

Abstract
Abdominal aortic aneurysms (AAAs) are a chronic inflammatory vascular disease for which pharmacological treatments are not available. Gadd153 is closely associated with the onset of vascular smooth muscle cells (VSMCs) apoptosis. However, a role for Gadd153 in AngII-induced AAA formation is currently unknown. In our study, lentiviral-mediated silencing of Gadd153 through small RNA interference was performed in mice, which was further used for the establishment of mouse experimental AAA induced by infusion of angiotensin II (AngII). We found that Gadd153 deficiency prevented AngII-induced AAA formation in mice 14 days post perfusion compared with wild-type control mice. Moreover, Gadd153 deficiency significantly reduced lesion macrophage and CD4+ T-cell content, T-cell proliferation, SMC apoptosis, and matrix metalloproteinase expression. In vitro studies revealed that Gadd153 deficiency regulated microvessel growth and monocyte migration. In addition, Gadd153 deficiency also affected AAA lesion Mac-3 macrophage accumulation or CD31 microvessel numbers. In conclusion, our study demonstrates that Gadd153 plays an essential role in AngII-induced AAA formation by promoting inflammatory cells proliferation and vascular SMC apoptosis affecting MMPs expression.
Keywords: Gadd153, angiotensin II, abdominal aortic aneurysms, matrix metalloproteinases
Introduction
Abdominal aortic aneurysm (AAA) is one of leading causes of mortality worldwide, which might be associated with male sex, advanced age, hypertension, hypercholesterolemia, coronary artery disease, atherosclerosis, and cigarette smoking [1-3]. Considered that surgical treatment is associated with high postoperative mortality (up to 6%) [4], further epidemiologic and pathologic studies are urgently needed to reveal the causes of AAA development, progression, and ultimate rupture.
Although the causes of AAA are not completely understood, it is widely known that pathologic features of aneurysm contribute to its pathogenesis, including local inflammation, increased oxidative stress, significant matrix degradation and smooth muscle cell apoptosis [4,5]. Apoptosis of VSMCs is significantly increased, accompanied by reduced VSMC density within the medial layer of aneurysmal aortic tissue. The increased degree of VSMC apoptosis has already been widely found in AAA, which might be closely triggered by local inflammation and increased oxidative stress [6,7]. Structural degeneration of aortic tissue at the cellular level contributes to aneurysmal formation. For example, overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms [8]. To explore the underlying pathways involved in AAA, many studies were widely performed. For example, a recent study has demonstrated store operated calcium entry (SOCE) can modulate cell behavior through Orai1 in AAA VSMC apoptosis [9]. These results strongly suggest a better understanding of the mechanisms involved in AAA may identify new targets that could be manipulated pharmacologically or biologically to halt disease progression.
The growth arrest and DNA damage inducible gene 153 (GADD153) encodes Gadd153 protein (also called CHOP-10), which belongs to a member of the CCAAT/enhancer-binding protein (C/EBP) family of transcriptional factors [10]. The expression levels of Gadd153 are very low in normal growing cells and are highly induced in response to a variety of cellular stresses, including glucose deprivation, exposure to alkylating agents, oxidative stress, and other growth-arresting situations [11-13]. Furthermore, microinjection of Gadd153 into NIH-3T3 fibroblasts induces G1 arrest [14], and the transient expression of Gadd153 into different tumor cell lines also leads to growth arrest and apoptosis [15]. Thus, a number studies have proved that Gadd153 is closely associated with the onset of vascular smooth muscle cells (VSMCs) apoptosis, regulated by local inflammation and oxidative stress [16], indicating a potential role of Gadd153 in AAA pathogenesis. But to date no study has addressed the relationship between expression of Gadd153 and AAA progression. Therefore, we hypothesized that altering expression of Gadd153 participates directly in AAA pathogenesis and examined our hypothesis by using Gadd153-deficient mice induced by lentiviral-mediated silencing of SOD1 through small RNA interference and angiotensin II-induced experimental AAA models, therebypotentially offer a novel molecular therapy for AAA treatment.
Materials and methods
Lentiviral vector production
The full length of human H1-RNA promoter was cloned into pBluescript SK (+) by using the primers (forward) and CATACAGAGCGACAATCTTACTTGAGACTATGTCTT (reverse). The design of the reverse primer incorporates BglII and HindIII sites juxtaposed on the transcriptional start site into which DNA sequences containing siRNA hairpins can be cloned [17]. siRNA oligonucleotides were designed that contained a sense strand of 22 (Gadd153) nucleotide sequences followed by a short spacer (TTCAAGAGA), the reverse complement of the sense strand, and five thymidines as an RNA polymerase III transcriptional stop signal. Oligos were annealed and cloned into the BglII-HindIII site. For cloning into lentivectors, the complete human H1-RNA promoter plus the siRNA cassette was PCR-amplified by introducing XbaI sites both upstream and downstream of the sequence and cloned into a unique NheI site of the 3’LTR of a lentiviral vector containing cytomegalovirus (CMV)-Gadd153. This CMV-Gadd153 cassette was deleted to generate LV-siGadd153.
293T cells were cotransfected with a plasmid expressing Gadd153 together with a plasmid expressing siRNA specific for Gadd153 (siGadd153). Recombinant lentiviruses were produced by transient transfection in 293T cells using the calcium-phosphate method as described [18,19]. Infectious lentiviruses were harvested at 48 and 72 h post transfection and filtered through 0.22-μm-pore cellulose acetate filters. Recombinant lentiviruses were concentrated by ultracentrifugation (2 h at 50,000 × g) and subsequently purified on a sucrose 20% gradient (2 h at 46,000 × g) as described [18,19].
Animal surgery
All surgical procedures were performed as previously described [20]. Briefly, we first anesthetized 40-d-old mice with 4% isoflurane and maintained them on 1.5% isoflurane anesthesia. Following laminectomy, lentiviral vectors were bilaterally injected at two sites separated by 2 mm in the lumbar L3-L4 region using a stereotaxic frame. Using a 5-µl Hamilton syringe with a 34-gauge needle, we injected 1.5 µl of concentrated viral solutions (60,000 ng of p24 antigen/ml) per site (0.75 mm below dura) with a rate of 0.5 µl/min. The needle was then left in place for an additional 5 min and gently withdrawn. After surgery, we injected animals subcutaneously with a single dose of carprofen (5 mg/kg) to limit inflammatory reaction resulting from the surgery.
PCR detection
Viral and siRNA integration were detected by PCR analysis. Fifty to 100 ng of DNA were used in a 25-µl reaction. Primers spanning the H1-siGadd153 cassette were U3 forward (5’-GGGCAGCTGTTCCAGACAACTTA-3’) and U3 reverse (5’-GCTTGTCTTTTGCGTGATGGGA-3’). U3-H1 primers, which amplify the H1 portion of the H1-siRNA cassette, were U3 forward in combination with H1 promoter internal primer H1 reverse 5’-CGTACGGGCCCGTGGTCTCATACAGAACTT-3’. The PCR conditions were 94°C denaturation for 3 min followed by 40 cycles of 94°C for 30 sec, 55°C for 40 sec, and 72°C for 50 sec. The Gadd153 primers were Gadd153 forward (5’-CCGAAGTTCATCCACTGCA-3’) and Gadd153 reverse (5’-GCGTCCTTGAAGGGTAAGAT-3’).
Mouse AAA model and lesion characterization
Wild-type (Gadd153+/+) and lentiviral-mediated Gadd153-deficient (Gadd153-/-) male mice at 20 weeks of age were infused with angiotensin II (Ang II; 0.8 mg/kg per day, up to 14 days) to produce experimental AAA mouse models, as previously described [21]. The aortic tissues were harvested from 10 mice each group at 7 and 14 days postperfusion. The presence of an AAA and the scoring of AAA pathology were determined using a classification scheme described previously [21]. Two researchers measured aortic diameters independently. On determination of the AAA incidence and classification, another investigator matched the scored AAAs to the genotypes of the mice. Studies were performed by the approval of the Animal Care and Use Committees in The First Affiliated Hospital of Harbin Medical University.
SMC apoptosis
SMC apoptosis was performed using primary cultured Gadd153-/- and wild type mouse aortic SMCs on an 8-well chamber slide. The apoptosis of confluent SMCs was induced by treatment of 60 mol/L pyrrolidine dithiocarbamate for 24 h. Apoptotic cells were detected with In Situ Cell Death Detection Kit according to the manufacturer’s instructions.
Mean blood pressure measurements
Mean systolic blood pressures were measured in conscious mice using a computerized tail-cuff method (BP-2000 Visitech Systems). Mice were acclimatized to the system for 1 week prior to the initiation of studies and systolic blood pressure was measured 5 days per week until study termination.
Cell proliferation assay and transmigration assay
CD4+T cells were purified from splenocytes by depleting major histocompatibility complex class II-positive cells and CD8+T cells using anti-mouse I-Ab and CD8 monoclonal antibodies (BD Biosciences), followed by complement depletion, as described previously [22]. Monocytes were isolated from peripheral blood by Percoll (Sigma, CA, USA) gradient centrifugation. T-cell and monocyte proliferation were assessed with the Cell Titer 96AQ Assay kit, according to the manufacturer’s instructions. T-cell and monocyte transmigration assay was performed on a type I collagen (100 ng/25 µl per well in a pH7.0 HEPES buffer)-precoated 96-well chemotaxis plate, according to the manufacturer’s instructions.
Aortic ring assay
A 96-well plate was coated with 50 µl of Matrigel. A 1-mm long mouse aortic ring from WT mouse or Gadd153-/-mice mouse was laid on top of the solidified Matrigel and covered with 100 µl of Matrigel. After solidification at room temperature, 150 µl of RPMI (with 10% FBS) was added to each well. After 7 to 10 days of culture, the aortas were photographed, and the endothelial outgrowth was analyzed using ImagePro Plus software and presented as square millimeters. Basic fibroblast growth factor (bFGF) (10 ng/ml) was used as a positive control.
Histology and immunohistochemistry
Mice were euthanized at the designated time points (7 or 14 days), and abdominal aortic sections were stained with hematoxylin and eosin or Verhoeff-Van Geison (elastin) stain for histological analysis or with antibodies against macrophage marker protein Mac-3 or CD4+ for immunohistochemical analysis. Quantitation of immunohistochemistry was performed by determining the ratio of the number of Mac-3 or CD4+ positive cells to the total number of hematoxylin-positive nuclei per field (at × 400 magnification).
Quantitation of mRNA expression
Real-time quantitative polymerase chain reaction analysis for COX-2, CD68, cathepsins B and cathepsins K, MMP2, and MMP9 was performed using TaqMan gene expression assays and was analyzed by the ÄÄCt method with GADPH as the endogenous control.
Statistical analyses
For comparing two groups on a continuous response variable, a two-sample Student’s t-test was used after verifying that data met constraints of normality and equivalence of variance to permit parametric analysis. A one-way ANOVA followed by Tukey’s multiple comparison test was used to compare more than two groups on a continuous response variable. Percent incidence of AAAs was analyzed by Fisher’s exact test. P values < 0.05 were considered to be statistically significant. All data are represented as mean ± SEM.
Results
Gadd153 deficiency attenuates the incidence and severity of Ang II-induced AAAs
We first determined the effect of Gadd153 deficiency on AAA development in mice treated with lentiviral-mediated silencing of Gadd153 through small RNA interference (Gadd153-/-mice). At the 7-day time point, no significant differences were observed in aortic diameters between Gadd153-/-mice and wild type (WT) mice before and immediately after Ang II infusion. At 7 days postperfusion, Gadd153-/-mice and WT mice also showed no significant differences in aortic diameters, and none of these mice developed AAA, as defined by a 100% increase in aortic diameter (Figure 1A). In Gadd153-/-mice used for the 14-day time point, both preperfusion and immediate postperfusionaortic diameters were larger than those from WT mice. At 14 days postperfusion, however, aortic diameters from WT mice were not significantly different from those immediately postperfusionbut were significantly larger than those from Gadd153-/-mice (P < 001) (Figure 1B). In addition, at 14-day time point, all WT mice (10 of 10) developed AAA, but not all Gadd153-/-mice (3 of 10) formed AAA, indicating that Gadd153 plays an essential role in AAA formation.
Figure 1.
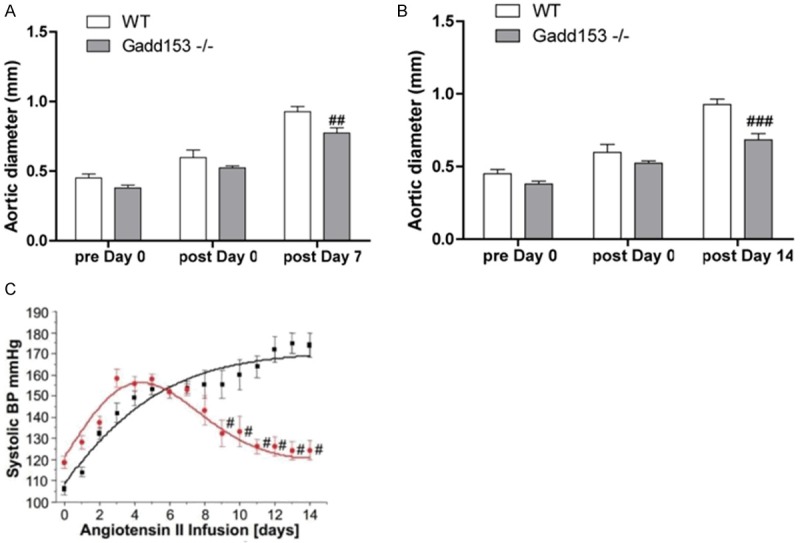
Reduced abdominal aortic aneurysm (AAA) formation in Gadd153-/-mice. A, B. Aortic diameters in both Gadd153-/-mice and WT mice were measured preperfusion, immediately postperfusion, and at 7 and 14 days post perfusion. C. Changes in mean blood pressure (MBP) in AngII-infused Gadd153-/-mice (Red) and WT mice (WT). The MBP was monitored by an intracarotid-telemetry method continuously for 14 days. Data are mean ± SEM. P < 0.05 is considered statistically significant, #indicates compared to WT mice.
The MBP was measured by an intracarotid telemetry method. We found that MBP of Gadd153-/-mice was slightly higher than that of WT mice at baseline (i.e., day 0 Ang II infusion; Figure 1C). Moreover, MBP was increased in both groups up to day 14 during Ang II infusion (WT: 109-147 mmHg; Gadd153-/-: 118-150 mmHg; Figure 1C). Thereafter, MBP of Gadd153-/-mice was reduced due to Gadd153 deficiency compared with continued increasing MBP in WT mice (Figure 1C).
Effects of Gadd153 deficiency on inflammatory cells in AAA lesions
Inflammatory cell infiltration is a crucial event contributing to AAA development. As showed in Figure 2A, a significantly higher levels of lesion macrophage contents was found in WT mice than Gadd153-/-mice at both 7-day and 14-day time point (Figure 2A). Moreover, we showed that AAA lesions from WT mice contained more higher numbers of CD4+ T cells than that of Gadd153-/-AAA lesions at 14 days postperfusion (Figure 2B), suggesting that Gadd153 activity might be involved in the migration or proliferation of macrophage and T-cell.
Figure 2.
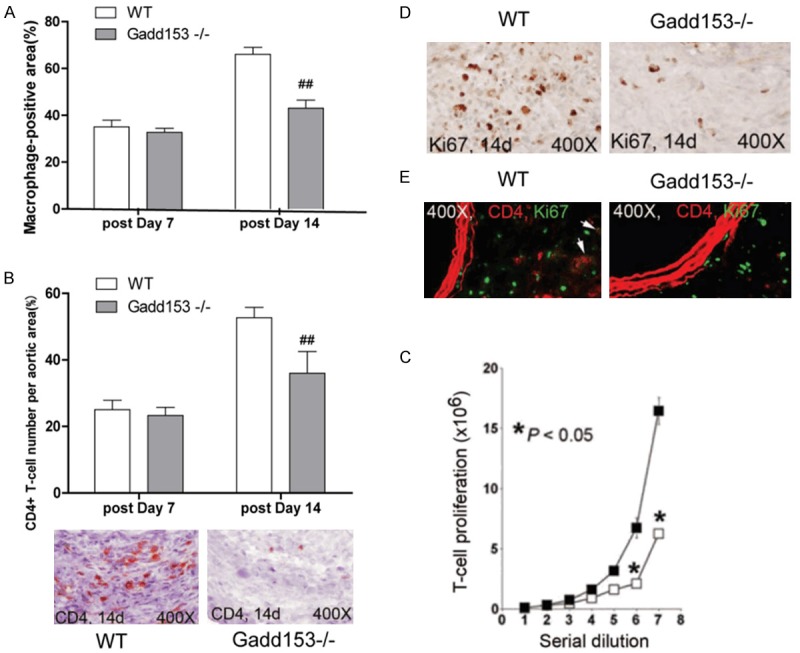
Gadd153 activities on inflammatory cells proliferation. Deficiency of Gadd153 reduced abdominal aortic aneurysm (AAA) lesion Mac-3+ macrophage-positive area (A) and CD4+ T-cell numbers (B) in the adventitia at 14 days post perfusion. In vitro cell proliferation assay showed that Gadd153 deficiency impaired CD4+ T-cell proliferation (C). In AAA lesion adventitia, numbers of Ki67-positive cells (D) and percentage of CD4+ T-cells among Ki67-positive cells (E; arrows indicate Ki67+; CD4+ T cells) were also reduced in Gadd153-/-mice at 14 days post perfusion. The number of mice per group is indicated in each bar. All data are mean ± SE. P < 0.05 is considered statistically significant; Mann-Whitney U test.
To test these possibilities, we performed Boyden chamber cell transmigration assay by using stromal cell-derived factor-1 as a chemoattractant. As showed in Figure 2C, both macrophage and CD4+ T cells from WT mice proliferated much faster than those Gadd153-/-mice. Furthermore, the number of proliferating Ki67+ cells were significantly reduced in Gadd153-/-mice, compared with those in WT mice at 14 days postperfusion (Figure 2D). To further determine the number of CD4+ T and Ki67+ cells directly, we performed the co-immunostaining by using anti-CD4 and anti-Ki67 monoclonal antibody in AAA lesions. Among all Ki67+ proliferating cells, the percentages of CD4+T cells were significantly lower in Gadd153-/-mice than WT mice (Figure 2E), confirming a crucial of Gadd153 in macrophage and T-cell proliferation in vivo.
Effects of Gadd153 deficiency on Ang II-induced vascular cell apoptosis in AAA lesions
Apoptosis of SMC is a key factor that contributes tunica media thinning in AAA lesions, whereas infiltrating leukocytes release apoptotic stimuli to promote vascular cell apoptosis[23]. The deficiency of Gadd153 inhibited Ang II-induced cell apoptosis significantly AAA lesion. As showed in Figure 3A, the number of terminal deoxynucleotidyl transferase UTP nick-end labeling (TUNEL)-positive cells were reduced in whole AAA lesions and in the media (Figure 3B, mainly SMCs) from Gadd153-/-mice compared with those in WT mice at 14 days postperfusion. Consistent with this observation, medial SMC loss in AAA lesions from Gadd153-/-mice also was significantly impaired at this time point (Figure 3C), although both lesion cell apoptosis and medial SMC loss were not significantly different between both groups at the 7-day time point (Figure 3A and 3C). To examine the contribution of Gadd153 to SMC apoptosis, we induced SMC apoptosis with pyrrolidine dithiocarbamate. The SMCs from WT mice was stimulated to apoptosis by treatment with pyrrolidine dithiocarbamate, which was on the contrast to SMC from Gadd153-/-mice, suggesting a protective role of Gadd153 in SMCs (Figure 3D).
Figure 3.
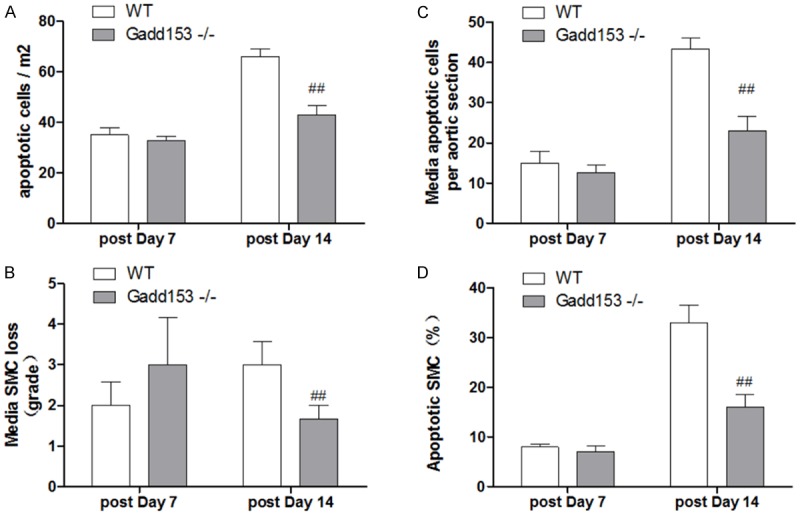
Gadd153 activities on lesion cell apoptosis. Abdominal aortic aneurysm (AAA) lesion adventitia (A) and medial (B) terminal deoxynucleotidyl transferased UTP nick-end labeling (TUNEL)-positive cell numbers were significantly lower in Gadd153-/-mice than in WT mice at 14 days post perfusion. At the same time point, lesion medial smooth muscle cell (SMC) loss was reduced in Gadd153-/-mice (C). The number of mice per group is indicated in each bar. Aortic SMCs from Gadd153-/-mice were resistant to pyrrolidine dithiocarbamate (PDTC)-induced apoptosis (D).
Effects of Gadd153 deficiency on Ang II-induced angiogenesis in AAA lesions
To investigate the role of Gadd153in angiogenesis, AAA lesions were collected from WT mice and Gadd153-/-mice. We found that numbers of CD31+ microvessels in Gadd153-/-mice was reduced by Gadd153 deficiency at 14 days postperfusion and of proangiogenic laminin-5 γ2+ vessels at both 7 days and 14 days postperfusion, compared with WT mice (Figure 4A, 4B). In vitro aortic ring angiogenesis assay showed defects of microvessels sprouting from aortic rings from Gadd153-/-mice with or without angiogenic factor bFGF (Figure 4C). To assess whether the absence of Gadd153 affected the expression or activities of other proteases, we performed RT-PCR in MHEC from Gadd153-/-mice and Gadd153+/+mice demonstrated that the absence of Gadd153 reduced cathepsin S, cathepsin B and cathepsin K mRNA levels in MHEC (Figure 4D), suggesting that reduced angiogenesis in Gadd153-/-mice was caused in part by reduced EC protease expression and activities.
Figure 4.
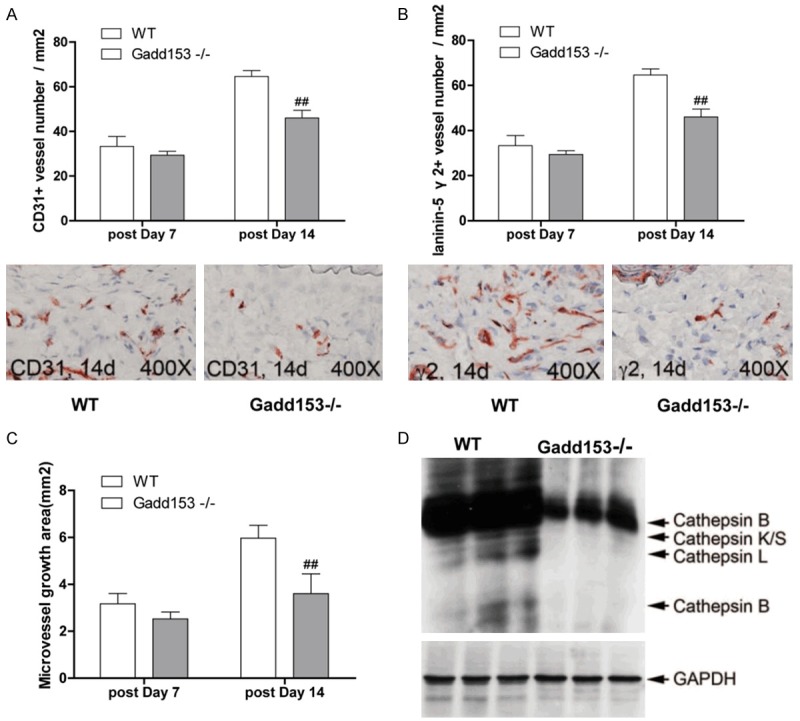
Gadd153 function in neovascularization in abdominal aortic aneurysms (AAA) lesions. CD31+ (A) and proangiogenic laminin-5 fragment γ2+ (B) microvessel numbers were reduced in AAA lesions from Gadd153-/-mice. The number of mice per group is indicated in each bar. Both measurements are from the entire lesion including adventitia and media. Aortic ring assay in vitro demonstrated impaired microvessel sprouting from Gadd153-/-mouse aortic rings with or without angiogenic factor bFGF (C). RT-PCR showed reduced mRNA levels of cathepsins S, B and K in microvessel endothelial cells (MHEC) from Gadd153-/-mice compared with those from WT mice (D).
Effects of Gadd153 deficiency on Ang II-induced MMP2 and MMP9 expression in AAA pathogenesis
MMPs have been known to be mechanistically implicated in the pathogenesis of AAAs, and MMP2 and MMP9, in particular, are identified a concerted role in AAA initiation and progression [24]. As shown in Figure 5A and 5B, Ang II treatment significantly increased MMP2 mRNA expression at 14-day in both group of mice. However, in Gadd153-/-mice, the increase in MMP2 expression was significantly lower when analyzed after Ang II infusion compared with WT mice (Figure 5A). However, after 14 days of Ang II infusion, MMP9 expression was significantly increased in WT mice, which was reversed by Gadd153 deficiency (Figure 5A). Increased MMP expression has been associated with the proteolytic degradation and elastin breakage that occur during aneurysmal expansion. Verhoeff-van Gieson staining of abdominal aortic segments from Gadd153-/-mice after 14 days of Ang II infusion showed regions of breakage and discontinuity of the medial elastin layer (Figure 5B).
Figure 5.
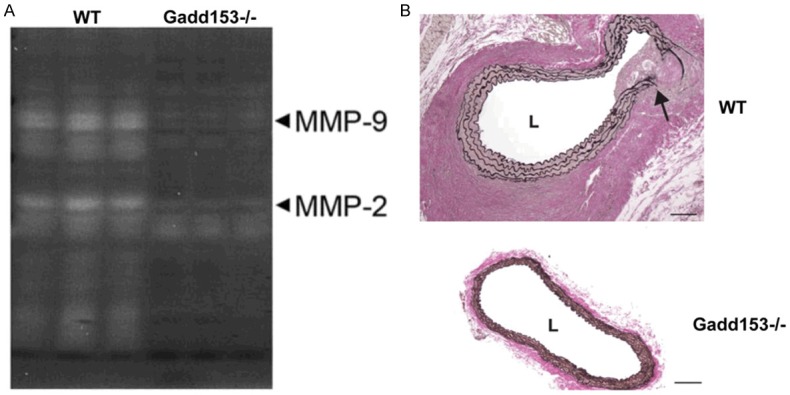
Matrix metalloproteinase (MMP) expression is attenuated in the aortas of Gadd153-/-mice. Quantitative polymerase chain reaction (PCR) analysis of MMP2 mRNA expression in the abdominal aortas of Gadd153-deficient mice at A. 14 days after angiotensin II (AngII) infusion. B. Representative images of Verhoeff-van Geison-stained abdominal aortic sections from Ang II-treated Gadd153-/-mice. Arrow indicates breakage in the elastin layer.
Discussion
The pathological mechanism involving in the development and progression of aneurysm is still unclear. Thus, identification of potential causes and medical treatment is a major challenge in modern vascular medicine. Our study identified the essential role of Gadd153 in Ang II-induced AAA development and we provided first evidence that Gadd153 contributed to AAA formation by regulating monocyte and T-cell recruitment, lesion cell proliferation, angiogenesis and MMP expression, suggesting that Gadd153 deficiency exerts protective roles in Ang II-induced experimental AAA mice models.
A number of studies have already proved that Ang II infusion results in the formation of AAAs in mice [25,26], due to its multiple bioactive effects, including medial degeneration, inflammation, thrombus formation, and rupture of the abdominal aorta, therefore acting as a critical factor in Ang II-induced AAA development [26]. In addition, our studies also showed that Ang II infusion caused an abnormal rapid rise in blood pressure in the Gadd153-/-mice and WT mice. However, blood pressure of Gadd153-/-mice began to progressively fall after day 7 and 14 of Ang II infusion. In contrast, blood pressure of WT mice increased continuously throughout the course of Ang II infusion, indicating a protective role of Gadd153 in Ang II infusion-induced AAA development.
The inflammatory cell population in AAA lesions is another crucial factor that contributes to Ang II-induced AAA mice models [27]. After treated with Ang II up to 14 days, macrophage content and T-cell number in lesions from Gadd153-/-mice, were both significantly reduced compared with WT mice. Moreover, the transmigration of monocytes or T cells from Gadd153-/-mice was higher than that of WT mice, which may explain reduced T cells and macrophages in AAA lesions from Gadd153-/-mice, although we found that the deficiency of Gadd153 has inhibited Ang II-induced monocytes and T-cell proliferation. Here, we demonstrated that Gadd153 contributed to AAA lesion inflammatory cell accumulation.
Next, we assessed the effects of Gadd153 in PDTC-induced SMC apoptosis. The apoptosis of aortic SMC in vitro from Gadd153-/-mice was markedly reduced compared with cells from WT mice, similar results of trend was found AAA lesion cell apoptosis measured by TUNEL, indicating that Gadd153 is essential to SMC apoptosis and to AAA lesion cell apoptosis, which is consistent with previous reports of Gadd153-induced SMC apoptosis [28]. For example, a number studies have proved that Gadd153 is closely associated with the onset of vascular smooth muscle cells (VSMCs) apoptosis, regulated by local inflammation in AAA pathogenesis [16]. Next, in vitro studies of aortic ring assay and immunostaining of AAA lesion sections have revealed that Gadd153 deficiency resulted in the reduction of microvessel or CD31+ microvessel numbers in AAA lesions from Gadd153-/-mice. Consistently, the number of CD31+ or laminin-5 γ2+ microvessel was significantly fewer in Gadd153-/-mice AAA lesions than in WT mouse AAA lesions, suggesting the effects of Gadd153 in angiogenesis. Finally, we investigated how Gadd153 regulated AAA expansion. Considered that multiple proteases in AAA lesions are responsible for the extracellular elastin degradation [29], we examined the expression of cysteinyl cathepsins and matrix metallopreteinases. As a result, cathepsins K, S and L, MMP2 and 9 were significant by absence of Gadd153. A mechanism research has revealed that CatL and other cathepsins (e. g., CatS and CatK.) formed sequestered and acidic environments into in cultured human monocyte derived macrophages with water-insoluble elastin fibers [30,31]. Thus, we demonstrated that Gadd153 deficiency degraded extracellular elastin in the aortic wall and led to AAA expansion by down-regulation of most cathepsins and MMPs expressions in MHEC.
Taken together, our data from experimental AAA models of Gadd153-/-mice reveals the essential role of Gadd153 in AAA formation and development. Thus, pharmacological inhibitors for selective inhibition of Gadd153 might benefit patients with AAA, atherosclerosis, and other cardiovascular diseases. Therefore, Gadd153 will be of an interesting target in the future studies of cardiovascular diseases.
Disclosure of conflict of interest
None.
References
- 1.Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8:92–102. doi: 10.1038/nrcardio.2010.180. [DOI] [PubMed] [Google Scholar]
- 2.Blanchard JF, Armenian HK, Friesen PP. Risk factors for abdominal aortic aneurysm: results of a case-control study. Am J Epidemiol. 2000;151:575–583. doi: 10.1093/oxfordjournals.aje.a010245. [DOI] [PubMed] [Google Scholar]
- 3.Lederle FA, Nelson DB, Joseph AM. Smokers’ relative risk for aortic aneurysm compared with other smoking-related diseases: a systematic review. J Vasc Surg. 2003;38:329–334. doi: 10.1016/s0741-5214(03)00136-8. [DOI] [PubMed] [Google Scholar]
- 4.Poldermans D, Bax JJ, Kertai MD, Krenning B, Westerhout CM, Schinkel AF, Thomson IR, Lansberg PJ, Fleisher LA, Klein J, van Urk H, Roelandt JR, Boersma E. Statins are associated with a reduced incidence of perioperative mortality in patients undergoing major noncardiac vascular surgery. Circulation. 2003;107:1848–1851. doi: 10.1161/01.CIR.0000066286.15621.98. [DOI] [PubMed] [Google Scholar]
- 5.Rowe VL, Stevens SL, Reddick TT, Freeman MB, Donnell R, Carroll RC, Goldman MH. Vascular smooth muscle cell apoptosis in aneurysmal, occlusive, and normal human aortas. J Vasc Surg. 2000;31:567–576. [PubMed] [Google Scholar]
- 6.Lopez-Candales A, Holmes DR, Liao S, Scott MJ, Wickline SA, Thompson RW. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol. 1997;150:993–1007. [PMC free article] [PubMed] [Google Scholar]
- 7.Henderson EL, Geng YJ, Sukhova GK, Whittemore AD, Knox J, Libby P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99:96–104. doi: 10.1161/01.cir.99.1.96. [DOI] [PubMed] [Google Scholar]
- 8.Parastatidis I, Weiss D, Joseph G, Taylor WR. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2013;33:2389–2396. doi: 10.1161/ATVBAHA.113.302175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey M, Young R, Rode B, Foster R, Li J, Beech D. Significance of store operated calcium entry in human abdominal aortic aneurysm vascular smooth muscle cells (1057.3) FASEB J. 2014;28:1057–1053. [Google Scholar]
- 10.Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5:1538–1552. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- 11.Mohan C, Sathyamurthy M, Lee GM. A role of GADD153 in ER stress-induced apoptosis in recombinant Chinese hamster ovary cells. Biotechnol Bioprocess Eng. 2012;17:446–455. [Google Scholar]
- 12.Weng TI, Wu HY, Chen BL, Jhuang JY, Huang KH, Chiang CK, Liu SH. C/EBP homologous protein deficiency aggravates acute pancreatitis and associated lung injury. World J Gastroenterol. 2013;19:7097–7105. doi: 10.3748/wjg.v19.i41.7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guyton KZ, Xu Q, Holbrook NJ. Induction of the mammalian stress response gene GADD153 by oxidative stress: role of AP-1 element. Biochem J. 1996;314:547–554. doi: 10.1042/bj3140547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oyadomari S, Mori M. Roles of CHOPGADD153 in endoplasmic reticulum stress. Cell Death Differ. 2003;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 15.Maytin EV, Ubeda M, Lin JC, Habener JF. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp Cell Res. 2001;267:193–204. doi: 10.1006/excr.2001.5248. [DOI] [PubMed] [Google Scholar]
- 16.Tang JR, Nakamura M, Okura T, Takata Y, Watanabe S, Yang ZH, Liu J, Kitami Y, Hiwada K. Mechanism of oxidative stress-induced GADD153 gene expression in vascular smooth muscle cells. Biochem Biophys Res Commun. 2002;290:1255–1259. doi: 10.1006/bbrc.2002.6336. [DOI] [PubMed] [Google Scholar]
- 17.Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, Szulc J, Henderson CE, Aebischer P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 18.Cockrell AS, Kafri T. Gene delivery by lentivirus vectors. Mol Biotechnol. 2007;36:184–204. doi: 10.1007/s12033-007-0010-8. [DOI] [PubMed] [Google Scholar]
- 19.Dykxhoorn DM, Novina CD, Sharp PA. Killing the messenger: short RNAs that silence gene expression. Nat Rev Mol Cell Biol. 2003;4:457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 20.Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, Lee DC, Wong LF, Bilsland LG, Greensmith L, Kingsman SM, Mitrophanous KA, Mazarakis ND, Azzouz M. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- 21.Gitlin JM, Trivedi DB, Langenbach R, Loftin CD. Genetic deficiency of cyclooxygenase-2 attenuates abdominal aortic aneurysm formation in mice. Cardiovasc Res. 2007;73:227–236. doi: 10.1016/j.cardiores.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 22.Shi GP, Villadangos JA, Dranoff G, Small C, Gu L, Haley KJ, Riese R, Ploegh HL, Chapman HA. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity. 1999;10:197–206. doi: 10.1016/s1074-7613(00)80020-5. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Bockler D, Ryschich E, Klemm K, Schumacher H, Schmidt J, Allenberg JR. Impaired Fas-induced apoptosis of T lymphocytes in patients with abdominal aortic aneurysms. J Vasc Surg. 2007;45:1039–1046. doi: 10.1016/j.jvs.2006.12.055. [DOI] [PubMed] [Google Scholar]
- 24.Wang YX, Martin-McNulty B, da Cunha V, Vincelette J, Lu X, Feng Q, Halks-Miller M, Mahmoudi M, Schroeder M, Subramanyam B, Tseng JL, Deng GD, Schirm S, Johns A, Kauser K, Dole WP, Light DR. Fasudil, a Rho-kinase inhibitor, attenuates angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice by inhibiting apoptosis and proteolysis. Circulation. 2005;111:2219–2226. doi: 10.1161/01.CIR.0000163544.17221.BE. [DOI] [PubMed] [Google Scholar]
- 25.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler. Thromb Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 27.Tang EH, Shvartz E, Shimizu K, Rocha VZ, Zheng C, Fukuda D, Shi GP, Sukhova G, Libby P. Deletion of EP4 on bone marrow-derived cells enhances inflammation and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler. Thromb Vasc Biol. 2011;31:261–269. doi: 10.1161/ATVBAHA.110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng WP, Hung HF, Wang BW, Shyu KG. The molecular regulation of GADD153 in apoptosis of cultured vascular smooth muscle cells by cyclic mechanical stretch. Cardiovasc Res. 2008;77:551–559. doi: 10.1093/cvr/cvm057. [DOI] [PubMed] [Google Scholar]
- 29.Sun J, Zhang J, Lindholt JS, Sukhova GK, Liu J, He A, Abrink M, Pejler G, Stevens RL, Thompson RW, Ennis TL, Gurish MF, Libby P, Shi GP. Critical role of mast cell chymase in mouse abdominal aortic aneurysm formation. Circulation. 2009;120:973–982. doi: 10.1161/CIRCULATIONAHA.109.849679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy VY, Zhang QY, Weiss SJ. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte-derived macrophages. Proc Natl Acad Sci U S A. 1995;92:3849–3853. doi: 10.1073/pnas.92.9.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Punturieri A, Filippov S, Allen E, Caras I, Murray R, Reddy V, Weiss SJ. Regulation of elastinolytic cysteine proteinase activity in normal and cathepsin K-deficient human macrophages. J Exp Med. 2000;192:789–799. doi: 10.1084/jem.192.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]