

Abstract
Rheumatoid arthritis (RA) is an immune-mediated disease that causes chronic inflammation of the joints and involves CD4+ T cell activation. In RA, CD4+ T cells are the main drivers of disease initiation and the perpetuation of the damaging inflammatory process. In the present study, we investigated the role of Lysine-specific histone demethylase 1 (LSD1) in RA. The frequency of LSD1-positive CD4+ T cells in the synovial fluid (SF) of active RA patients was higher than that of inactive RA patients. In CD4+ T cells isolated from SF of active RA patients, LSD1 downregulation significantly increased cell proliferation, as shown by MTT assay. LSD1 knockdown also significantly increased the production of IFN-γ and IL-17, and increased that of IL-10, as determined by ELISA and qRT-PCR aasay. In CD4+ T cells isolated from SF of inactive RA patients, LSD1 was overexpressed by LSD1 plasmid transfection. As expected, LSD1 overexpression resulted in an opposite effect on cell proliferation and the production of cytokines, including IFN-γ, IL-17 and IL-10. LSD1 was downregulated in RA mouse by lenti-vector infection. As expected, LSD1 knockdown in vivo significantly alleviated the disease severity and increased the levels of anti-collagen II antibodies. LSD1 downregulation in the early stage was more effective to ameliorate disease severity. Our data suggested the potential therapeutic role of LSD1 in RA patients.
Keywords: Rheumatoid arthritis, LSD1, synovial fluid, CD4+ T cell
Introduction
Rheumatoid arthritis (RA) is the most frequent autoimmune chronic inflammatory disease of the joints that causes significant morbidity and mortality. It is characterized as a symmetric polyarticular arthritis that affects multiple joints [1]. CD4 T cells play a vital role in the pathogenesis of RA by secreting pro-inflammatory cytokines [2,3]. Interferon-γ (IFN-γ)-producing T helper cell 1 (Th1) and IL-17-producing T helper cell 17 (Th17) promote synovial inflammation and osteoclast forma-tion that eventually lead to joint destruction [4].Insight into the mechanisms of immunopathogenesis has also been gained through the characterization of the cytokine profiles of infiltrating T cells. Type 1 T cells produce IFN-γ and IL-17 whereas a type 2 response is characterized by the production of IL-10, and IL-13 [5]. High levels of and IL-17 presented in the synovial tissues and fluids of RA patients are culprits leading to bone and cartilage destruction [6]. The balance between these two subsets regulates the choice between inflammatory cellular and Ab-mediated immune responses while strongly polarized responses can also promote immunopathological reactions.
Lysine-specific demethylase 1 (LSD1; also known as KDM1) was the first histone demethylase discovered and belonged to the superfamily of the flavin adenine dinucleotide (FAD)-dependent amine oxidases [7]. Histone demethylases contribute to the regulation of the steady-state levels of histone methylation and are thus critical for many chromatin-based cellular processes [8]. The mechanisms that control this dual specificity of demethylation seem to be modulated by other proteins associated with LSD1 and by other histone marks displayed on the histone tail [9]. LSD1 regulates different physiological processes including hematopoiesis, adipogenesis [10], developmental processes, maintenance of the DNA methylation and tumorigenesis [11-14]. It has been reported that Gfi1 exerts its role as a transcriptional repressor by interacting with a number of histone modification enzymes including LSD1 [9]. It is well established that Gfi1 regulates Th2 cell expansion through the enhancement of Stat5 activity [15]. More recently, the Gfi1-mediated inhibition of Th17 and iTreg cell development has been reported. Gfi1 also seems to suppress IFN-γ production [16]. However, the role of LSD1 in the pathogenesis of RA is still unknown.
In the present study, we explored the role of LSD1 in CD4+ T cells isolated from the SF of RA in vitro. We also investigated the effect of LSD1 on disease severity in a RA mouse model. This suggests that the effectiveness of LSD1 inhibition is due to inhibition of multiple cytokine pathways such as IFN-γ and IL-17. Taken together, the results of this study provide new insights into the key regulatory elements of CD4+ T cells in RA.
Materials and methods
Patients
Twenty-three active RA patients (15 females, 8 males) and 10 age-matched inactive RA patients (7 females, 3 males) from the participated in this study. All of the patients fulfilled the American College of Rheumatology criteria for RA and did not show symptoms any other autoimmune disease. RA disease activity was measured using the DAS28. All of these patients had not received any immunomodulatory drugs for at least six weeks before the time of synovial fluid (SF) collection. Written informed consent was obtained from all of the patients. The Ethics Committee of approved the study.
CD4+ T cell isolation
SF samples obtained from patients with RA were treated with hyaluronidase (Sigma-Aldrich, St. Louis, MO) before then being washed in phosphate buffered saline (PBS). Next, mononuclear cells in the SF samples were isolated using Ficoll (Sigma-Aldrich) density gradient centrifugation. CD4+ T cells in mononuclear cells were isolated using a CD4+ T Cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). For CD4+ T cell isolation from the peripheral blood of mice, a Dynabeadss Mouse CD4 kit (Invitrogen, Carlsbad, CA, USA) was used.
Flow cytometry
To determine the LSD1 expression on the CD4+ T cells, the CD4+ T cells isolated from the SF were incubated with PE conjugated anti-human LSD1 antibodies (Abcam) for 30 min at 4°C. After washing twice with 2% fetal calf serum in PBS, the cells were analyzed using the LSRII flow cytometer (BD Biosciences, San Diego, CA). A PE conjugated mouse IgG1 (Abcam) was used as an isotype control.
LSD1 overexpression and knockdown
LSD1 overexpression recombined plasmids, EF1a-LV6-LSD1, was purchased from GenePharma Biotechnology (GenePharma, Shanghai). The EF1a-LV6-LSD1 was transfected into CD4+ T cells using the Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA). The empty vector was used as a negative control.
LSD1 expression in CD4+ T cells was knocked down by small interfering RNA (siRNA) transfection. The siRNA targeting LSD1 and scramble negative control siRNA were both purchased from GenePharma Biotechnology (GenePharma, Shanghai). Transfection was performed using the Lipofectamine 2000 transfection reagent (Invitrogen).
Cell proliferation assay
The proliferation of stimulated CD4+ T cells was measured using the MTT assay. Briefly, the culture medium was replaced with fresh medium containing MTT (5 mg/ml, Sigma). The cells were incubated for an additional 4 h at 37°C. The supernatants were removed and the formazan crystals were dissolved in 150 ml of dimethylsulfoxide (Sigma) for 10 min. Finally, the absorbance at 490 nm was determined. Each cell assay was performed in quadruplicate and repeated three times.
Cell stimulation
The collected CD4+ T cells (1×105/well) were cultured in RPMI 1640 completed medium containing 10 mM HEPES, 2 mM L-glutamine, 50 μg/ml of gentamycin and 10% fetal bovine serum (Gibco, Rockville, MD) in 48-well tissue culture plates (Nunc, Roskilde, Denmark) at 37°C. For activation, the CD4+ T cells were stimulated with plate-bound anti-CD3ε (5 μg/ml, Abcam) and anti-CD28 antibodies (2.5 μg/ml, Abcam) in the presence of agonistic anti-LSD1 at 37°C for 48 h. Then, cell proliferation and cytokines levels were determined.
ELISA assay
For the cytokine production analysis in CD4+ T cells, the supernatants were collected from the stimulated CD4+ T cell culture. An enzyme-linked immunosorbent assay (ELISA) was performed using a human ELISA Set specific for IFN-γ, IL-17 and IL-10 (BD Biosciences, San Jose, CA). For analysis of anti-type II collagen IgG production, serum samples collected from RA mouse models were added into the ELISA plates precoated with bovine type II collagen (Chondrex, Redmond, WA). The peroxidaseconjugated rabbit antimouse IgG antibodies (Abcam) were used to reflect the levels of anti-type II collagen IgG.
Quantitative real-time PCR
The total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA, USA) from collected CD4+ T cells. Reverse transcription was performed using a PrimeScript RT reagent kit (Takara, Dalian, China) with oligo-dT primers as per the manufacturer’s instructions. Quantitative RT-PCR (qRT-PCR) was carried out on an ABI Prism 7500 System (Applied Biosystems Inc., USA) in a 25 μl reaction system. The PCR procedure was as follows: 50°C for 2 min, 95°C for 10 min and 40 cycles at 95°C for 15 s and 60°C for 1 min. All reactions were performed in triplicate. The results were represented as relative mRNA levels calculated according to the 2-ΔΔCT method.
Western blotting
The CD4+ T cells were harvested and lysed using RIPA lysis buffer (Beyotime, Nantong, China) for total protein preparation. Protein concentration was determined using a micro-BCA protein assay kit (Pierce, Rockford, IL, USA). Equal amount proteins (25 mg per lane) were separated on 12% SDS-PAGE and transferred onto a nitrocellulose membrane (Amersham Pharmacia, Germany). After a 2 h incubation with 5% non-fat milk at room temperature, the membranes were incubated with rabbit anti-human LSD1 polyclonal antibody (Abcam) or mouse anti-human β-actin monoclonal antibody (Abcam) for human samples. For mice samples, the rabbit anti-mouse LSD1 polyclonal antibody (Antibodies-online.com, Aachen, Germany) and goat anti-mouse β-actin monoclonal antibody (Abcam) were used. The membranes were then washed five times with a PBST buffer and incubated with HRPconjugated corresponding secondary antibodies (Sigma) for 1 h at room temperature. ECL kit (Pierce Chemical, Rockford, IL) was used for development. All experiments were performed in triplicate, and the results were normalized according to β-actin.
Collagen-induced RA mouse models and lentivector infection
Six-week-old female BALB/c mice were used in the study. All mice were kept at a constant temperature (23 ± 1°C) with a 12 h light-dark cycle and were allowed free access to water and food.
RA mouse models were induced using type II collagen according to a previous report using minor modifications. Then, the RA mouse models were divided into three groups: the untreated control group, the group infected with empty lentivector on day 0 post initial immunization, the group of lentivector-inhibited LSD1 infection on day 0 post initial immunization, and the group with LSD1-LV infection on day 14 post initial immunization. Every group contained at least 12 female mice and all experiments were performed three times. For lentiviral infection, 1010 pfu lentivector was injected into the mouse via the tail vein. Meanwhile, the mouse models also received a bilateral intraarticular injection at the ankle joints (5×108 pfu) and the knee joints (109 pfu), and bilateral periarticular injection at the tarsal joints (5×108 pfu). The mice were scored weekly for three weeks after the primary immunization to monitor the severity of the mice’s arthritis. On days 28, 42 and 56, serum levels of anti-collagen II IgG were determined using an ELISA test.
Statistical analysis
Each experiment was performed at least 3 times, on independent passages, usually in triplicates. Data were analyzed by Newman-Keuls test using Statistica software as indicated and are presented as mean ± SEM. P<0.05 was considered statistically significant. Results of time lapse microscopy experiments were analyzed with Wilcoxon test in R software.
Results
LSD1 expression on SF CD4+ T cells was increased in active RA patients
CD4+ T cells isolated from the SF of 23 active RA patients and 10 age-matched inactive RA patients were used for a flow cytometry assay. As shown in Figure 1A and 1B, the frequency of LSD1-positive CD4+ T cells was significantly higher in the SF from active RA patients than that of the inactive RA patients (P<0.05). The qRT-PCR assay also indicated that the expression levels of LSD1 were significantly increased in SF CD4+ T cells from active RA patients (P<0.05, Figure 1C).
Figure 1.
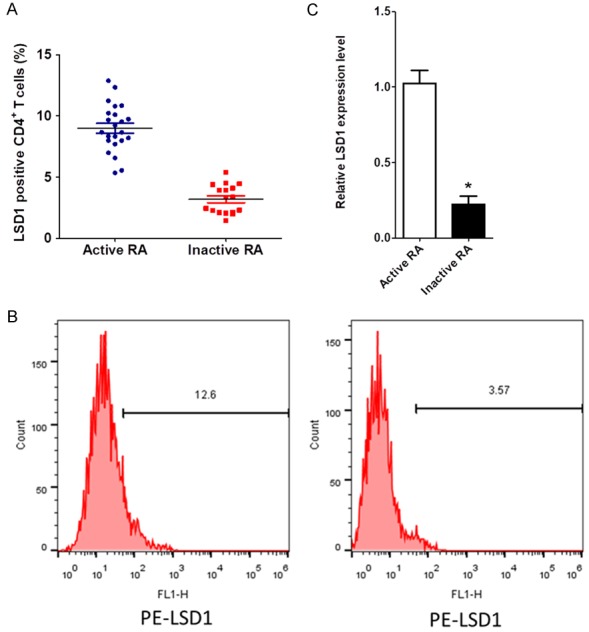
LSD1 expression on CD4+ T cells. CD4+ T cells were isolated from the SF of 23 active RA patients and 10 inactive RA patients. The representative charts of LSD1 expression on CD4+ T cells determined by flow cytometry and the quantitative data of the percentage of LSD1 positive CD4+ T cells (A, B). The mRNA levels of LSD1 on SF CD4+ T cells were determined using qRT-PCR (C). Data were normalized based on GAPDH levels and were expressed as mean ± SEM. *P<0.05.
LSD1 knockdown diminished the function of CD4+ T cells from patients with RA
As LSD1 expression on CD4+ T cells was significantly upregulated in SF from active RA patients, we investigated whether the pro-inflammatory property of CD4+ T cells could be impaired by LSD1 downregulation. CD4+ T cells isolated from SF of 5 active RA patients was transfected with siRNA targeting LSD1. As shown in Figure 2A and 2B, the mRNA and protein levels of LSD1 were significantly decreased by siRNA transfection, compared to the control group (P<0.05). The MTT assay showed that LSD1 knockdown significantly inhibited the proliferation of CD4+ T cells (P<0.05, Figure 2C). We also demonstrated that the production of IFN-γ and IL-17 were both significantly decreased by LSD1 knockdown in CD4+ T cells (P<0.05, Figure 2D). An obvious increase of IL-10 expression was observed in the LSD1 knockdown group, compared to the control group (P<0.05, Figure 2D). The mRNA levels also demonstrated that IL-17 and IFN-γ were significantly inhibited but IL-10 was significantly enhanced in the LSD1 knockdown group compared to the control group (P<0.05, Figure 2E-G). These data suggested that LSD1 knockdown could inhibit the function of CD4+ T cells in the SF from RA patients.
Figure 2.

The effect of LSD1 Knockdown on CD4+ T cells. CD4+ T cells isolated from the SF of 5 active RA patients were transfected with siRNA targeting LSD1. The scrambled siRNA was used as negative control. An empty vector was used as negative control. After 24 h, the mRNA (A) and protein levels (B) of LSD1 were determined using qRT-PCR and Western blot, respectively. Data were normalized based on GAPDH levels and were expressed as the mean fold of the 5 patients ± SEM. *P<0.05. (C) 48 h after cell activation, cell proliferation was determined using a MTT assay. Data were expressed as the mean fold of the 5 patients ± SEM. *P<0.05. (D) The production of IFN-γ, IL-17 and IL-10 in the supernatant was determined using ELISA kits. Data of every patient were expressed as mean ± SEM. *P<0.05. (E-G) The mRNA levels of IL-17, IFN-γ and IL-10 were determined using qRT-PCR. Data were normalized based on GAPDH levels and were expressed as the mean fold of the 5 patients ± SEM. *P<0.05. PA indicated active RA patient.
LSD1 overexpression promotes the proliferation and inflammatory cytokine production of CD4+ T cells
To further verify the role of LSD1, CD4+ T cells with lower LSD1 expression isolated from SF of an inactive RA patient was transfected with targeting LSD1. As shown in Figure 3A and 3B, the mRNA and protein expression of LSD1 was significantly increased by LSD1 plasmid transfection (P<0.05). The proliferation of CD4+ T cells was promoted by LSD1 overexpression, as demonstrated by the MTT assay (P<0.05, Figure 3C). As expected, LSD1 overexpression significantly promoted the production of IFN-γ and IL-17, and decreased the expression of IL-10 (P<0.05, Figure 3D and 3E).
Figure 3.

LSD1 overexpression promoted the proliferation and inflammatory cytokine production by CD4+ T cells. The CD4+ T cells isolated from the SF of 5 inactive RA patients were transfected with LV3-sh-LSD1. An empty vector was used as negative control. After 24 h, the mRNA (A) and protein levels (B) of LSD1 were determined using qRT-PCR and Western blot, respectively. Data were normalized based on GAPDH levels and were expressed as the mean fold of the 5 patients ± SEM. *P<0.05. (C) 48 h after cell activation, cell proliferation was determined using a MTT assay. Data were expressed as the mean fold of the 5 patients ± SEM. *P<0.05. (D) The production of IFN-γ, IL-17 and IL-10 in the supernatant was determined using ELISA kits. Data of every patient were expressed as mean ± SEM. *P<0.05. (E-G) The mRNA levels of IL-17, IFN-γ and IL-10 were determined using qRT-PCR. Data were normalized based on GAPDH levels and were expressed as the mean fold of the 5 patients ± SEM. *P<0.05. PI indicated inactive RA patient.
Mice with LSD1 knockdown were resistant to collagen induced RA
To evaluate the role of LSD1 in vivo, a type II collagen induced RA mouse model was developed. Three days after lentivector infection, the peripheral blood was obtained and the CD4+ T cells were isolated. The protein and mRNA levels of LSD1 were determined using western blot and RT-PCR, respectively. It was demonstrated the LSD1-LV infection significantly increased the expression of LSD1 on CD4+ T cells (P<0.05, Figure 4A and 4B). As shown in Figure 4C, the severity of RA was gradually increased over time. LSD1-LV infection on both day 0 and day 28 post initial immunization significantly alleviated the severity of RA (P<0.05, Figure 4C). The therapeutic effect was more obvious when LSD1-LV was infected on day 0 post initial immunization. ELISA assay showed that the LSD1-LV infection significantly reduced the levels of anti-collagen II antibodies (P<0.05, Figure 4D). No obvious difference was observed in the two LSD1-LV infection groups at different time points (P40.05, Figure 4D). These data demonstrated that LSD1 downregulation ameliorated the severity of RA in mice and reduced the production of anti-collagen II antibodies. LSD1 downregulation in the early stage of RA progression was more effective for alleviating the disease severity.
Figure 4.
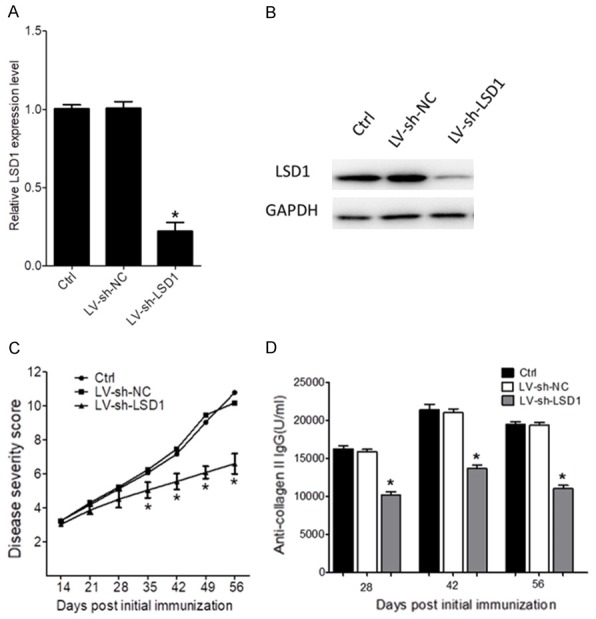
Mice with LSD1 knockdown were resistant to collagen induced RA. The collagen-induced RA mouse models were infected with sh-lentivector-inhibited LSD1 on days 0 initial immunization. The control group indicated collagen-induced RA mice received no further treatment. The empty LV group indicate collagen-induced RA mice infected with empty lentivector on day 0 post initial immunization. (A) Three days after sh-lentivector infection, the peripheral blood was obtained and the CD4+ T cells were isolated. The protein levels of LSD1 were determined using western blot, and (B) the mRNA levels of LSD1 were determined using RT-PCR. (C) The severity score of the RA mouse. Each paw was scored on a graded scale from 0 to 3:0, normal paw; 1, swelling and/or redness of one toe or finger joint; 2, swelling of two or more toes or joints, or increased swelling; 3, severe swelling and/or ankylosis throughout the entire paw. The four scores were summed such that the maximal score per mouse was 12. Every group contained at least 12 female mice and all experiments were performed three times. (D) The serum levels of anti-collagen II IgG were determined using an ELISA test on 28, 42 and 56 days post initial immunization. Data are expressed as mean ± SEM. *P<0.05 versus control group.
Discussion
Rheumatoid arthritis (RA) is an immune-mediated disease that is characterised by chronic inflammation in the joints and synovial hyperplasia, leading to cartilage and bone loss [17]. Earlier studies in the joints of patients with RA identified the presence of T cells in the synovium [18]. T cell receptor (TCR) signal is crucial for T cell activation [19]; however, ultimate effector function and differentiation such as Th lineage commitment are determined by co-stimulatory signals and cytokines via activation of the specific master transcriptional regulators [20]. Previously study shown that the activation of the ERK/MAPK pathway by the TCR-mediated stimulation induces Gfi1 expression in CD4 T cells [21]. Gfi-1mediated inhibition of histone H3K4 methylation at the Th1-related transcription factor gene loci seems to be important for the repression of the Th1 programme in activated CD4 T cells [9]. The pharmacological inhibition of LSD1 is sufficient to induce Th1 cell differentiation in activated CD4 T cells. Knockdown of LSD1 on CD4+ T cell inhibits its function in vitro and ameliorates aplastic anemia [11]. In the present study, we found that the frequency of LSD1-positive CD4+ T cells in SF was significantly reduced in active RA patients compared to inactive ones. By down-regulation of LSD1 on CD4+ T cells isolated from the SF of active RA patients, cell proliferation and the secretion of pro-inflammatory cytokines were inhibited. In contrast, LSD1 overexpression on CD4+ T cells isolated from the SF of inactive RA patients enhanced cell proliferation and the secretion of pro-inflammatory cytokines. These results were consisted with the data of LSD1 on CD4+ T cells isolated from the sera of aplastic anemia patients.
Inflammatory responses require the activation of a complex gene expression program that involves the inducible transcription of hundreds of genes whose products restrain microbial colonization, recruit and activate leukocytes, increase vascular permeability, amplify the immune response, and protect inflammatory and tissue cells from apoptosis. Changes in lysine methylation were detected at inflammatory gene promoters upon stimulation indicating a possible role for histone demethylases in gene regulation [22-24]. The fact that LSD1 might activate the alternative and MBL pathway while repressing the classical and vice versa offers the interesting possibility that LSD1 is a balance between the different complement pathways [9]. In the type II collagen induced RA mouse model, we demonstrated that LSD1 knockdown by lentivector infection ameliorated disease severity and the levels of sera anti-collagen II IgG. However, several items still need to be elucidated. In the present study, the LSD1 was not specially overexpressed in CD4+ T cells, so it was difficult to assess the role of LSD1 overexpressed CD4+ T cells in vivo. Treg cells overexpressing the LSD1 might also contribute to the alleviation of RA, as Treg expressing LSD1 selectively inhibits proinflammatory Th1 and Th17 cell responses [25]. To more precisely verify the role of LSD1 on CD4+ T cells in vivo, we will establish a mouse model in which LSD1 is specifically overexpressed on Th1 cells. A mouse with LSD1 deficiency in the Th1 cells was also needed to investigate whether it is more susceptible to type II collagen induced RA. LSD1 plays a pivotal role in the inhibition of Th1-type immune responses in developing Th2 cells. It is possible that LSD1 inhibits the induction of the Th1 programme by the stabilization of Gfi1 in activated CD4 T cells.
Taken together, we found that LSD1 knockdown in early stages of RA pathogenesis more efficiently eased the disease severity. We explored the function of LSD1 on CD4+ T cells in SF of RA patients and in a RA mouse model. Our data suggested the potential therapeutic role of LSD1 in RA patients. These findings demonstrate a novel regulatory role of LSD1 in the regulation of the Th1-type immune response.
Acknowledgements
This work was supported by grants from the Jiangsu Provincial Six Talent Peaks Foundation (2015-WSN-096), Science and Technology Program of Nantong (MS12015102), and Jiangsu Provincial Young Medical Talent Foundation (QNRC2016411).
Disclosure of conflict of interest
None.
References
- 1.Van Boxel JA, Paget SA. Predominantly Tcell infiltrate in rheumatoid synovial membranes. N Engl J Med. 1975;293:517–520. doi: 10.1056/NEJM197509112931101. [DOI] [PubMed] [Google Scholar]
- 2.Ahmad SF, Zoheir KM, Abdel-Hamied HE, Ashour AE, Bakheet SA, Attia SM, Abd-Allah AR. Amelioration of autoimmune arthritis by naringin through modulation of T regulatory cells and Th1/Th2 cytokines. Cell Immunol. 2014;287:112–120. doi: 10.1016/j.cellimm.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Baba N, Rubio M, Sarfati M. Interplay between CD45RA + regulatory T cells and TNFalpha in the regulation of human Th17 differentiation. Int Immunol. 2010;22:237–244. doi: 10.1093/intimm/dxp130. [DOI] [PubMed] [Google Scholar]
- 4.Lee EY, Seo M, Juhnn YS, Kim JY, Hong YJ, Lee YJ, Lee EB, Song YW. Potential role and mechanism of IFN-gamma inducible protein-10 on receptor activator of nuclear factor kappa-B ligand (RANKL) expression in rheumatoid arthritis. Arthritis Res Ther. 2011;13:R104. doi: 10.1186/ar3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Hamburg JP, de Bruijn MJ, Ribeiro de Almeida C, van Zwam M, van Meurs M, de Haas E, Boon L, Samsom JN, Hendriks RW. Enforced expression of GATA3 allows differentiation of IL-17-producing cells, but constrains Th17-mediated pathology. Eur J Immunol. 2008;38:2573–2586. doi: 10.1002/eji.200737840. [DOI] [PubMed] [Google Scholar]
- 6.Nakajima A, Aoki Y, Sonobe M, Watanabe F, Takahashi H, Saito M, Nakagawa K. Relative expression and correlation of tumor necrosis factor-alpha, interferon-gamma, and interleukin-17 in the rheumatoid synovium. Clin Rheumatol. 2016;35:1691–1697. doi: 10.1007/s10067-016-3249-2. [DOI] [PubMed] [Google Scholar]
- 7.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 8.Rudolph T, Beuch S, Reuter G. Lysine-specific histone demethylase LSD1 and the dynamic control of chromatin. Biol Chem. 2013;394:1019–1028. doi: 10.1515/hsz-2013-0119. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki J, Maruyama S, Tamauchi H, Kuwahara M, Horiuchi M, Mizuki M, Ochi M, Sawasaki T, Zhu J, Yasukawa M, Yamashita M. Gfi1, a transcriptional repressor, inhibits the induction of the T helper type 1 programme in activated CD4 T cells. Immunology. 2016;147:476–487. doi: 10.1111/imm.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan Z, Zarebski A, Montoya-Durango D, Grimes HL, Horwitz M. Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol Cell Biol. 2005;25:10338–10351. doi: 10.1128/MCB.25.23.10338-10351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Musri MM, Carmona MC, Hanzu FA, Kaliman P, Gomis R, Parrizas M. Histone demethylase LSD1 regulates adipogenesis. J Biol Chem. 2010;285:30034–30041. doi: 10.1074/jbc.M110.151209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell. 2007;27:562–572. doi: 10.1016/j.molcel.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L, Li R, Li Y, Zhang Y, Li Q, Yi X, Shang Y. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660–672. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 15.Soliera AR, Mariani SA, Audia A, Lidonnici MR, Addya S, Ferrari-Amorotti G, Cattelani S, Manzotti G, Fragliasso V, Peterson L, Perini G, Holyoake TL, Calabretta B. Gfi-1 inhibits proliferation and colony formation of p210BCR/ABL-expressing cells via transcriptional repression of STAT 5 and Mcl-1. Leukemia. 2012;26:1555–1563. doi: 10.1038/leu.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furusawa J, Mizoguchi I, Chiba Y, Hisada M, Kobayashi F, Yoshida H, Nakae S, Tsuchida A, Matsumoto T, Ema H, Mizuguchi J, Yoshimoto T. Promotion of expansion and differentiation of hematopoietic stem cells by interleukin-27 into myeloid progenitors to control infection in emergency myelopoiesis. PLoS Pathog. 2016;12:e1005507. doi: 10.1371/journal.ppat.1005507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity. 2017;46:183–196. doi: 10.1016/j.immuni.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh AK, Misra R, Aggarwal A. Th-17 associated cytokines in patients with reactive arthritis/undifferentiated spondyloarthropathy. Clin Rheumatol. 2011;30:771–776. doi: 10.1007/s10067-010-1646-5. [DOI] [PubMed] [Google Scholar]
- 19.Flerin NC, Chen H, Glover TD, Lamothe PA, Zheng JH, Fang JW, Ndhlovu ZM, Newell EW, Davis MM, Walker BD, Goldstein H. T-cell receptor (TCR) clonotype-specific differences in inhibitory activity of HIV-1 Cytotoxic T-cell clones is not mediated by TCR alone. J Virol. 2017:91. doi: 10.1128/JVI.02412-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hillen KM, Gather R, Enders A, Pircher H, Aichele P, Fisch P, Blumenthal B, Schamel WW, Straub T, Goodnow CC, Ehl S. T cell expansion is the limiting factor of virus control in mice with attenuated TCR signaling: implications for human immunodeficiency. J Immunol. 2015;194:2725–2734. doi: 10.4049/jimmunol.1400328. [DOI] [PubMed] [Google Scholar]
- 21.Shinnakasu R, Yamashita M, Kuwahara M, Hosokawa H, Hasegawa A, Motohashi S, Nakayama T. Gfi1-mediated stabilization of GATA3 protein is required for Th2 cell differentiation. J Biol Chem. 2008;283:28216–28225. doi: 10.1074/jbc.M804174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Gazzar M, Yoza BK, Hu JY, Cousart SL, McCall CE. Epigenetic silencing of tumor necrosis factor alpha during endotoxin tolerance. J Biol Chem. 2007;282:26857–26864. doi: 10.1074/jbc.M704584200. [DOI] [PubMed] [Google Scholar]
- 23.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 24.Saccani S, Natoli G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002;16:2219–2224. doi: 10.1101/gad.232502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ERnegative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31:512–520. doi: 10.1093/carcin/bgp324. [DOI] [PubMed] [Google Scholar]