

Abstract
Glucocorticoids have been shown to induce apoptosis in different cell types. Recent studies have indicated that apoptosis may not be the only form of death that is activated in osteoblasts in response to glucocorticoids. The aim of this study was to investigate whether necrostatin-1 could protect osteoblasts from glucocorticoid-induced cell death. Dexamethasone could induce both apoptotic and necrotic cell death in MC3T3-E1 cells, in a dose- and time-dependent manner. Necrotic cell death was induced by dexamethasone in MC3T3-E1 cells and was characterized by caspase independence, delayed externalization of phosphatidylserine, cellular swelling and plasma membrane disruption. Blockages of necroptotic induction by a special inhibitor (Necrostatin-1) succeed to protect cells against dexamethasone induced cell death. The levels of RIP-1 production and loss of mitochondrial membrane potential were also determined to assess the effects of dexamethasone. This study showed, for the first time, that high-doses of dexamethasone can induce necrotic-like cell death in osteoblastic MC3T3-E1 cells, and this induction could be inhibited by necrostatin-1.
Keywords: Glucocorticoids, dexamethasone, necroptosis, apoptosis, necrostatin-1, MC3T3-E1
Introduction
Glucocorticoids (GCs) have been used as chemotherapeutic and anti-inflammatory agents for many years. However, the long-term administration of GCs can induce skeletal complications such as osteoporosis and hip fracture [1]. GCs exert their injurious effects, to a great extent, by affecting the proliferation and function of bone cells, including osteoblasts, osteoclasts and osteocytes. All these effects result in the disequilibrium between bone formation and absorption. It has been shown that GCs can induce apoptosis in osteoblasts and osteocytes, which could be an important factor in the complications of prolonged GC treatment, as mentioned above. Prednisone treatment significantly increases the rate of apoptosis in both osteoblasts and osteocytes in adult mice [2]. Additionally, corticosterone may induce apoptosis in rat and mouse osteoblasts by decreasing the Bcl2/Bax ratio [3].
Although several studies have focused on the apoptosis of osteoblasts and osteocytes caused by GCs, neither the changes in bone metabolism and bone architecture nor the modest amount of apoptosis in osteoblasts and osteocytes can explain the elevated bone fragility that has been observed in GC-treated patients [4]. Additionally, a previous study showed that less than 4% of the total osteocytes were apoptotic in a prednisolone-treated mouse model [5], and treatment with different concentrations of dexamethasone has been shown to suppress apoptosis in osteoblasts [6]. These results suggest that GCs must have another effect, in addition to apoptosis, on bone cells.
Degterev et al. reported a new basic cell death pathway termed ‘necroptosis’, this pathway shares many characteristics with both necrosis and apoptosis [7]. Necroptotic cells are morphologically similar to necrotic cells and exhibit characteristics such as nuclear condensation, organelle swelling and loss of plasma membrane integrity; additionally, necroptosis is caspase-independent and can be triggered by death receptors. Receptor Interacting Protein 1 (RIP-1) and Receptor- Interacting Protein 3 (RIP-3) are activated to form the necrosome complex, and necroptosis results in elevated reactive oxygen species (ROS) production and a decrease in the mitochondria membrane potential (MMP). Most importantly, necroptosis can be specifically inhibited by a small molecule, necrostatin-1 (Nec-1), which targets the death domain of RIP-1 [8]. Necroptosis has been shown to occur in many different cell lines and tissues [9-13]. We have found the number of osteoblasts was significantly increased after treated with Nec-1 in glucocorticoid-induced osteoporosis (GIOP) [14]; however, whether necroptosis is involved in the effect of GCs on osteoblasts in vitro is currently unknown.
In this study, we observed that dexamethasone treatment induced as different type of cell death in osteoblasts in a dose-dependent manner, and treatment with necrostatin-1, the specific inhibiter of necroptosis, could significantly inhibit the necrotic-like cell death induced by dexamethasone. We also showed that the mechanism of Nec-1 protection against GC-induced cell death may be mediated by caspase-independent pathways involving RIP-1 and RIP-3, ultimately resulting in a reduction of the MMP.
Materials and methods
Reagents
The mouse osteoblastic cell line MC3T3-E1 was obtained from the American Type Culture Collection (ATCC, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), ascorbic acids, non-essential amino acid, penicillin/streptomycin, fetal bovine serum (FBS), and trypsin/EDTA were purchased from Gibco (USA). Carbobenzoxy-valyl-analyl-aspartyl [O-methyl]-fluoromethylk-etone (zVAD-fmk) and necrostatin-1 (Nec-1) were obtained from Enzo (USA). Dexamethasone (Dex) and DMSO were purchased from Sigma (USA). The RIP-1 and RIP-3 antibodies were obtained from Abcam (USA), The caspase-8 activity assay kit, DCFH-DA, WST-1 assay kit, Annexin V/PI double staining kit and MMP kit were purchased from Beyotime (China). All reagents used in this study were of trace element analysis grade.
Cell lines and cell culture
MC3T3-E1 cells, which were derived from mouse calvarias were cultured in α-modified Eagle’s minimum essential medium (α-MEM) supplemented with 10% FBS and 1% penicillin/streptomycin, as previously described [15]. The cells were incubated in a humidified incubator at 37°C with 95% O2 and 5% CO2 and were maintained in a sub-confluent state unless otherwise indicated. The medium was changed twice a week, and the cells were subcultured using 0.05% trypsin with 0.01% EDTA.
Induction of cell death
Different concentrations (10-5, 10-6 and 10-7 M) of Dex were added to the cells, and the cells were incubated for the indicated time periods.
Cell viability assay
Cell viability was determined for each concentration of Dex using the WST-1 assay, as previously described [16]. Briefly, the MC3T3-E1 cells were planted at a density of 2×104 cells per well in 96-well plates. To assess the effects of Dex on cell proliferation, the cells were incubated in growth medium or in conditioned medium for 0, 12, 24, 48 or 72 h at concentrations of 10-5, 10-6 and 10-7 M. After exposure to the various concentrations of Dex for the indicated times, the cells were exposed to 10 mL of WST-1 reagent for 1 to 2 h at 37°C. The absorbance was measured at 450 nm using a microplate reader. Alternatively, the MC3T3-E1 cells were plated in a 6-well plate and incubated overnight at 37°C. After Dex treatment, the number of viable cells was counted. The cells in each sample were quantified according to the instructions.
Measurement of caspase-8 activity
Caspase-8 activity was measured using a Caspase-8/PP32 Colorimetric Assay kit according to the manufacturer’s instructions. Briefly, 1.0×106 cells were lysed, and the lysate was incubated at 37°C with 200 μmol/L DEVD-UNA substrate. The samples were read at 400 nm using a model ELX800 Micro Plate Reader and the data were expressed as the fold increase over the control DMSO-treated cells.
Annexin V/PI double staining
The cells were incubated with 10-5 M Dex for 0, 6, 12, 24, 48 or 72 h. The cells were then trypsinised and harvested by centrifugation before being incubated with Annexin V and PI for 15 min at room temperature. Necrosis was examined, and the rate of cell death was analyzed by flow cytometry using the Annexin V-FITC/PI kit. Annexin V-FITC binds to necrotic and apoptotic cells that have exposed phosphatidylserine on the cell surface, thus allowing the quantification of the percentage of necrotic cells was determined [17].
Electron microscopy
Cultured cells were washed once in PBS and fixed for 30 min in 2.5% glutaraldehyde in 0.1 M sodium-phosphate buffer (pH 7.4). The samples were treated with 1.5% osmium tetroxide, dehydrated with acetone and embedded in Durcupan resin. Thin sections were poststained with lead citrate and examined using a 7650 electron microscope (HITACHI, Japan) at 80 kV.
Experimental exposures
Once the Dex concentration that led to intermediate levels of cell death was determined, the cells were treated with either 40 μM z-VAD-fmk, a specific inhibitor of the caspases involved in classical apoptosis, or 10 μM necrostatin-1, a specific inhibitor of necroptosis, or they were pretreated with the inhibitors for 2 h prior to treatment with 10-5 M Dex. The impact of these inhibitors on cell viability and cell death-related parameters in dexamethasone exposed cells was evaluated after 72 h.
All of the drugs were diluted in medium containing 2% FBS immediately before the drug treatments, and the final concentration of DMSO was diluted to 0.1% or less to eliminate the influence of the DMSO [18]. No significant effects on thymidine incorporation or LDH leakage were observed in the medium containing 0.1% DMSO. Therefore, the control cultures were cultivated in medium without drugs or DMSO.
Production of reactive oxygen species (ROS)
Reactive oxygen species production was detected using H2DCFDA, as previously described [19]. MC3T3-E1 cells were treated with 10 μmol/L Nec-1 or 10 μmol/L z-VAD-fmk for 72 h. Subsequently, 2 μmol/L H2DCF-DA was added to the cells during dissociation with trypsin. After incubation for 10 min at 37°C, the cells were washed and resuspended in 1 mL of phenol red-free DMEM supplemented with 2% dialyzed fetal bovine serum and 8 μg/mL propidium iodide. The data were collected using a FAC-SCalibur fluorescence cell scanner (BD Biosciences, San Jose, CA, USA) and the CEllQuest data acquisition program. Fluorescence data were collected at an excitation wave length of 475 nm and an emission wave length of 525 nm. Ten thousand cells were analyzed per sample, and the propidium iodide fluorescence was used to exclude dead cells. The dichlorofluorescein (DCF) data were plotted as histograms and was analyzed using the CellQuestTM software to determine the differences between the groups using a non-parametric statistical method (Kolmogorov-Smirnov test). Five samples in each group were analyzed.
Determination of mitochondrial membrane potential
The JC-1 mitochondrial membrane potential assay kit was used to evaluate the changes in the mitochondrial membrane potential (MMP) in the cells. JC-1 is a lipophilic and cationic dye that permeates the plasma and mitochondrial membranes. The dye fluoresces red when it aggregates in healthy mitochondria with high membrane potential; on the other hand, it appears in monomeric form and fluoresces green in mitochondria with diminished membrane potential. The cells were incubated with the MMP sensitive fluorescent dye JC-1 for 20 min at 37°C, washed twice with PBS, and the “red” (excitation 550 nm, emission 600 nm) and “green” (excitation 485 nm, emission 535 nm) fluorescence intensities were measured using a fluorescence microplate reader (Molecular Devices). Mitochondrial depolarisation (i.e., the loss of MMP) manifests itself as a decrease in the red/green fluorescence ratio.
Western blot analysis
The cells were lysed in a lysis buffer containing 625 mmol/L Tris-HCl (pH 6.8), 10% SDS, 25% glycerol, 5% β-mercaptoethanol, and 0.015% bromophenol blue, followed by sonication and heat denaturation. The protein were applied to a 10%-15% SDS-polyacrylamide gel, transferred to a nitrocellulose membrane, and then detected using the proper primary and secondary antibodies before visualization with a chemiluminescence kit (Pierce). β-actin served as a loading control.
Statistics
All data shown are the mean ± SE of at least three independent experiments, except when otherwise indicated. Significant differences were evaluated using an ANOVA, followed by the appropriate post hoc tests, or by the Mann-Whitney U-test. A P value <0.05 was considered significant.
Results
High concentrations of dexamethasone induced cell death through a necrotic and caspase-independent pathway
Dex reduced cell viability is shown in Figure 1A. At a concentration of 10-5 M, the percentage of viable cells was obviously decreased but was not significantly different from that of the other concentrations. The Annexin-V/PI assay indicated that cell death was due to necrosis (Figure 1B), as the apoptotic population was very small. TEM showed some necrotic changes in MC3T3-E1 cells treated with 10-5 M Dex: cell swelling and cytoplasmic vacuolation (a), numerous swollen mitochondria and dilation of endoplasmic reticulum (b), plasma membrane rupture (c), severe damage of mitochondria with disrupted internal structures (d) (Figure 1C). Dex-induced cell death was caspase-independent (Figure 1D).
Figure 1.

High concentrations of dexamethasone induced cell death through a necrotic and caspase-independent pathway. A. Cell viability in response to different concentration of Dex. B. AV/PI double stainning. C. The ultrastructural changes of MC3T3-E1 cells induced by 10-5 M Dex for 72 hours. D. The caspase-3 activity of dexamethasone induced MC3T3-E1 cell death. Each point is mean ± S.D. of three experiments. *P<0.05; **P<0.01.
Nec-1 protected MC3T3-E1 cells against necrotic cell death induced by 10-5 M dexamethasone
To quantify the protection provided by Nec-1, MC3T3-E1 cells were exposed to 10-5 M Dex in the presence of Nec-1 for 72 h. The caspase inhibitor, z-VAD-fmk, was used for comparison. In our experiments, Nec-1 significantly reduced the Dex induced cell death (P<0.01), whereas z-VAD-fmk had a smaller but still significantly affect (P<0.05) (Figure 2A). The AV/PI staining showed that more cells showed PI-positive when exposed to 10-5 M Dex compared to the control (P<0.05) (Figure 2B). Nec-1 significantly decreased the PI-positive cells (P<0.01) while z-VAD-fmk increased (P<0.001) (Figure 2C). Nec-1 also attenuated the lesion of mitochondria induced by Dex from the TEM (Figure 2D).
Figure 2.
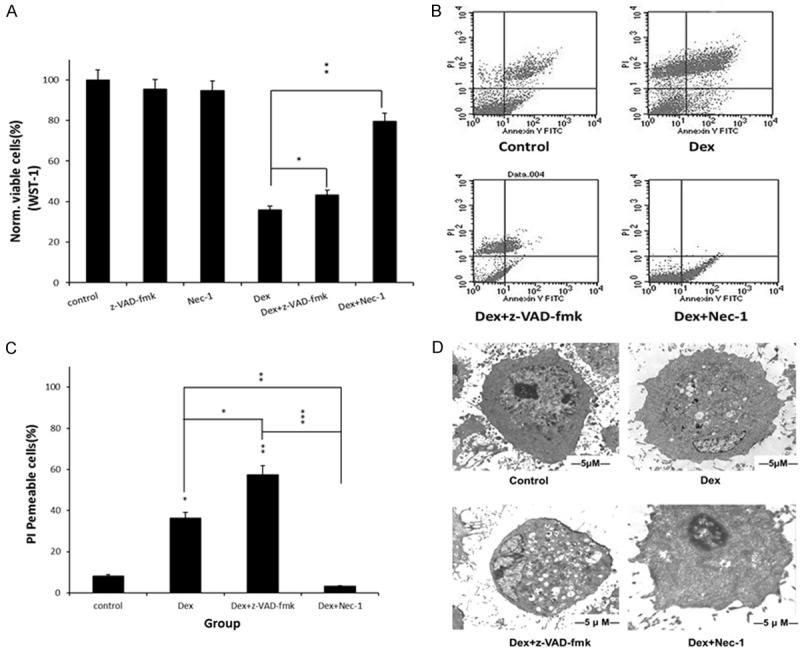
Nec-1 protected MC3T3-E1 cells against necrotic cell death induced by 10-5 M Dex. A. Nec-1 decreased cell viability compared with z-VAD-fmk. B, C. Nec-1 decreased the PI-positive cells while z-VAD-fmk increased it. D. TEM showed cell protection of Nec-1 from 10-5 M Dex. *P<0.05; **P<0.01.
Nec-1 increased the MMP but had no effect on the production of reactive oxygen species (ROS)
To further investigate the effect of Nec-1 on mitochondrial membrane function, we examined the production of ROS and the MMP level in the different groups. Our data demonstrated that, dex was found to reduce the MMP; however, treatment with Nec-1 significantly prevented the Dex-induced loss of MMP, while z-VAD-fmk had no effect (Figure 3A). In the ROS assay, 10-5 M Dex significantly increased the intracellular ROS levels (P<0.01) (Figure 3B). Interestingly, Nec-1 had no effect on the ROS production of cells treated with Dex alone (P>0.05).
Figure 3.
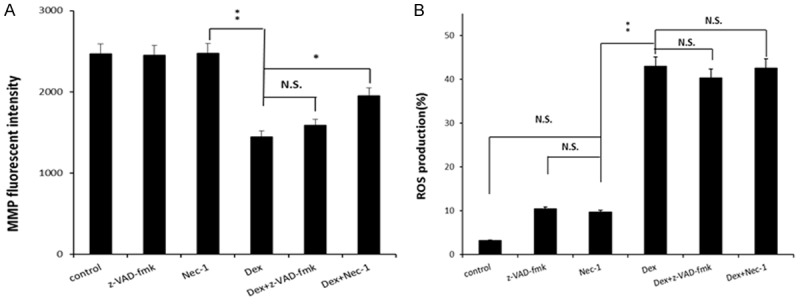
Nec-1 increased the MMP (A) but had no effect on the production of ROS (B). Data represent mean ± S.D. of three experiments. N.S. = Not significant. *P<0.05; **P<0.01.
Nec-1 decreased the levels of activated RIP-1 dexamethasone treatment
To further explore the mechanism of Nec-1 protection, we examined the activity of RIP-1 and RIP-3. Western blot analysis showed that treatment with 10-5 M Dex for 72 h markedly increased the levels of RIP-1 and RIP-3 (P<0.01) (Figure 4). Importantly, as shown in Figure 4, Nec-1 inhibited the activation of RIP-1 but had no effect on RIP-3 (P<0.01).
Figure 4.
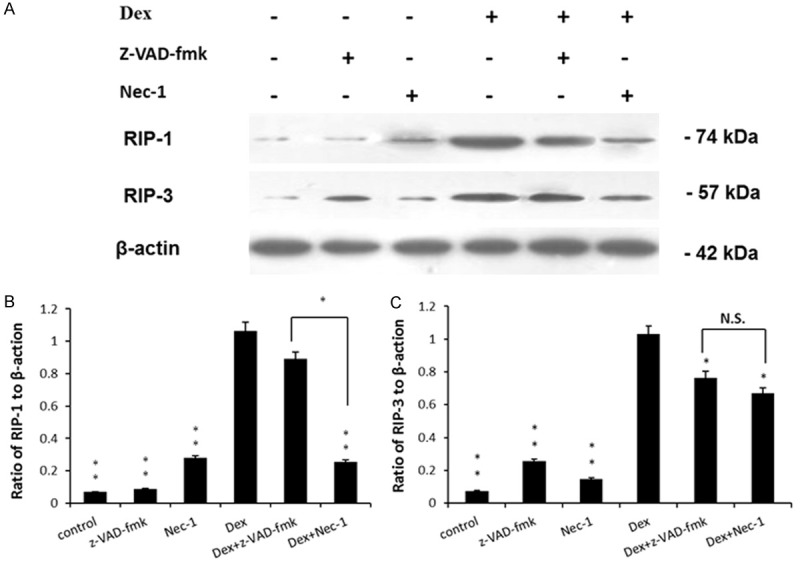
Nec-1 decreases the level of activated RIP-1 but had no effect on RIP-3 after dexamethasone treatment. A. Representative western blots of RIP-1, RIP-3 and β-action were shown. B. Quantitative analysis of RIP-1. C. Quantitative analysis of RIP-3. n=6 for each group. N.S. = Not significant. *P<0.05; **P<0.01.
Discussion
Previous investigations have focused on the apoptotic effects of GCs on osteoblasts, while less attention has been paid to the necrotic death induced by GCs. Our research confirmed that the necrotic-like cell death induced by 10-5 M dexamethasone in MC3T3-E1 cells was caspase-independent. We also found that necrostatin-1 could immediately interrupt this mechanism of cell death through the down regulation of RIP-1 and the restoration of MMP.
Previous studies have reported that GCs can promote [20] or protect osteoblast from apoptosis [21] in vitro. Interestingly, in MC3T3-E1 osteoblasts, Dex inhibited the TNF-α-induced apoptosis by up to 50% and led to a decrease in the activities of caspase-3, cyt-c, and NF-κB [22]. GCs promote apoptosis in vitro when culture conditions (cell density, differentiation promoting agents, serum concentration) do not fully support differentiation [23]. However, Weinstein demonstrated that GC-induced bone cell apoptosis in humans is rare [24]. A study of ischemic osteonecrosis demonstrated osteocyte and osteoblast apoptosis 12 h after the insult, whereas no evidence for apoptosis remained after 96 h, at that time, only empty lacunae were detected [25]. Our previous studies also confirmed that both apoptosis and necroptosis were participating in the pathological mechanism of GIOP rats [14]. These conflicts suggested that there may be another mechanism, such as necroptosis, occurring in osteoblasts and osteocytes that may account for the necrotic changes observed during GCs treatment. Using the WST-1 assay, we found that GCs induced MC3T3-E1 cell death in a time- and dose-dependent manner, and the Annexin-V/PI double staining showed that the number of necrotic-like cells synchronously increased with the concentration of dexamethasone. In partial agreement with our findings, others have reported that glucocorticoid- induced apoptosis was dependent on the concentration of glucocorticoids, with a maximum effect observed at a concentration of 10-7 M after 72 h in osteoblast culture [3]. However, the author did not discuss why higher concentrations of GCs led to a decrease in the number of apoptotic osteoblast cells. All these findings suggested that there may be another cell death mechanism active in osteoblasts after GC treatment, and this new cell death mechanism was caspase-independent.
Necroptosis can be triggered when caspase activity of caspase is inhibited. In our study, we also found that the activity of the caspases was significantly decreased after treatment with 10-5 M Dex for extended periods of time. Although the mechanism of caspase- depression is still undefined, the cell culture conditions, stage of differentiation and concentrations of stimulant may be involved [26]. The critical upstream components of the pathway include RIP-1 and RIP-3, while ROS production and mitochondrion function are the downstream effectors [27]. The kinase function of RIP-1 was essential for programmed necrosis but was dispensable for NF-κB activation [28,29], suggesting that programmed necrosis might be regulated at the level of RIP-1 activation. RIP-3 had been identified as a crucial upstream activating kinase that regulates RIP1-dependent programmed necrosis. Both RIP-3 and the kinase activity of RIP-1 are essential for the stable formation of the RIP-1-RIP-3 pro-necrotic complex, which critically controls downstream reactive oxygen species (ROS) production [30]. Our western blot analysis showed that the osteoblasts displayed high levels of RIP-1 and RIP-3 after treatment with 10-5 M Dex for 3 days.
The function of the mitochondria is essential for necroptosis. Using transmission electron microscopy, we found that the mitochondria of MC3T3-E1 cells treated with dexamethasone were obviously swollen and vacuolisation had occurred, indicating the disruption of mitochondrial function. There was also a remarkably elevated level of MMP in MC3T3-E1 cells under our treatment regime, which indicated a negative effect on mitochondrial function. Additionally, the mitochondria are also the principal producers of ROS in cells [31]. It has been previously demonstrated that TNF-α-induced necroptosis requires the participation of ROS [32]. We also observed an exaggerated production of ROS following Dex treatment. Although the exact mechanism that triggers ROS production remains poorly understood, RIP-3 accelerates mitochondrial ROS production and mitochondrial metabolism through the activation of a series of metabolism-related enzymes in 293T cells [33].
Nec-1 was first identified as an antagonist of RIP-1, which, in combination with RIP-3, activates necroptosis [7]. In our study, the cell viability assay showed that the number of osteoblasts was reduced after treatment with 10-5 M Dex for 72 h, and this reduction was almost completely reversed by pre-treatment with Nec-1, pre-treatment with the caspase-inhibitor z-VAD-fmk had very little effect. The effect of z-VAD-fmk could be attributed to the slight apoptotic effect induced by 10-5 M Dex, which was also observed by Annexin-V/PI staining. We also confirmed that Nec-1 restored the MMP level, but it had no effects on the Dex-induced ROS production. Therefore, it is tempting to speculate that the production of ROS was regulated by RIP-3 or other downstream kinases, which were mentioned above. It was confirmed that Nec-1 protected the cells from necroptosis by blocking RIP-1. In our study, we demonstrated that the expression of RIP-1 significantly decreased when the cells were pre-treated with Nec-1.
In summary, necrostatin-1 could significantly inhibit the cell death induced by high doses and long-term treatment with dexamethasone. Interestingly, necroptosis has also been reported to be involved in cell protection [34]. In certain circumstances, necroptosis and apoptosis can occur simultaneously in cells [35]. Future studies are required to fully unravel the detailed mechanisms regulating the development and progression of cell death in response to GC treatment.
Acknowledgements
This study was supported by the National Science Foundation of China (81672173; 81271964).
Disclosure of conflict of interest
None.
References
- 1.Bollet AJ, Black R, Bunim JJ. Major undesirable side-effects resulting from prednisolone and prednisone. J Am Med Assoc. 1955;158:459–463. doi: 10.1001/jama.1955.02960060017005. [DOI] [PubMed] [Google Scholar]
- 2.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–282. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gohel A, McCarthy MB, Gronowicz G. Estrogen prevents glucocorticoid-induced apoptosis in osteoblasts in vivo and in vitro. Endocrinology. 1999;140:5339–5347. doi: 10.1210/endo.140.11.7135. [DOI] [PubMed] [Google Scholar]
- 4.Che M, Ettinger B, Nguyen M, Pressman A, Johnston J. High-dose corticosteroid exposure and osteoporosis intervention in adults. Ann Allergy Asthma Immunol. 2006;97:497–501. doi: 10.1016/S1081-1206(10)60941-6. [DOI] [PubMed] [Google Scholar]
- 5.Lane NE, Yao W, Balooch M, Nalla RK, Balooch G, Habelitz S, Kinney JH, Bonewald LF. Glucocorticoid-treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or estrogen-deficient mice. J Bone Miner Res. 2006;21:466–476. doi: 10.1359/JBMR.051103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakashima T, Sasaki H, Tsuboi M, Kawakami A, Fujiyama K, Kiriyama T, Eguchi K, Ichikawa M, Nagataki S. Inhibitory effect of glucocorticoid for osteoblast apoptosis induced by activated peripheral blood mononuclear cells. Endocrinology. 1998;139:2032–2040. doi: 10.1210/endo.139.4.5932. [DOI] [PubMed] [Google Scholar]
- 7.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 8.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krumschnabel G, Ebner HL, Hess MW, Villunger A. Apoptosis and necroptosis are induced in rainbow trout cell lines exposed to cadmium. Aquat Toxicol. 2010;99:73–85. doi: 10.1016/j.aquatox.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6:1641–1649. doi: 10.1158/1535-7163.MCT-06-0511. [DOI] [PubMed] [Google Scholar]
- 11.Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert SJ, Singhrao SK, Khan IM, Gonzalez LG, Thomson BM, Burdon D, Duance VC, Archer CW. Enhanced tissue integration during cartilage repair in vitro can be achieved by inhibiting chondrocyte death at the wound edge. Tissue Eng Part A. 2009;15:1739–1749. doi: 10.1089/ten.tea.2008.0361. [DOI] [PubMed] [Google Scholar]
- 14.Feng M, Zhang R, Gong F, Yang P, Fan L, Ni J, Bi W, Zhang Y, Wang C, Wang K. Protective effects of necrostatin-1 on glucocorticoid-induced osteoporosis in rats. J Steroid Biochem Mol Biol. 2014;144:455–462. doi: 10.1016/j.jsbmb.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Hanazawa S, Ohmori Y, Amano S, Miyoshi T, Kumegawa M, Kitano S. Spontaneous production of interleukin-1-like cytokine from a mouse osteoblastic cell line (MC3T3-E1) Biochem Biophys Res Commun. 1985;131:774–779. doi: 10.1016/0006-291x(85)91306-3. [DOI] [PubMed] [Google Scholar]
- 16.Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol. 1998;143:1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Han W, Gu Y, Qiu S, Lu Q, Jin J, Luo J, Hu X. Honokiol induces a necrotic cell death through the mitochondrial permeability transition pore. Cancer Res. 2007;67:4894–4903. doi: 10.1158/0008-5472.CAN-06-3818. [DOI] [PubMed] [Google Scholar]
- 18.Chang JK, Li CJ, Wu SC, Yeh CH, Chen CH, Fu YC, Wang GJ, Ho ML. Effects of anti-inflammatory drugs on proliferation, cytotoxicity and osteogenesis in bone marrow mesenchymal stem cells. Biochem Pharmacol. 2007;74:1371–1382. doi: 10.1016/j.bcp.2007.06.047. [DOI] [PubMed] [Google Scholar]
- 19.Sagara Y, Ishige K, Tsai C, Maher P. Tyrphostins protect neuronal cells from oxidative stress. J Biol Chem. 2002;277:36204–36215. doi: 10.1074/jbc.M203895200. [DOI] [PubMed] [Google Scholar]
- 20.Chen HL, Demiralp B, Schneider A, Koh AJ, Silve C, Wang CY, McCauley LK. Parathyroid hormone and parathyroid hormone-related protein exert both pro- and anti-apoptotic effects in mesenchymal cells. J Biol Chem. 2002;277:19374–19381. doi: 10.1074/jbc.M108913200. [DOI] [PubMed] [Google Scholar]
- 21.Davies JH, Evans BA, Jenney ME, Gregory JW. In vitro effects of combination chemotherapy on osteoblasts: implications for osteopenia in childhood malignancy. Bone. 2002;31:319–326. doi: 10.1016/s8756-3282(02)00822-0. [DOI] [PubMed] [Google Scholar]
- 22.Chae HJ, Chae SW, Kang JS, Bang BG, Cho SB, Park RK, So HS, Kim YK, Kim HM, Kim HR. Dexamethasone suppresses tumor necrosis factor-alpha-induced apoptosis in osteoblasts: possible role for ceramide. Endocrinology. 2000;141:2904–2913. doi: 10.1210/endo.141.8.7604. [DOI] [PubMed] [Google Scholar]
- 23.Pereira RM, Delany AM, Canalis E. Cortisol inhibits the differentiation and apoptosis of osteoblasts in culture. Bone. 2001;28:484–490. doi: 10.1016/s8756-3282(01)00422-7. [DOI] [PubMed] [Google Scholar]
- 24.Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab. 2000;85:2907–2912. doi: 10.1210/jcem.85.8.6714. [DOI] [PubMed] [Google Scholar]
- 25.Sato M, Sugano N, Ohzono K, Nomura S, Kitamura Y, Tsukamoto Y, Ogawa S. Apoptosis and expression of stress protein (ORP150, HO1) during development of ischaemic osteonecrosis in the rat. J Bone Joint Surg Br. 2001;83:751–759. doi: 10.1302/0301-620x.83b5.10801. [DOI] [PubMed] [Google Scholar]
- 26.Zalavras C, Shah S, Birnbaum MJ, Frenkel B. Role of apoptosis in glucocorticoid-induced osteoporosis and osteonecrosis. Crit Rev Eukaryot Gene Expr. 2003;13:221–235. [PubMed] [Google Scholar]
- 27.Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 29.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 30.Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS, Tran JH, Nedospasov SA, Liu ZG. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem. 2004;279:10822–10828. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 31.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jezek P, Hlavata L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol. 2005;37:2478–2503. doi: 10.1016/j.biocel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 33.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK, Schafer BW, Schrappe M, Stanulla M, Bourquin JP. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest. 2010;120:1310–1323. doi: 10.1172/JCI39987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galluzzi L, Joza N, Tasdemir E, Maiuri M, Hengartner M, Abrams J, Tavernarakis N, Penninger J, Madeo F, Kroemer G. No death without life: vital functions of apoptotic effectors. Cell Death Differ. 2008;15:1113–1123. doi: 10.1038/cdd.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]