

Abstract
This study aimed to elucidate the potential role of long non-coding RNA PlncRNA-1 in the septic acute kidney injury (AKI). The expression of PlncRNA-1 in the serum of patients with septic AKI patient was detected. We then established lipopolysaccharide (LPS)-induced septic AKI model in NRK-52E cells to investigate the effects of the overexpression of PlncRNA-1 on cell proliferation, apoptosis, and autophagy. In addition, the regulatory relationship between PlncRNA-1 and B-cell lymphoma 2 (BCL2) was explored to further elucidate the regulatory mechanism of PlncRNA-1 in septic AKI. PlncRNA-1 is downregulated in the serum of patients with septic AKI and in LPS-induced septic AKI cells. The overexpression of PlncRNA-1 considerably increases proliferation and inhibits apoptosis and autophagy of LPS-induced septic AKI cells. In addition, PlncRNA-1 can promote BCL2 expression, and the overexpression of BCL2 enhances proliferation and inhibits apoptosis and autophagy of LPS-induced septic AKI cells. Our findings reveal that the overexpression of PlncRNA-1 may promote cell proliferation and inhibit apoptosis and autophagy in septic AKI by regulating BCL2 expression. PlncRNA-1 may serve as a potential biomarker or target for the diagnosis and treatment of septic AKI.
Keywords: Sepsis, acute kidney injury, PlncRNA-1, B-cell lymphoma 2
Introduction
Sepsis is a frequently fatal condition that is characterized by a whole-body inflammatory response caused by an uncontrolled and harmful host response to infection [1]. Severe sepsis is the leading cause of death among critically ill patients in non-coronary intensive care units, and limited treatment options are available for it [2,3]. Acute kidney injury (AKI) is a common and serious complication of sepsis that leads to a high morbidity and mortality rate [4,5]. The increasing incidence of AKI has become a severe public health problem [6]. However, the pathogenesis of AKI in sepsis is still unclear. Therefore, great significance exists to further elucidate the mechanisms involved in septic AKI.
Long non-coding RNAs (lncRNAs) are transcribed but not translated RNA segments with more than 200 bases in length, which exhibit diverse functions in various physiological and pathological processes [7-9]. LncRNAs are being identified as key regulators that are involved in sepsis and AKI [10,11]. For example, the lncRNA plasmacytoma variant translocation 1 can promote inflammatory response in lipopolysaccharide (LPS)-induced septic AKI cells [12], and circulating lncRNA TapSAKI is considered an independent predictor of mortality in critically ill patients with AKI [13]. PlncRNA-1 has been found to initiate malignancy in a variety of cancers, such as esophageal squamous carcinoma [14], prostate cancer [15], and hepatocellular carcinoma [16]. Moreover, PlncRNA-1 regulates the function of intestinal epithelial barrier in inflammatory bowel disease [17]. However, the role of PlncRNA-1 in septic AKI has not been documented.
In this study, the expression of lncRNA (PlncRNA-1) in the serum of patients with septic AKI was detected. In addition, the administration of LPS produced by gram-negative bacteria reproduces many of the clinical features of sepsis, including AKI [18]. We thus established LPS-induced septic AKI model in NRK-52E cells to investigate the effects of the overexpression of PlncRNA-1 on cell proliferation, apoptosis, and autophagy. Furthermore, the regulatory relationship between PlncRNA-1 and B-cell lymphoma 2 (BCL2) was studied to further elucidate the regulatory mechanism of PlncRNA-1 in septic AKI cells. All efforts of this study aimed to provide the theoretical basis for the treatment of septic AKI in clinics.
Materials and methods
Serum sampling
From March 2016 to April 2017, six patients with septic AKI and six normal subjects were enrolled in this study. After all, the patients were admitted to the hospital, 5 mL of venous blood was extracted on the first day. The blood was collected in the coagulation tube and centrifuged at low temperature for 10 min (3000 rpm/min). The supernatant was then collected and frozen at -80°C in a refrigerator to further detect the expression of PlncRNA-1 and BCL2.
Cell culture and treatment
Rat renal proximal tubular cell line NRK-52E (The Chinese Academy of Sciences, Shanghai, China) was grown at 37°C in Dulbecco’s modified Eagle (DMEM) medium with F12 (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (Gibco). The NRK-52E cells were digested using trypsin, seeded in 6-well plates, and grown until confluent. The day before stimulation with LPS (0.1 µg/mL), the NRK-52E culture medium was abandoned and replaced with fresh medium without serum. The cells were incubated with LPS for 24 h. All experiments were at least conducted three times independently.
Cell transfection
After incubation with LPS for 24 h, the NRK-52E cells were then divided into five groups: control (without LPS stimulation and any transfection), LPS (LPS-treated cells without transfection), LPS + blank control (LPS-treated cells transfected with blank vector as a control), LPS + PlncRNA-1 (LPS-treated cells transfected with pcDNA3.1-PlncRNA-1), and LPS + BCL2 (LPS-treated cells transfected with pcDNA3.1BCL2). As per the manufacturer’s instructions, pcDNA3.1-PlncRNA-1, pcDNA3.1-BCL2, or blank control were transfected into the NRK-52E cells using Lipofectamine 2000 (Life Technologies Corporation, Carlsbad, CA). The medium was replaced at 6 h after transfection, and the cells were harvested at 48 h of post-transfection for conducting subsequent experiments.
MTT assay
The cell viability was determined using MTT assay (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium bromide; Beyotime, Shanghai, China). NRK-52E cells were adjusted to 5 × 104 cells/mL in the logarithmic growth phase. These cells were then added to a 96-well plate (100 μL per well) and allowed to attach to surfaces for overnight. After transfection for 24, 48, and 72 h, 10 μL MTT buffer was added to each well. Subsequently, the 96-well plate was incubated at 37°C for 4 h, after which 150 μL dimethyl sulfoxide was added to each well for 15 min. The optical density (OD) of each cell was then measured at 490 nm using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). The OD of LPS, LPS + blank control, or LPS + PlncRNA-1 groups was compared with that of the control group to indicate cell viability of each group. All experiments were repeated for at least three times.
Immunofluorescence
For immunofluorescence analysis, NRK-52E cells in different treated groups were seeded on coverslips until reaching 40% confluence and were harvested. The cells were then fixed with 4% paraformaldehyde (Sangon Biotech, Shanghai, China), washed three times with phosphate buffer saline (PBS), and permeabilized with 2.5% Triton X-100 (Sangon Biotech). After being blocked with 10% goat serum in PBS, the cells were incubated with primary rabbit anti-mouse polyclonal antibody against LC3B (1:400; Cell Signaling Technology, Shanghai, China) for overnight at 4°C in a humidified chamber. Subsequently, the cells were incubated with donkey anti-rabbit IgG (H+L), which was Alexa Fluor 488-conjugated secondary antibody (1:400; Invitrogen Life Technologies, Carlsbad, CA), and rhodamine-labeled phalloidin (1:1,000; Invitrogen Life Technologies) at room temperature for 1 h. The cells were stained with 4’,6-diamino-2-phenylindole for visualization of the nuclei of the cells. Finally, the coverslips were mounted on glass slides. The immunofluorescence images were captured by a microscope (Nikon), and further, the number of green fluorescent puncta in 50 cells per group were counted.
Colony formation
NRK-52E cells in different treated groups were harvested and cultured in a 6-well plate with 500 cells per well, each group had was repeated twice in parallel. The cells were incubated at 37°C in a humidified incubator with 5% CO2 for 14 days. After incubation, the cells were washed two times with PBS and stained using crystal violet staining solution. The number of colonies containing more than 50 cells was counted under a microscope, and the value was used to calculate the plate clone formation efficiency using the formula: plate clone formation efficiency = (number of colonies/number of cells inoculated) × 100%.
Flow cytometry
NRK-52E cells were harvested, seeded in 6-well plates to obtain 4 × 105 cells per well in a fresh medium without antibiotic, and transfected with PlncRNA-1, BCL2, or blank control using Lipofectamine 2000. After 24 h, NRK-52E cells were washed, harvested, and stained with annexin V-FITC and propidium iodide (BD Bioscience, San Diego, CA). Subsequently, apoptosis percentage was determined by flow cytometry (Beckman Coulter, Miami, USA). All experiments were repeated in triplicate.
Western blot
The total proteins of different treated groups were extracted by radioimmunoprecipitation assay buffer on ice. Following the removal of cellular debris by centrifugation at 13,000 rpm for 15 min, the protein concentration was determined using bicinchoninic acid assay kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. For western blot analysis, 20 μg of protein from each sample was loaded in each lane for sodium dodecyl sulfate with polyacrylamide gel electrophoresis, and then electrophoretically transferred to polyvinylidene fluoride membranes. After being blocked in 5% skim milk for 2 h at room temperature, the membranes were incubated for overnight at 4°C with primary antibodies against caspase-3, cleaved caspase-3, LC-3, Beclin-1, or BCL2 (1:1000, Abcam, Cambridge, UK). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal standard to normalize loading protein. The membranes were subsequently washed three times with PBS and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies. After extensive washes with TBS containing 0.1% X-100 (TBST), the enhanced chemiluminescence was performed to visualize the immunoreactive protein bands. The densitometric analyses of protein bands were carried out by NIH Imaging software packages.
qPCR
Total RNA was extracted from tissue samples or cell lines using TRIzol Reagent (Invitrogen, Burlington, Canada) according to the manufacturer’s recommended protocol. RNA was reverse transcribed into cDNAs using the iScript cDNA Synthesis kit (Bio-Rad Laboratories, USA). The expression of PlncRNA-1 was detected by SYBR green-based qRT-PCR. The primers used for PlncRNA-1 amplification are as follows: forward 5’-CAGTGGGGAACTCTGACTCG-3’ and reverse 5’-GTGCCTGGTGCTCTCTTACC-3’. The primers used for BCL2 amplification are as follows: forward 5’-CGATTGTGGCAGTCCCTTA-3’ and reverse 5’-CAGGATGAAGTGCTCAGGTG-3’. GAPDH (forward 5’-GTCAACGGATTTGGTCTGTATT-3’ and reverse 5’-AGTCTTCTGGGTGGCAGTGAT-3’) was used as the internal control, and the relative expression of mRNA was calculated using the 2-ΔΔCT method.
Statistical analysis
All data obtained from triplicate experiments were presented as mean ± standard deviation. The data were analyzed using the Student’s t-test. Statistical analyses were then performed using a one-way analysis of variance in SPSS 20 statistical software (Chicago, IL, USA). A p-value of < 0.05 was considered to indicate a statistically significant difference.
Results
PlncRNA-1 was downregulated in serum of patients with septic AKI and in cell model
The expression of PlncRNA-1 was detected by qPCR in the serum of normal individuals and patients with septic AKI (Figure 1A). The expression of PlncRNA-1 in patients with septic AKI was markedly lower than that in the normal group (P < 0.05). In addition, the expression level of PlncRNA-1 in NRK-52E cells was measured before and after LPS treatment (Figure 1B). The level was considerably lower after LPS treatment than that in the control group (P < 0.05). Moreover, the expression of PlncRNA-1 in LPS-treated NRK-52E cells was considerably increased at 48 h compared with that in blank control after transfection of pcDNA3.1-PlncRNA-1 (P < 0.05), indicating that PlncRNA-1 was successfully overexpressed in LPS-treated NRK-52E cells.
Figure 1.
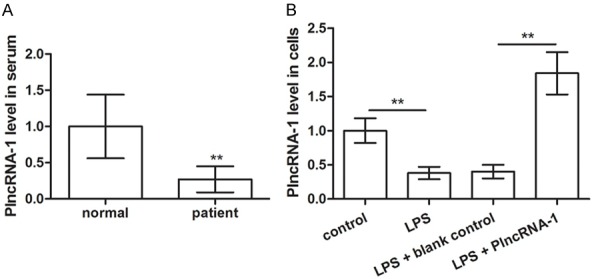
PlncRNA-1 downregulated in the serum of patients with septic acute kidney injury (AKI) (A) and lipopolysaccharide (LPS)-induced septic AKI model in NRK-52E cells (B). **P < 0.01 compared with corresponding controls.
Overexpression of PlncRNA-1 promoted proliferation and inhibited apoptosis and autophagy of LPS-treated cells
Cell viability was detected in control, LPS, LPS + blank control, and LPS + PlncRNA-1 groups by MTT assay (Figure 2A). The results showed that LPS treatment decreased cell viability compared with that of the control group (P < 0.05). However, in comparison with LPS + blank control group, the overexpression of PlncRNA-1 led to an increase in cell viability after LPS treatment and transfection of pcDNA3.1-PlncRNA-1 (P < 0.05). Cell proliferation was detected by plate cloning method (Figure 2B). The number of clone formation decreased after LPS treatment compared to that of the control group (P < 0.05), and the overexpression of PlncRNA-1 increased the clone formation after LPS treatment and the transfection of pcDNA3.1-PlncRNA-1 (P < 0.05). We subsequently characterized the role of PlncRNA-1 in cell apoptosis by flow cytometry (Figure 2C). The percentage of apoptotic cells increased after LPS treatment compared to that of the control group (P < 0.05), and the overexpression of PlncRNA-1 decreased the percentage of apoptotic cells after the LPS treatment and transfection of pcDNA3.1-PlncRNA-1 (P < 0.05). The Western blot technique was used to detect the expression of active caspase-3 and -9, which was consistent with the results of flow cytometry (Figure 2D). LC3 was labeled by cell immunofluorescence assay (Figure 2E). LPS promoted autophagy formation compared to the control group (P < 0.05), whereas the overexpression of PlncRNA-1 inhibited autophagy formation after LPS treatment and transfection of pcDNA3.1-PlncRNA-1 (P < 0.05). Furthermore, the expression of autophagy factor LC3-II/I and Beclin 1 was detected by western blot analysis, which was consistent with the results of immunofluorescence assay (Figure 2F). These results suggest that PlncRNA-1 alleviates LPS-induced cell proliferation, apoptosis, and autophagy and promotes proliferation and inhibits apoptosis and autophagy.
Figure 2.

The overexpression of PlncRNA-1 increases proliferation and inhibits apoptosis and autophagy in LPS-induced septic AKI cells. NRK-52E cells were treated with LPS to establish the septic AKI model. LPS-induced septic AKI cells were transfected with pc-PlncRNA-1 and blank. A. MTT assay showing cell viability of different treated groups. B. Colony formation assay showing the colony number in different treated groups. C. Flow cytometry showing cell apoptosis in the different treated groups. D. Western blot showing the expression levels of apoptosis-related proteins (active caspase-3 and -9) in the different transfected groups. E. Immunofluorescence assay showing the positive cells with LC3 puncta in the different transfected groups. F. Western blot showing the expression levels of autophagy-related proteins (LC3-II/I ratio and Beclin-1) in the different transfected groups. *P < 0.05, **P < 0.01, ***P < 0.001 compared with corresponding controls.
Overexpression of PlncRNA-1 promoted BCL2 expression
The western blot analysis showed that the expression of BCL2 in patients with septic AKI was considerably lower than that in the normal group (P < 0.05) (Figure 3A). The expression of BCL2 and its mRNA was markedly reduced in LPS-treated cells compared to that in the untreated cells (P < 0.05) (Figure 3B and 3C). However, the overexpression of PlncRNA-1 after LPS treatment and transfection of pcDNA3.1-PlncRNA-1 can promote the expression of BCL2 and its mRNA both (P < 0.05; Figure 3B and 3C). This indicates that PlncRNA-1 promotes BCL2 expression.
Figure 3.
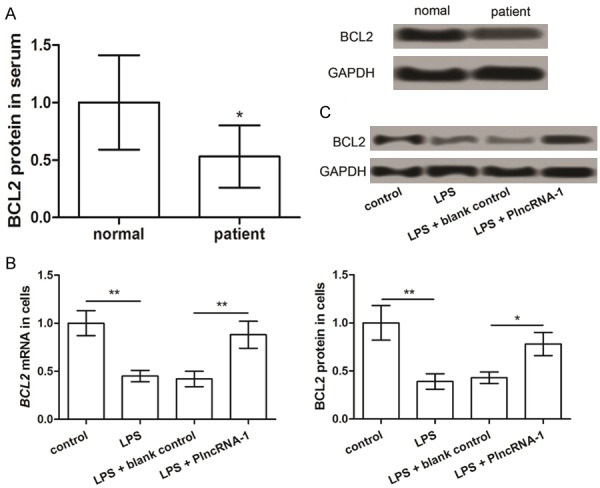
The expression of BCL2 in the serum of patients with septic AKI (A) and LPS-induced septic AKI model in NRK-52E cells (B and C). *P < 0.05, **P < 0.01 compared with corresponding controls.
Overexpression of BCL2 promoted proliferation and inhibited apoptosis and autophagy of LPS-treated cells
BCL2 expression in NRK-52E cells after LPS treatment and transfection of pcDNA3.1-BCL2 was considerably increased compared with that in LPS + blank control group (P < 0.05) (Figure 4A). On comparing the ability of clone formation of the two groups, we noted that the overexpression of BCL2 promotes clone formation (P < 0.05; Figure 4B). The results of flow cytometry (Figure 4C) and the western blot analysis of the expression of the apoptotic factors, active caspase-3 and -9 (Figure 4D), showed that the overexpression of BCL2 after LPS treatment and transfection of pcDNA3.1-BCL2 considerably inhibits cell apoptosis (P < 0.05). Immunofluorescence results showed that the overexpression of BCL2 after LPS treatment and transfection of pcDNA3.1-BCL2 markedly inhibit the formation of autophagy (P < 0.05) (Figure 4E) and expression of autophagy factors, LC3-II/I and Beclin-1 (P < 0.05) (Figure 4F). These results suggest that BCL2 regulates cell proliferation, apoptosis, and autophagy after LPS treatment, which may be one of the regulatory mechanisms of PlncRNA-1.
Figure 4.
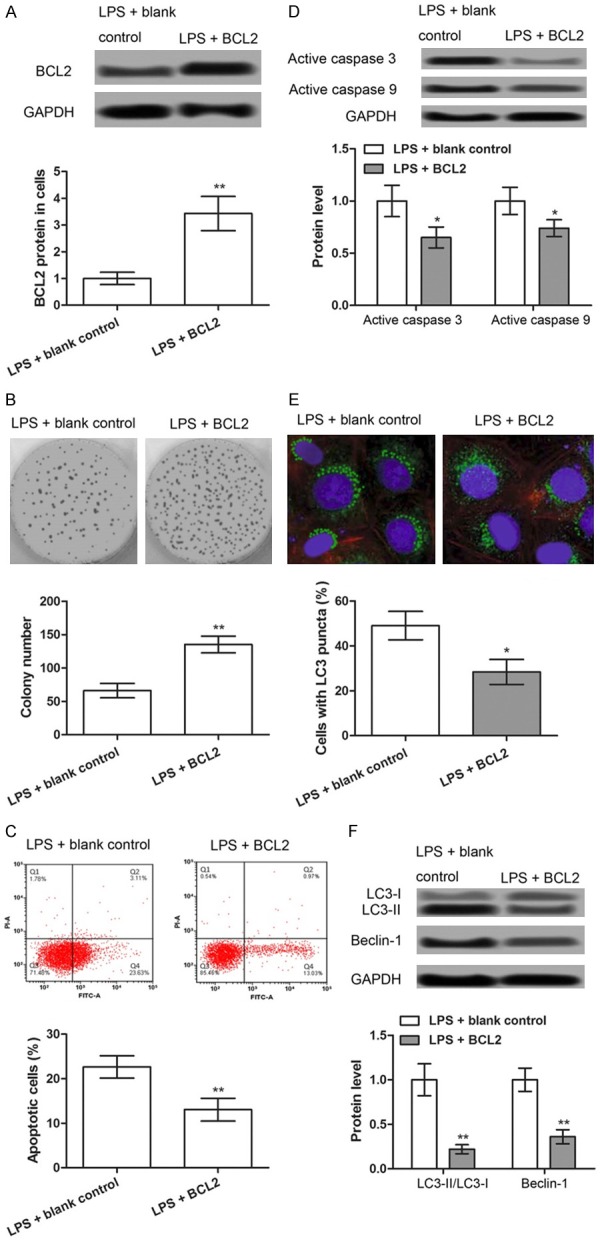
The overexpression of BCL2 increases proliferation and inhibits apoptosis and autophagy in LPS-induced septic AKI cells. NRK-52E cells were treated with LPS to establish the septic AKI model. LPS-induced septic AKI cells were transfected with pc-BCL2 and blank. A. Western blot showing the expression levels of BCL2 in different treated groups. B. Colony formation assay showing the colony number in the different treated groups. C. Flow cytometry showing cell apoptosis in the different treated groups. D. Western blot showing the expression levels of apoptosis-related proteins (active caspase-3 and -9) in the different treated groups. E. Immunofluorescence assay showing the positive cells with LC3 puncta in the different treated groups. F. Western blot showing the expression levels of autophagy-related proteins (LC3-II/I ratio and Beclin-1) in the different treated groups. *P < 0.05, **P < 0.01 compared with corresponding controls.
Discussion
In patients with severe sepsis, AKI is considered as a leading cause of morbidity and mortality [19]. LncRNAs have been identified as key regulators that are involved in septic AKI [11]. Therefore, the identification of key lncRNAs implicated in septic AKI is necessary. In this study, we found that PlncRNA-1 was downregulated in the serum of patients with septic AKI and LPS-induced cell model of septic AKI. The overexpression of PlncRNA-1 increased proliferation and inhibited apoptosis and autophagy of LPS-induced septic AKI cells. In addition, PlncRNA-1 promoted BCL2 expression, and the overexpression of BCL2 enhanced proliferation and inhibited apoptosis and autophagy of LPS-induced septic AKI cells. This indicates that the key role of PlncRNA-1 in the development of septic AKI is mediated through BCL2.
Recently, the key role of BCL2 in regulating cell proliferation has been confirmed by several studies. For example, the overexpression of BCL2 promotes myocyte proliferation [20]. BCL2 proteins determine cell viability in the pathologic circumstances of AKI [21]. In this study, the results showed that the overexpression of PlncRNA-1 or BCL2 considerably promoted the proliferation of LPS-induced septic AKI cells. In addition, PlncRNA-1 promoted BCL2 expression. These data prompt us to speculate that PlncRNA-1 may control cell proliferation.
Furthermore, apoptosis is a programmed cell death, which maintains the balance of survival and death in living organisms [22,23]. BCL2 as an anti-apoptosis protein plays a vital role in the apoptosis pathway, and both apoptosis mechanism and BCL2 are found to be key regulators in AKI [24]. Frank demonstrated that two SNPs (rs8094315 and rs12457893) in the BCL2 gene are associated with decreased risk of AKI, confirming the key role of apoptosis in AKI [25]. In addition, caspase-3 was found to be a key mediator of mitochondrial events of apoptosis [26]. The activation of caspase-3 is a crucial mechanism for the induction of apoptosis in human lung cancer A549 cells [27]. In sepsis, caspase-3 has been shown to mediate in part hippocampal apoptosis [28]. Moreover, caspase-9 is involved in sepsis-induced lymphocyte apoptosis [29]. In this study, we found that the overexpression of PlncRNA-1 or BCL2 considerably inhibited the apoptosis of LPS-induced septic AKI cells by inhibiting caspase-3 and -9. Considering the regulatory relationship between PlncRNA-1 and BCL2, we hypothesize that PlncRNA-1 medicates cell apoptosis in septic AKI by interacting with BCL2 to regulate the expression of caspase-3 and -9.
In addition, autophagy is a catabolic process that is essential for cellular homeostasis [30]. Moreover, autophagy is a key mechanism that regulates the pathogenesis of AKI [31]. It has been reported that BCL2 can interact with the autophagy-related protein, Beclin 1, thus facilitating in maintaining autophagy at levels that match cell survival [32]. Lee demonstrated that carbon monoxide plays a protective role in sepsis by enhancing Beclin 1-dependent autophagy [33]. Moreover, renal LC3-II and urinary LC3 proteins have been suggested as promising biomarkers for autophagy in AKI [34]. In our study, the overexpression of PlncRNA-1 or BCL2 considerably inhibited the autophagy of LPS-induced septic AKI cells by regulating LC3-II/I ratio and Beclin 1. Although the role of PlncRNA-1 in septic AKI cells has not been fully explored, we speculate that PlncRNA-1 regulates the autophagy of septic AKI cells by interacting with BCL2 to regulate LC3-II/I ratio and Beclin 1.
In conclusion, our findings reveal that the overexpression of PlncRNA-1 may promote cell proliferation and inhibit apoptosis and autophagy in septic AKI by regulating BCL2 expression, which will provide important implications for better understanding of the pathogenesis and therapy of septic AKI.
Acknowledgements
This study was supported by the Natural Foundation of Jiangsu Province (Grants No.BK20140735).
Disclosure of conflict of interest
None.
References
- 1.Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A, Brenner T, Uhle F, Iwamoto Y, Robbins CS. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science. 2015;347:1260–1265. doi: 10.1126/science.aaa4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence. 2014;5:4–11. doi: 10.4161/viru.27372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. The new normal: immunomodulatory agents against sepsis immune suppression. Trends Mol Med. 2014;20:224–233. doi: 10.1016/j.molmed.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoyanoff TR, Todaro JS, Aguirre MV, Zimmermann MC, Brandan NC. Amelioration of lipopolysaccharide-induced acute kidney injury by erythropoietin: involvement of mitochondria-regulated apoptosis. Toxicology. 2014;318:13–21. doi: 10.1016/j.tox.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- 6.Bilgili B, Haliloğlu M, Cinel İ. Sepsis and acute kidney injury. Turk J Anaesthesiol Reanim. 2014;42:294. doi: 10.5152/TJAR.2014.83436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boon RA, Jaé N, Holdt L, Dimmeler S. Long noncoding RNAs. J Am Coll Cardiol. 2016;67:1214–1226. doi: 10.1016/j.jacc.2015.12.051. [DOI] [PubMed] [Google Scholar]
- 9.Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21:1253. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 10.Zhou P, Chen Z, Zou Y, Wan X. Roles of noncoding RNAs in acute kidney injury. Kidney Blood Press Res. 2016;41:757–769. doi: 10.1159/000450566. [DOI] [PubMed] [Google Scholar]
- 11.Chun-Mei H, Qin-Min G, Shu-Ming P, Xiang-Yang Z. Expression profiling and ontology analysis of circulating long non-coding RNAs in septic acute kidney injury patients. Clin Chem Lab Med. 2016;54:e395–e399. doi: 10.1515/cclm-2015-1281. [DOI] [PubMed] [Google Scholar]
- 12.Huang W, Lan X, Li X, Wang D, Sun Y, Wang Q, Gao H, Yu K. Long non-coding RNA PVT1 promote LPS-induced septic acute kidney injury by regulating TNFα and JNK/NF-κB pathways in HK-2 cells. Int Immunopharmacol. 2017;47:134–140. doi: 10.1016/j.intimp.2017.03.030. [DOI] [PubMed] [Google Scholar]
- 13.Lorenzen JM, Schauerte C, Kielstein JT, Hübner A, Martino F, Fiedler J, Gupta SK, Faulhaber-Walter R, Kumarswamy R, Hafer C. Circulating long noncoding RNA TapSAKI is a predictor of mortality in critically ill patients with acute kidney injury. Clin Chem. 2015;61:191–201. doi: 10.1373/clinchem.2014.230359. [DOI] [PubMed] [Google Scholar]
- 14.Wang CM, Wu QQ, Li SQ, Chen FJ, Tuo L, Xie HW, Tong YS, Ji L, Zhou GZ, Cao G. Upregulation of the long non-coding RNA PlncRNA-1 promotes esophageal squamous carcinoma cell proliferation and correlates with advanced clinical stage. Dig Dis Sci. 2014;59:591–597. doi: 10.1007/s10620-013-2956-7. [DOI] [PubMed] [Google Scholar]
- 15.Cui Z, Ren S, Lu J, Wang F, Xu W, Sun Y, Wei M, Chen J, Gao X, Xu C, Mao JH, Sun Y. The prostate cancer-up-regulated long noncoding RNA PlncRNA-1 modulates apoptosis and proliferation through reciprocal regulation of androgen receptor. Urol Oncol. 2013;31:1117–1123. doi: 10.1016/j.urolonc.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 16.Dong L, Ni J, Hu W, Yu C, Li H. Upregulation of long non-coding RNA PlncRNA-1 promotes metastasis and induces epithelial-mesenchymal transition in hepatocellular carcinoma. Cell Physiol Biochem. 2016;38:836–846. doi: 10.1159/000443038. [DOI] [PubMed] [Google Scholar]
- 17.Chen T, Xue H, Lin R, Huang Z. MiR-34c and PlncRNA1 mediated the function of intestinal epithelial barrier by regulating tight junction proteins in inflammatory bowel disease. Biochem Biophys Res Commun. 2017;486:6–13. doi: 10.1016/j.bbrc.2017.01.115. [DOI] [PubMed] [Google Scholar]
- 18.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. 2009;119:2868. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22:999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 20.Limana F, Urbanek K, Chimenti S, Quaini F, Leri A, Kajstura J, Nadal-Ginard B, Izumo S, Anversa P. bcl-2 overexpression promotes myocyte proliferation. Proc Natl Acad Sci U S A. 2002;99:6257–6262. doi: 10.1073/pnas.092672899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borkan SC. The role of BCL-2 family members in acute kidney injury. Semin Nephrol. 2016;36:237–250. doi: 10.1016/j.semnephrol.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Hassan M, Watari H, Abualmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. 2014;2014:150845–150845. doi: 10.1155/2014/150845. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Sankari SL, Masthan KM, Babu NA, Bhattacharjee T, Elumalai M. Apoptosis in cancer--an update. Asian Pac J Cancer Prev. 2012;13:4873–4878. doi: 10.7314/apjcp.2012.13.10.4873. [DOI] [PubMed] [Google Scholar]
- 24.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80:29–40. doi: 10.1038/ki.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frank AJ, Sheu CC, Zhao Y, Chen F, Su L, Gong MN, Bajwa E, Thompson BT, Christiani DC. BCL2 genetic variants are associated with acute kidney injury in septic shock. Crit Care Med. 2012;40:2116. doi: 10.1097/CCM.0b013e3182514bca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lakhani SA, Masud A, Kuida K, Porter GA, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu HF, Chen YS, Yang JS, Chen JC, Lu KW, Chiu TH, Liu KC, Yeh CC, Chen GW, Lin HJ. Gypenosides induced G0/G1 arrest via inhibition of cyclin E and induction of apoptosis via activation of caspases-3 and-9 in human lung cancer A-549 cells. In Vivo. 2008;22:215–221. [PubMed] [Google Scholar]
- 28.Comim CM, Barichello T, Grandgirard D, Dal-Pizzol F, Quevedo J, Leib SL. Caspase-3 mediates in part hippocampal apoptosis in sepsis. Mol Neurobiol. 2013;47:394–398. doi: 10.1007/s12035-012-8354-x. [DOI] [PubMed] [Google Scholar]
- 29.Oberholzer C, Tschoeke SK, Moldawer LL, Oberholzer A. Local thymic caspase-9 inhibition improves survival during polymicrobial sepsis in mice. J Mol Med. 2006;84:389–395. doi: 10.1007/s00109-005-0017-1. [DOI] [PubMed] [Google Scholar]
- 30.Luo M, Zhao X, Song Y, Cheng H, Zhou R. Nuclear autophagy: an evolutionarily conserved mechanism of nuclear degradation in the cytoplasm. Autophagy. 2016;12:1973–1983. doi: 10.1080/15548627.2016.1217381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livingston MJ, Dong Z. Autophagy in acute kidney injury. Semin Nephrol. 2014;34:17–26. doi: 10.1016/j.semnephrol.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Lee S, Lee SJ, Coronata AA, Fredenburgh LE, Su WC, Perrella MA, Nakahira K, Ryter SW, Choi AM. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal. 2014;20:432–442. doi: 10.1089/ars.2013.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa S, Nishihara K, Inui KI, Masuda S. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur J Pharmacol. 2012;696:143–154. doi: 10.1016/j.ejphar.2012.09.010. [DOI] [PubMed] [Google Scholar]