

Abstract
Acute lung injury (ALI), which is an excessive uncontrolled inflammatory response in the lung, is mediated by several pro-inflammatory mediators. Recent evidence has implicated microRNAs (miRNAs) in regulation of inflammation in different diseases. However, the roles and underlying molecular mechanism of miRNAs in ALI have not been adequately elucidated. Thus, the aim of the present study was to investigate the possible regulatory mechanism of miRNAs in ALI. In this study, microRNA microarray analysis showed that 48 miRNAs were differentially expressed in lung tissues of an ALI model induced by LPS. Downregulation of miR-27a, played a key role in the regulation of the inflammatory response and protection from traumatic injury. Functional analyses indicated that overexpression of miR-27a using miR-27a agomir (agomiR-27a) protected the animals from LPS-induced ALI through decreased pulmonary inflammation, decreased wet-to-dry weight ratio, and ameliorated lung histopathological changes. In addition, agomiR-27a also decreased production of inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and interleukin-1β (IL-1β) in bronchoalveolar lavage fluid (BALF). Moreover, transforming growth factor β-activated kinase 1 binding protein 3 (TAB3), as an activator of NF-κB, was confirmed as a direct target of miR-27a. Further study showed that the anti-inflammatory mechanism of miR-27a is exerted via suppression NF-κB signaling by inhibiting expression of TAB3 in LPS-induced ALI mice. Taken together, these data define the protective mechanism of miR-27a via inhibition of the inflammatory response through blocking NF-κB pathway. Therefore, miR-27a/TAB3/NF-κB axis may be therapeutically targeted to repress inflammation following ALI in the future.
Keywords: ALI, inflammation, microRNA-27a, TAB3, nuclear factor-κB (NF-κB)
Introduction
Inflammation is a type of nonspecific immune reaction that may be initiated by a complex process involving leukocytes and inflammatory mediators such as cytokines [1]. However, inflammation can often lead to the development of auto-immune diseases and organ dysfunction [2]. An important inflammatory lung disease of rapid onset is acute lung injury (ALI), which causes a high morbidity and mortality rate worldwide [3]. ALI is characterized as an acute diffuse, inflammatory lung injury, leading to increased pulmonary vascular permeability, decreased lung compliance, and loss of ae rated lung tissue leading to severe perturbations in gas exchange [4]. Although great advances in understanding the pathophysiology of ALI had been achieved, the available therapies remain unsatisfactory [5,6]. It is an urgent need to find a new therapeutic strategy for the treatment of ALI. Thus, increased understanding of molecular mechanism involved in ALI formation and development is important.
MicroRNAs (miRNAs) are highly conserved, small, noncoding RNAs that regulate protein coding gene expression by binding to the 3’-untranslated regions (UTRs) of mRNAs [7]. A large body of evidence has indicated that miRNAs are involved in the regulation of immune system development, differentiation of B and T cells, proliferation of monocytes and neutrophils, antibody production, release of inflammatory mediators and certain inflammatory lung diseases [8,9]. Recent studies show that miR-27a attenuates ischemia reperfusion-induced inflammatory damage and LPS-induced production inflammatory cytokines in microglia [10-12]. Additionally, miR-27a has been reported to play a protective role in a variety of tissue injuries [13-15]. However, whether miR-27a protects ALI by inhibiting inflammatory responses remains unclear.
Nuclear factor-kappaB (NF-κB) is a family of transcription factors that play an important role in the regulation of inflammatory processes and inhibition of NF-κB function may be useful for protection of ALI [16-19]. For example, Liao et al. reported that a novel DHA-derived mediator resolvin D1 (RvD1) attenuates lung inflammation of LPS-induced acute lung injury by suppressing NF-κB activation [20]. Furthermore, it has been reported that apigenin shows anti-inflammatory effects against LPS-induced ALI by suppressing NF-κB pathway [21]. To date, there have been no reports disclosing a regulatory mechanism of miR-27a inhibiting inflammatory responses through the NF-κB pathway in ALI. Therefore, we hypothesized that miR-27a may negatively regulate of inflammatory response via NF-κB signaling pathway in LPS-induced ALI mice model.
In the present study, we evaluate the potential role of miR-27a against ALI using a LPS-induced mice model, and we found that miR-27a significantly reduced LPS-induced ALI through inhibition of pulmonary inflammation. Moreover, miR-27a suppresses NF-κB signaling by targeting TAB3 in LPS-induced ALI mice. Therefore, these data suggest that miR-27a/TAB3/NF-κB signaling pathway may be a potential therapeutic target for ALI.
Materials and methods
Animals and ALI model
Healthy male BALB/c mice (8-10 weeks old, 18-20 g each) were purchased from Shanghai Laboratory Animal Co Ltd (SLAC, Shanghai, China). They were kept in plastic cages at 22°C with free access to pellet food and water on a 12 h light/dark cycle. Mice allowed to acclimate for 3 days before experimentation. Animal welfare and experimental procedures were strictly in accordance with the Guide for the Care and Use of Laboratory Animals. All animal experiments were approved by the Animal Care and Use Committee of the Second Affiliated Hospital of Kunming Medical University. The murine model of LPS-induced ALI was established as previous reported [29]. Briefly, all mice were randomly divided into different groups: a control group with intra-tracheal instillation of 1.5 mg/kg normal saline (NS) and ALI was induced in mice by intra-tracheal instillation of LPS. Mice were anesthetized by an intraperitoneal injection of 10% chloral hydrate and kept in a supine position while spontaneous breathing was monitored. Mice were sacrificed at the indicated time after injury. ALI induction was verified by pathological examination of the lung.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA from tissue samples, bronchoalveolar lavage fluid and cells were extracted using an RNeasy mini kit (Qiagen, Japan) for both miR-27a and TAB3 mRNA analyses according to the manufacturer’s instructions. RNA quality was measured using the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). cDNA synthesis was performed using PrimeScript RT reagent Kit (Takara, China) according to the manufacturer’s instructions. For detection of miR-27a and TAB3 mRNA expression, qPCR was performed using the miScriptSYBR®green PCR Kit (Qiagen, USA) according to the manufacturer’s protocol. The relative expression levels of genes of interest were calculated by the 2-ΔΔCt method. β-actin were used as internal controls for miRNAs. the primer for miR-27a were: 5’-ACA CTC CAG CTG GGT TCA CAG TGG CTA AG-3’ (sense) and 5’-TGG TGT CGT GGA GTC G-3’ (antisense), and their reverse primer was the universal primer supported by the miScriptSYBR®green PCR Kit (Qiagen, USA). The TAB3 mRNA forward primer was 5’-CAG CCC ACA GCT TGA TAT TC-3’ and the reverse primer was 5’-CAT GAC TTT GCC CGA GTT AG-3’. The β-actin primer forward primer was 5’-CGA GCG GGC TAC AGC TTC-3’ and the reverse primer was 5’-GTC ACG CAC GAT TCC CTC T-3’.
Microarray analysis
Total RNA was extracted from lung tissues of mice with or without LPS treatment using a miRNAeasy mini kit (Qiagen). Purity and quantity of total RNA were assessed by NanoDrop ND-1000 Spectrophotometry (Thermo Scientific, USA) and Agilent’s 2100 Bioanalyzer. The miRCURYHy3/Hy5Power labeling kit (Exiqon, Vedbaek, Denmark) was used according to the manufacturer’s guideline for miRNA labeling. Total RNA (200 ng) was labeled using the miRCURYHy3/Hy5Power labeling kit (Exiqon, Vedbaek, Denmark) according to the manufacturer’s guideline, and the Hy3TM-labeled samples were hybridized on the miRCURYTM LNA Array (v.16.0) (Exiqon) according to the manufacturer’s instruction. The feature extraction software (Agilent Technologies) was used to quantify the fluorescent intensity of each spot of microarray images, and signal intensities >10 were considered positive expression. The statistical significance of upregulated or downregulated miRNAs was analyzed by t-test. MEV software (v4.6, TIGR) was used to perform hierarchical clustering.
Cell culture and transfection
293T cells were purchased from the China Cell Culture Center (Shanghai, China) and were cultured in RPMI 1640 (Hyclone, USA) medium, supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin) (Invitrogen, China). The 293T cells were kept in an incubator in a humidified atmosphere with 5% CO2 at 37°C. miR-27a mimic and corresponding mimic negative control (mimic NC), miR-27a inhibitor and corresponding inhibitor negative control (inhibitor NC) were designed and purchased from GenePharma (Shanghai, China). These molecular products were transiently transfected into 293T cells using Lipofectamine 2000 Reagent (Invitrogen, USA) according to the manufacturer’s protocol.
Myeloperoxidase (MPO) activity assay
After BALF collection, the left upper lobe was removed, washed and kept in -80°C. Then, after weighing, the lungs were homogenized in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid containing 0.5% cetyltrimethyl ammonium bromide and subjected to three freeze-thaw cycles. The homogenate was then centrifuged (4°C, 8000 × g for 20 min). MPO activity was assayed using a commercially available ELISA kit (Invitrogen, USA). The samples were diluted in phosphate citrate buffer (pH 5.0) and the absorbance was measured at 460 nm with a microplate reader (Model 550, Bio-Rad Laboratories, USA). Results are expressed as units of MPO activity per gram of lung tissue.
Lung wet/dry weight ratio
The severity of pulmonary edema was assessed by the wet to dry ratio (W/D ratio) by the wet/dry ratio from the initial weight of the right lung middle lobe (wet weight) to its weight after desiccation at 70°C for 24 h (dry weight).
Histological examination
The left lower lung from each mouse was fixed in 10% formalin, imbedded in paraffin and sliced. Following H&E staining, pathological changes of lung tissues were observed under a light microscope (BXFM, Japan). The standard lung injury score performed by a blinded pathologist to objectively quantify the lung injury.
Cytokine assay
To obtain the BALF, ice-cold PBS was infused into the lungs two times and withdrawn each time using a tracheal cannula. The total leukocyte count was determined using a hemocytometer. BALF samples were centrifuged at 2000 rpm for 10 min at 4°C, the supernatants were stored in -80°C for analysis of cytokine concentrations. Levels of TNF-α, IL-6, IL-1β and IFN-γ in BALF were determined by ELISA kits according to the instructions recommended by the manufacturers. The optical density of each well was read at 450 nm.
Western blot analysis
Lung tissue homogenate samples and cells were harvested and were separated on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels and then transferred to polyvinylidene fluoride membranes. The membranes were blocked with 5% non-fat milk for 1 hr and then incubated overnight at 4°C with the following specific primary antibodies. The primary antibodies used include p65, IκB-α, TAB3 and β-actin (Santa Cruz Biotechnology, USA). After three times washing, the membranes were incubated in horseradish peroxidase-conjugated secondary antibody (Amersham Biosciences, USA) for 1 hr at room temperature. Then, the protein band was visualized using the ECL Western blotting substrate (Promega, USA).
Vector construction and luciferase activity assay
For luciferase reporter experiments, luciferase reporter assays were performed in 293T cells. Cells were co-transfected with psiCheck-2 reporter plasmid (Promega, USA) containing the wild type or mutant type of TAB3 3’UTR, along with mimic negative control (NC), miR-27a mimic, inhibitor NC and miR-27a inhibitor by the Lipofectamine 2000 (Invitrogen, USA). The cells were harvested 24 h after transfection, and luciferase activity was measured with a dual luciferase reporter assay kit (Promega, USA) on a luminometer (Lumat LB9507, Germany).
Statistical analysis
GraphPad Prism 5.0 software and the SPSS 16.0 software was used to conduct all the statistical analyses. The results are represented as mean ± SD (standard deviation) of at least three independent experiments. The differences between two experimental conditions were compared on a one-to-one basis using two-tailed Student’s test. Values of P<0.05 was considered to be statistically significant.
Results
miRNA expression signatures in an ALI model induced by LPS
To determine the potential involvement of miRNAs in the ALI mice after LPS-induced, we used microarray analysis to determine miRNAs levels in the lung tissues. The results showed that compared with the normal group, 48 miRNAs were identified as differentially expressed within the ALI group induced by LPS. Among them, 25 miRNAs were up-regulated, whereas 23 miRNAs were down-regulated in ALI groups. miR-27a which is one of the most dysreregulated miRNAs and plays key roles in the regulation of the inflammatory response and protection of traumatic injury [10-15]. Therefore, it was selected for further studies (Figure 1A). To verify microRNA microarray analysis findings, the abnormal expression of miR-27a was detected in mouse lung tissue, bronchoalveolar lavage fluid (BALF), and splenocytes by qRT-PCR assay. We found that the expression level of miR-27a was significantly decreased in the lung tissue of ALI mice (Figure 1B, P<0.01). Furthermore, miR-27a expression was also down-regulated in BALF and splenocytes of ALI mice (Figure 1C and 1D, P<0.01). Taken together, these findings may indicate an important role for miR-27a in the progression of ALI.
Figure 1.
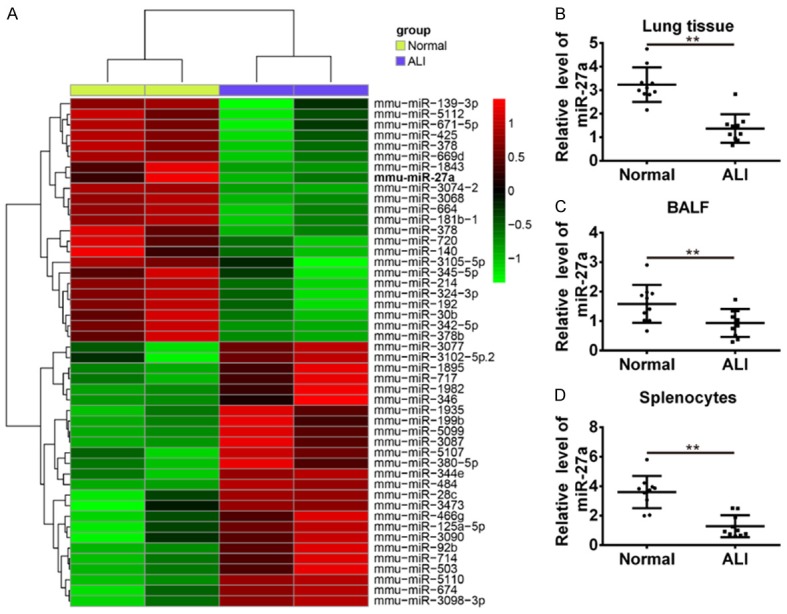
Screening of differentially expressed miRNAs in the murine ALI model. A. microRNA microarray was conducted with the paired lung samples from four mice of ALI group and normal group. Both down-regulated (green) and up-regulated (red) miRNAs are shown. B. Levels of miR-27a were determined by qRT-PCR in lung tissues of LPS-induced ALI mice or normal mice. C. Levels of miR-27a in BLAF was determined in LPS-induced ALI mice or normal mice. D. Levels of miR-27a were determined by qPCR in splenocytes of LPS-induced ALI mice or normal mice. Data are showed as mean ± SD (n = 3). **P<0.01 vs. normal group.
agomiR-27a attenuates LPS-induced lung injury
To access the effect of miR-27a-based strategy on the treatment of ALI, groups of mice were challenged with LPS and agomiR-27a. To assess the pathological changes, HE staining and lung injury score system were used in our study. In the LPS group, HE staining of lung tissue showed significant inflammatory exudates, fibroblastic foci and distortion of the structural damage of lung tissue. However, after agomiR-27a treatment, the lung injury scores were significantly reduced than LPS group (Figure 2A). We also investigated the effect of overexpression of miR-27a on lung vascular permeability. As determined by Evans blue dye extravasation, LPS challenged mice showed the significant increase in lung microvascular permeability compared with the control group. However, such increase was significantly blocked by agomiR-27a treatment, evidenced by reduced Evans blue content in lung tissue compared with LPS group (Figure 2B). Subsequently, the lung wet/dry weight ratio in the lung tissues was analyzed. The lung wet/dry ratio in the lung tissues were significantly increased after the LPS challenge compared with the control group. However, agomiR-27a post-treatment group obviously attenuated the wet/dry ratio than LPS group in lung tissues (Figure 2C). In addition, we found that MPO activity in the ALI model group was markedly higher than control group. However, agomiR-27a post-treatment group obviously reduced the MPO activity ratio than LPS group in lung tissues (Figure 2D). These data suggest that overexpression of miR-27a reduced the ALI damage to the lung tissue.
Figure 2.
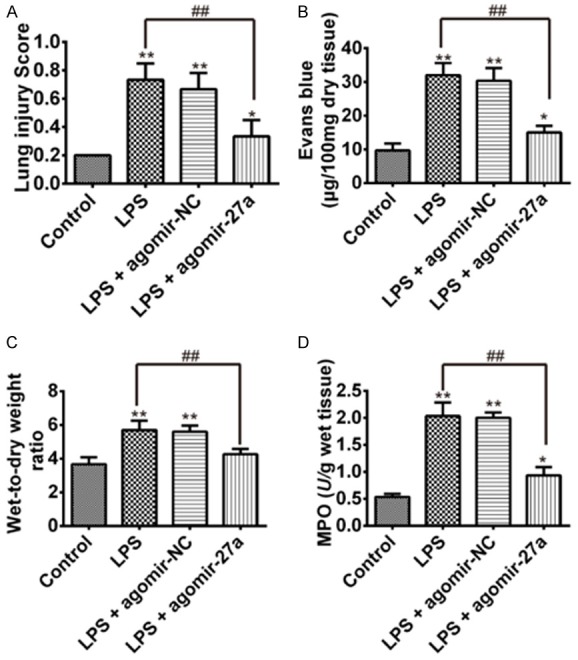
Effects of miR-27a on LPS-induced ALI. Three days after LPS injection with agomiR-27a or agomiR-NC treatments, mice were sacrificed and their lungs were removed, lung injury score, Evans blue (EB), wet-to-dry ratio and MPO activity was detected. A. Effects of agomiR-27a on lung injury score was measured via HE stain. B. Effects of agomiR-27a on EB content in lungs. C. Effects of agomiR-27a on lung wet-to-dry ratio in lungs. D. Effects of agomiR-27a on MPO activity in lungs. Data are shown as mean ± SD (n = 3). *P<0.05 **P<0.01 vs. Control group; ##P<0.01 vs. LPS-treated mice group.
agomiR-27a down-regulated the LPS-induced production of pro-inflammatory cytokines in BALF
To examine whether miR-27a plays a role in the LPS-induced inflammatory response, we further detected the pro-inflammatory cytokines including TNF-α, IL-6, IL-1β and IFN-γ in BALF by ELISA. We found that TNF-α, IL-6, IL-1β and IFN-γ in BALF in the mice with LPS injection were markedly higher than the mice in control group. However, up-regulation of miR-27a resulted in reduced the TNF-α, IL-6, IL-1β and IFN-γ level in BALF in the LPS-induced ALI mice with agomiR-27a injections (Figure 3A-D). These results suggest that agomiR-27a treatment attenuates LPS induced inflammation response in lung.
Figure 3.
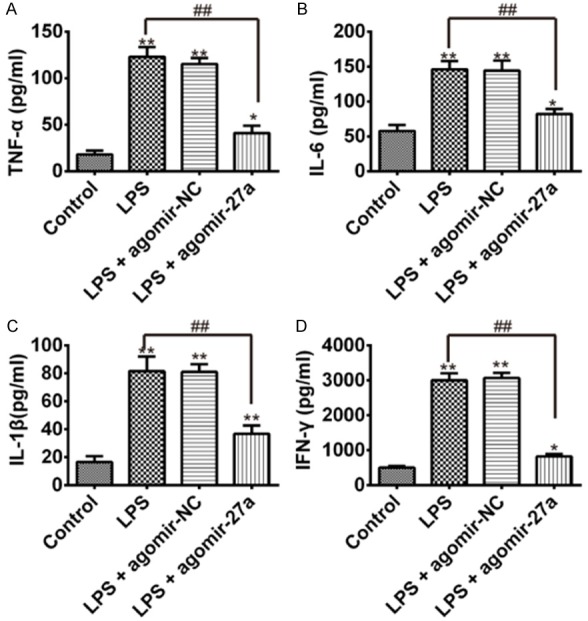
agomiR-27a down-regulates TNF-α, IL-6, IL-1β and IFN-γ in BALF. Three days after LPS injection with agomir-NC or agomiR-27a treatments, mice were sacrificed, their lungs were lavaged, and the BALF were collected. TNF-α, IL-6, IL-1β and IFN-γ were detected by ELISA. A. TNF-α concentration in BALF. B. IL-6 concentration in BALF. C. IL-1β concentration in BALF. D. IFN-γ concentration in BALF. Data are showed as mean ± SD (n = 3). *P<0.05 **P<0.01 vs. Control group; ##P<0.01 vs. LPS alone group.
TAB3 is a target of miR-27a
To explore the underlying mechanism of miR-27a in regulating inflammation response in ALI mice, the predicted target genes of miR-27a were screened by TargetScan and RNAhybrid algorithms assay, and we found that TAB3, the upstream positive regulator of the NF-κB pathway [22], is a potential target for miR-27a since there was a putative miR-27a binding sites within the 3’UTR of TAB3 mRNA (Figure 4A). To test whether TAB3 is a target of miR-27a, we constructed luciferase reporter plasmids with WT/Mut TAB3 3’UTR. Next, these constructs were transfected into 293T cells with miR-27a mimic or miR-27a inhibitor. Luciferase activity indicated that transfection of miR-27a mimics in the wild-type group efficiently reduced expression of the luciferase reporter compared with transfection of mimics NC, but transfection of miR-27a inhibitor in the wild-type group efficiently increased the expression of the luciferase reporter compared with transfection of inhibitor NC. However, there was no effect was observed in the mut-type group, suggesting that miR-27a efficiently controls TAB3 expression direct targeting the 3’UTR of TAB3 mRNA (Figure 4B). Furthermore, qRT-PCR results showed that transfected with miR-27a mimics could decrease TAB3 mRNA level compared with control and mimics NC group, in contrast, miR-27a inhibitor could increase TAB3 mRNA level compared with inhibitor NC group (Figure 4C). Western blotting results showed that transfected with miR-27a mimics could decrease TAB3 expression on protein level compared with mimics NC group, in contrast, transfected with miR-27a inhibitor could increase TAB3 expression on protein level compared with inhibitor NC group (Figure 4D). These data show that miR-27a can downregulate TAB3 expression by directly targeting its 3’UTR, suggesting miR-27a may inhibit inflammatory responses through TAB3/NF-κB signaling.
Figure 4.
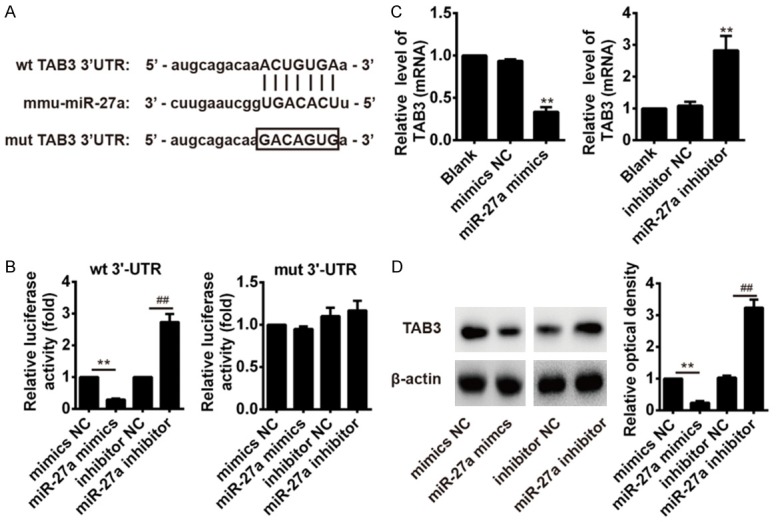
miR-27a targets NF-κB signaling pathway member TAB3. A. The target regions of the miR-27a in TAB3 was predicted using Targetscan. B. A luciferase reporter plasmid containing wild TAB3 3’UTR or mutant TAB3 3’UTR was transfected into 293T cells alone, together with miR-27a mimic, inhibitor or controls and luciferase activity was measured. C. qRT-PCR detection of TAB3 mRNA expression after administration of miR-27a mimics or miR-27a inhibitor. D. Western blot detection of TAB3 protein expression after administration of miR-27a mimics or miR-27a inhibitor in. Data are shown as mean ± SD (n = 3). **P<0.01 vs. mimics control group; ##P<0.01 vs. inhibitor control group.
Effects of miR-27a on activation of NF-κB in LPS-induced ALI mice
As mentioned above, TAB3 is the upstream positive regulator of the NF-κB pathway, and NF-κB signaling pathway plays an important role in the regulation of inflammatory cascade. We therefore examined the effects of miR-27a on the activation of NF-κB signaling pathways which mediated immune responses in LPS-induced ALI mice. Western blot was performed to investigate the levels of TAB3, NF-κB p65 and IκB-α in mouse lung tissues. As shown in Figure 5, after LPS administration, the expression of TAB3 and NF-κB p65 in lung tissues markedly increased compared with control group, in contrast, LPS markedly decreased the protein expression of IκB-α compared with control group. However, the pre-treatment with agomiR-27a significantly decreased TAB3 and NF-κB p65 expression in lung tissues compared with LPS + agomiR-NC group, in contrast, significantly increased the protein expression of IκB-α compared with LPS + agomiR-NC group. Thus, miR-27a suppresses NF-κB signaling by inhibiting the expression of TAB3 in LPS-induced ALI mice model. These data suggested that the protective mechanism of miR-27a may be attributed partly to decreased production of pro-inflammatory cytokines through the TAB3/NF-κB signaling pathway.
Figure 5.
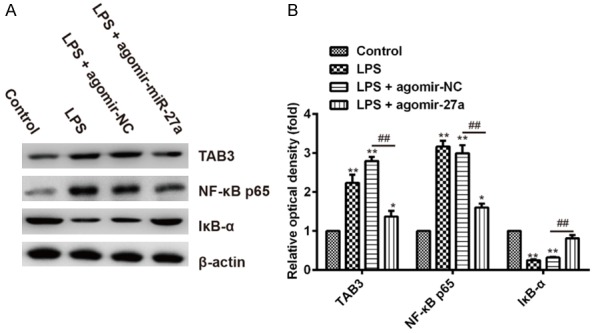
miR-27a suppressed NF-κB pathway in lung tissues of LPS-induced ALI mice. A. The protein levels of TAB3, NF-κB p65 and IκB-α were assessed by Western blotting. β-actin were used as internal control. B. Quantification of protein expression was normalized to internal control using a densitometer. Data are shown as mean ± SD (n = 3). *P<0.05 **P<0.01 vs. Control group; ##P<0.01 vs. LPS + agomir-NC group.
Discussion
ALI is a severe inflammatory disease with high morbidity and mortality rates worldwide, however there are no effective drugs in the clinic treatment [3]. Therefore, find out a novel approach to understand the molecular mechanism involved in ALI is in urgent need. It is widely known that an imbalance of inflammatory responses plays an important role in the development of ALI. An increase in the levels of pro-inflammatory cytokines such as TNF-α, IL-6, IL-1β and IFN-γ have been documented to aggravate ALI severity [23,24]. However the underlying mechanisms of excessive inflammatory response during the progression of ALI are unknown.
As a newly discovered gene regulator, microRNAs have drawn increasing attention for possible mediation of immune response in the ALI progression. For example, miR-155 has been identified as a pro-inflammatory factor in LPS-induced ALI mice, and overexpression of miR-155 remarkably exaggerated LPS-induce acute lung injury [25]. It has been reported that miR-19 inhibited the LPS-induced inflammatory response in mice via targeting p47phox which is an important molecule in ROS-derived activating of inflammatory response [26]. In the present study, microRNA microarray revealed a multitude of microRNAs differentially expressed in LPS-induced ALI mice, among them, miR-27a was the most obvious down-regulated miRNA. In recent studies, miR-27a was found to be linked to cellular immunity in various diseases. For instances, miR-27a significantly decreased the production of inflammatory mediators during LPS-induced microglial activation, suggesting miR-27a may be an appropriate therapeutic target for various neuronal damage disease [10]. A previous study revealed an important role of miR-27a on relieving pulmonary inflammation and promoting survival rate in septic mice [27]. Xie et al. reported that miR-27a negatively regulates inflammatory response of LPS-stimulated macrophages by targeting IL-10 [28]. However, a functional role and the underlying molecular mechanism of miR-27a in ALI have never been reported. In the present study, we found that treatment with agomiR-27a could decrease the LPS-induced pulmonary inflammation, wet to dry weight ratio, and MPO activity. Furthermore, we also found that treatment with agomiR-27a significant attenuated TNF-α, IL-6, IL-1β and IFN-γ expression in BALF after LPS challenge. These results suggested that the protective effects of agomiR-27a on LPS-induced ALI are exerted through anti-inflammatory response, suggesting that agomiR-27a treatment may serve as a new therapeutic approach in human ALI.
To assess the anti-inflammatory mechanism of miR-27a in LPS-induced ALI, the target genes of miR-27a was predicted using bioinformatics methods. We found that TAB3 a constituent of the NF-κB pathway is a potential target gene of miR-27a. It is well known that NF-κB is an important transcription factor in inflammatory responses through regulating the production of pro-inflammatory cytokines [16-19]. Previous studies have demonstrated that NF-κB pathway played a pivotal role in ALI disease and restraining NF-κB pathway is a potential strategy for ALI treatment. For example, Li et al. reported that pretreatment with ulinastatin significantly attenuated LPS induced ALI in mice. The protective mechanism of ulinastatin may be attributed to decreased production of pro-inflammatory cytokines through blocking NF-κB signaling pathway [29]. Zhu et al. reported that andrographolide treatment reduced the severity of LPS-induced ALI by virtue of andrographolide-mediated inhibition of IKKβ/NF-κB activation [30]. Since TAB3 is essential for the activation of the NF-κB pathway, we suspected that the anti-inflammatory effects of agomiR-27a in LPS-induced ALI mice are exerted through inhibition NF-κB activity via targeting TAB3. Further, our results confirmed that NF-κB signaling is activated in LPS-induced ALI mice. However, the pre-treatment with agomiR-27a significantly decreased TAB3 and NF-κB p65 expression in lung tissues, suggesting blocked the NF-κB pathway. These data suggest that the protective mechanism of miR-27a may be attributed partly to decreased production of pro-inflammatory cytokines through the TAB3/NF-κB signaling pathway.
In summary, the present study identified a list of differentially expressed microRNAs that may contribute to LPS-induced ALI. Among these identified microRNAs, miR-27a functions as an anti-inflammatory factor in LPS-induced ALI via suppressing the TAB3/NF-κB pathway. Our findings could expand our understanding of the effect of miR-27a on the development of ALI, and lead to miR-27a treatment as a novel approach for ALI patients.
Acknowledgements
This work was supported by the Research Institute Project of Yunnan Province (No. 2017NS291).
Disclosure of conflict of interest
None.
References
- 1.Feghali CA, Wright TM. Cytokines in acute and chronic inflammation. Front Biosci. 1997;2:d12–26. doi: 10.2741/a171. [DOI] [PubMed] [Google Scholar]
- 2.Schottenfeld D, Beebe-Dimmer J. Chronic inflammation: a common and important factor in the pathogenesis of neoplasia. CA Cancer J Clin. 2006;56:69–83. doi: 10.3322/canjclin.56.2.69. [DOI] [PubMed] [Google Scholar]
- 3.Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, Guest CB, Mazer CD, Mehta S, Stewart TE, Kudlow P, Cook D, Slutsky AS, Cheung AM Canadian Critical Care Trials Group. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. 2011;364:1293–1304. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- 4.Tsushima K, King LS, Aggarwal NR, De Gorordo A, D’Alessio FR, Kubo K. Acute lung injury review. Intern Med. 2009;48:621–630. doi: 10.2169/internalmedicine.48.1741. [DOI] [PubMed] [Google Scholar]
- 5.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 6.Mendez JL, Hubmayr RD. New insights into the pathology of acute respiratory failure. Curr Opin Crit Care. 2005;11:29–36. doi: 10.1097/00075198-200502000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 8.Zhou T, Garcia JG, Zhang W. Integrating microRNAs into a system biology approach to acute lung injury. Transl Res. 2011;157:180–190. doi: 10.1016/j.trsl.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- 10.Lv YN, Ou-Yang AJ, Fu LS. MicroRNA-27a negatively modulates the inflammatory response in lipopolysaccharide-stimulated microglia by targeting TLR4 and IRAK4. Cell Mol Neurobiol. 2017;37:195–210. doi: 10.1007/s10571-016-0361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li XQ, Lv HW, Wang ZL, Tan WF, Fang B, Ma H. MiR-27a ameliorates inflammatory damage to the blood-spinal cord barrier after spinal cord ischemia: reperfusion injury in rats by downregulating TICAM-2 of the TLR4 signaling pathway. J Neuroinflammation. 2015;12:25. doi: 10.1186/s12974-015-0246-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng Y, Du L, Jiao H, Zhu H, Xu K, Guo S, Shi Q, Zhao T, Pang F, Jia X, Wang F. Mmu-miR-27a-5p-Dependent upregulation of MCPIP1 inhibits the inflammatory response in LPSInduced RAW264.7 macrophage cells. Biomed Res Int. 2015;2015:607692. doi: 10.1155/2015/607692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai Q, Wang T, Yang WJ, Fen X. Protective mechanisms of microRNA-27a against oxygenglucose deprivation-induced injuries in hippocampal neurons. Neural Regen Res. 2016;11:1285–1292. doi: 10.4103/1673-5374.189194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L, Zhao M, Wang Y, Liu A, Lv M, Li Y, Yang X, Wu Z. Neuroprotective effects of miR-27a against traumatic brain injury via suppressing FoxO3a-mediated neuronal autophagy. Biochem Biophys Res Commun. 2017;482:1141–1147. doi: 10.1016/j.bbrc.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Z, Wan J, Hou X, Geng J, Li X, Bai X. MicroRNA-27a promotes podocyte injury via PPARgamma-mediated beta-catenin activation in diabetic nephropathy. Cell Death Dis. 2017;8:e2658. doi: 10.1038/cddis.2017.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumazawa Y, Kawaguchi K, Takimoto H. Immunomodulating effects of flavonoids on acute and chronic inflammatory responses caused by tumor necrosis factor alpha. Curr Pharm Des. 2006;12:4271–4279. doi: 10.2174/138161206778743565. [DOI] [PubMed] [Google Scholar]
- 17.Jing W, Chunhua M, Shumin W. Effects of acteoside on lipopolysaccharide-induced inflammation in acute lung injury via regulation of NF-kappaB pathway in vivo and in vitro. Toxicol Appl Pharmacol. 2015;285:128–135. doi: 10.1016/j.taap.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 18.Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L622–L645. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 20.Liao Z, Dong J, Wu W, Yang T, Wang T, Guo L, Chen L, Xu D, Wen F. Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARgamma/NF-kappaB pathway. Respir Res. 2012;13:110. doi: 10.1186/1465-9921-13-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Liu YT, Xiao L, Zhu L, Wang Q, Yan T. Anti-inflammatory effects of apigenin in lipopolysaccharide-induced inflammatory in acute lung injury by suppressing COX-2 and NF-κB pathway. Inflammation. 2014;37:2085–2090. doi: 10.1007/s10753-014-9942-x. [DOI] [PubMed] [Google Scholar]
- 22.Cheung PC, Nebreda AR, Cohen P. TAB3, a new binding partner of the protein kinase TAK1. Biochem J. 2004;378:27–34. doi: 10.1042/BJ20031794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strieter RM, Kunkel SL. Acute lung injury: the role of cytokines in the elicitation of neutrophils. J Investig Med. 1994;42:640–651. [PubMed] [Google Scholar]
- 24.Shinbori T, Walczak H, Krammer PH. Activated T killer cells induce apoptosis in lung epithelial cells and the release of pro-inflammatory cytokine TNF-alpha. Eur J Immunol. 2004;34:1762–1770. doi: 10.1002/eji.200425097. [DOI] [PubMed] [Google Scholar]
- 25.Wang W, Liu Z, Su J, Chen WS, Wang XW, Bai SX, Zhang JZ, Yu SQ. Macrophage micro-RNA-155 promotes lipopolysaccharide-induced acute lung injury in mice and rats. Am J Physiol Lung Cell Mol Physiol. 2016;311:L494–506. doi: 10.1152/ajplung.00001.2016. [DOI] [PubMed] [Google Scholar]
- 26.Wang T, Liu YP, Wang T, Xu BQ, Xu B. ROS feedback regulates the microRNA-19-targeted inhibition of the p47phox-mediated LPS-induced inflammatory response. Biochem Biophys Res Commun. 2017;489:361–368. doi: 10.1016/j.bbrc.2017.05.022. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Ruan Z, Mao Y, Dong W, Zhang Y, Yin N, Jiang L. miR-27a is up regulated and promotes inflammatory response in sepsis. Cell Immunol. 2014;290:190–195. doi: 10.1016/j.cellimm.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 28.Xie N, Cui H, Banerjee S, Tan Z, Salomao R, Fu M, Abraham E, Thannickal VJ, Liu G. miR-27a regulates inflammatory response of macrophages by targeting IL-10. J Immunol. 2014;193:327–334. doi: 10.4049/jimmunol.1400203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W, Qiu X, Jiang H, Zhi Y, Fu J, Liu J. Ulinastatin inhibits the inflammation of LPS-induced acute lung injury in mice via regulation of AMPK/NF-kappaB pathway. Int Immunopharmacol. 2015;29:560–567. doi: 10.1016/j.intimp.2015.09.028. [DOI] [PubMed] [Google Scholar]
- 30.Zhu T, Wang DX, Zhang W, Liao XQ, Guan X, Bo H, Sun JY, Huang NW, He J, Zhang YK, Tong J, Li CY. Andrographolide protects against LPS-induced acute lung injury by inactivation of NF-kappaB. PLoS One. 2013;8:e56407. doi: 10.1371/journal.pone.0056407. [DOI] [PMC free article] [PubMed] [Google Scholar]