data obtained from cultured cells and mice were analyzed with Student's t-test and one way ANOVA to determine the significance of difference between two groups and are presented as means ± SD. Western blotting analyses were repeated three times with similar results quantified using the ImageJ software (github.com/imagej/imagej1; NIH, Rockville, MD, USA). Statistical analysis was carried out using the SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). A value of P < 0.05 was considered significant.
3. Results
3.1. GSTpi suppresses VE-cadherin phosphorylation, internalization and related increase of monolayer HUVEC permeability
The VE-cadherin-based junctional structure is particularly important for maintaining endothelial barrier homeostasis [33]. VE-cadherin dynamcis acts as a key in controlling EC barrier function, the subtle changes such as VE-cadherin phosphorylation and internalization may result in modification of junctional architecture and increased endothelium permeability without frank appearance of intercellular gaps [32,37,38]. TNF-α stimulation showed markedly endothelial permeability increase in time and dose dependently manner, whereas GSTpi-overexpression reversed this effect at an early phase (60 min 50 ng/ml TNF-α stimulaiton) (Fig. 1A and B). We treated HUVECs with TNF-α (50 ng/mL) for 0–60 min and found that VE-cadherin phosphorylation at Y685 increased apparently in cells 5 min after TNF-α stimulation and kept at high levels within the 60 min observation (Fig. 1A). To evaluate the effect of GSTpi on VE-cadherin phosphorylation, HUVECs were transfected with pcDNA3.1, Flag-GSTpi, NC-siRNA or GSTpi-siRNA. As shown in Fig. 1A, GSTpi overexpression attenuated TNF-α-induced VE-cadherin phosphorylation, whereas GSTpi-RNAi enhanced phosphorylation of VE-cadherin (Fig. 1H). The immunofluorescence observation demonstrated that in confluent endothelial monolayers, VE-cadherin showed a linear staining pattern at the cell borders under control conditions, but incubation of cells with TNF-α for 30 min resulted in irregular or increase of VE-cadherin internalization, no obvious cell size change (Fig. 1B–I). GSTpi overexpression significantly decreased whereas GSTpi-RNAi enhanced TNF-α-induced VE-cadherin loss from the cell borders (Fig. 1B–I). In parallel, Western blot assay showed that GSTpi overexpression significantly suppressed TNF-α-induced membrane VE-cadherin reduction and cytoplasmic VE-cadherin increase, whereas disruption of GSTpi expression enhanced such effect of TNF-α. However the total VE-cadherin level was not affected by Flag-GSTpi or GSTpi-siRNA transfection (Fig. 1C and J). Further FACS detection showed that TNF-α-induced reduction of cell surface staining of PE-conjugated anti-CD144 (VE-cadherin) was apparently inhibited by GSTpi overexpression but enhanced by GSTpi-RNAi (Fig. 1D–K). Altogether, above results demonstrated that GSTpi protected HUVECs against TNF-α-induced VE-cadherin phosphorylation and internalization.
Fig. 1.

GSTpi inhibited TNF-α-induced VE-cadherin internalization and monolayer permeability increases in human umbilical vein endothelial cell (HUVEC). HUVECs were transfected with pcDNA3.1 or Flag-GSTpi and further treated with TNF-α (50 ng/mL). (A) Levels of phospho-VE-cadherin was measured and calculated. VE-cadherin internalization was measured by (B) immunofluorescence (scale bar = 25 μm, the yellow arrowheads indicate the internalization of VE-cadherin), (C) analysis of cell membrane and cytoplasmic VE-cadherin protein levels using immunoblot and (D) flow cytometry (TNF-α (50 ng/mL) for 30 min). (E) The cell indexes of HUVECs were recorded and calculated Endothelial cell permeability was measured, (F) endothelial cell permeability and (G) transepithelial electrical resistance (TEER) were measured and calculated (TNF-α (50 ng/mL) for 1 h). *, compared with the pcDNA3.1 group; #, compared with the pcDNA3.1 + TNF-α group). HUVECs were transfected with NC-siRNA or GSTpi-siRNA and treated with TNF-α (50 ng/mL). (H) Levels of phospho-VE-cadherin was measured and calculated. VE-cadherin internalization was measured by (I) immunofluorescence (scale bar = 25 μm, the yellow arrowheads indicate the internalization of VE-cadherin), (J) analysis of cell membrane and cytoplasmic VE-cadherin protein levels by immunoblot and (K) flow cytometry (TNF-α (50 ng/mL) for 30 min). (L) Cell indexes, (M) endothelial cell permeability and (N) TEER were measured and calculated (TNF-α (50 ng/mL) for 1 h). *, compared with the NC-siRNA group; #, compared with the NC-siRNA + TNF-α group). Data are expressed as means ± SDs (n = 3). Data were analyzed by unpaired Student's t-tests. * or #, P < 0.05; ** or ##, P < 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
We then used a real-time iCELLigence system to record the dynamic changes of monolayer HUVEC barrier function under the condition as same as above experiments. HUVEC indexes began to reduce 10 min after cells being treated with TNF-α and continued to reduce along with 60 min TNF-α stimulation, which was inhibited by GSTpi overexpression and in contract, was enhanced by GSTpi downregulation (Fig. 1E–L). Moreover, when we used transwell culture plates to detect the EC permeability and the transepithelial electrical resistance (TEER) we founded that TNF-α-induced EC permeability increase and TEER reduction were suppressed significantly by GSTpi overexpression (Fig. 1F–G) and were enhanced by GSTpi downregulation (Fig. 1M and 1N). Also, overexpression of GSTpi inhibited EC permeability increase and TEER decrease induced by both VEGF and thrombin as well (Figs. S2A–D). These results indicated that short period of TNF-α stimulation induced apparent impairment of HUVEC monolayer barrier function, which was related with phosphorylation and internalization of VE-cadherin. GSTpi significantly inhibited permeability increase in HUVECs by inhibiting phosphorylation and internalization. Notably, overexpression of GSTA1, GSTM1, and GSTT1 did not alter EC permeability and TEER changes induced by TNF-α (Figs. S2G and 1H) suggesting the specific effect of GSTpi.
3.2. GSTpi downregulates activation of Src/VE-cadherin pathway
As above experiments indicated that GSTpi inhibited TNF-α-induced VE-cadherin phosphorylation and internalization, we then explored how GSTpi regulated this process. The binding of VE-cadherin/catenins is essential for VE-cadherin to stabilize and retain at the cell membrane. Our co-immunoprecipitation examinations showed that GSTpi overexpression significantly inhibited TNF-α-induced dissociation of VE-cadherin from β-catenin and p120-catenin (Fig. 2A–B), whereas downregulation of GSTpi promoted dissociation of VE-cadherin from β-catenin and p120-catenin (Fig. 2C–D), suggesting that GSTpi strengthened the binding of VE-cadherin/catenins against the pro-inflammatory stress. The phosphorylation of VE-cadherin, β-catenin and p120-catenin is essential for the disassociation of the complex of VE-cadherin, β-catenin and p120-catenin, and thus results in the increase of vascular permeability [39,40]. GSTpi inhibited TNF-α-induced phosphorylation in Y685 of VE-cadherin, phosphorylation of P120 at Y228 and β-catenin at Y142, whereas GSTpi-RNAi enhanced phosphorylation of VE-cadherin, P120 and β-catenin (Fig. 2E–F), which suggested that GSTpi inhibited disruption of VE-cadherin/catenin binding through reducing the phosphorylation level of VE-cadherin, P120 and β-catenin.
Fig. 2.

Effects of GSTpi on TNF-α-induced Src/VE-cadherin pathway activation. (A, B) The pcDNA3.1 and Flag-GSTpi groups were treated with TNF-α for 30 min, and co-immunoprecipitation of p120 and β-catenin and immunoblotting analysis were performed to assess the binding of VE-cadherin (IP results come from different blots with same samples. *, compared with the pcDNA3.1 group; #, compared with the pcDNA3.1 + TNF-α group). (C, D) The NC-siRNA and GSTpi-siRNA groups were treated with TNF-α for 30 min, and co-immunoprecipitation of p120 and β-catenin and immunoblotting analysis were performed to assess the binding of VE cadherin (IP results come from different blots with same samples. *, compared with the NC-siRNA group; #, compared with the NC-siRNA + TNF-α group). HUVECs were transfected with pcDNA3.1 or Flag-GSTpi and further treated with TNF-α (50 ng/mL). (E) Levels of phospho-p120, phospho-β-catenin, phospho-SHP-2, phospho-Src (Tyr416), and phospho-Src (Tyr527) were measured and calculated (*, compared with the pcDNA3.1 group; #, compared with the pcDNA3.1 + TNF-α group). (F) HUVECs were transfected with NC-siRNA or GSTpi-siRNA and treated with TNF-α (50 ng/mL). Level of phospho-p120, phospho-β-catenin, phospho-Src (Tyr416) and phospho-Src (Tyr527) were measured and calculated (*, compared with the NC-siRNA group; #, compared with the NC-siRNA + TNF-α group). HUVECs were transfected with pcDNA3.1, Flag-GSTpi, or Myc/Src and further treated with TNF-α (50 ng/mL). (G) Levels of phospho-VE-cadherin, phospho-p120, phospho-β-catenin, and phospho-Src (Tyr416) were measured and calculated (TNF-α (50 ng/mL) for 30 min). (H) Cell membrane and cytoplasmic VE-cadherin protein levels in HUVECs were measured and calculated (TNF-α (50 ng/mL) for 1 h). (I) Endothelial cell permeability by detecting the FITC-Dextran 40 fluorescence intensity level and TEER were measured and calculated (TNF-α (50 ng/mL) for 1 h *, compared with the Flag-GSTpi group; #, compared with the Flag-GSTpi + TNF-α group). Data are expressed as means ± SDs (n = 3). Data were analyzed by unpaired Student's t-tests. * or #, P < 0.05; ** or ##, P < 0.01.
We then performed further experiments to analyze the mechanism by which GSTpi inhibited the phosphorylation of VE-cadherin, P120 and β-catenin. Src-induced phosphorylation of VE-cadherin and other proteins may alter endothelial barrier function [39,[41], [42], [43]], and Src-specific inhibitor PP2 protects against TNF-α-induced phosphorylation of VE-cadherin and opening of the paracellular pathway of endothelium [44]. As shown in Fig. 2E, TNF-α induced a significant activation of Src. GSTpi overexpression significantly inhibited phosphorylation of Src induced by TNF-α (Fig. 2E) whereas GSTpi RNAi enhanced Src phosphorylation (Fig. 2F). Src is an SHP-2 effector and SHP-2 counteracts the effect of Src on protein phosphorylation [39]. GSTpi overexpression enhanced phosphorylation of SHP-2 induced by TNF-α (Fig. 2E), and GSTpi RNAi reduced phosphorylation of SHP-2 (Fig. 2F). As shown in Figs. S2E–F, GSTpi also significantly inhibited phosphorylation of Src and VE-cadherin induced by VEGF and thrombin. These data demonstrated that GSTpi significantly inhibited Src activation induced by TNF-α, VEGF and thrombin. To confirm the inhibitory effect of GSTpi on Src phosphorylation was essential to VE-cadherin internalization and the increase of EC permeability, we co-transfected HUVECs with Myc-Src and Flag-GSTpi and then treated cells with TNF-α for 30 min. Overexpression of Myc-Src reversed the inhibitory effects of GSTpi on TNF-α-induced the phosphorylation of VE-cadherin, P120, β-catenin and Src (Fig. 2G). More important, overexpression of Myc-Src also reversed inhibitory effects of GSTpi on TNF-α-induced plasma membrane VE-cadherin reduction and cytoplasmic VE-cadherin increase (Fig. 2H) as well as the increase of EC permeability and TEER decrease (Fig. 2I). All these results suggested that GSTpi inhibited TNF-α-induced activation of the Src/VE-cadherin pathway in HUVECs. It has been reported that eNOS-derived nitric oxide and AMP-activated protein kinase (AMPK) may regulate endothelial barrier function [[45], [46], [47]]. Our results showed that both TNF-α and GSTpi did not alter phosphorylation of eNOS, total eNOS, phosphorylation of AMPK and total AMPK level (Fig. S2I). RhoA has been studied as a key regulator of vascular leakage [48]. GSTpi overexpression only slightly influenced the activation of RhoA induced by TNF-α (Fig. S2J). Altogether, these results showed that GSTpi inhibited TNF-α-induced phosphorylation and dissociation of the VE-cadherin/catenin complex through regulating activation of Src.
The glutathione transferase activity is essential for GSTpi to inhibit Src/VE-cadherin pathway activation.
The seventh tyrosine (Tyr7) in GSTpi structure is essential for its glutathione transferase activity and when Tyr7 is replaced by Phe, the transferase activity of GSTpi decreases by 55-fold [26]. GSTpi not only acts as a Phase II metabolic enzyme, but also plays an important role in modulating kinase activities through nonenzymatic protein-protein interactions. We thus evaluated whether the transferase activity was involved in the effect of GSTpi on EC permeability and Src/VE-cadherin activation. We transfected the GSTpi enzyme mutant GSTpi (Y7F) into HUVECs and found that overexpression of GSTpi (Y7F) did not affect TNF-α-induced EC permeability increase, TEER decrease and cell index decrease in HUVECs (Fig. 3A–C). The results both from flow cytometry and immunofluorescence microscopy showed that GSTpi (Y7F) did not inhibit TNF-α-induced cell membrane VE-cadherin loss (Fig. 3, Fig. 4E) and Western blot analysis also showed that GSTpi (Y7F) did not affect the cell membrane and cytoplasmic levels of VE-cadherin (Fig. 3F). Consistent with these results, GSTpi (Y7F) did not alter TNF-α-induced Src/VE-cadherin pathway activation (Fig. 3G). These resulted indicated that glutathione transferase activity was necessary for GSTpi to inhibit TNF-α-induced VE-cadherin phosphorylation and internalization of VE-cadherin and increase of EC permeability. TNF-α stimulation may lead to ROS generation, and the increase of ROS in endothelial cells accompanied with the elevation of glutathione S-transferase activity [49]. In the present study, although GSTpi overexpression did not suppressed TNF-α-induced increase of mitochondrial superoxide radical and total ROS levels, disruption of GSTpi expression did elevate the levels of mitochondrial superoxide radical and cellular ROS (Figs. S3A and B), which further indicated that the inhibitory effects of GSTpi on the impairment of EC barrier function and Src/VE-cadherin activation were related with its glutathione transferase activity and changes in intracellular redox state.
Fig. 3.

GSTpi enzyme mutants failed to protect against TNF-α induced HUVEC monolayer permeability increase. HUVECs were transfected with pcDNA3.1 or Flag-GSTpi (Y7F) and further treated with TNF-α (50 ng/mL) for 30 min. (A–C) The cell indexes of HUVECs, endothelial cell permeability and TEER were measured and calculated (TNF-α (50 ng/mL) for 1 h). (D–F) VE-cadherin internalization were measured by analysis of cell membrane and cytoplasmic VE-cadherin protein levels, flow cytometry and immunofluorescence (scale bar = 25 μm, the yellow arrowheads indicate the internalization of VE-cadherin) (TNF-α (50 ng/mL) for 30 min). (G) Levels of phospho-VE-cadherin, phospho-p120, phospho-β-catenin, phospho-SHP-2, phospho-Src (Tyr416) and phospho-Src (Tyr527) were measured and calculated (*, compared with the pcDNA3.1 group; #, compared with the pcDNA3.1 + TNF-α group). Data are expressed as means ± SDs (n = 3). Data were analyzed by unpaired Student's t-tests. * or #, P < 0.05; ** or ##, P < 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Fig. 4.

GSTpi inhibited TNF-α induced Src phosphorylation through Selective S-glutathionylation of Src kinase. HUVECs were transfected with pcDNA3.1 or Flag-GSTpi and further treated with TNF-α (50 ng/mL). (A) Co-immunoprecipitation of Src and GSH was performed, and relative Src glutathionylation levels were measured and calculated (Up panel: IP results come from different blots with same samples; low panel: IP results come from same blot. *, compared with the pcDNA3.1 group; #, compared with the pcDNA3.1 + TNF-α group.). (B) Immunofluorescence was used to detect colocalization of Src and protein S-glutathionylation (scale bar = 25 μm). HUVECs were transfected with NC-siRNA or GSTpi-siRNA and treated with TNF-α (50 ng/mL) for 30 min. (C) Co-immunoprecipitation of Src and GSH was performed, and relative Src glutathionylation levels were measured and calculated (Up panel: IP results come from different blots with same samples; low panel: IP results come from same blot. *, compared with the NC-siRNA group; #, compared with the NC-siRNA + TNF-α group). (D) Immunofluorescence was used to detect the colocalization of Src and protein S-glutathionylation. (E) HUVECs were transfected with pcDNA3.1 or Flag-GSTpi (Y7F) and further treated with TNF-α (50 ng/mL) for 30 min. Co-immunoprecipitation of Src and GSH was performed, and relative Src glutathionylation levels were measured and calculated (Up panel: IP results come from different blots with same samples; low panel: IP results come from same blot. *, compared with the pcDNA3.1 group; #, compared with the pcDNA3.1 + TNF-α group.). (F) HUVECs were transfected with pcDNA3.1, Flag-GSTpi, or Myc/Src and further treated with TNF-α (50 ng/mL) for 30 min. Co-immunoprecipitation of Myc and GSH was performed, and relative Myc glutathionylation levels were measured and calculated (Up panel: IP results come from different blots with same samples; low panel: IP results come from same blot. *, compared with the Flag-GSTpi group; #, compared with the Flag-GSTpi + TNF-α group). Data are expressed as means ± SDs (n = 3). Data were analyzed by unpaired Student's t-tests. * or #, P < 0.05; ** or ##, P < 0.01.
3.3. GSTpi selectively induces Src S-glutathionylation
S-glutathionylation is a reversible oxidative modification of protein and is emerging as an important redox signaling paradigm in cardiovascular physiopathology including vascular barrier dysfunction [1]. When cells are exposed to ROS, S-glutathionylation can protect proteins against irreversible oxidative damage. As GSTpi catalyzes S-glutathionylation of cysteine residues to alter structure/function of certain targeted proteins, we probe if GSTpi regulated S-glutathionylation of proteins in Src/VE-cadherin pathway. The mass spectrometry analysis and co-immunoprecipitation demonstrated that GSTpi overexpression induced S-glutathionylation of Src kinase (Fig. S3C and Fig. 4A). Immunofluorescence observation showed that GSTpi overexpression increased colocalization of Src and GSH, which confirmed the effect of GSTpi on promoting S-glutathionylation of Src protein (Fig. 4B). However, GSTpi did not induce S-glutathionylation of VE-cadherin, β-catenin, P-120, SHP-2 and CSK (Fig. S3D), strongly suggesting that GSTpi selectively promoted Src S-glutathionylation. Additionally, TNF-α stimulation slightly increased Src S-glutathionylation and overexpression of GSTpi further enhanced S-glutathionylation of Src (Fig. 4A). When we co-transfected HUVECs with Myc-Src and Flag-GSTpi, and then stimulated cells with TNF-α, exogenous Myc-Src also was S-glutathionylation by TNF-α and further enhanced by overexpression of GSTpi (Fig. 4F). In contrary, both co-immunoprecipitation and immunofluorescence detection showed that downregulation of GSTpi decreased Src S-glutathionylation levels (Fig. 4C and D). As the glutathione transferase activity is essential for GSTpi to promote S-glutathionylation [22], it was not surprised that overexpression of enzyme mutant GSTpi (Y7F) did not increase the level of Src S-glutathionylation (Fig. 4E) and the co-localization of Src and GSH (Fig. S4A). The NBDEX, a GSTpi enzyme inhibitor, also did not increase Src S-glutathionylation (Figs. S4B–D). Comparably, overexpression of GSTA1, GSTM1, and GSTT1 did not alter Src S-glutathionylation (Fig. S4E).
We then examined the S-glutathionylation site in Src. Results from the molecular docking analysis showed that peptides containing Cys185, Cys245, and Cys400 might be S-glutathionylated (Fig. 5A). The experiment results through co-immunoprecipitation and MS detection confirmed that GSTpi modified Src by S-glutathionylation at Cys185, Cys245, and Cys400 (Fig. 5B). We then constructed pcDNA3.1-Myc-Src(C185A), pcDNA3.1-Myc-Src(C245A) and pcDNA3.1-Myc-Src(C400A) in which Cys185, Cys245 and Cys400 were replaced by Ala respectively. HUVECs were co-transfected with Flag-GSTpi and wild type Myc-Src, Myc-Src (C185A), Myc-Src (C245A), or Myc-Src (C400A) and S-glutathionylation levels of Src were evaluated by co-immunoprecipitation and immunobloting detection. Compared with the wild type Myc-Src transfected cell, S-glutathionylation levels of Src decreased in those cells transfected with Src mutants including Myc-Src (C185A), Myc-Src (C245A) and Myc-Src (C400A) (Fig. 5C–D). The results also showed that the S-glutathionylation level decreased to a greater extent in cells transfected with Myc-Src (C185A) than in cells transfected with Myc-Src (C245A) or Myc-Src (C400A) (Fig. 5C–D). Moreover, immunofluorescence microscopy observation showed that in cells transfected with Myc-Src (C185A), the colocalization of Myc-Src and GSH diminished (Fig. 5E). All these data indicated that GSTpi promoted Src S-glutathionylation at Cys185, Cys245, and Cys400, and Cys185 was more important for Src S-glutathionylation.
Fig. 5.

GSTpi inhibited Src Tyr416 phosphorylation through Cys185, Cys245, and Cys400 S-glutathionylation. (A) The crystal structure of Src in complex with S-glutathionylated Cys185, Cys245, and Cys400. (B) Matrix-assisted laser desorption/ionization mass spectrometric analysis showed that peptides containing Cys185, Cys245, and Cys400 were S-glutathionylated. HUVECs were transfected with Flag-GSTpi, Myc/Src, Myc/Src (C185A), Myc/Src (C245A), or Myc/Src (C400A) and further treated with TNF-α (50 ng/mL) for 30 min. (C, D) Co-immunoprecipitation of Myc and GSH was performed, and relative Myc glutathionylation levels were measured and calculated (Up panel: IP results come from different blots with same samples; low panel: IP results come from same blot). (E) Immunofluorescence was performed to detect the colocalization of Src and protein S-glutathionylation (scale bar = 25 μm). *, compared with the Myc/Src group; #, compared with the Myc/Src + TNF-α group. Data are expressed as means ± SDs (n = 3). Data were analyzed by unpaired Student's t-tests. * or #, P < 0.05; ** or ##, P < 0.01.
3.4. GSTpi-induced Src S-glutathionylation is essential for the inhibition of Src activity
Cys185, Cys245, and Cys400 are the evolutionarily conserved cysteine residues in Src, suggesting that it played a key role in the regulation of Src activity and function (Fig. 6A). Above results demonstrated that GSTpi inhibited Src phosphorylation and enhanced Src protein S-glutathionylation simultaneously, which suggested that GSTpi inhibited activity through enhancing Src protein S-glutathionylation. Further in vitro experiments by using GSTpi, Src and GSH proteins demonstrated that while GSTpi significantly increased Src protein S-glutathionylation level (Fig. 6B) and decreased Src kinase activity (Fig. 6C). Fig. 6D–E showed that as long with Src protein S-glutathionylation being increased by GSTpi in a dose dependent manner, the Src kinase activity reduced as a result. Since GSTpi promoted Src S-glutathionylation in HUVECs, thus it was not surprised that GSTpi significantly decreased TNF-α-induced the increase of endogenous Src kinase activity in HUVECs both in GSTpi overexpression and RNAi experiments (Fig. 6F and G), but GSTpi(Y7F) did not altered the Src kinase activity (Fig. 6H). Besides, in Flag-GSTpi and myc-Src co-transfected HUVECs, GSTpi also significantly decreased TNF-α-induced the increase of exogenous Src kinase activity (Fig. 6I). These results demonstrated that GSTpi-induced inhibition of Src phosphorylation and its kinase activity was related with the S-glutathionylation of Src protein. We then applied an in vitro experiment to analyze if Src protein S-glutathionylation could influence Src and SHP-2 interaction. The result showed that Src protein S-glutathionylation significantly decreased the binding of Src and SHP-2 (Fig. 6J). In vivo experiments also showed that GSTpi overexpresstion decreased the binding level of Src and SHP-2 in HUVECs (Fig. 6K and L). These data demonstrated that GSTpi inhibited Src activation by promoting Src protein S-glutathionylation.
Fig. 6.

In Vitro S-glutathionylation of Src. (A) Amino acid sequence alignments of full-length Src. Green highlight, conserved cysteines in Src protein; underline, cysteine-containing peptides detected in MS analysis. His-tag-purified Src was incubated with 10 μM H2O2, 250 μM glutathione and 100 ng GSTpi. (B) Immunoblots of glutathione-modified human Src with glutathionylation antibody. Total protein loading was evaluated with Src -specific antibodies. (C) Src kinase assay of S-glutathionylated Src protein. His-tag-purified Src was incubated with 10 μM H2O2, 250 μM glutathione, different contents GSTpi. (D) Immunoblots of glutathione-modified human Src with glutathionylation antibody. Total protein loading was evaluated with Src -specific antibodies. (E) Src kinase assay of S-glutathionylated Src protein. (F–I) Src kinase assay of Flag-GSTpi, GSTpi-siRNA, Flag-GSTpi (Y7F) and myc-Src, Flag-GSTpi transfected HUVEC cells. (J) His-tag-purified Src was incubated with 10 μM H2O2, 250 μM glutathione, 100 ng GSTpi. Further co-immunoprecipitation of SHP-2 and Src was performed, and relative SHP-2 levels were measured and calculated (IP results come from different blots with same samples). (K–L) HUVECs were transfected with pcDNA3.1 or Flag-GSTpi and further treated with TNF-α (50 ng/mL) for 30 min. Co-immunoprecipitation of SHP-2 and Src was performed, and relative Src and SHP-2 levels were measured and calculated (IP results come from different blots with same samples. *, compared with the pcDNA3.1 group; #, compared with the pcDNA3.1 + TNF-α group). Data are expressed as means ± SDs (n = 3). Data were analyzed by unpaired Student's t-tests. * or #, P < 0.05; ** or ##, P < 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.5. GSTpi regulates mouse pulmonary microvascular permeability in acute lung injury model
Pulmonary capillary leak often results in the impairment of gas exchange and respiratory failure [50]. We then evaluated the effect of GSTpi on the permeability of human pulmonary microvascular endothelial cells (HPMECs). Consistent with the data in HUVECs, the results from both GSTpi overexpression and GSTpi RNAi demonstrated that GSTpi inhibited TNF-α-induced increase of endothelial permeability and TEER (Fig. 7A and B, S5A and S5B), and also attenuated TNF-α-induced VE-cadherin internalization in HPMECs (Fig. 7C and D, S5C and S5D). As similar as in HUVECs, GSTpi inhibited TNF-α-triggered Src/VE-cadherin pathway activation and promoted S-glutathionylation of Src simultaneously in HPMECs (Fig. 7E and F, S5E and S5F).
Fig. 7.

GSTpi inhibits the destabilization of membrane VE-cadherin in human pulmonary microvascular endothelial cells (HPMECs). HPMECs were transfected with pcDNA3.1, Flag-GSTpi or Flag-GSTpi(Y7F) and further treated with TNF-α (50 ng/mL). (A–B) Endothelial cell permeability and TEER were measured and calculated (TNF-α (50 ng/mL) for 1 h). VE-cadherin internalization was measured by (C) immunofluorescence (scale bar = 25 μm, the yellow arrowheads indicate the internalization of VE-cadherin) and (D) analysis of cell membrane and cytoplasmic VE-cadherin protein levels (TNF-α (50 ng/mL) for 30 min). (E) Level of phospho-VE-cadherin, phospho-p120, phospho-β-catenin and phospho-Src (Tyr416) were measured and calculated (TNF-α (50 ng/mL) for 30 min). (F) Co-immunoprecipitation of Src and GSH was performed, and relative Src glutathionylation levels were measured and calculated (Up panel: IP results come from different blots with same samples; low panel: IP results come from same blot. TNF-α (50 ng/mL) for 30 min). *, compared with the pcDNA3.1 group; #, compared with the pcDNA3.1 + TNF-α group. Data are expressed as means ± SDs (n = 3). Data were analyzed by unpaired Student's t-tests. * or #, P < 0.05; ** or ##, P < 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Increased vascular permeability is the key feature in ALI. We used lipopolysaccharide (LPS)-induced mouse ALI model to explore whether GSTpi regulated inhibited the increase of pulmonary vascular permeability in vivo. C57BL/6 mice challenged with LPS were treated with AAV-GSTpi or AAV-GSTpi-shRNA respectively (Fig. 8A and G), and lung injury was evaluated 24 h after LPS administration. The extravasation of albumin-bound Evans blue dye, an indicator of vascular leakage injected intravenously into mice, into lung tissue was detected. The results from AAV-GSTpi or AAV-GSTpi-shRNA treatment experiments showed that GSTpi inhibited albumin-bound Evans blue increase in mouse lung tissues, which indicated that GSTpi prevented LPS-induced vascular leak (Fig. 8B and H). Coincidently, GSTpi suppressed LPS-induced polymorphonuclear neutrophil (PMN) infiltration and protein exudation in bronchoalveolar lavage fluid (Fig. 8C and D and 8I-J). Furthermore, the lung histological observation also demonstrated a significant reduction of infiltration caused by GSTpi (Fig. 8E and K). As myeloperoxidase (MPO) activity increase in the pulmonary parenchyma reflects neutrophil infiltration into the lung tissues [51], we measured the MPO activities to evaluate PMN infiltration in above experiments (Fig. 8F and L).
Fig. 8.

The barrier-protective effects of GSTpi in the model of lipopolysaccharide (LPS)–induced lung injury. Mice pre-treated with AAV-mCherry or AAV-GSTpi, (A) The lung tissues were collected and lysised for protein analyzed of GSTpi level. Mice pre-treated with AAV-mCherry or AAV-GSTpi were challenged with LPS (5 mg/kg, IP), bronchoalveolar lavage (BAL) was performed after LPS injection. (B) Evans blue accumulation in the lung parenchyma. (C) BAL polymorphonuclear neutrophil (PMN) count and (D) protein content in mice. (E) Hematoxylin and eosin staining of sections of lungs. Right, Representative lung histology. Magnification, 20 × . (F) After LPS/saline administration, lungs were used to measure MPO activity, expressed as change in absorbance (ΔA) (*, compared with the AAV-mCherry group; #, compared with the AAV-mCherry + LPS group.). Mice pre-treated with AAV–NC–shRNA or AAV-GSTpi-shRNA, (G) The lung tissues were collected and lysised for protein analyzed of GSTpi level. Mice pre-treated with AAV–NC–shRNA or AAV-GSTpi-shRNA were challenged with LPS. (H) Evans blue accumulation in the lung parenchyma. (I) BAL polymorphonuclear neutrophil (PMN) count and (J) protein content in mice. (K) Hematoxylin and eosin staining of sections of lungs. Right, Representative lung histology. Magnification, 20 × . (L) Lungs were used to measure MPO activity, expressed as change in absorbance (ΔA) (*, compared with the AAV–NC–shRNA group; #, compared with the AAV–NC–shRNA + LPS group). Data are expressed as means ± SDs (n = 5). Data were analyzed by unpaired Student's t-tests. * or #, P < 0.05; ** or ##, P < 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
Endothelial integrity and the function of the endothelial barrier are important for blood vessel and organ homeostasis and adaptation [52]. The endothelial permeability is often affected by a variety of chemical and biological factors, thus appropriate and dynamic regulation is required during normal physiological processes. In the present study, we first found that GSTpi negatively regulated VE-cadherin internalization in HUVECs and HPMECs, and inhibited cytokine-induced endothelial cell hyperpermeability. These results are consistent with our previous findings that GSTpi inhibited inflammatory responses in mouse inflammation models [29,53]. Although the function of GSTpi in cancer cells has been reported widely, the effect of GSTpi in endothelial cells has been seldomly reported. We have found that GSTpi prevented Ang II-stimulated proliferation and migration of vascular smooth muscle cells (Chen et al., 2014), and GSTpi suppressed TNF-α-induced EC actin remodeling and stress fiber formation by blocking the p38 MAPK/heat-shock protein 27 signaling pathway [28]. In our previous study, we noticed that 6h after TNF-α stimulation, the stress fiber formed and EC permeability increased [50]. However, phosphorylation of VE-cadherin and other junctional proteins may result in increased permeability without actin polymerization and frank appearance of intercellular gaps [32], it is needed to investigate if GSTpi might regulate EC permeability through affecting VE-cadherin. In the present study, we used a real-time iCELLigence system to record the dynamic processes of monolayer HUVEC barrier function and found that EC indexes began to decrease 10 min after stimulation of TNF-α and continued to reduce within 1h period observation. GSTpi inhibited TNF-α-induced decreases of EC indexes suggesting that GSTpi protected EC from the impairment of barrier function induced by early stage of stimulation of TNF-α. Following transwell culture detection demonstrated that GSTpi inhibited the increase of EC permeability induced by 1h treatment of TNF-α, VEGF and thrombin. These findings demonstrate that GSTpi acts as an important regulator in maintaining endothelial barrier function and vascular homeostasis at the early stage of pro-inflammatory stimulation conditions.
The rapid endocytosis of VE-cadherin may disrupt the endothelial barrier function after ECs being stimulated by inflammatory mediators and growth factors [13]. This early changes of endothelial barrier function may not be related with the actin remodeling and stress fiber formation but the dissociation of the complex of VE-cadherin/catenins (including β-catenin γ-catenin, and p120-catenin). In the present study, we found that GSTpi significantly inhibit the dissociation of the complex of VE-cadherin/catenins and VE-cadherin internalization, which suggested the important mechanism that GSTpi regulated EC barrier function through affecting VE-cadherin internalization. Phosphorylation of VE-cadherin and catenins resulted in the complex degradation and VE-cadherin internalization [8]. Our findings demonstrated that all the phosphorylation of VE-cadherin, p120 and β-catenin induced by TNF-α was inhibited by GSTpi strongly suggesting that GSTpi prevented VE-cadherin internalization through inhibiting VE-cadherin/catenins phosphorylation. We further investigated how GSTpi affected VE-cadherin/catenins phosphorylation. The activation of Src family kinases has been described as a critical step in the subsequent VE-cadherin phosphorylation and the induction of hyperpermeability of vascular endothelium [37,38,54,55]. All Src family nonreceptor tyrosine kinases have a single tyrosine residue in the activation loop at the position corresponding to Tyr416 of Src and phosphorylation of Src at Tyr416 results in its kinase activation [56]. Interestingly, our results showed that GSTpi inhibited TNF-α-induced Tyr416 phosphorylation of Src and the activation of Src/VE-cadherin pathway.
As a glutathione transferase, GSTpi can add GSH to electrophilic substances with various chemical structures and can transfer GSH to cysteine residue of proteins to lead to S-glutathionylation of these proteins [57]. Although protein S-glutathionylation can occur spontaneously, the reaction rate and magnitude are greatly enhanced by GSTpi [58]. When we decreased mutant catalytic activity of GSTpi by substituting Phe for Tyr7, the above effects of GSTpi in vascular endothelial cells attenuated, which indicated that the glutathione transferase activity was necessary for GSTpi to inhibit Src/VE-cadherin/catenin phosphorylation. This result provided a possibility that GSTpi might potentiate S-glutathionylation of the proteins in Src/VE-cadherin pathway. S-glutathionylation is a reversible post-translational modification, which protects the cysteine against further oxidative damage and creates distinct structural and functional changes in the target protein [58]. It has been reported that GSTpi potentiates S-glutathionylation reactions following oxidative and nitrosative stress in vitro and in vivo [22]. ROS and the adhesion receptors are critical mediators of the transient disruption of endothelial barriers in response to TNF-α [39]. In this study, we found that GSTpi selectively S-glutathionylated Src kinase but not VE-cadherin, p120 and β-catenin. Previous studies show that TNF-α-induced ROS increase promotes oxidation of Cys245 and Cys487 (respectively localized within SH2 and kinase domain) of Src kinase and leads to a conformational change that accelerates the activation of Src family kinases (SFKs) [[59], [60], [61]]. Our study demonstrated that GSTpi induced S-glutathionylation of Src at Cys185, Cys245 and Cys400, which first displayed the effect of GSTpi on Src S-glutathionylation though GSTpi has been reported to lead to S-glutathionylation of other proteins. S-glutathionylation of Src at Cys245 may inhibit the disulfide bridges formation of Cys245 and Cys487, releasing Src from the active conformation. In the inactive conformation, Cys185 is located in the SH2 domain of Src kinase and close to the Src Tyr527 site, thus, S-glutathionylation of Src at Cys185 may block the binding of Src to SH2-containing protein tyrosine phosphatases, thereby inhibiting Src kinase [62]. S-glutathionylation of Src at Cys400 may prevent the binding of Src with downstream effectors, and form an inhibition feedback loop to decrease Src phosphorylation at Tyr416. When we used Src mutants including Myc-Src (C185A), Myc-Src (C245A) and Myc-Src (C400A) to replace wide type Myc-Src, GSTpi-induced S-glutathionylation of Src was inhibited. Catalytically, GSTpi binds GSH and lowers the pKa of the cysteine residue of GSH through abstraction of the proton by Tyr7, resulting in a thiolate anion (GS−) at the active site [63]. To explore whether GSTpi induced S-glutathionylation of Src decreased Src phosphorylation at Tyr416, we performed both in vitro and in vivo experiments. Our results demonstrated that GSTpi-induced S-glutathionylation of Src and decreased Src phosphorylation at Tyr416 occurred simultaneously. When GSTpi-induced Src S-glutathionylation was inhibited the Src phosphorylation at Tyr416 and Src kinase activity increased. Further experiment showed that Src protein S-glutathionylation significantly decreased the binding of Src and SHP-2. As Src is an SHP-2 effector and SHP-2 counteracts the effect of Src on protein phosphorylation [39], above findings strongly suggested that GSTpi inhibited Src activation through promoting Src S-glutathionylation. We also observed the effects of GSTpi on vascular endothelial barrier function in mouse ALI model. As expected, GSTpi reduced vascular leakage, and alleviated LPS-induced ALI.
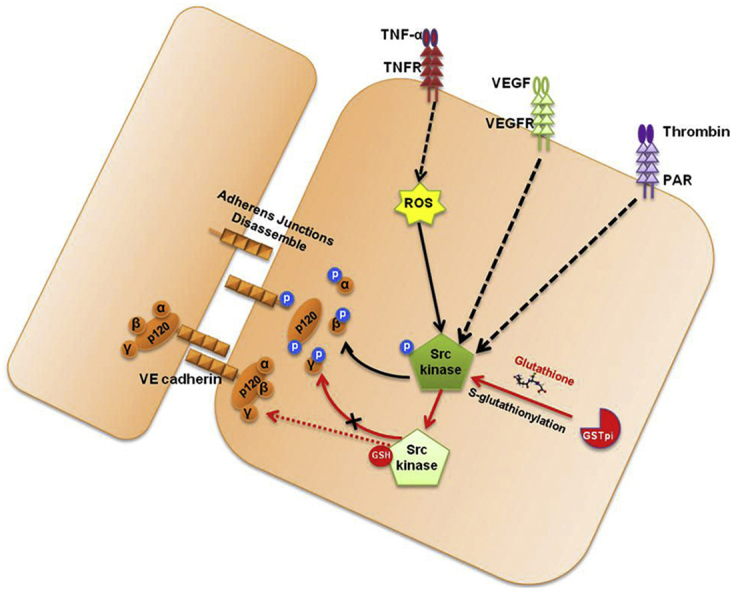
In summary, our findings in the present study provided a novel mechanism underlying the effects of GSTpi on regulation of vascular endothelial barrier function in endothelial cells and animal models. The S-glutathionylation of Src by GSTpi is necessary for GSTpi to inhibit Src activation and Src/VE-cadherin signaling, thereby to prevent VE-cadherin internalization. Thus, GSTpi may act as an important negative regulator in maintaining suitable vascular endothelial permeability and integrity of vascular endothelial barrier function to prevent inflammation-related endothelial barrier disruption (Fig. 9).
Fig. 9.

Proposed pathway for inhibition of increases in GSTpi-mediated monolayer permeability via Src/VE-cadherin signaling. GSTpi inhibited TNF-α-induced endothelial permeability by S-glutathionylation of Src kinase, thereby decreasing downstream phospho-VE-cadherin, phospho-p120, and phospho-β-Catenin levels and inhibiting vascular permeability.
Online supplemental material
Fig. S1 describes the GSTpi inhibits TNF-α induced endothelial permeability in a time dependently manner. Fig. S2 shows the effect of GSTpi in endothelial permeability and related signal pathway. Fig. S3 presents GSTpi selectively induces Src kinase S-glutathionylation. Fig. S4 shows the Glutathione transferase activity is required for GSTpi induced Src kinase S-glutathionylation. Fig. S5 shows downregulation of GSTpi increased TNF-α-induced VE-cadherin internalization and monolayer permeability increases in human pulmonary microvascular endothelial cells (HPMECs).
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information File or from the corresponding authors upon request.
Authorship contributions
YY: Procurement of funding, conception and design, acquisition of data, analysis and interpretation of data, writing of the manuscript. XD: Conception and design, interpretation of data, revision of the manuscript. SZ, JS: Analysis and interpretation of data. JC, JY: Acquisition of data, analysis and interpretation of data. YF: Acquisition of data, analysis and interpretation of data, proof-reading of the manuscript. XD: Development of methodology, revision of the manuscript. BZ, YY: Technical support, analysis and interpretation of data. SZ: Statistical analysis. ZY, PC, LL: Procurement of funding, conception and design, study supervision, revision of the manuscript. All authors read and approved the final manuscript.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially supported by grants from the Natural Science Foundation of China (Nos. 31571166, 81913498, 81602733, 81973498, 81771703, 81902706, 81471557 and 81671565), the Outstanding Youth Fund of Jiangsu Province (No. BK20140049), and the Priority Academic Program Development of Jiangsu Higher Education Institution (PADD) to Z. Yin.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101416.
Contributor Information
Zhimin Yin, Email: yinzhimin@njnu.edu.cn.
Peng Cao, Email: cao_peng@njucm.edu.cn.
Lan Luo, Email: lanluo@nju.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Han J., Weisbrod R.M., Shao D., Watanabe Y., Yin X., Bachschmid M.M., Seta F., Janssen-Heininger Y.M.W., Matsui R., Zang M., Hamburg N.M., Cohen R.A. The redox mechanism for vascular barrier dysfunction associated with metabolic disorders: glutathionylation of Rac1 in endothelial cells. Redox. Biol. 2016;9:306–319. doi: 10.1016/j.redox.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reglero-Real N., Colom B., Bodkin J.V., Nourshargh S. Endothelial cell junctional adhesion molecules: role and regulation of expression in inflammation. Arterioscler. Thromb. Vasc. Biol. 2016;36:2048–2057. doi: 10.1161/ATVBAHA.116.307610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klinghammer L., Urschel K., Cicha I., Lewczuk P., Raaz-Schrauder D., Achenbach S., Garlichs C.D. Impact of telmisartan on the inflammatory state in patients with coronary atherosclerosis--influence on IP-10, TNF-alpha and MCP-1. Cytokine. 2013;62:290–296. doi: 10.1016/j.cyto.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Willerson J.T., Ridker P.M. Inflammation as a cardiovascular risk factor. Circulation. 2004;109:II2–10. doi: 10.1161/01.CIR.0000129535.04194.38. [DOI] [PubMed] [Google Scholar]
- 5.Xiao N., Yin M., Zhang L., Qu X., Du H., Sun X., Mao L., Ren G., Zhang C., Geng Y., An L., Pan J. Tumor necrosis factor-alpha deficiency retards early fatty-streak lesion by influencing the expression of inflammatory factors in apoE-null mice. Mol. Genet. Metab. 2009;96:239–244. doi: 10.1016/j.ymgme.2008.11.166. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y., Yang X., Bian F., Wu P., Xing S., Xu G., Li W., Chi J., Ouyang C., Zheng T., Wu D., Zhang Y., Li Y., Jin S. TNF-alpha promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: crosstalk between NF-kappaB and PPAR-gamma. J. Mol. Cell. Cardiol. 2014;72:85–94. doi: 10.1016/j.yjmcc.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Bazzoni G., Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 8.Dejana E. Endothelial cell-cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 9.Dejana E., Tournier-Lasserve E., Weinstein B.M. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev. Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Oda H., Takeichi M. Evolution: structural and functional diversity of cadherin at the adherens junction. J. Cell. Biol. 2011;193:1137–1146. doi: 10.1083/jcb.201008173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallez Y., Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta. 2008;1778:794–809. doi: 10.1016/j.bbamem.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Alcaide P., Martinelli R., Newton G., Williams M.R., Adam A., Vincent P.A., Luscinskas F.W. p120-Catenin prevents neutrophil transmigration independently of RhoA inhibition by impairing Src dependent VE-cadherin phosphorylation, American journal of physiology. Cell. Physiol. 2012;303:C385–C395. doi: 10.1152/ajpcell.00126.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gavard J., Gutkind J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 14.Vandenbroucke St Amant E., Tauseef M., Vogel S.M., Gao X.P., Mehta D., Komarova Y.A., Malik A.B. PKCalpha activation of p120-catenin serine 879 phospho-switch disassembles VE-cadherin junctions and disrupts vascular integrity. Circ. Res. 2012;111:739–749. doi: 10.1161/CIRCRESAHA.112.269654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corada M., Mariotti M., Thurston G., Smith K., Kunkel R., Brockhaus M., Lampugnani M.G., Martin-Padura I., Stoppacciaro A., Ruco L., McDonald D.M., Ward P.A., Dejana E. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. U. S. A. 1999;96:9815–9820. doi: 10.1073/pnas.96.17.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adler V., Yin Z., Fuchs S.Y., Benezra M., Rosario L., Tew K.D., Pincus M.R., Sardana M., Henderson C.J., Wolf C.R., Davis R.J., Ronai Z. Regulation of JNK signaling by GSTp. EMBO J. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mileo A.M., Abbruzzese C., Mattarocci S., Bellacchio E., Pisano P., Federico A., Maresca V., Picardo M., Giorgi A., Maras B., Schinina M.E., Paggi M.G. Human papillomavirus-16 E7 interacts with glutathione S-transferase P1 and enhances its role in cell survival. PLoS One. 2009;4 doi: 10.1371/journal.pone.0007254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tew K.D., Townsend D.M. Glutathione-s-transferases as determinants of cell survival and death. Antioxidants Redox Signal. 2012;17:1728–1737. doi: 10.1089/ars.2012.4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y., Fan Y., Xue B., Luo L., Shen J., Zhang S., Jiang Y., Yin Z. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene. 2006;25:5787–5800. doi: 10.1038/sj.onc.1209576. [DOI] [PubMed] [Google Scholar]
- 20.Manevich Y., Feinstein S.I., Fisher A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. U. S. A. 2004;101:3780–3785. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ralat L.A., Manevich Y., Fisher A.B., Colman R.F. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry. 2006;45:360–372. doi: 10.1021/bi0520737. [DOI] [PubMed] [Google Scholar]
- 22.Townsend D.M., Manevich Y., He L., Hutchens S., Pazoles C.J., Tew K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 2009;284:436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye Z.W., Zhang J., Ancrum T., Manevich Y., Townsend D.M., Tew K.D. Glutathione S-transferase P-mediated protein S-glutathionylation of resident endoplasmic reticulum proteins influences sensitivity to drug-induced unfolded protein response. Antioxidants Redox Signal. 2017;26:247–261. doi: 10.1089/ars.2015.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong X., Yang Y., Zhou Y., Bi X., Zhao N., Zhang Z., Li L., Hang Q., Zhang R., Chen D., Cao P., Yin Z., Luo L. Glutathione S-transferases P1 protects breast cancer cell from adriamycin-induced cell death through promoting autophagy. Cell Death Differ. 2019;26:2086–2099. doi: 10.1038/s41418-019-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chauhan A.K., Mittra N., Singh B.K., Singh C. Inhibition of glutathione S-transferase-pi triggers c-jun N-terminal kinase-dependent neuronal death in Zn-induced Parkinsonism. Mol. Cell. Biochem. 2019;452:95–104. doi: 10.1007/s11010-018-3415-8. [DOI] [PubMed] [Google Scholar]
- 26.Chen D., Liu J., Rui B., Gao M., Zhao N., Sun S., Bi A., Yang T., Guo Y., Yin Z., Luo L. GSTpi protects against angiotensin II-induced proliferation and migration of vascular smooth muscle cells by preventing signal transducer and activator of transcription 3 activation. Biochim. Biophys. Acta. 2014;1843:454–463. doi: 10.1016/j.bbamcr.2013.11.024. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh Dastidar S., Jagatheesan G., Haberzettl P., Shah J., Hill B.G., Bhatnagar A., Conklin D.J. Glutathione S-transferase P deficiency induces glucose intolerance via JNK-dependent enhancement of hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metabol. 2018;315:E1005–E1018. doi: 10.1152/ajpendo.00345.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y., Yin F., Hang Q., Dong X., Chen J., Li L., Cao P., Yin Z., Luo L. Regulation of endothelial permeability by glutathione S-transferase pi against actin polymerization, cellular physiology and biochemistry. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018;45:406–418. doi: 10.1159/000486918. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Y., Cao X., Yang Y., Wang J., Yang W., Ben P., Shen L., Cao P., Luo L., Yin Z. Glutathione S-transferase pi prevents sepsis-related high mobility group box-1 protein translocation and release. Front. Immunol. 2018;9:268. doi: 10.3389/fimmu.2018.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan G.L., Holz D., Yamashiro S., Taniguchi D., Watanabe N., Vavylonis D. Cell protrusion and retraction driven by fluctuations in actin polymerization: a two-dimensional model. Cytoskeleton (Hoboken) 2017;74:490–503. doi: 10.1002/cm.21389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corada M., Zanetta L., Orsenigo F., Breviario F., Lampugnani M.G., Bernasconi S., Liao F., Hicklin D.J., Bohlen P., Dejana E. A monoclonal antibody to vascular endothelial-cadherin inhibits tumor angiogenesis without side effects on endothelial permeability. Blood. 2002;100:905–911. doi: 10.1182/blood.v100.3.905. [DOI] [PubMed] [Google Scholar]
- 32.Andriopoulou P., Navarro P., Zanetti A., Lampugnani M.G., Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler. Thromb. Vasc. Biol. 1999;19:2286–2297. doi: 10.1161/01.atv.19.10.2286. [DOI] [PubMed] [Google Scholar]
- 33.Xiao K., Allison D.F., Buckley K.M., Kottke M.D., Vincent P.A., Faundez V., Kowalczyk A.P. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J. Cell. Biol. 2003;163:535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong W., He B., Qian H., Liu Q., Wang D., Li J., Wei Z., Wang Z., Xu Z., Wu G., Qian G., Wang G. RAB26-dependent autophagy protects adherens junctional integrity in acute lung injury. Autophagy. 2018;14:1677–1692. doi: 10.1080/15548627.2018.1476811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kilkenny C., Browne W.J., Cuthill I.C., Emerson M., Altman D.G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Osteoarthr. Cartil. 2012;20:256–260. doi: 10.1016/j.joca.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 36.Huang X., Dai Z., Cai L., Sun K., Cho J., Albertine K.H., Malik A.B., Schraufnagel D.E., Zhao Y.Y. Endothelial p110gammaPI3K mediates endothelial regeneration and vascular repair after inflammatory vascular injury. Circulation. 2016;133:1093–1103. doi: 10.1161/CIRCULATIONAHA.115.020918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Angelini D.J., Hyun S.W., Grigoryev D.N., Garg P., Gong P., Singh I.S., Passaniti A., Hasday J.D., Goldblum S.E. TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;291:L1232–L1245. doi: 10.1152/ajplung.00109.2006. [DOI] [PubMed] [Google Scholar]
- 38.Orsenigo F., Giampietro C., Ferrari A., Corada M., Galaup A., Sigismund S., Ristagno G., Maddaluno L., Koh G.Y., Franco D., Kurtcuoglu V., Poulikakos D., Baluk P., McDonald D., Grazia Lampugnani M., Dejana E. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 2012;3:1208. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marcos-Ramiro B., Garcia-Weber D., Millan J. TNF-induced endothelial barrier disruption: beyond actin and Rho. Thromb. Haemost. 2014;112:1088–1102. doi: 10.1160/TH14-04-0299. [DOI] [PubMed] [Google Scholar]
- 40.Oas R.G., Nanes B.A., Esimai C.C., Vincent P.A., Garcia A.J., Kowalczyk A.P. p120-catenin and beta-catenin differentially regulate cadherin adhesive function. Mol. Biol. Cell. 2013;24:704–714. doi: 10.1091/mbc.E12-06-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ikezoe T., Yang J., Nishioka C., Umezawa K., Yokoyama A. Thrombomodulin blocks calcineurin inhibitor-induced vascular permeability via inhibition of Src/VE-cadherin axis. Bone Marrow Transplant. 2017;52:245–251. doi: 10.1038/bmt.2016.241. [DOI] [PubMed] [Google Scholar]
- 42.Lambeng N., Wallez Y., Rampon C., Cand F., Christe G., Gulino-Debrac D., Vilgrain I., Huber P. Vascular endothelial-cadherin tyrosine phosphorylation in angiogenic and quiescent adult tissues. Circ. Res. 2005;96:384–391. doi: 10.1161/01.RES.0000156652.99586.9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weis S., Shintani S., Weber A., Kirchmair R., Wood M., Cravens A., McSharry H., Iwakura A., Yoon Y.S., Himes N., Burstein D., Doukas J., Soll R., Losordo D., Cheresh D. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J. Clin. Investig. 2004;113:885–894. doi: 10.1172/JCI20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nwariaku F.E., Liu Z., Zhu X., Turnage R.H., Sarosi G.A., Terada L.S. Tyrosine phosphorylation of vascular endothelial cadherin and the regulation of microvascular permeability. Surgery. 2002;132:180–185. doi: 10.1067/msy.2002.125305. [DOI] [PubMed] [Google Scholar]
- 45.Di Lorenzo A., Lin M.I., Murata T., Landskroner-Eiger S., Schleicher M., Kothiya M., Iwakiri Y., Yu J., Huang P.L., Sessa W.C. eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J. Cell Sci. 2013;126:5541–5552. doi: 10.1242/jcs.115972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kronstein R., Seebach J., Grossklaus S., Minten C., Engelhardt B., Drab M., Liebner S., Arsenijevic Y., Taha A.A., Afanasieva T., Schnittler H.J. Caveolin-1 opens endothelial cell junctions by targeting catenins. Cardiovasc. Res. 2012;93:130–140. doi: 10.1093/cvr/cvr256. [DOI] [PubMed] [Google Scholar]
- 47.Xing J., Wang Q., Coughlan K., Viollet B., Moriasi C., Zou M.H. Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am. J. Pathol. 2013;182:1021–1030. doi: 10.1016/j.ajpath.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szulcek R., Beckers C.M., Hodzic J., de Wit J., Chen Z., Grob T., Musters R.J., Minshall R.D., van Hinsbergh V.W., van Nieuw Amerongen G.P. Localized RhoA GTPase activity regulates dynamics of endothelial monolayer integrity. Cardiovasc. Res. 2013;99:471–482. doi: 10.1093/cvr/cvt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Millar T.M., Phan V., Tibbles L.A. ROS generation in endothelial hypoxia and reoxygenation stimulates MAP kinase signaling and kinase-dependent neutrophil recruitment. Free Radic. Biol. Med. 2007;42:1165–1177. doi: 10.1016/j.freeradbiomed.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 50.Aman J., Weijers E.M., van Nieuw Amerongen G.P., Malik A.B., van Hinsbergh V.W. Using cultured endothelial cells to study endothelial barrier dysfunction: challenges and opportunities. Am. J. Physiol. Lung Cell Mol. Physiol. 2016;311:L453–L466. doi: 10.1152/ajplung.00393.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haegens A., Heeringa P., van Suylen R.J., Steele C., Aratani Y., O'Donoghue R.J., Mutsaers S.E., Mossman B.T., Wouters E.F., Vernooy J.H. Myeloperoxidase deficiency attenuates lipopolysaccharide-induced acute lung inflammation and subsequent cytokine and chemokine production. J. Immunol. 2009;182:7990–7996. doi: 10.4049/jimmunol.0800377. [DOI] [PubMed] [Google Scholar]
- 52.Alimperti S., Mirabella T., Bajaj V., Polacheck W., Pirone D.M., Duffield J., Eyckmans J., Assoian R.K., Chen C.S. Three-dimensional biomimetic vascular model reveals a RhoA, Rac1, and N-cadherin balance in mural cell-endothelial cell-regulated barrier function. Proc. Natl. Acad. Sci. U. S. A. 2017;114:8758–8763. doi: 10.1073/pnas.1618333114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo L., Wang Y., Feng Q., Zhang H., Xue B., Shen J., Ye Y., Han X., Ma H., Xu J., Chen D., Yin Z. Recombinant protein glutathione S-transferases P1 attenuates inflammation in mice. Mol. Immunol. 2009;46:848–857. doi: 10.1016/j.molimm.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 54.Cain R.J., Vanhaesebroeck B., Ridley A.J. The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J. Cell. Biol. 2010;188:863–876. doi: 10.1083/jcb.200907135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corcoran A., Cotter T.G. Redox regulation of protein kinases. FEBS J. 2013;280:1944–1965. doi: 10.1111/febs.12224. [DOI] [PubMed] [Google Scholar]
- 56.Cabail M.Z., Chen E.I., Koller A., Miller W.T. Auto-thiophosphorylation activity of Src tyrosine kinase. BMC Biochem. 2016;17:13. doi: 10.1186/s12858-016-0071-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Townsend D.M., S-glutathionylation Indicator of cell stress and regulator of the unfolded protein response. Mol. Interv. 2007;7:313–324. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J., Ye Z.W., Singh S., Townsend D.M., Tew K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018;120:204–216. doi: 10.1016/j.freeradbiomed.2018.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Giannoni E., Chiarugi P. Redox circuitries driving Src regulation. Antioxidants Redox Signal. 2014;20:2011–2025. doi: 10.1089/ars.2013.5525. [DOI] [PubMed] [Google Scholar]
- 60.Giannoni E., Buricchi F., Raugei G., Ramponi G., Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 2005;25:6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saksena S., Gill R.K., Tyagi S., Alrefai W.A., Ramaswamy K., Dudeja P.K. Role of Fyn and PI3K in H2O2-induced inhibition of apical Cl-/OH- exchange activity in human intestinal epithelial cells. Biochem. J. 2008;416:99–108. doi: 10.1042/BJ20070960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu W., Doshi A., Lei M., Eck M.J., Harrison S.C. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 63.Hill B.G., Bhatnagar A. Protein S-glutathiolation: redox-sensitive regulation of protein function. J. Mol. Cell. Cardiol. 2012;52:559–567. doi: 10.1016/j.yjmcc.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information File or from the corresponding authors upon request.