

Abstract
To investigate the role of toll like receptors (TLRs) 2 and 4 in rhabdomyolysis (RM)-related acute kidney injury (AKI). Wild-type (WT) mice and TLR2 knockout (TLR2-/-) or TLR4 knockout (TLR4-/-) mice were injected with either saline (sham) or glycerin (to induce RM-related AKI). Samples were collected for detection of 0 h 24 h (Cr) creatinine, urea nitrogen (BUN), creatine kinase (CK), and PAS staining of renal tissues. Serum Cr and BUN level was significantly increased in TLR2-/- and TLR4-/-AKI groups more than those in the control group and the WT mice in AKI group. TLR4-/-AKI group Cr, BUN level, and the pathological damage was lightest. The expression levels of signal transduction proteins in TLR2-/- and TLR4-/-AKI group were higher than in the control group, but was lower than that in the wild AKI group, with the TLR4-/-AKI group having the lowest levels. The expression level of inflammatory factor mRNA in TLR2-/- and TLR4-/-AKI groups was higher than that in control group, but was lower than that in wild AKI group, with TLR4-/-AKI group displaying lowest levels. Knockout of TLRs 2 and 4 decreased kidney inflammation and improved RM-related AKI.
Keywords: Toll like receptor 4, toll like receptor 2, acute kidney injury, rhabdomyolysis, inflammation
Introduction
Rhabdomyolysis (RM) is a life-threatening syndrome resulting from the breakdown of striated muscle cell membrane in cases of muscle trauma, ischemia, inflammation, metabolic abnormalities, and systemic poisoning [1,2]. During RM, large quantities of muscular cell constituents, which can lead to persistent renal injury and intractable inflammatory conditions, are released into the extracellular fluid and the circulation [3,4]. Today, RM muscle cell leakage has been identified as one of the leading causes of acute kidney injury (AKI) [5]. It is reported that 13-50% of the RM patients develop AKI [6]. In the United States, 7-10% of the AKI are caused by RM [7]. These RM-related AKI patients have a mortality of approximately 20% [8,9]. According to Sever et al. (2006), after both natural disasters (e.g. earthquakes and hurricanes) and man-made catastrophes (e.g. wars and mining accidents), crush syndrome-induced RM-related AKI, apart from direct trauma, is the most frequent cause of death [3]. Therefore, understanding the pathogenic mechanism of RM-related AKI is important for early intervention and appropriate treatment.
In retrospect, the muscle leakage of myoglobin, an 18,800-Dalton oxygen carrier that can cause glomeruli obstruction and renal dysfunction, has been proposed as a major contributor to the development of RM-related AKI [10,11]. Interestingly, in recent years, more and more evidence indicates that, apart from myoglobin-induced renal injury, inflammation also plays a key role in the development and progression of RM-related AKI [12-14]. After RM, the released cell constituents activate pattern recognition receptor (e.g. TOLL like receptors, TLRs) and cause immediate and significant inflammatory responses [2]. The inflammatory activation, which can cause endothelial injury, induce leukocyte entrapment and decrease microvascular blood flow. This causes damage to both the medulla and the cortex of the kidney [15-18]. Since TLRs 2 and 4, which are constitutively expressed on renal epithelium, can recognize endogenous ligands during tissue injury and trauma [19,20], it would be interesting to investigate the roles of TLRs 2 and 4 in the development of RM-related AKI. Indeed, studies have shown that TLRs are involved in the progress of ischemia-reperfusion injury (IRI) in heart, lung, brain, and liver [21,22]. Chen et al. [23] reported that the TLRs contribute to renal injury during ischemic-induced AKI.
In this study, the role of TLRs 2 and 4 in RM-related AKI was investigated using developing RM-related AKI in wildtype mice as well as in mice with TLR2 or 4 knockout.
Materials and methods
All of the procedures conducted in this study were approved by the Animal Ethical Committee of Chinese PLA General Hospital (Beijing, China).
Mice model of RM-related AKI
Wildtype (WT, n = 10), TLR2 knockout (TLR2-/-, n = 10) and TLR4 knockout (TLR4-/-, n = 10) mice (all males, 8-week-old, with body weights of 20-25 g) were obtained from Nanjing University Model Animal Research Center (Nanjing, China). Mice from each of these three groups were equally divided into two treatments: sham and RM-related AKI. For sham mice, physiological saline was injected through thigh muscle (both side) at a dose of 8 ml/kg body weight. For RM-related AKI development, mice were injected with 50% glycerin solution (purchased from US Sigma-Aldrich Corporation, 8 ml/kg body weight) in the same way as the sham group. All of the mice were raised in a SPF grade animal house under controlled temperature (22 ± 2°C) and lighting (12:12-h light/dark) with free access to food and water during this study. Twenty-four hours after injection, 10 mice from each treatment were randomly selected and carefully anesthetized (2% pentobarbital) and euthanized (pick eyeball blood after anesthesia) for serum isolation [blood collected via eye bleeding, centrifuged and stored at -80°C for the determination of creatinine (Cr), urea nitrogen (BUN) and creatine kinase (CK) levels] and kidney (fixed using 10% formalin for Schiff’s staining) samples. Twenty-four hours after injection, 10 mice from each treatment were sampled for serum and kidney (stored at -80°C for Western Blot and RT-PCR analysis).
The evaluation of RM and AKI status
Serum levels of Cr, BUN and CK were determined by commercial ELISA kits according to manufacturer’s instructions. The mice were considered as RM when the serum CK level was increased to 5 times of the WT-sham treatment. The mice were considered as RM-related AKI when the serum Cr level was increased to 1.5 times of the WT-sham treatment.
Formalin fixed kidney samples were subjected to Schiff’s (periodic acid-Schiff PAS) staining. An optical microscope (Olympus Optical Company, Japan) was used to examine the stained slides and results were expressed as photograph objects. The kidney necrosis (ATN) score was determined as previously described [24].
Western blot
Western blot was conducted as previously described [25]. The primary rabbit anti-mouse TLR2, TLR4, myeloid differentiation factor 88 (MYD88, lot number), tumor necrosis factor receptor-associated factor 3 (TRAF-3, please add lot number), interferon regulatory factor (IRF-3, please add lot number), TRAF-6 (please add lot number), activating protein-1 (AP-1, please add lot number), nuclear factor kappa B (NF-KB, please add lot number) and β-actin (please add lot number) antibodies were purchased from American Abcam company (Cambridge, MA, USA). Horseradish peroxidase-labeled goat anti-rabbit secondary antibodies (please add lot number) were purchased from Abcam company. The band density was quantified using Bandscan analysis software and then normalized to β-actin content, because the levels of β-actin did not differ between experimental groups.
RT-PCR
The sequences of endogenous interferon-β (INF-β), macrophage inflammatory protein-1α (MIP-1α), interleukin 6 (IL-6), tumor necrosis factor-α (TNF-α) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, used as an internal reference) were obtained from GenBank. Primer premier 5 software was used to design primers for RT-PCR. Primers (Table 1) were synthesized by Huada Gene Company (Shenzhen, Guangdong, China). RT-PCR was performed using TaqMan probes (sample was analyzed in triplicate). The relative mRNA expression was calculated using the 2-ΔΔCT method and the results were expressed as fold-change relative to GAPDH.
Table 1.
Primer sequences for RT-PCR
| Gene | Forward Primers (5’-3’) | Reverse Primer (3’-5’) |
|---|---|---|
| INF-β | CGTGGGAGATGTCCTCAACT | AGATCTCTGCTCGGACCACC |
| MIP-1α | TGCCAAGTAGCCACATCGAG | GAGATGGGGGTTGAGGAACG |
| IL-6 | CAACGATGATGCACTTGCAGA | CAACGATGATGCACTTGCAGA |
| TNF-α | AGGCACTCCCCCAAAAGATG | CCCTCACACTCACAAACCAC |
Statistical analysis
Data were analyzed using SPSS 16 software (IBM Corp., Chicago, IL). Data are shown as mean ± standard error of the mean (SEM). Two-tailed independent Student’s t-test was conducted to compare the differences between two groups. Significant differences were considered at P ≤ 0.05.
Results
Establishment of RM-related AKI
Our results found that at 24 h in the WT AKI group, TLR2-/-AKI group and TLR4-/-AKI group, serum creatinine, urea nitrogen and creatine kinase values were significantly higher than those in the control group. Meanwhile, rhabdomyolysis and acute kidney injury were observed in mice at after 24 h injection, suggesting successful establishment of rhabdomyolysis acute renal injury model mice. The levels of serum creatinine and urea nitrogen in mice from TLR2-/-AKI group and TLR4-/-AKI group were significantly lower than those in group WT and group 24 h, indicating that the degree of acute kidney injury in the TLR2-/-AKI group was lower than that in WT AKI group, with the TLR4-/-AKI group displaying lower kidney injury compared with TLR2-/-AKI group at 24 h. The level of serum creatinine and urea nitrogen in TLR4-/-AKI group was significantly lower than that in TLR2-/-AKI group, indicating that the degree of 24 h acute renal injury in TLR4-/-AKI group was the lightest among these groups. However, WT AKI group, TLR2-/-AKI group, and TLR4-/-AKI group mice in 24 h displayed no significant difference of serum creatine kinase in the degree of injury to the three groups of striated muscle, rhabdomyolysis, consistent with no different effects on acute kidney injury induced by rhabdomyolysis degree (Table 2).
Table 2.
Serum levels of Cr, BUN and CK in different time points of mice with rhabdomyolysis
| Mice | Treatment | Time | Cr (μmol/L) | BUN (mmol/L) | CK (U/L) |
|---|---|---|---|---|---|
| WT | N = 10 | Sham | 12.6 ± 0.73 | 8.23 ± 0.39 | 439.57 ± 102.74 |
| AKI | 24 H | 212.95 ± 22.15* | 68.7 ± 1.14* | 2615.15 ± 292.25* | |
| AKI | 48 H | 105.92 ± 20.01 | 54.27 ± 13.4 | 538.58 ± 127.55 | |
| TLR2-/- | N = 10 | Sham | 13.87 ± 1.39 | 8.23 ± 1.61 | 468.3 ± 101.56 |
| AKI | 24 H | 155.49 ± 30.42*,& | 51.57 ± 8.64*,& | 2637.49 ± 166.59*,& | |
| AKI | 48 H | 28.96 ± 12.76 | 14.73 ± 9.24 | 869.44 ± 189.74 | |
| TLR4-/- | N = 10 | Sham | 13.15 ± 1.25 | 8.10 ± 1.24 | 459.15 ± 154.35 |
| AKI | 24 H | 63.76 ± 33.01*,&,$ | 36.02 ± 8.54*,&,$ | 2564.6 ± 203.53*,& | |
| AKI | 48 H | 14.09 ± 2.66 | 8.27 ± 1.09 | 790.53 ± 141.93 |
Note: compare with WT sham;
P < 0.05.
Compare with WT AKI group 24 h;
P < 0.05 (Cr, BUN);
P > 0.05 (CK).
Compare with TLR2-/-AKI group;
P < 0.05.
As shown in Figure 1, under light microscope, the structure of glomerular and renal tubules in WT-sham mice was intact 24 hour after saline injection. However, in WT-AKI mice, the renal tubular epithelial cells were edematous with vacuolar and granular degeneration, the brush border of the proximal renal tubule was detached, and the basement membrane of renal tubules was exposed. Compared to WT-AKI mice, the TLR2-/--AKI and TLR4-/--AKI mice had decreased epithelial cells edema, decreased vacuolar degeneration, and granular degeneration, as well as decreased basement membrane exposed. No renal ATN was scored in WT-sham mice. However, an average ATN score of 8 was observed in WT-AKI mice. The TLR2-/--AKI (lower) and TLR4-/--AKI (lowest) mice had decreased renal ATN scores compared with WT-AKI mice.
Figure 1.

Schiff’s staining and renal ATN score 24 h after injection. Compared with WT AKI group, *P < 0.05; compared with TLR2-/-AKI group, &P < 0.05.
Renal protein expression of MYD88, TRAF-6, NF-kB, AP-1, TRAF-3, and IRF-3
In the study of dependence on the MyD88 channel, we found that compared with WT sham group, the TLR2-/-AKI group and TLR4-/-AKI group presented significantly increased MyD88, TRAF-6, AP-1, NF-kappa B protein expression, suggesting MyD88 dependent downstream signal transduction protein expression was increased. TLR2 and TLR4 are inflammatory signal transduction mediators through their MYD88 dependent pathways and might be involved in skeletal muscle during the dissolution of acute kidney injury. However, compared with the WT AKI group, the TLR2-/-AKI and TLR4-/-AKI groups displayed significantly decreased MYD88, TRAF-6, AP-1, and NF-kappa B protein levels, indicating that TLR2 and TLR4 might be involved in the signal transduction pathway of MYD88 dependent inflammation. Interestingly, compared with the TLR2-/-AKI group, expression of MYD88, TRAF-6, and AP-1 expression in TLR4-/-AKI group was significantly decreased (Figure 2).
Figure 2.
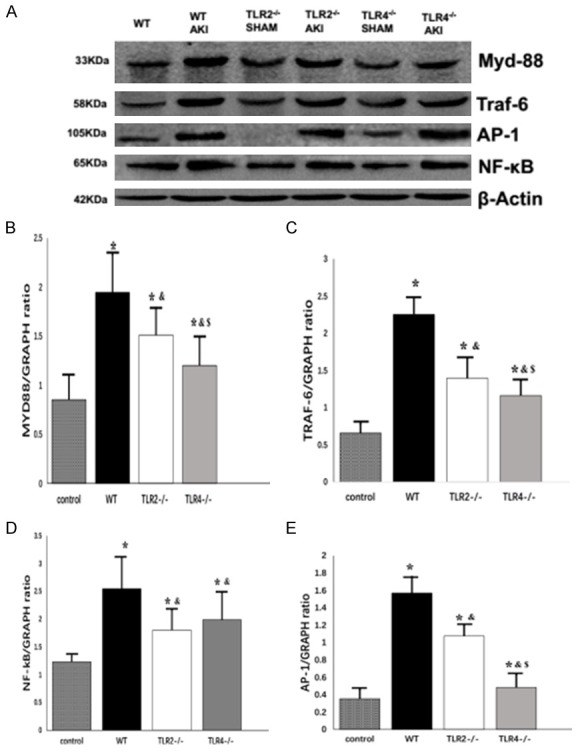
Renal protein expression of MYD88, TRAF-6, NF-kB and AP-1. Compared with WT SHAM group, *P < 0.05; compared with WT AKI group, &P < 0.05; compared with TLR2-/-AKI group, $P < 0.05.
In a study of IRF-3 dependent channels we found that, compared with the WT sham group, the WT AKI group and TLR2-/-AKI group showed significantly increased TRAF-3 and IRF-3 protein expression. However, no expression of TRAF-3 and IRF-3 was observed in the TLR4-/-AKI group, suggesting that TLR4 might transduce inflammatory signals through IRF-3, while TLR2 does not depend on the IRF-3 signaling pathway. Compared with the WT AKI group, expression levels of TRAF-3 and IRF-3 in TLR2-/-AKI group were decreased, indicating that the expression level of proteins downstream of IRF-3 signaling pathway are down-regulated after knockdown of TLR2 (Figure 3).
Figure 3.

Renal protein expression of TRAF-3 and IRF-3. Compared with WT SHAM group, *P < 0.05; compared with WT AKI group, &P < 0.05.
Renal mRNA expression of INF-β, MIP-1α, IL-6 and TNF-α
Analysis of the expression of inflammatory factors found that compared with WT sham group, the WT AKI group and TLR2-/-AKI group showed significantly increased IL-6, inflammatory factor IFN-β MIP-1 and TNF-alpha mRNA levels and the TLR4-/-AKI group displayed significantly increased IFN-β and IL-6 levels, indicating TLR2 and TLR4 are involved in the injury mediated by rhabdomyolysis after. Compared with the WT AKI group, the TLR2-/-AKI and TLR4-/-AKI groups, presented significantly reduced IL-6, inflammatory factor IFN-β, MIP-1α and TNF-α mRNA expression levels, indicating knockdown of TLR2 or TLR4 could downregulate the secretions of inflammatory factors, reduced the degree of inflammation of kidney. Compared with TLR2-/-AKI group, the expression levels of IFN-β, IL-6, MIP-1, TNF-α in TLR4-/- and AKI groups were significantly lower, indicating that TLR4 had a stronger effect on the inhibition of the expression of inflammatory cytokines than TLR2 (Figure 4).
Figure 4.

Renal mRNA expression of INF-β, IL-6, TNF-α and MIP-1α. Compared with WT SHAM group, *P < 0.05; compared with WT AKI group, &P < 0.05; compared with TLR2-/-AKI group, $P < 0.05.
Discussion
In this study, RM-related AKI was successfully developed in WT, TLR2-/- and TLR4-/- mice 24 hours after glycerin injection. However, while all the mice experienced the same level of RM (using serum CK level as an indicator), TLR2-/- and TLR4-/- mice had improved AKI status (using serum Cr as an indicator) when compared to WT mice. Similarly, Schiff’s staining and renal ATN scores indicate that, at the same time (24 hours after glycerin injection), WT mice experienced more severe kidney injury compared with TLR2-/- and TLR4-/- mice. These results clearly demonstrate that TLR 2 and 4 contribute to the development and progression of glycerin-induced RM-related AKI. In recent years, RM-induced inflammation response has been proposed as a major cause of AKI [10,13]. Since TLR2 and TLR4 are key pattern recognition receptors for endogenous ligands recognition [15,16], the current results were as expected. In a study conducted by Rusai et al. (2010), knockout of TLR2 or/and TLR4 could protect mice from renal ischemia/reperfusion injury which was basically characterized as inflammation and cell death [26].
Of particular interest in this study, is that within the TLR knockout AKI groups, TLR4-/--AKI mice had improved AKI status (indicated by serum Cr level, epithelial cell edema, vacuolar and granular degeneration, tubular basement membrane explosion, and kidney ATN score) compared with TLR2-/--AKI mice. These results suggest that TLR2 and TLR4 play different roles in RM-induced inflammatory response. Basically, TLR signaling pathway can be divided into two sub-pathways: MYD88-dependent (both TLR2 and TLR4) and MYD88-independent (TLR4 but not TLR2). In the MYD88-dependent pathway, TLRs-induced MyD88 dimerization and interleukin-1 receptor-associated kinase (IRAK) activation initiates recruitment of TRAF-6 and TRAF-3 [27]. TRAF-6 activates transcription factors like NF-kB and AP-1. TRAF-3 is instrumental for the recruitment of TRAF family member-associated-binding kinase 1 (TBK-1) and the production of type I IFNs and IL-10. In the MYD88-independent pathway, TLR 3 and 4 can activate transcription factor IRF-3 and thereafter induce the production of IFN-β [28]. In this study, glycerin injection increased the renal expression of proteins (MYD88, TRAF-6, NF-kB, and AP-1) in the MYD88-dependent pathway in WT, TLR2-/- and TLR4-/- mice. These results suggest significant inflammatory responses were triggered in the kidney after the development of RM. Interestingly, compared with WT-AKI mice, renal expression of MYD88-dependent pathway proteins were decreased in both TLR2-/--AKI and TLR4-/--AKI mice. Thus, it would be safe to hypothesize that knockout of TLR 2 and 4 reduce RM-induced renal inflammatory response and therefore alleviate RM-related AKI. In addition, TLR4-/--AKI mice had lower levels of MYD88 pathway protein (MYD88, TRAF-6 and AP-1) expression compared with TLR2-/--AKI mice, suggesting TLR4 plays major roles in the RM-induced MYD88-dependent kidney inflammation. As expected, glycerin-induced RM-related AKI also increased the renal expressions of TRAF-3 and IRF-3 in WT and TLR2-/- but not TLR4-/- mice. These results are consistent with the current understanding that TLR4 plays key roles in the recruitment and/or activation of TRAF-3 and IRF-3 [28]. Interestingly, TLR2-/--AKI mice had decreased renal TRAF-3 and IRF-3 expression compared with WT-AKI mice, indicating that TLR2 also participates in the development of RM-related AKI through MYD88-independent pathway. In the literature, the MYD88-independent pathway (or the IRF-3 pathway) has been proposed as unique to TLR 3 and 4 [28]. However, similar to our results, Shigeoka et al. (2007) reported that TLR2 participated in ischemic renal injury through both MyD88-dependent and -independent pathways [29]. Future investigation is required to confirm the roles of TLR2 in the MYD88-independent pathway.
Glycerin injection up-regulated mRNAs expression of INF-β, IL-6, TNF-α and MIP-1α in the kidney of both WT and TLR2-/--mice. However, in TLR4-/--mice, glycerin induced increased INF-β and IL-6 expression and decreased TNF-α and MIP-1α expression.
In summary, TLR 4 and 2 involve in the renal inflammatory response in the development of glycerin-induced RM-related AKI through MYD88-dependent and -independent pathways. Targeting TLR4 and TLR2 may provide novel therapies for the treatment of RM-related AKI.
Acknowledgements
Supported in part by the National Natural Science Foundation of China (81170643); the Fund of Chinese PLA 12th Five-year plan for Medical Science (BWS14J040).
Disclosure of conflict of interest
None.
References
- 1.Bosch X. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361:1416–1416. doi: 10.1056/NEJMra0801327. [DOI] [PubMed] [Google Scholar]
- 2.Vanholder R, Sever MS, Erek E, Lameire N. Rhabdomyolysis. J Am Soc Nephrol. 2000;11:1553–1561. doi: 10.1681/ASN.V1181553. [DOI] [PubMed] [Google Scholar]
- 3.Sever MS, Vanholder R, Lameire N. Medical progress-management of crush-related injuries after disasters. N Engl J Med. 2006;354:1052–1063. doi: 10.1056/NEJMra054329. [DOI] [PubMed] [Google Scholar]
- 4.Zager RA. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int. 1996;49:314–326. doi: 10.1038/ki.1996.48. [DOI] [PubMed] [Google Scholar]
- 5.Bedry R, Baudrimont I, Deffieux G, Creppy EE, Pomies JP, Ragnaud JM, Dupon M, Neau D, Gabinski C, De Witte S, Chapalain JC, Godeau P. Wild-mushroom intoxication as a cause of rhabdomyolysis. N Engl J Med. 2001;345:798–802. doi: 10.1056/NEJMoa010581. [DOI] [PubMed] [Google Scholar]
- 6.Melli G, Chaudhry V, Cornblath DR. Rhabdomyolysis-an evaluation of 475 hospitalized patients. Medicine. 2005;84:377–385. doi: 10.1097/01.md.0000188565.48918.41. [DOI] [PubMed] [Google Scholar]
- 7.Bagley WH, Yang H, Shah KH. Rhabdomyolysis. Intern Emerg Med. 2007;2:210–218. doi: 10.1007/s11739-007-0060-8. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Fu P, Wang L, Cai GY, Zhang L, Chen DZ, Guo DY, Sun XF, Chen FQ, Bi WH, Zeng XJ, Li HY, Liu ZH, Wang Y, Huang SM, Chen XM. The clinical features and outcome of crush patients with acute kidney injury after the Wenchuan earthquake: differences between elderly and younger adults. Injury. 2012;43:1470–1475. doi: 10.1016/j.injury.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 9.Mohaupt MG. Rhabdomyolysen. Ther Umsch. 2003;60:391–397. doi: 10.1024/0040-5930.60.7.391. [DOI] [PubMed] [Google Scholar]
- 10.Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66:480–485. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 11.Holt S, Moore K. Pathogenesis of renal failure in rhabdomyolysis: the role of myoglobin. Exp Nephrol. 2000;8:72–76. doi: 10.1159/000020651. [DOI] [PubMed] [Google Scholar]
- 12.Bonventre JV. Pathophysiology of AKI: injury and normal and abnormal repair. Contrib Nephrol. 2010;165:9–17. doi: 10.1159/000313738. [DOI] [PubMed] [Google Scholar]
- 13.Ramesh G, Reeves WB. Inflammatory cytokines in acute renal failure. Kidney Int Suppl. 2004;66:S56–S61. doi: 10.1111/j.1523-1755.2004.09109.x. [DOI] [PubMed] [Google Scholar]
- 14.Belliere J, Casemayou A, Ducasse L, Zakaroff-Girard A, Martins F, Iacovoni JS, Guilbeau-Frugier C, Buffin-Meyer B, Pipy B, Chauveau D, Schanstra JP, Bascands JL. Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J Am Soc Nephrol. 2015;26:1363–1377. doi: 10.1681/ASN.2014040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medzhitov R, PrestonHurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 16.wasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 17.Linas SL, Shanley PF, Whittenburg D, Berger E, Repine JE. Neutrophils accentuate ischemia-reperfusion injury in isolated perfused rat kidneys. Am J Physiol. 1988;255:F728–735. doi: 10.1152/ajprenal.1988.255.4.F728. [DOI] [PubMed] [Google Scholar]
- 18.Willinger CC, Schramek H, Pfaller K, Pfaller W. Tissue distribution of neutrophils in postischemic acute renal failure. Virchows Arch B Cell Pathol Incl Mol Pathol. 1992;62:237–243. doi: 10.1007/BF02899687. [DOI] [PubMed] [Google Scholar]
- 19.Wolfs TG, Buurman WA, van Schadewijk A, de Vries B, Daemen MA, Hiemstra PS, van’t Veer C. In vivo expression of toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J Immunol. 2002;168:1286–1293. doi: 10.4049/jimmunol.168.3.1286. [DOI] [PubMed] [Google Scholar]
- 20.Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arumugam TV, Okun E, Tang SC, Thundyil J, Taylor SM, Woodruff TM. Toll-like receptors in ischemia-reperfusion injury. Shock. 2009;32:4–16. doi: 10.1097/SHK.0b013e318193e333. [DOI] [PubMed] [Google Scholar]
- 22.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, John R, Richardson JA, Shelton JM, Zhou XJ, Wang Y, Wu QQ, Hartono JR, Winterberg PD, Lu CY. Toll-like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int. 2011;79:288–299. doi: 10.1038/ki.2010.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El-Shahawy MA, Agbing LU, Badillo E. Severity of illness scores and the outcome of acute tubular necrosis. Int Urol Nephrol. 2000;32:185–191. doi: 10.1023/a:1007177130883. [DOI] [PubMed] [Google Scholar]
- 25.Benderro GF, LaManna JC. Kidney EPO expression during chronic hypoxia in aged mice. Adv Exp Med Biol. 2013;765:9–14. doi: 10.1007/978-1-4614-4989-8_2. [DOI] [PubMed] [Google Scholar]
- 26.Ha T, Hu Y, Liu L, Lu C, McMullen JR, Kelley J, Kao RL, Williams DL, Gao X, Li C. TLR2 ligands induce cardioprotection against ischaemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Cardiovasc Res. 2010;87:694–703. doi: 10.1093/cvr/cvq116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 29.Shigeoka AA, Holscher TD, King AJ, Hall FW, Kiosses WB, Tobias PS, Mackman N, McKay DB. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol. 2007;178:6252–6258. doi: 10.4049/jimmunol.178.10.6252. [DOI] [PubMed] [Google Scholar]