

Abstract
Myocardial fibrosis is an important pathological feature of diabetic cardiomyopathy (DCM) and endothelial-to-mesenchymal transition (EndMT) is an essential process for myocardial fibrosis. Recent studies have demonstrated an association between miRs and DCM. Therefore, the aim of this study is to investigate the role and the mechanism of miRNAs in the process of EndMT. We simulated the conditions occurring in EndMT by application of high glucose in primary human aortic endothelial cells (HAECs). Firstly, we compared the expression profiles of miRNAs in HAECs with or without HG treatment using microarray. Then, after addition of miR-142-3p mimics, the expression levels of EndMT markers were assessed by qRT-PCR and Western Blot. Moreover, bioinformatics analysis and luciferase assay were used to confirm the direct regulation of miR-142-3p to TGF-β1. Furthermore, the role of TGF-β1 in the inhibitory effect of miR-142-3p on EndMT was evaluated. In addition, the expressions of TGF-β1/Smad signaling signatures were measured by Western Blot. MiR-142-3p screened by miRNA microarray was significantly down-regulated in HAECs under HG stimulation in a dose and time dependent manner. Subsequently, we found that overexpression of miR-142-3p could inhibit HG-induced EndMT, as evidenced by decreased α-SMA and vimentin expression, and increased CD31 and VE-cadherin expression. Of note, transforming growth factor beta 1 (TGF-β1), one of the molecular mediators implicated in the progression of EndMT, was confirmed to be downstream target gene of miR-142-3p in HAECs. Moreover, TGF-β1 overexpression remarkably abolished the inhibitory effects of miR-142-3p overexpression on HG induced EndMT. Finally, miR-142-3p also mediated its anti-EndMT action by inactivation of TGF-β1/Smad pathway, as demonstrated by downregulation of TGF-β1, phospho-Smad2 and phospho-Smad2. Our findings demonstrated that miR-142-3p could attenuate HG-induced EndMT in HAECs, the mechanism of which may be at least partly through blocking TGF-β1/Smad signaling pathway. This might provide a potential therapeutic target for DCM in future.
Keywords: Diabetic cardiomyopathy, EndMT, miR-142-3p, TGF-β1/Smad signaling pathway
Introduction
Diabetic cardiomyopathy (DCM) is a serious cardiac dysfunction occurring in the absence of coronary artery disease and hypertension, which is characterized by dilatation and hypertrophy of the left ventricle, fetal gene reactivation, and lipid accumulations in cardiac cells [1]. There is growing evidence indicated that myocardial fibrosis is an important pathological process of DCM [2,3]. Of note, endothelial-to-mesenchymal transition (EndMT) has been shown to contribute significantly to myocardial fibrosis and remodeling [4-6]. The prevention of EndMT could therefore serve a promising new therapeutic strategy for DCM. However, the mechanisms of regulating EndMT are not currently fully understood and remain to be elucidated.
MicroRNAs (miRNAs) is a group of short noncoding RNAs, which inhibit or destabilize translation of the transcripts by binding to the 3’-untranslated region (3’-UTR) of target gene mRNA [7,8]. They have critical roles in a wide range of biological processes, such as cellular proliferation, apoptosis, cell cycle control and metabolism [9]. And, kinds of miRNAs have been confirmed to participate in the pathogenesis of DCM. For example, Li et al. showed that miR-30d regulated cardiomyocyte pyroptosis by directly targeting foxo3a in DCM [10]. Feng et al. showed that miR-200b overexpression also prevented diabetes-induced cardiac functional and structural changes [11]. Recently, aberrant expression of miRNAs was found to be involved in EndMT-related diseases [12]. For example, miR-21 was found to prevent TGF-β-induced EndMT via the Akt pathway in cardiac fibrosis [13]. However, to date there have only been preliminary studies on the role of miRNAs in regulating EndMT in DCM.
In the present study, we explored the expression and function of miRNAs in HG-induced EndMT cell model. Moreover, the interactions among miR-142-3p and TGF-β1/Smad signaling were also studied in order to reveal the underlying mechanisms of miR-142-3p in the participation of HG-induced EndMT process. Our findings may provide a novel target for the treatment of DCM.
Materials and methods
Cell culture and treatment
HAECs were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). HAECs were cultured in Endothelial Cell Growth Medium BulletKit-2 (EGM-2, Lonza, USA) containing 10% fetal bovine serum (FBS; Life Technologies, Inc.) and 1% penicillin/streptomycin mix (Gibco; Life Technologies, Lofer, AU) at 37°C. Cells grown overnight in medium containing 2% FBS were used for experiments. HAECs were exposed to normal glucose (5 mM) and high glucose [14] (10, 20, 30 and 40 mM) for 0, 1, 2, 3, 4, and 5 days, respectively. Some of the cells that were exposed to HG (30 mM) were incubated for 48 h for further expriments.
Microarray analysis
Total RNA was extracted from HAECs with or without 48 h HG treatment using a miRNAeasy mini kit (Qiagen). Purity and quantity of total RNA were assessed by NanoDrop ND-1000 Spectrophotometry (Thermo Scientific, USA) and Agilent’s 2100 Bioanalyzer. Total RNA (200 ng) was labeled using the miRCURYHy3/Hy5Power labeling kit according to the manufacturer’s guideline, and the Hy3TM-labeled samples were hybridized on the miRCURYTM LNA Array (v.16.0) (Exiqon) according to the manufacturer’s instruction. Data was analyzed using Genespring software (Agilent Technologies, USA).
Observations with adjusted P-values ≥ 0.05 were removed, and thus excluded from further analysis. The heat map of the 53 microRNAs most obvious differences was created using a method of hierarchical clustering by GeneSpring GX, version 7.3 (Agilent Technologies, California, United Stages).
Quantitative RT-PCR analyses
MiRNA was prepared using a miRNAeasy mini kit (Qiagen) and total RNA was prepared using Trizol Reagent (Life Technologies) according to the manufacturer’s protocol. For miRNA reverse transcription, cDNA was synthesized using a miRNA reverse transcription kit (Qiagen, Valencia, CA, USA). For mRNA reverse transcription, cDNA was synthesized using the Takara PrimeScriptTM First Strand cDNA Synthesis kit (Takara Bio, Inc., Dalian, China). Real-time PCR for miRNA and mRNA were performed on an ABI Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.) with SYBR Green Real-time PCR Master Mix (Toyobo, Japan). Relative quantification was determined by normalization to U6 or GAPDH. Real-time PCR primers used for GAPDH are (forward: 5’-AGGTCGGTG TGAACGGATTTG-3’, reverse: 5’-TGTAGACCATGTAGTTGAGGTCA-3’); α-SMA (forward: 5’-ATAACATCAAGCCCAAATCTGC-3’, reverse: 5’-TTCCTTTTTTCTTTCCCAACA-3’); Vimentin (forward: 5’-ACCGCAACAACGCCATCTATG-3’, reverse: 5’-GCACTGCTTCCCGAATGTC-3’); CD31 (forward: 5’-ACCGGGTGCTGTTCTATAAGG-3’, reverse: 5’-CACCTTGGGCTTGGATACGC-3’); VE-cadherin (forward: 5’-CGAGGACAGCAACTTCACCC-3’, reverse: 5’-CTCCCGATTAAACTGCCCAT-3’); miR-142-3p (forward: 5’-CGCCGTGTAGTGTTTCCTAC-3’, reverse: 5’-GTTTAATTTATCTTTTCCACCCA-3’); U6 (forward: 5’-CTCGCTTCGGCCGT-3’). The qRT-PCR assays were performed in triplicate and the change in expression level was calculated using the 2-ΔΔCt method [15].
Cell transfection
The miR-142-3p mimics, mimics negative control (mimics NC), miR-142-3p inhibitor, and inhibitor NC were bought from GenePharm (Shanghai, China). In addition, the coding domain sequence of TGF-β1 mRNA was amplified by PCR, and inserted into pcDNA 3.0 vector to enhance its expression (Invitrogen, Grand Island, NY, USA), named as pcDNA-TGF-β1. Transfection was performed using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) following manufacturer’s instructions. The transfected HAECs were then stimulated with HG (30 mM) for 24 h in replaced EGM-2 medium and were supplemented with 10% FBS. Culture supernatants were collected and then indicated for Western Blot and qRT-PCR.
Luciferase reporter assay
3’-UTR of TGF-β1 and the mutated sequence were inserted into the pGL3 control vector (Promega Corporation, Madison, WI, USA) to construct wt TGF-β1-3’-UTR vector and mutant TGF-β1-3’-UTR vector, respectively. For luciferase reporter assay, HEK293 cells were transfected with the corresponding vectors; 48 hours after transfection, the dual-luciferase reporter assay system (Promega, Shanghai, the People’s Republic of China) were used to measure the luciferase activity. All experiments were performed in triplicate.
Western blot
Total protein was extracted using radio immunoprecipitation assay (RIPA) lysis buffer (Beyotime Biotechnology, Shanghai, China). Concentrations of total cellular protein were determined using a BCA assay kit (Pierce, Rockford, IL, USA). Total protein samples (40 μg) were analyzed by 8% SDS-PAGE gel and transferred to polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Freiburg, DE) by electroblotting. Primary antibodies against TGF-β1 (Santa Cruz Biotechnology, 1:1000 dilution), fibronectin (Abcam, 1:1,000 dilution), fibroblast-specific protein-1 (FSP-1) (Abcam, 1:1,000 dilution), VE-cadherin (Cell Signaling Technology, 1:1,000 dilution), CD31 (Abcam, 1:1,000 dilution), α-smooth muscle actin (Thermo Fisher Scientific, 1:1,000 dilution), vimentin (Santa Cruz Biotechnology, 1:2,000 dilution), phospho-Smad2 (Cell Signaling Technology, 1:1,000 dilution), Smad2 (Cell Signaling Technology, 1:1,000 dilution), phospho-Smad3 (Cell Signaling Technology, 1:1,000 dilution), Smad3 (Cell Signaling Technology, 1:1,000 dilution) and β-actin (Santa Cruz Biotechnology, 1:2000 dilution) were probed with proteins on the membrane at 4°C overnight. After incubating with secondary antibodies (1:10000, Cell Signaling Technology, Danvers, MA), Bands were detected by enhanced chemiluminescence (ECL) kit (GE Healthcare, Freiburg, DE). The intensity of the bands of interest was analyzed by ImageJ software (Rawak Software, Inc. Munich, Germany).
Statistical analyses
Statistical analyses were performed with SPSS 13.0 software (SPSS, Chicago, IL, USA). Data are expressed as the mean ± standard deviation of 3 independent experiments. One-way analysis of variance (ANOVA) or two-tailed Student’s t-test was used for comparisons between groups. P < 0.05 was considered to indicate a statistically significant difference.
Results
miR-142-3p was involved in HG induced EndMT in HAECs
As already known, HAECs is a commonly used cell model that simulated the conditions occurring in EndMT during myocardial fibrosis [16-18]. In the present study, we first established the cell model in accordance with previous descriptions [16]. As shown in Figure 1A, HAECs gained a fibroblast-like phenotype after treatment with HG for 5 days. We also observed that HAECs became polygonal or short type and the cells had cobble-like arrangement under the optical inverted microscope. It is well known that EndMT is characterized by loss of endothelial markers and acquisition of mesenchymal markers [4]. Western Blot analysis showed that HG induced EndMT in HAECs, as evidenced by the increased expression of mesenchymal markers (α-SMA, FSP-1, vimentin, and FN protein) and decreased expression of endothelial markers (CD31 and VE-cadherin protein) (Figure 1B).
Figure 1.
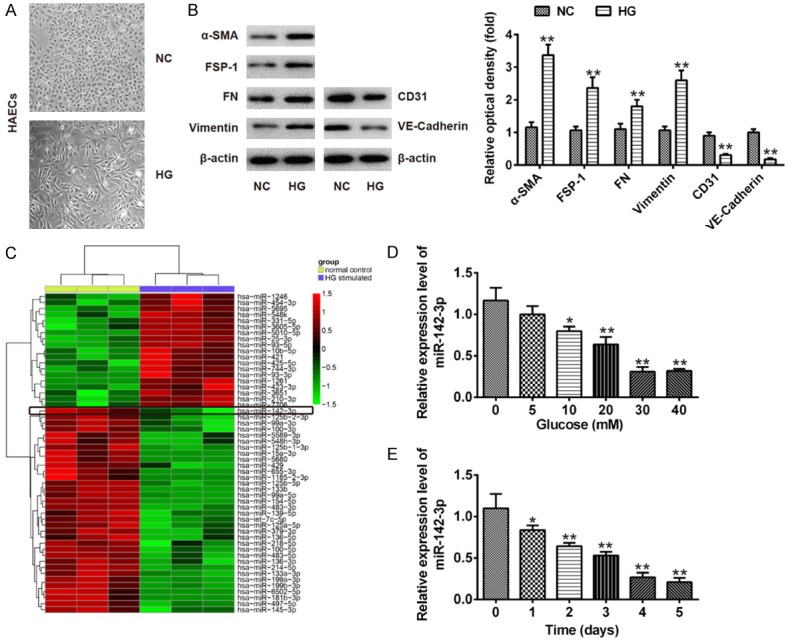
miR-142-3p was involved in HG induced EndMT in HAECs. A. Morphological changes in HAECs after HG treatment. B. The protein expression of mesenchymal markers (α-SMA, FSP-1, vimentin, FN) and endothelial markers (CD31 and VE-cadherin) were determined by Western Blot. Data represent the mean ± SD of three independent experiments. **P < 0.01 vs. NC group. NC: 5 mmol/l glucose, HG: 30 mmol/l glucose. C. Heatmap of normalized expression levels of miRNAs in HAECs treated with/without HG. Green indicates low expression levels; red indicates high expression levels. D. qRT-PCR was performed to determine the expression levels of miR-142-3p in HAECs pre-treated for 5 days with 0, 10, 20, 30, or 40 mM HG. Data represent the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01. E. qRT-PCR was performed to determine the expression levels of miR-142-3p in HAVECs pre-treated with 30 mM HG for 1, 2, 3, 4 or 5 days. Data represent the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01.
To explore the role of miRNAs in the process of EndMT, we analyzed the expression profiles of miRNAs in HAECs with or without HG treatment using microarray. As shown in Figure 1C, we found that the expression levels of 53 miRNAs potentially involved in EndMT were dysregulated in the HG group, including 34 down-regulated miRNAs and 19 up-regulated miRNAs. MiR-142-3p was identified as being one of the most down-regulated miRNAs among these aberrantly expressed miRNAs. It has previously been reported that miR-142-3p was downregulated in the animal model for DCM [19], which is consistent with our results. For these reasons, miR-142-3p was chosen as the candidate for further study.
To clarify the expression of miR-142-3p in HG-induced EndMT in HAECs, HAECs were stimulated with HG at different concentrations (5-40 mM) for different times (1-5 days). The results of qRT-PCR revealed that the expression of miR-142-3p was decreased in a dose- and time-dependent manner (Figure 1D, 1E). All data indicate that miR-142-3p may involve in HG-induced EndMT process.
Overexpression of miR-142-3p inhibited HG-induced EndMT in HAECs
To explore the effect of miR-142-3p on HG-induced EndMT, HAECs were transfected with miR-142-3p mimics and mimics NC for 24 h, and then incubated with HG for 5 days, after which the mRNA expression levels of mesenchymal markers (α-SMA, and vimentin) and endothelial markers (CD31 and VE-cadherin) were measured by qRT-PCR. Compared with NC group, HG significantly increased the expression levels of α-SMA and vimentin, but decreased the expression levels of CD31 and VE-cadherin, confirming EndMT. However, miR-142-3p overexpression attenuated the promoting effects of HG on the expression of α-SMA and vimentin and reversed the inhibitory effect of HG on the expression of CD31 and VE-cadherin (Figure 2A-D). The protein levels of α-SMA and vimentin, as well as of CD31 and VE-cadherin showed similar changes (Figure 2E). In addition, we observed that HAECs gained a fibroblast-like phenotype after treatment with HG, whereas the cells kept a cobble stone-like pattern when the cells were transfection with miR-142-3p mimics, followed by HG stimulation (Data not shown). These results suggest that overexpression of miR-142-3p inhibited HG-induced EndMT in HAECs.
Figure 2.
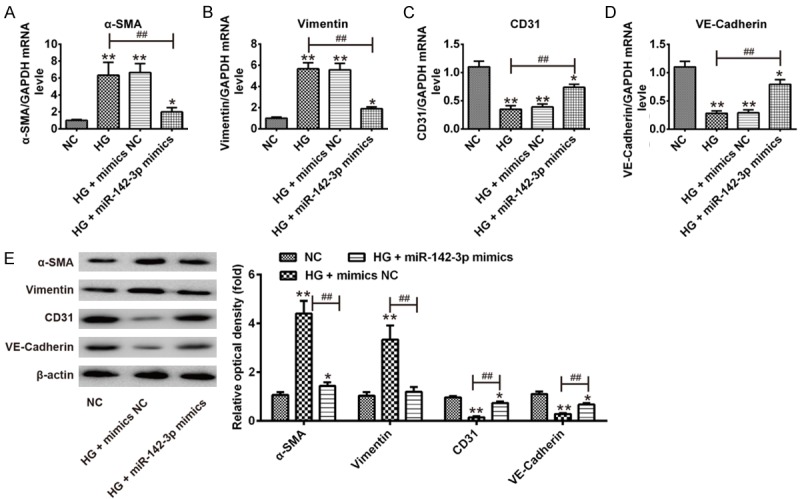
Overexpression of miR-142-3p inhibited HG-induced EndMT in HAECs. HAECs were transfected with miR-142-3p mimics and mimics NC for 24 h, and then incubated with HG for 5 days, after which the cells were harvested for subsequent experiment. A-D. The mRNA expression levels of α-SMA, vimentin, CD31 and VE-cadherin were measured by qRT-PCR. E. The protein expression levels of α-SMA, vimentin, CD31 and VE-cadherin were measured by Western Blot. Data represent the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 vs. NC group. ##P < 0.01 vs. HG group.
TGF-β1 was a direct target of miR-142-3p
To elucidate the underlying mechanism by which miR-142-3p inhibited HG-induced EndMT in HAECs, we performed bioinformatic analysis (Tagetscan, miRanda and PicTar) to predicate the putative targets of miR-142-3p. Our analysis revealed that TGF-β1 was a potential target of miR-142-3p and the target site located in the 3’-UTR of TGF-β1 (Figure 3A). To determine whether miR-142-3p directly targeted TGF-β1, a luciferase reporter assay was conducted. We observed that overexpression of miR-142-3p decreased the relative luciferase activities in the presence of the wild-type 3’-UTR, whereas knockdown of miR-142-3p increased the relative luciferase activities (Figure 3B). Likewise, the luciferase activity did not change significantly when the targeted sequence of TGF-β1 was mutated in the miR-142-3p-binding site. To further confirm that TGF-β1 is negatively regulated by miR-142-3p, TGF-β1 mRNA and protein expression level were analyzed by qRT-PCR and Western blot analysis. We found that miR-142-3p mimics decreased, while miR-142-3p inhibitor enhanced TGF-β1 expression at mRNA and protein levels in HAECs (Figure 3C-E). Taken together, miR-142-3p inhibited the expression of TGF-β1, suggesting that miR-142-3p/TGF-β1 axis may be involved in HG-induced EndMT in HAECs.
Figure 3.
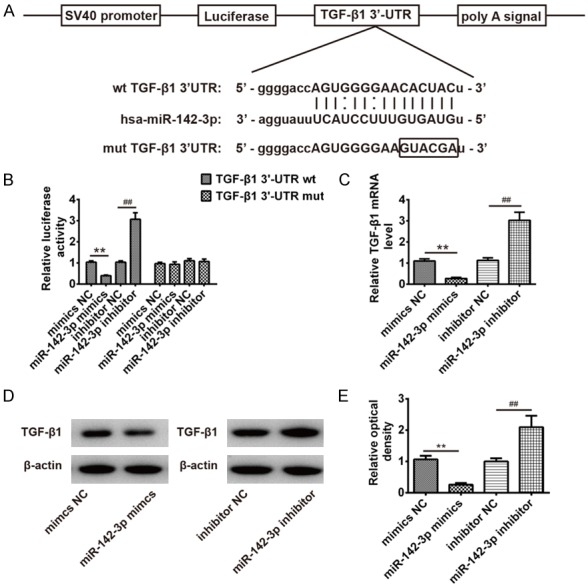
TGF-β1 was a direct target of miR-142-3p. A. The putative binding site of miR-142-3p and TGF-β1 is shown. B. Luciferase assay of HEK293 cells co-transfected with firefly luciferase constructs containing the TGF-β1 wild-type or mutated 3’-UTRs and miR-142-3p mimics, mimics NC, miR-142-3p inhibitor or inhibitor NC, as indicated (n = 3). Data represent the mean ± SD of three independent experiments. **P < 0.01 vs. mimics NC, ##P < 0.01 vs. inhibitor NC. C. The expression of TGF-β1 mRNA after transfection with miR-142-3p mimic or miR-142-3p inhibitor were measured by Western Blot. D. The expression of TGF-β1 protein after transfection with miR-142-3p mimic or miR-142-3p inhibitor were measured by Western Blot. E. The bands were semi-quantitatively analyzed by using Image J software, normalized to β-actin density. Data represent the mean ± SD of three independent experiments. **P < 0.01 vs. mimics NC, ##P < 0.01 vs. inhibitor NC.
miR-142-3p inhibited HG-induced EndMT by targeting TGF-β1
It has been reported that TGF-β1 plays an important role in fibrosis in DCM [20,21]. To further investigate whether miR-142-3p inhibited HG-induced EndMT through its target TGF-β1, pcDNA-TGF-β1 and miR-142-3p mimics were co-transfected into HAECs, followed by HG treatment, and then the mRNA and protein expression levels of α-SMA, vimentin, CD31 and VE-cadherin were measured by qRT-PCR and Western Blot. As shown in Figure 4A-D, TGF-β1 overexpression reversed the inhibitory effects of miR-142-3p mimics on the expression of α-SMA and vimentin and attenuated the promoting effects of miR-142-3p mimics on the expression of CD31 and VE-cadherin. The protein levels of α-SMA and vimentin, as well as of CD31 and VE-cadherin showed similar changes (Figure 4E). Additionally, we observed that HAECs regained a fibroblast-like phenotype when the cells were co-transfection with miR-142-3p mimics and pcDNA-TGF-β1, followed by HG stimulation (Data not shown). These data indicate that miR-142-3p overexpression inhibits HG-induced EndMT by downregulating TGF-β1.
Figure 4.

miR-142-3p inhibited HG-induced EndMT by targeting TGF-β1. pcDNA-TGF-β1 and miR-142-3p mimics were co-transfected into HAECs, followed by HG treatment for 5 days, after which the cells were harvested for subsequent experiment. A-D. The mRNA expression levels of α-SMA, vimentin, CD31 and VE-cadherin were measured by qRT-PCR. E. The protein expression levels of α-SMA, vimentin, CD31 and VE-cadherin were measured by Western Blot. Data represent the mean ± SD of three independent experiments. ##P < 0.01 vs. HG + mimics group.
miR-142-3p inhibited HG-induced EndMT through blocking TGF-β1/Smad signaling pathway
It is reported that TGF-β1/Smad signaling pathway plays a vital role in the progression of myocardial fibrosis in DCM [23]. Thus, we sought to determine whether miR-142-3p modulates the TGF-β1/Smad signaling pathway in the process of HG-induced EndMT. Western blot results indicated that the level of TGF-β1, phospho-Smad2 and phospho-Smad3 in HG group were significantly increased compared to NC group, while the level of TGF-β1, phospho-Smad2 and phospho-Smad3 in HG + mimics group were significantly decreased compared to HG group. Moreover, overexpression of TGF-β1 attenuated the inhibitory effect of miR-142-3p on these proteins expression (Figure 5A, 5B). These results indicated miR-142-3p overexpression inhibited the activation of TGF-β1/Smad signaling pathway which activating by HG-inducing, thus improving HG-induced EndMT.
Figure 5.
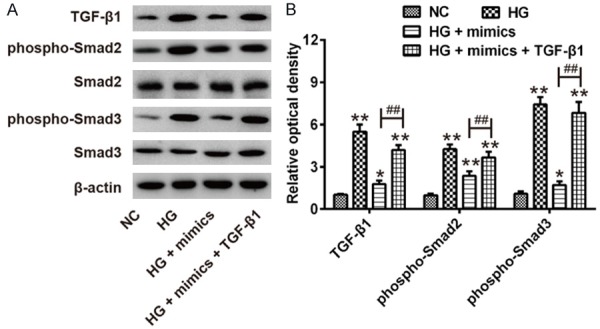
miR-142-3p improved HG-induced EndMT through TGF-β1/Smad signaling pathway. pcDNA-TGF-β1 and miR-142-3p mimics were co-transfected into HAECs, followed by HG treatment for 5 days, after which the cells were harvested for subsequent experiment. A. The protein levels of TGF-β1, Smad2, phospho-Smad2, Smad2 and phospho-Smad3 were measured by Western Blot. B. The bands were semi-quantitatively analyzed by using Image J software, normalized to β-actin density. Data represent the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 vs. NC group. ##P < 0.01 vs. HG + mimics group.
Discussion
In the present study, we found that miR-142-3p was significantly downregulated in HG-treated HAECs and further studies showed that miR-142-3p inhibited HG-induced EndMT by inactivating TGF-β1/Smad signaling pathway. Our results suggest that targeting miR-142-3p/TGF-β1/Smad axis may be a potential therapeutic target for DCM.
Increasing evidences suggest that miRNAs play important roles in the pathogenesis of DCM [24-27]. For example, Chen et al. showed that miR-133a overexpression in the heart protected cardiac tissue from undergoing fibrosis during hyperglycaemia [28]. Duan et al. found that miR-150 regulated high glucose-induced cardiomyocyte hypertrophy by targeting the transcriptional co-activator p300 [29]. Li et al. demonstrated that miR-30d regulated cardiomyocyte pyroptosis by directly targeting foxo3a in DCM [10]. In this study, we established an EndMT cell model by application of HG in HAECs to explore the role of miRNAs in EndMT and DCM. The expression of the endothelial markers CD31 and VE-Cadherin was reduced, and the expression of the mesenchymal markers α-SMA, FSP-1, and FN was increased after HG treatment, demonstrating that HAECs acquired mesenchymal phenotype through an EndMT process. During EndMT, miR-142-3p was screened by miRNA microarray and differentially down-regulated in the process of EndMT. Our data suggest that miR-142-3p may play an important role in HG-induced EndMT.
Previous studies have focused on the role of miR-142-3p in human cancers. For example, miR-142-3p was found to be significantly downregulated in the cervical cancer tissue and low miR-142-3p expression level was closely associated with clinical features and poorer overall survival [30]. Deng et al. found that miR-142-3p inhibited cell proliferation and invasion of cervical cancer cells by targeting Frizzled7 receptor (FZD7) [31]. Recently, miR-142-3p has previously been reported to be associated with diabetes mellitus. For example, miR-142-3p was proved to be downregulated in peripheral blood mononuclear cell of type 1, type 2, and gestational diabetes mellitus patients [32]. Another study from Chavali V et al. showed that miR-142-3p was downregulated in the animal model of DCM [19]. However, whether miR-142-3p plays a role in HG-induced EndMT has never previously been studied, to the best of our knowledge. In the study, we found that overexpression of miR-142-3p rescued the protein level of the endothelial markers VE-cadherin and CD31 and inhibited HG-induced EndMT in HAECs. All data indicated that miR-142-3p inhibited the process of HG-induced EndMT.
TGF-β1 has been found to be one of the molecular mediators involved in the progression of EndMT [33-35]. In this study, we confirmed that TGF-β1 was a downstream target of miR-142-3p and overexpression of TGF-β1 could abrogate the inhibitory effects of miR-142-3p mimics on HG-induced EndMT. These data strongly suggested that miR-142-3p has important functions in HG-induced EndMT by downregulating the expression of TGF-β1. It is well known that Smad2 and Smad3 are the two important downstream mediators of TGF-β1/Smad signaling pathway. Of note, previous research has found that ectopic expression of miR-20a in endothelial cells blocks the activation of TGF-β1/Smad signaling pathway and protects the endothelium from EndMT [36]. Based on these findings, we speculated that miR-142-3p may inhibit HG-induced EndMT through TGF-β1/Smad signaling pathway. In this study, we found that overexpression of miR-142-3p inhibited TGF-β1 expression and reduced Smad2/3 phosphorylation in HG-treated HAECs. All data suggest that overexpression of miR-142-3p inhibits TGF-β1 expression, which suppresses the activation of TGF-β1/Smad signaling, and subsequently inhibits HG-induced EndMT in HAECs.
In summary, our findings indicate that miR-142-3p inhibits HG-induced EndMT in HAECs through modulation of TGF-β1/Smad signaling pathway. Such findings open up the possibility of future microRNA-based therapeutic strategies for DCM.
Acknowledgements
This work was supported by the Scientific Research Foundation of Chongqing Medical University (KY-2017-10) and the National Nature Science Foundation of China (Grant No. 81170723).
Disclosure of conflict of interest
None.
References
- 1.Aneja A, Tang WH, Bansilal S, Garcia MJ, Farkouh ME. Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am J Med. 2008;121:748–757. doi: 10.1016/j.amjmed.2008.03.046. [DOI] [PubMed] [Google Scholar]
- 2.Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57:660–671. doi: 10.1007/s00125-014-3171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asbun J, Villarreal FJ. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;47:693–700. doi: 10.1016/j.jacc.2005.09.050. [DOI] [PubMed] [Google Scholar]
- 4.Medici D, Potenta S, Kalluri R. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J. 2011;437:515–520. doi: 10.1042/BJ20101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Wu X, Li Y, Zhang H, Li Z, Zhang Y, Zhang L, Ju J, Liu X, Chen X, Glybochko PV, Nikolenko V, Kopylov P, Xu C, Yang B. Endothelial to mesenchymal transition contributes to arsenic-trioxide-induced cardiac fibrosis. Sci Rep. 2016;6:33787. doi: 10.1038/srep33787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 7.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 9.Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Li X, Du N, Zhang Q, Li J, Chen X, Liu X, Hu Y, Qin W, Shen N, Xu C, Fang Z, Wei Y, Wang R, Du Z, Zhang Y, Lu Y. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014;5:e1479. doi: 10.1038/cddis.2014.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S. miR-200b mediates endothelial-to-mesenchymal transition in diabetic cardiomyopathy. Diabetes. 2016;65:768–779. doi: 10.2337/db15-1033. [DOI] [PubMed] [Google Scholar]
- 12.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 13.Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park DH, Thum T. Transforming growth factor-beta-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler Thromb Vasc Biol. 2012;32:361–369. doi: 10.1161/ATVBAHA.111.234286. [DOI] [PubMed] [Google Scholar]
- 14.Tiedt S, Prestel M, Malik R, Schieferdecker N, Duering M, Kautzky V, Stoycheva I, Bock J, Northoff BH, Klein M, Dorn F, Krohn K, Teupser D, Liesz A, Plesnila N, Holdt LM, Dichgans M. RNA-Seq identifies circulating miR-125a-5p, miR-125b-5p, and miR-143-3p as potential biomarkers for acute ischemic stroke. Circ Res. 2017;121:970–980. doi: 10.1161/CIRCRESAHA.117.311572. [DOI] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Geng H, Guan J. MiR-18a-5p inhibits endothelial-mesenchymal transition and cardiac fibrosis through the Notch2 pathway. Biochem Biophys Res Commun. 2017;491:329–336. doi: 10.1016/j.bbrc.2017.07.101. [DOI] [PubMed] [Google Scholar]
- 17.Tang R, Li Q, Lv L, Dai H, Zheng M, Ma K, Liu B. Angiotensin II mediates the high-glucose-induced endothelial-to-mesenchymal transition in human aortic endothelial cells. Cardiovasc Diabetol. 2010;9:31. doi: 10.1186/1475-2840-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Hu J, Liu L. MiR-200a modulates TGF-beta1-induced endothelial-to-mesenchymal shift via suppression of GRB2 in HAECs. Biomed Pharmacother. 2017;95:215–222. doi: 10.1016/j.biopha.2017.07.104. [DOI] [PubMed] [Google Scholar]
- 19.Chavali V, Tyagi SC, Mishra PK. Differential expression of dicer, miRNAs, and inflammatory markers in diabetic Ins2+/- Akita hearts. Cell Biochem Biophys. 2014;68:25–35. doi: 10.1007/s12013-013-9679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujio K, Komai T, Inoue M, Morita K, Okamura T, Yamamoto K. Revisiting the regulatory roles of the TGF-beta family of cytokines. Autoimmun Rev. 2016;15:917–922. doi: 10.1016/j.autrev.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Zhang F, Dang Y, Li Y, Hao Q, Li R, Qi X. Cardiac contractility modulation attenuate myocardial fibrosis by inhibiting TGF-beta1/Smad3 signaling pathway in a rabbit model of chronic heart failure. Cell Physiol Biochem. 2016;39:294–302. doi: 10.1159/000445624. [DOI] [PubMed] [Google Scholar]
- 22.Ma Z, Zhu L, Liu Y, Wang Z, Yang Y, Chen L, Lu Q. Lovastatin alleviates endothelial-to-mesenchymal transition in glomeruli via suppression of oxidative stress and TGF-beta1 signaling. Front Pharmacol. 2017;8:473. doi: 10.3389/fphar.2017.00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F, Ma H, Liang WJ, Yang JJ, Wang XQ, Shan MR, Chen Y, Jia M, Yin YL, Sun XY, Zhang JN, Peng QS, Chen YG, Liu LY, Li P, Guo T, Wang SX. Lovastatin upregulates microRNA-29b to reduce oxidative stress in rats with multiple cardiovascular risk factors. Oncotarget. 2017;8:9021–9034. doi: 10.18632/oncotarget.14462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X, Liu S. Role of microRNAs in the pathogenesis of diabetic cardiomyopathy. Biomed Rep. 2017;6:140–145. doi: 10.3892/br.2017.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Figueira MF, Monnerat-Cahli G, Medei E, Carvalho AB, Morales MM, Lamas ME, da Fonseca RN, Souza-Menezes J. MicroRNAs: potential therapeutic targets in diabetic complications of the cardiovascular and renal systems. Acta Physiol (Oxf) 2014;211:491–500. doi: 10.1111/apha.12316. [DOI] [PubMed] [Google Scholar]
- 26.Diao X, Shen E, Wang X, Hu B. Differentially expressed microRNAs and their target genes in the hearts of streptozotocin-induced diabetic mice. Mol Med Rep. 2011;4:633–640. doi: 10.3892/mmr.2011.489. [DOI] [PubMed] [Google Scholar]
- 27.Rawal S, Ram TP, Coffey S, Williams MJ, Saxena P, Bunton RW, Galvin IF, Katare R. Differential expression pattern of cardiovascular microRNAs in the human type-2 diabetic heart with normal ejection fraction. Int J Cardiol. 2016;202:40–43. doi: 10.1016/j.ijcard.2015.08.161. [DOI] [PubMed] [Google Scholar]
- 28.Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW 2nd, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med. 2014;18:415–421. doi: 10.1111/jcmm.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duan Y, Zhou B, Su H, Liu Y, Du C. miR-150 regulates high glucose-induced cardiomyocyte hypertrophy by targeting the transcriptional co-activator p300. Exp Cell Res. 2013;319:173–184. doi: 10.1016/j.yexcr.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 30.Li M, Li BY, Xia H, Jiang LL. Expression of microRNA-142-3p in cervical cancer and its correlation with prognosis. Eur Rev Med Pharmacol Sci. 2017;21:2346–2350. [PubMed] [Google Scholar]
- 31.Deng B, Zhang Y, Zhang S, Wen F, Miao Y, Guo K. MicroRNA-142-3p inhibits cell proliferation and invasion of cervical cancer cells by targeting FZD7. Tumour Biol. 2015;36:8065–8073. doi: 10.1007/s13277-015-3483-2. [DOI] [PubMed] [Google Scholar]
- 32.Collares CV, Evangelista AF, Xavier DJ, Rassi DM, Arns T, Foss-Freitas MC, Foss MC, Puthier D, Sakamoto-Hojo ET, Passos GA, Donadi EA. Identifying common and specific microRNAs expressed in peripheral blood mononuclear cell of type 1, type 2, and gestational diabetes mellitus patients. BMC Res Notes. 2013;6:491. doi: 10.1186/1756-0500-6-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez L, Munoz-Durango N, Riedel CA, Echeverria C, Kalergis AM, Cabello-Verrugio C, Simon F. Endothelial-to-mesenchymal transition: cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017;33:41–54. doi: 10.1016/j.cytogfr.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Choi HY, Lee HG, Kim BS, Ahn SH, Jung A, Lee M, Lee JE, Kim HJ, Ha SK, Park HC. Mesenchymal stem cell-derived microparticles ameliorate peritubular capillary rarefaction via inhibition of endothelial-mesenchymal transition and decrease tubulointerstitial fibrosis in unilateral ureteral obstruction. Stem Cell Res Ther. 2015;6:18. doi: 10.1186/s13287-015-0012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shu Y, Liu Y, Li X, Cao L, Yuan X, Li W, Cao Q. Aspirin-triggered resolvin D1 inhibits TGF-beta1-induced EndMT through increasing the expression of Smad7 and is closely related to oxidative stress. Biomol Ther (Seoul) 2016;24:132–139. doi: 10.4062/biomolther.2015.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee FT, Mountain AJ, Kelly MP, Hall C, Rigopoulos A, Johns TG, Smyth FE, Brechbiel MW, Nice EC, Burgess AW, Scott AM. Enhanced efficacy of radioimmunotherapy with 90Y-CHX-A’’-DTPA-hu3S193 by inhibition of epidermal growth factor receptor (EGFR) signaling with EGFR tyrosine kinase inhibitor AG1478. Clin Cancer Res. 2005;11:7080s–7086s. doi: 10.1158/1078-0432.CCR-1004-0019. [DOI] [PubMed] [Google Scholar]