

Abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis, has led to many clinical disorders and remains a major global health problem, leading to great morbidity and mortality worldwide. Previous studies reported that Mycobacterium tuberculosis (M.tb) has evolved to utilize various mechanisms to evade or attenuate the immune response, such as dysregulation of miRNAs. However, reports concerning the role of miRNAs in M.tb infection are limited. Here, we report that a host microRNA, miR-26b, was significantly down-regulated by M.tb infection in THP-1 cells. Subsequently, our results of in vitro experiments demonstrate that miR-26b is a negative regulator of the NF-κB pathway by directly targeting TAK1, resulting in the inhibition of immune response, and promotion of M.tb replication and gene expression. Taken together, our findings provide detailed molecular mechanisms for how miR-26 inhibits inflammatory cytokine secretion during M.tb infection in THP-1 cells, and these results suggest anti-M.tb as a promising therapy.
Keywords: Mycobacterium tuberculosis, miR-26b, TAK1, NF-κB
Introduction
Tuberculosis (TB) is a leading global health problem, caused by the intracellular pathogen Mycobacterium tuberculosis (M.tb), with an estimated one-third of the global population latently infected and an estimated 2 million deaths per year [1,2]. It has been reported that M. tuberculosis subverts the innate immune defense of the host to promote infection and replication and that it can outmaneuver these mechanisms to infect and persist within mammalian tissues [3-5]. Upon phagocytosis of M. tuberculosis, a serial of antimicrobial mechanisms would be activated by macrophages, the main host cells for mycobacteria, to control intracellular replication of the bacilli [6]. During these responses, the production of various cytokines, including tumor necrosis factor alpha (TNF-α), is critical for defense against mycobacterial infection, and it has been shown that it could alter expression of host immune-related genes [7,8]. Indeed, efforts have been made to better elucidate the pathogenic mechanism of M.tb and underlying molecular mechanisms of M.tb infection. Previous studies reported that virulent M.tb strains could induce macrophage necrosis and inhibit apoptosis to spread infection [9], suggesting that macrophage necrosis is a host innate defense mechanism against TB. However, the current understanding of the immune mechanism of macrophages infected with M.tb is poor.
Among strategies to evade the immune response, M.tb usually utilize viral products or host-cellular components to regulate a subsequent complex network to favor its expression and replication. As firmly established regulators of immune homeostatic mechanisms, microRNAs (miRNAs) play an important role in this process. Previous studies have shown that dysfunction or dysregulation of miRNAs is involved in many bacterial infections [10]. For example, it has been shown that inhibition of miR-210 expression increased proliferation of gastric epithelium during chronic Helicobacter pylori infection by activating STMN1 and DIMT1 [11]. Jin T et al. reported that overexpression of miR-24 could inhibit the effects of Staphylococcus aureus in Osteomyelitis Caused by S. aureus [12]. MicroRNA-302b is also shown to enhance host defense to bacteria by regulating inflammatory responses via feedback to TLR/IRAK4 circuits upon Gram-negative bacterium Pseudomonas aeruginosa infection [13]. As with M.tb, although several miRNAs have been identified as regulators that play important roles to up-regulate or down-regulate innate immune response in the regulation of M.tb replication and infection [7,14-16], the relationship among miRNAs, M.tb and macrophage necrosis remains poorly understood, and thus should be further investigated to better elucidate the underlying details.
Nuclear factor κB (NF-κB) is a family of transcription factors that are involved in cellular processes, such as inflammation, carcinogenesis, and chemoresistance [17,18]. Among the five structurally related proteins of this family, RelA (p65), NF-κB-1 (p105/p50), NF-κB-2 (p100/p52), RelB, and c-Rel, the p50 and p65 subunits constitute the predominant forms of NF-κB in mammals. Usually, NF-κB is mainly localized in the cytoplasm in an inactive form bound to IκB proteins, which function as the inhibitors of NF-κB [19]. Upon stimulation by tumor necrosis factor alpha (TNFα) or interleukin-1β (IL1β), TGFβ-activated kinase-1 (TAK1) and its adaptors TAB2/3 would be recruited to the receptor proximal signaling complex, resulting in the activation of IκB kinase (IKK) and further phosphorylation, ubiquitination, and degradation of IκB, which leads to the disassociation of p50/p65 heterodimer. Then, the released p65 and p50 protein would be translocated into the nucleus to bind to sequence-specific DNA to activate the transcription of many immune-related genes, such as TNF-α, IL-6, and IL-12 [20,21]. These cytokines are crucial for inflammation and other kinds of immune process, such as the host response to pathogens infection.
In the present study, we analyzed the gene expression profiles of M.tb infected patients with normal controls downloaded from GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52819) and chose a significant decreased miRNA-miR-26b for further investigation. Then, we found that the expression of miR-26b was markedly decreased in M.tb infected THP-1 cells. Through further exploration, we found that miR-26b, through targeting the 3’UTR of TAK1, modulates NF-κB and inflammatory cytokine expression, leading to the attenuation of immune response. As NF-κB the pathway is commonly activated in bacteria-host interactions, the roles and mechanisms revealed in this study may be also applied in infections of other bacteria. Together, miR-26b could represent a novel potential anti-M.tb therapeutic target and could be exploited for antibacterial development.
Material and methods
Cells and transfection
Human THP-1 macrophages were obtained from ATCC and grown in RPMI-1640 media (Hyclone/Thermo Fisher) supplemented with 10% fetal bovine serum (FBS, Gibco/Life Technology) and penicillin-streptomycin (100 g/ml) at 37°C in a humidified atmosphere with 5% CO2. The cell concentration was adjusted to 1.0 × 106/mL for further study. Transfection was done in antibiotic-free Opti-MEM medium (Invitrogen) with lipofectamine 2000 reagent (Invitrogen).
Mycobacterial and infection
M. tuberculosis strain H37Rv (ATCC) was grown in Middlebrook 7H9 broth medium supplemented with 10% OADC (Becton, Dickinson and Company, Franklin Lakes, NJ). Cells were infected at a multiplicity of infection of 10 bacteria per cell (MOI 10:1). After 6 h of incubation, the infected cells were washed 6 times with RPMI 1640 to remove any extracellular bacteria, and then incubated in fresh medium to different time points.
MiR-26b mimics and inhibitor
MiR-26b mimics, mimics NC, miR-26b inhibitor, and inhibitor NC were purchased from the Shanghai GenePharma Company (Shanghai, China). THP-1 cells were transfected with miR-26b mimics or miR-26b inhibitor using lipofectamine 2000 (Invitrogen, USA) according to manufacturer’s instruction.
ELISA assay
IFN-γ, IL-1β, IL-6, and TNF-α in supernatant were measured by ELISA kit (all from R&D Systems) according to manufacturer. THP-1 cells were transfected with miR-26b mimics and mimics NC or left untransfected. Cells were infected with H37Rv at MOI 10 at 24 h after transfection. Supernatant were harvest at 12 h, 24 h, and 48 h post-infection to perform ELISA assay.
Real-time PCR
To quantify the level of gene expression, total RNAs were extracted using TRIzol (Invitrogen), according to the manufacturer’s instruction. cDNA was reverse transcribed from 1 μg total RNA by SuperScript III Reverse Transcriptase (Invitrogen). Real-time PCR was carried out with PrimeScript RT reagent kit (Takara) on ABI 7900HT Fast Real-Time PCR System. Relative expression of target genes was quantitatively normalized against the expression of GAPDH using ΔΔCt method. Primer sequences were designed as follows:
miR-26b forward: 5’-CCGGGACCCAGTTCAAGTA-3’; miR-26b reverse: 5’-CCGGTCCCCGTGCCTTGTAA-3’; miR-124 forward: 5’-AATGATACGGCGACCACCGAACACTCTTTCCCTACACGACG-3’; miR-124 reverse: 5’-GGCATTCACCGCGTGCCT-3’; miR-16 forward: 5’-UAGCAGCACGUAAAUAUUGGCG-3’; miR-16 reverse: 5’-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCGCCAATA-3’; miR-451 forward: 5’-AAACCGTTACCATTACTGAGTT-3’; miR-451 reverse: 5’-CGCTACGTAACGGCATGACAGTG-3’.
Luciferase assay
Luciferase activity of promoter was evaluated by Dual-Luciferase Reporter Assay System (Promega). To quantitatively examine NF-κB activity, THP-1 cells were first transfected with miR-182 mimics or NC in a 24-well plate for 24 hours, followed by co-transfection with 10 ng pRL-TK (Promega, USA) and 50 ng pNF-κB-Luc (Clontech, USA) for 32 hours, then remained untreated or treated with 20 ng/ml TNFα (cat. 210-TA-010, R&D Systems) for 4 hours before luciferase activity analysis. Cells were harvested for protein and 50 μl of each sample extract was used to detect luciferase activity. To measure luciferase activity of TAK1, THP-1 cells were transfected with TAK1 WT or TAK1 mutant luciferase reporter vector (100 ng), miR-26b mimics, or NC mimics, miR-26b inhibitor, or NC inhibitor. Total protein was prepared after 30 hr post-transfection. The 50 μl of each sample extract was used to detect luciferase activity.
Western blot analysis
Cell lysates were obtained with RIPA lysis buffer, and proteins were analyzed by western blot. β-actin serves as a control. The following antibodies were used: anti-p-IκBα (Ser32, cat. #2859, CST), anti- IκBα (cat. #4814, CST), anti-p-p65 (cat. #3033, CST), anti-p65 (cat. #4814, CST), anti-TAK1 (cat. #5206, CST), anti-β-actin (cat. BM0627, Boster, Wuhan, China) and goat anti-rabbit, goat anti-mouse (Tianjin Saier Biotech, China).
Identification of differentially expressed miRNA
The gene expression profiles of M.tb patients with normal controls were downloaded from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) with accession number of GSE52819. The original datasets included 40 genechips, and we extracted 22 of them for further analysis. During processing, the probe-level data was transferred into expression measures. Then, the background was corrected and quartile data was subsequently normalized. Finally, we choose the file in the platform annotation files resulting from the Affymetrix Company to map the relationship between the probes and gene symbols. To identify differentially expressed miRNA, the threshold of false discovery rate (FDR) 0.05 was applied.
Statistical analysis
All experiments were repeated at least three times with similar results. Statistics are represented as the mean and the standard error of the mean for each group. Statistical significance analysis was performed using paired Student’s t test (**P < 0.01, *P < 0.05).
Results
MiR-26b is markedly down-regulated in M.tb infected THP-1
In order to illustrate the role of microRNAs in M.tb infection, THP-1 cells were infected with M.tb H37Rv strain at MOI of 10. First, we tested the secretion of inflammatory cytokines in the medium of THP-1 cells at 12, 24, and 48-hour post-infection to confirm the efficacy of M.tb infection. ELISA results showed that production of IFN-γ, IL-1β, IL-6, and TNF-α were upregulated in M.tb infected cells at all timepoints compared with the Ctrl group (Figure 1A-D). Then, to identify miRNAs that may be involved in the regulation of immune response of M.tb infection, miRNA data was downloaded from GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52819) to screen differentially expressed miRNAs. As shown in Figure 1E, many miRNAs were down-regulated or up-regulated after M.tb infection. Among these miRNAs, miR-26b, miR-124, miR-16, and miR451 were chosen for further validation in THP-1 cells since their expression reduced significantly after infection. Except for miR-16, three other miRNAs were down-regulated after M.tb infection (Figure 1F-I). Specifically, expression of miR-26b and miR-124 dramatically decreased as early as 12 h post-infection and continued to decrease at 24 h and 48 h while the decrease of miR-451 was later compared with that of miR-26b and miR-124. A previous paper reported that miR-26b negatively regulates the NF-κB pathway by inhibiting expression of Rel A (p65) in breast cancer cells [22] and expression of TAK1 and TAB3 in hepatocellular carcinoma cells [23]. However, the role of miR-26b in host immune response to M.tb infection remains unclear. Since activation of the NF-κB pathway plays a pivotal role in early events of the immune response to M.tb infection, we suspected that miR-26b may be involved in the regulation of M.tb infection through controlling activation of the NF-κB pathway.
Figure 1.

miR-26b is markedly down-regulated in M.tb infected THP-1. (A-D) THP-1 cells were infected with H37Rv at MOI 10. Supernatant was harvested at different time point and ELISA assay was conducted for measuring IFN-γ (A), IL-1β (B), IL-6 (C) and TNF-α (D) secretion. (E) Heat-map of microRNA array. (F-I) Cells were treated as (A-D) and were harvested at indicated time for RNA extraction. Real-time PCR assay was conducted to measure the levels of miR-26b (F), miR-124 (G), miR-16 (H) and miR-451 (I). Results represent of three independent experiments (mean ± SD). *P < 0.05, **P < 0.01 vs uninfected groups.
Overexpression of miR-26b reduced inflammatory cytokines secretion in M.tb infection
In order to determine the biological relevance of downregulation of miR-26b in M.tb infection, THP-1 cells were transfected with miR-26b prior to infection. The efficiency of miR-26b mimics was confirmed through real-time PCR assay. MiR-26b was successfully overexpressed by mimics transfection (Figure 2A). ELISA assay was performed to assess the secretion of M.tb-induced cytokine secretion at 24 hr post-infection. Consistent with previous data, we found that H37Rv infection led to robust secretion of IFN-γ, IL-1β, IL-6, and TNF-α (Figure 2B-E). Conversely, ectopic expression of miR-26b greatly attenuated this effect upon H37Rv infection. These results indicate that over-expression of miR-26b inhibited inflammatory cytokines secretion upon M.tb infection.
Figure 2.
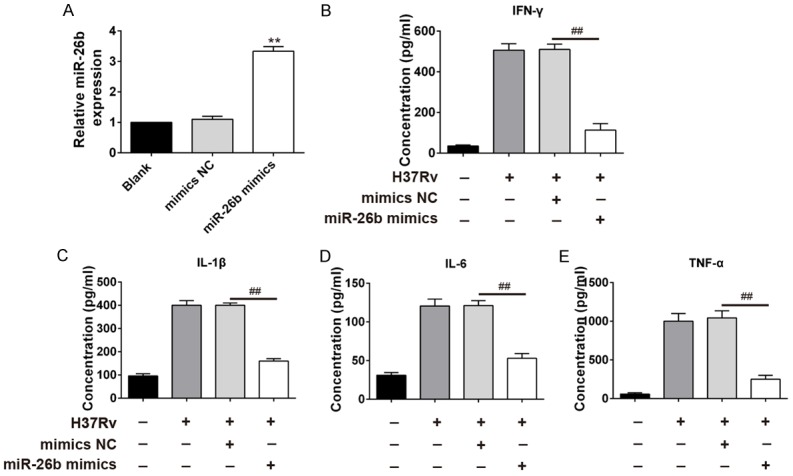
miR-26b suppresses M.tb induced inflammatory cytokines secretion. THP-1 cells were transfected with miR-26b mimics or mimics NC. 24 h post-transfection, cells were infected with M.tb H37Rv strain at MOI of 10. (A) The miR-26b expression levels at 48 hpi were quantified by qRT-PCR. (B-E) 24 hr post infection, supernatant was harvested. ELISA assay were performed to measure IFN-γ (B), IL-1β (C) IL-6 (D) and TNF-α (E) secretion. Results represent of three independent experiments (mean ± SD). **P < 0.01, ##P < 0.01 vs mimics NC groups.
MiR-26b suppresses the TNFα-induced NF-κB signaling in THP-1 cells
Next, we tried to determine the influence of miR-26b on NF-κB signaling. We co-transfected miR-26b mimics and its negative control (NC) or miR-26b inhibitor and its inhibitor NC with the luciferase reporter that contained NF-κB binding sites in its promoter. Compared with the mimics NC, miR-26b mimics-transfected THP-1 cells displayed a significantly lower response to TNFα, the classical NF-κB activator (Figure 3A). In contrast, down-regulation of miR-26b led to higher NF-κB reporter activity compared with those of inhibitor NC groups (Figure 3B). Next, the impact of miR-26b on the signaling molecules of the NF-κB pathway was investigated. As reported, TNFα treatment led to a significant increase in the phosphorylation level of IκBα and p65 in control cells (Figure 3C and 3D), indicating the activation of NF-κB signaling. Notably, phosphorylation of IκBα and p65 was much less evident in miR-26b overexpressed-cells, compared with the control cells (Figure 3C). Conversely, TNFα-induced NF-κB signaling was enhanced in miR-26b inhibitor transfected-cells. Collectively, these data indicate that miR-26b negatively regulates NF-κB signaling.
Figure 3.
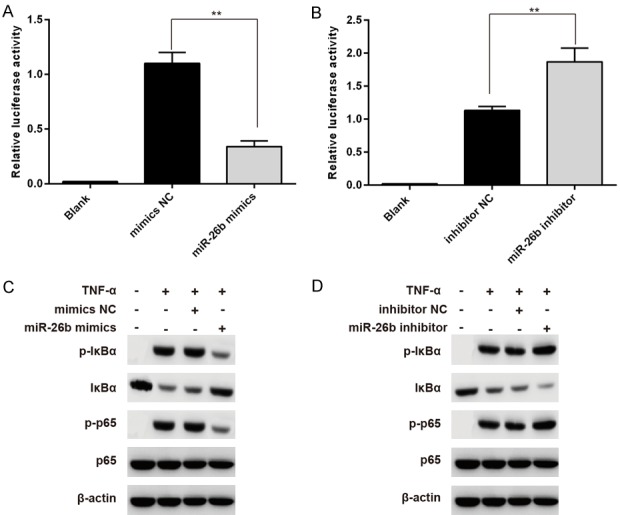
miR-26b suppresses TNFα-induced NF-κB signaling in THP-1 cell. (A, B) THP-1 cells were first transfected with mimics NC or miR-26b mimics (A), inhibitor NC or miR-26b inhibitor (B), followed by co-transfection of pRL-TK and pNF-κB-Luc, treatment with 20 ng/ml TNFα, and analysis for luciferase activity. **P < 0.01 vs mimics NC group or inhibitor NC group. (C, D) THP-1 cells transfected with mimics NC or miR-26b mimics (C), inhibitor NC or miR-26b inhibitor (D) were treated with 2 ng/ml TNFα for the 5 min. Cells were harvested for immunoblotting analysis. All data represent the mean value ± SD of at least three independent experiments.
TAK1 is directly targeted by miR-26b
To further illustrate the possible mechanism of miR-26b in regulation of the NF-κB pathway, it was necessary to identify the target genes of miR-26b. TargetScan Release 6.0 was subsequently used for prediction of miR-26b targets. TGFβ-activated kinase-1 (TAK1), an upstream positive regulator of NF-κB pathway, was found to have a putative miR-26b binding site within its 3’UTR (Figure 4A). To identify whether miR-26b directly binds the TAK1 3’UTR, the predicted target site in TAK1 was cloned into a firefly luciferase reporter vector, meanwhile, a mutant vector was constructed to eliminate the possible recognition by replacing seven seed nucleotides (UGAACUU to GUCGACG) (Figure 4A). Interestingly, in the presence of miR-26b mimics, the luciferase activity of TAK1 resulted in ~80% reduction compared to that of mimic NC, whereas blockage of endogenous miR-26b led to a 3.2-fold increase in luciferase activity compared to that of inhibitory NC (Figure 4B). However, all these effects mentioned above disappeared in cells transfected with the vectors bearing the mutant TAK1 3’-UTR (Figure 4B). Additionally, transfection of miR-26b mimics inhibited expression of TAK1 in THP-1 cells, whereas knockdown of miR-26b enhanced the expression of TAK1 (Figure 4C, 4D). Hence, it is clear that TAK1 is a direct target of miR-26b in THP-1 cells.
Figure 4.
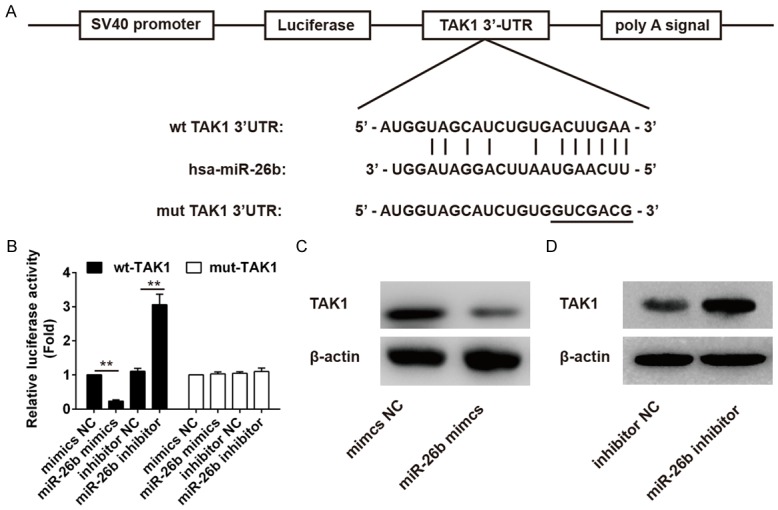
TAK1 is directly targeted by miR-26b. (A) Schematic diagram of the predicted target sites of miR-26b in TAK1 3’UTRs. The predicted target sites are underlined and mutated as indicated. (B) THP1 cells were co-transfected with TAK1 WT or TAK1 mutant luciferase reporter vector (100 ng), and miR-26b mimics or NC, miR-26b inhibitor or NC inhibitor for 36 h and then harvested for luciferase assay. **P < 0.01 vs mimics NC group or inhibitor NC group. (C, D) THP-1 cells were transfected with miR-26b mimics or NC mimics (C), miR-26b inhibitor or NC inhibitor (D) for 36 h and harvested for Western blot analysis of TAK1 expression. All data represent the mean value ± SD of at least three independent experiments.
MiR-26b regulated host immune response to M.tb through targeting TAK1
Finally, we examined the role of targeting TAK1 by miR-26b in host immune effect against M.tb. We co-transfected miR-26b mimics and an expressing vector containing the open reading frame (ORF) of TAK1 into THP-1 cells. As previous data, M.tb infection induced robust secretion of pro-inflammatory cytokines like IFN-γ, IL-1β, IL-6, and TNF-α, whereas, overexpression of miR-26b greatly abrogated secretion of these cytokines. However, this effect was restrained by co-transfecting pcDNA-TAK1 and miR-26b mimics (Figure 5A-D). Taken together, these data indicate that the host may modulate immune responses against M.tb infection through miR-26b targeting TAK1 expression.
Figure 5.

miR-26b regulates host immune response towards M.tb through targeting TAK1. THP-1 cells were transfected with miR-26b mimics, pcDNA-TAK1, or both. 24 hr after transfection, cells were infected with H37Rv at MOI 10. 24 hr post-infection, supernatant were harvested for ELISA assay to detect IFN-γ (A), IL-1β (B), IL-6 (C) and TNF-α (D) secretion. All data represent the mean value ± SD of at least three independent experiments. *P < 0.05, **P < 0.01 vs non-transfected groups; ##P < 0.01 vs miR-26b-transfected groups.
Discussion
Recently miRNAs have been reported to participate extensively in the complicated regulations of host-pathogen interactions and act as important regulators of immune homeostatic mechanisms [24-26]. It is firmly established that a number of bacteria could regulate the profile of bacterial or host cell miRNAs to favor their replication and expression [27-29]. In the current study, we found that expression of miR-26b was significantly decreased in M.tb infected THP-1 cells (Figure 1). Notably, overexpression of miR-26b markedly reduced expression of several inflammatory cytokines (Figure 2) and NF-κB signaling in THP-1 cells infected by M.tb (Figure 3). Consistent with the bioinformatics prediction, TAK1 was also identified as a direct target of miR-26 during M.tb infection by subsequent biochemical studies (Figure 4). Additionally, it is found that miR-26b could regulate host immune response to M.tb through directly targeting TAK1 to attenuate the activation of NF-κB and consequent immune responses (Figure 5).
MicroRNA-26b has been well known to be involved in the carcinogenesis and pathogenesis of various human cancers [30,31]. For example, overexpression of miR-26b can impair viability and trigger apoptosis of human breast cancer MCF7 cells by targeting MTDH and EZH2 [32]. It has been reported that miR-26b inhibits proliferation, migration, invasion, and vasculogenic mimicry of human glioma cells via targeting EphA2 [33]. As with its role in the interaction of host-pathogens, previous studies have reported that respiratory syncytial virus infection can increase the expression of miR-26b to inhibit TLR4 signaling to promote the viral replication and infection [34]. However, whether it participates in the immune response of host-bacteria or not remains unknown. In this study, we first demonstrated that up-regulation of miR-26b suppresses activation of NF-κB by directly targeting TAK1 to evade the immune response (Figures 3 and 5), suggesting that miR-26b may exert a similar function during the infection of the other bacteria.
Previous studies have revealed several important miRNAs that play important roles in M.tb infection [35-38]. Among the pathways involved in the complicated regulation networks, the NF-κB pathway has been extensively reported in host-pathogen regulation. It has been shown that microRNA let-7 modulates the immune response to Mycobacterium tuberculosis Infection via regulation of A20, which is an inhibitor of the NF-κB Pathway [36]. Interestingly, our results revealed that miR-26b can regulate the immune response in M.tb infection by control of TAK1 (Figures 4 and 5), one upstream kinase in the NF-κB pathway which is necessary for the activation of NF-κB. Based on these information, it is reasonable for us to confer that other members in the pathway may also be modulated by other micro-RNAs or some other mediators during the infection of M.tb.
Taken together, our study is the first to demonstrate that miR-26b facilitates M.tb replication and infection mainly by targeting TAK1 to down-regulate activation of NF-κB to suppress the immune response. Therefore, miR-26b is a promising anti-M.tb therapeutic target.
Acknowledgements
This study was supported by Shanghai Science and Technology Committee (Grant No.: 134119a9100).
Disclosure of conflict of interest
None.
References
- 1.Manson AL, Cohen KA, Abeel T, Desjardins CA, Armstrong DT, Barry CE 3rd, Brand J TBResist Global Genome Consortium. Chapman SB, Cho SN, Gabrielian A, Gomez J, Jodals AM, Joloba M, Jureen P, Lee JS, Malinga L, Maiga M, Nordenberg D, Noroc E, Romancenco E, Salazar A, Ssengooba W, Velayati AA, Winglee K, Zalutskaya A, Via LE, Cassell GH, Dorman SE, Ellner J, Farnia P, Galagan JE, Rosenthal A, Crudu V, Homorodean D, Hsueh PR, Narayanan S, Pym AS, Skrahina A, Swaminathan S, Van der Walt M, Alland D, Bishai WR, Cohen T, Hoffner S, Birren BW, Earl AM. Genomic analysis of globally diverse Mycobacterium tuberculosis strains provides insights into the emergence and spread of multidrug resistance. Nat Genet. 2017;49:395–402. doi: 10.1038/ng.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dye C, Williams BG. The population dynamics and control of tuberculosis. Science. 2010;328:856–861. doi: 10.1126/science.1185449. [DOI] [PubMed] [Google Scholar]
- 3.Krutzik SR, Modlin RL. The role of Toll-like receptors in combating mycobacteria. Semin Immunol. 2004;16:35–41. doi: 10.1016/j.smim.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Singh Y, Kaul V, Mehra A, Chatterjee S, Tousif S, Dwivedi VP, Suar M, Van Kaer L, Bishai WR, Das G. Mycobacterium tuberculosis controls microRNA-99b (miR-99b) expression in infected murine dendritic cells to modulate host immunity. J Biol Chem. 2013;288:5056–5061. doi: 10.1074/jbc.C112.439778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma F, Xu S, Liu XG, Zhang Q, Xu XF, Liu MF, Hua MM, Li N, Yao HP, Cao XT. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol. 2011;12:861–869. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
- 6.Rajaram MV, Ni B, Dodd CE, Schlesinger LS. Macrophage immunoregulatory pathways in tuberculosis. Semin Immunol. 2014;26:471–485. doi: 10.1016/j.smim.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ni B, Rajaram MV, Lafuse WP, Landes MB, Schlesinger LS. Mycobacterium tuberculosis decreases human macrophage IFN-gamma responsiveness through miR-132 and miR-26a. J Immunol. 2014;193:4537–4547. doi: 10.4049/jimmunol.1400124. [DOI] [PubMed] [Google Scholar]
- 8.Rojas M, Olivier M, Gros P, Barrera LF, Garcia LF. TNF-αlpha and IL-10 modulate the induction of apoptosis by virulent Mycobacterium tuberculosis in murine macrophages. J Immunol. 1999;162:6122–6131. [PubMed] [Google Scholar]
- 9.Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8:668–674. doi: 10.1038/nrmicro2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Staedel C, Darfeuille F. MicroRNAs and bacterial infection. Cell Microbiol. 2013;15:1496–1507. doi: 10.1111/cmi.12159. [DOI] [PubMed] [Google Scholar]
- 11.Kiga K, Mimuro H, Suzuki M, Shinozaki-Ushiku A, Kobayashi T, Sanada T, Kim M, Ogawa M, Iwasaki YW, Kayo H, Fukuda-Yuzawa Y, Yashiro M, Fukayama M, Fukao T, Sasakawa C. Epigenetic silencing of miR-210 increases the proliferation of gastric epithelium during chronic helicobacter pylori infection. Nat Commun. 2014;5:4497. doi: 10.1038/ncomms5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin T, Lu Y, He QX, Wang H, Li BF, Zhu LY, Xu QY. The role of MicroRNA, miR-24, and its target CHI3L1 in osteomyelitis caused by staphylococcus aureus. J Cell Biochem. 2015;116:2804–2813. doi: 10.1002/jcb.25225. [DOI] [PubMed] [Google Scholar]
- 13.Zhou X, Li X, Ye Y, Zhao K, Zhuang Y, Li Y, Wei Y, Wu M. MicroRNA-302b augments host defense to bacteria by regulating inflammatory responses via feedback to TLR/IRAK4 circuits. Nat Commun. 2014;5:3619. doi: 10.1038/ncomms4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ouimet M, Koster S, Sakowski E, Ramkhelawon B, van Solingen C, Oldebeken S, Karunakaran D, Portal-Celhay C, Sheedy FJ, Ray TD, Cecchini K, Zamore PD, Rayner KJ, Marcel YL, Philips JA, Moore KJ. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol. 2016;17:677–686. doi: 10.1038/ni.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwai H, Funatogawa K, Matsumura K, Kato-Miyazawa M, Kirikae F, Kiga K, Sasakawa C, Miyoshi-Akiyama T, Kirikae T. MicroRNA-155 knockout mice are susceptible to Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 2015;95:246–250. doi: 10.1016/j.tube.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Liu YH, Wang R, Jiang J, Yang BF, Cao ZH, Cheng XX. miR-223 is upregulated in monocytes from patients with tuberculosis and regulates function of monocyte-derived macrophages. Mol Immunol. 2015;67:475–481. doi: 10.1016/j.molimm.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012;12:121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 19.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 20.Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 22.Anbalagan D, Yap G, Yuan Y, Pandey VK, Lau WH, Arora S, Bist P, Wong JS, Sethi G, Nissom PM, Lobie PE, Lim LH. Annexin-A1 regulates microRNA-26b*and microRNA-562 to directly target NF-kappa B and angiogenesis in breast cancer cells. PLoS One. 2014;9:e114507. doi: 10.1371/journal.pone.0114507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao N, Wang R, Zhou L, Zhu Y, Gong J, Zhuang SM. MicroRNA-26b suppresses the NF-kappaB signaling and enhances the chemosensitivity of hepatocellular carcinoma cells by targeting TAK1 and TAB3. Mol Cancer. 2014;13:35. doi: 10.1186/1476-4598-13-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang GL, Li YX, Zheng SQ, Liu M, Li X, Tang H. Suppression of hepatitis B virus replication by microRNA-199a-3p and microRNA-210. Antiviral Res. 2010;88:169–175. doi: 10.1016/j.antiviral.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Yao YL, Li GX, Wu JH, Zhang X, Wang JJ. Inflammatory response of macrophages cultured with Helicobacter pylori strains was regulated by miR-155. Int J Clin Exp Pathol. 2015;8:4545–4554. [PMC free article] [PubMed] [Google Scholar]
- 26.Ru J, Sun HH, Fan HX, Wang CM, Li YX, Liu M, Tang H. MiR-23a facilitates the replication of HSV-1 through the suppression of interferon regulatory factor 1. PLoS One. 2014;9:e114021. doi: 10.1371/journal.pone.0114021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z, Wang TJ, Liu Z, Zhang GY, Wang JH, Feng SS, Liang JQ. Inhibition of autophagy by MiR-30A induced by mycobacteria tuberculosis as a possible mechanism of immune escape in human macrophages. Jpn J Infect Dis. 2015;68:420–424. doi: 10.7883/yoken.JJID.2014.466. [DOI] [PubMed] [Google Scholar]
- 28.Maudet C, Mano M, Sunkavalli U, Sharan M, Giacca M, Forstner KU, Eulalio A. Functional high-throughput screening identifies the miR-15 microRNA family as cellular restriction factors for Salmonella infection. Nat Commun. 2014;5:4718. doi: 10.1038/ncomms5718. [DOI] [PubMed] [Google Scholar]
- 29.Au KY, Pong JC, Ling WL, Li JC. MiR-1303 regulates mycobacteria induced autophagy by targeting Atg2B. PLoS One. 2016;11:e0146770. doi: 10.1371/journal.pone.0146770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin JX, Zhang L, Huang H, Huang YW, Huang L, Wang JH, Huang ST, He L, Zhou Y, Jia WH, Yun JP, Luo RZ, Zheng M. MiR-26b/KPNA2 axis inhibits epithelial ovarian carcinoma proliferation and metastasis through downregulating OCT4. Oncotarget. 2015;6:23793–23806. doi: 10.18632/oncotarget.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.John Clotaire DZ, Zhang B, Wei N, Gao R, Zhao F, Wang Y, Lei M, Huang W. MiR-26b inhibits autophagy by targeting ULK2 in prostate cancer cells. Biochem Biophys Res Commun. 2016;472:194–200. doi: 10.1016/j.bbrc.2016.02.093. [DOI] [PubMed] [Google Scholar]
- 32.Zhang B, Liu XX, He JR, Zhou CX, Guo M, He M, Li MF, Chen GQ, Zhao QA. Pathologically decreased miR-26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis. 2011;32:2–9. doi: 10.1093/carcin/bgq209. [DOI] [PubMed] [Google Scholar]
- 33.Wu N, Zhao XZ, Liu M, Liu HZ, Yao WC, Zhang YY, Cao SS, Lin XK. Role of microRNA-26b in glioma development and its mediated regulation on EphA2. PLoS One. 2011;6:e16264. doi: 10.1371/journal.pone.0016264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S, Gao L, Wang X, Xing Y. Respiratory syncytial virus infection inhibits TLR4 signaling via up-regulation of miR-26b. Cell Biol Int. 2015;39:1376–1383. doi: 10.1002/cbin.10518. [DOI] [PubMed] [Google Scholar]
- 35.Rivera-Marrero CA, Stewart J, Shafer WM, Roman J. The down-regulation of cathepsin G in THP-1 monocytes after infection with Mycobacterium tuberculosis is associated with increased intracellular survival of bacilli. Infect Immun. 2004;72:5712–5721. doi: 10.1128/IAI.72.10.5712-5721.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar M, Sahu SK, Kumar R, Subuddhi A, Maji RK, Jana K, Gupta P, Raffetseder J, Lerm M, Ghosh Z, van Loo G, Beyaert R, Gupta UD, Kundu M, Basu J. MicroRNA let-7 modulates the immune response to mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF-kappa B pathway. Cell Host Microbe. 2015;17:345–356. doi: 10.1016/j.chom.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 37.Wu Z, Lu H, Sheng J, Li L. Inductive microRNA-21 impairs anti-mycobacterial responses by targeting IL-12 and Bcl-2. FEBS Lett. 2012;586:2459–2467. doi: 10.1016/j.febslet.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Fu X, Zeng L, Liu Z, Ke X, Lei L, Li G. MicroRNA-206 regulates the secretion of inflammatory cytokines and MMP9 expression by targeting TIMP3 in Mycobacterium tuberculosis-infected THP-1 human macrophages. Biochem Biophys Res Commun. 2016;477:167–173. doi: 10.1016/j.bbrc.2016.06.038. [DOI] [PubMed] [Google Scholar]