

Abstract
Objective: Fusion gene detection is widely used in the diagnosis and treatment of leukemia. This study developed a rapid detection method of eight common pediatric leukemia fusion genes. Methods: In this study, one step multiplex RT-PCR assay was developed for the simultaneous detection of eight common leukemia fusion genes, including BCR-ABL, ETV6-RUNX1, MLL-AF4, E2A-PBX1, AML1-ETO, PML-RARα, CBFβ-MYH11 and SIL-TAL1. The single step RT-PCR approach is mediated by universal primers after obtaining total RNA from bone marrow specimens. The size of the amplified fragments were analyzed by capillary electrophoresis assay. A total of 122 patients with positive leukemia fusion genes were tested by real-time PCR. Results: Respectively, 21 cases were detected as CBRB-MYH11 fusion gene, 13 cases were detected as SIL-TAL1 fusion gene, 16 cases were detected as ETV6-RUNX1 fusion gene, 16 cases were detected as E2A-PBX1 fusion gene, 15 cases were detected as PML-RARα fusion gene, 14 cases were detected as AML1-ETO fusion gene, 13 cases were detected as MLL-AF4 fusion gene, except for 1 case where no fusion gene was detected. Conclusion: This method has a high accuracy and detection rate. Therefore, one step multiplex RT-PCR combined with a capillary electrophoresis analysis system can be used as an important tool for the clinical diagnosis, treatment and prognosis of pediatric leukemia.
Keywords: One-step multiplex RT-PCR, universal primer, leukemia fusion gene
Introduction
Leukemia is a malignant clonal disease originating from hematopoietic cells, where clonal cells lose the ability of further maturation and stagnate at different phases of cell development. Malignant hyperplasia in bone marrow and other hematopoietic tissues results in inhibition of normal hematopoiesis [1,2]. Cytogenetic changes of leukemia cells play an important role in the occurrence and development of leukemia [3,4]. First, fusion gene detection is widely used in the diagnosis and treatment of leukemia, because patients with the same fusion gene are treated with similar therapy and have comparable prognosis. Second, monitoring the expression of fusion genes also helps the clinician to further determine whether the patient has a complete remission at the molecular level after treatment. Adjusting the treatment regimen according to the detection result reduces the patient’s physical pain and financial burden. Third, regular detection of fusion gene expression in patients can predict the recurrence of leukemia and guide the clinical treatment [5]. Therefore, cellular and molecular genetic technologies gradually occupy more and more important positions in the diagnosis of leukemia.
To date, the number of reported leukemia-related chromosomal translocations and inversion fusion genes is up to 50. The common fusion genes in childhood leukemia are listed below: ETV6-RUNX1 fusion gene from t(12;21) translocation [6], E2A-PBX1 (TCF3-PBX1) fusion gene from t(1;19) translocation [7], MLL-AF4 fusion gene from t(4;11) translocation [8], m-BCR-ABL fusion gene from t(9;22) translocation [9], AML1-ETO (RUNX1-RUNX1T1) fusion gene from t(8;21) translocation [10], PML-RARα fusion gene from t(15;17) translocation [11], CBFβ-MYH11 (Inv(16)/t(16;16)) fusion gene from t(16;16) inversion [12]. Leukemia-associated fusion genes are specific molecular markers of leukemia, and comprise independent diagnostic criteria of childhood leukemia type [13].
At present, the commonly used methods of leukemia fusion gene detection include chromosome karyotype analysis, Fluorescent In Situ Hybridization (FISH), polymerase chain reaction (PCR) and DNA sequencing technology [14,15]. The positive rate of Chromosome G karyotype analysis is mainly dependent on the survival rate of tested leukemia cells, as well as technical conditions and artificial factors. Additionally, at least 10-30 homogeneous tumor cells in mitosis metaphase need to be analyzed in each test, so the researcher could not proceed with karyotype analysis when the cell volume is too small [16], especially in the situation of complex chromosome translocations. Although conventional cytogenetics is the standard method for identifying chromosomal translocation, it requires considerable expertise. This results either in false negatives or in no interpretable results in a large number of cases, especially in laboratories lacking experience in handling karyotype of malignant cells. FISH technology uses probes that span the chromosome break point to more accurately locate the rupture point and identify the fusion gene produced by chromosome rearrangement [17,18]. Expensive fluorescent probes, cumbersome experimental operation, and disability of batch sample detection restrict the application of FISH in clinical subject are limitations. Polymerase Chain Reaction (PCR) or real-time PCR are directly used to detect fusion genes with high sensitivity and specificity [19,20]. However, real-time PCR could be a good choice when detecting only 3-4 kinds target fusion genes [21]. The length of the fragment need to be broadened to cover as many fusions as possible, but the length of the PCR amplification fragment is preferably within 200 bp. Coupled with the high cost of fluorescent labeled probes and interference of large number probes in a tube, it is not suitable to detect large numbers of leukemia fusion genes by this technology. Fei et al. developed a multiplex RT-PCR method combined with liquid bead array cytometry for rapid detection of genetic alterations associated with leukemia [22]. The system involves multiplex RT-PCR amplification, capture of the fluorescent PCR products onto bead-probe complexes in solution, and fluorescence analysis on a flow cytometer. However, the procedure was streamlined for five to six hours at least [22]. In this study, we developed a multiplex RT-PCR method combined with capillary electrophoresis as a multiplex gene expression profiling analysis platform. PCR products were separated using capillary electrophoresis technology by QIAxcel Advanced system. The multiplex RT-PCR combined with capillary electrophoresis has been successfully used in the identification of different types of virus [23], with high sensitivity and specificity.
In this study, one step multiplex RT-PCR assay was developed for the simultaneous detection of eight common leukemia fusion genes, including BCR-ABL, ETV6-RUNX1, MLL-AF4, E2A-PBX1, AML1-ETO, PML-RARα, CBFβ-MYH11, and SIL-TAL1. The specificity and sensitivity of the assay were valuated. In total, 122 clinical samples were assayed and the detection results were compared with the results by mono real-time RT-PCR. This article proves that one step multiplex RT-PCR combined with capillary electrophoresis could be used in the detection of 8 common leukemia fusion genes.
Materials and methods
Patient population
The study contained a total of 122 patients newly diagnosed with childhood leukemia from a Han Chinese population between October 2014 to July 2016 at Children’s hospital of Nanjing medical university. The patients’ information could be identified after data collection. Childhood leukemia was diagnosed by Morphology, Immunology, Cytogenetics and Molecular Biology (MICM) according to the established guidelines for diagnosis and treatment of childhood leukemia in China (Society of Pediatrics, Chinese Medical Association, 2006). Subjects of negative results by real-time RT-PCR (n=20) were chosen as negative controls, which were consistent with metaphase cytogenetic or interphase FISH data and sequence data. Informed consent were performed before sample collection and the study was approved by the Institutional Ethics Committee of Children’s Hospital of Nanjing Medical University. Fresh bone marrow specimens of patients were taken at diagnosis and collected in EDTA tubes.
Nucleic acid extraction
Two milliliters of bone marrow were separated for multiplex RT-PCR study. Mononuclear cells from bone marrow specimens were isolated using Ficoll-hypaque density gradient ultracentrifugation. Total RNA was extracted from mononuclear cells using TRIzol reagent (Invitrogen, China) according to the manufacturer’s protocol. The RNA was suspended in a final volume of 60 μl elution buffer and separated for three parts and then stored at -80°C until use.
Primer design and validation
A multiplex PCR reaction for eight common fusion genes in pediatric leukemia and an endogenous control transcript (GAPDH) were performed in a single reaction system. This assay included one pair of universal primers, and ten pairs of chimeric primers (a fusion transcript specific sequence fused at the 5’ end to the universal sequence). Gene sequences of fusion transcripts and GAPDH were obtained from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/). Universal primers were designed based on a highly conserved region of bacteria sequences, distinguishable from human DNA sequences. All of the primers were designed using Primer Premier 5.0 software and evaluated using Primer-BLAST analysis on NCBI (NCBI, Bethesda, MD, USA). In order to separate the amplicons from each other, the length of all amplicons were designed different more than 50 bp. The primer pairs specific for BCR-ABL (p190) and BCR-ABL (p210) fusion transcripts were designed together in one reaction. The primer sequences, their target genes and the size of the resulting amplicons were summarized in Table 1. All primers were synthesized by Invitrogen Corporation (Shanghai, China). The in vitro transcribed RNA products were synthesized and sequenced by Invitrogen Corporation (Shanghai, China).
Table 1.
Primers for one step multiple RT-PCR combining capillary electrophoresis analysis system and amplicon size
| Fusion transcripts | Chimeric primer (5’-3’) | Size of PCR products (bp) | |
|---|---|---|---|
| CBFβ-MYH11 | F: AGGTGACACTATAGAATATGGGCTGTCTGGAGTTTGATG | 309 | |
| R: GTACGACTCACTATAGGGACTTGAGCGCCTGCATGTT | |||
| SIL-TAL1 | F: AGGTGACACTATAGAATAGCTCCTACCCTGCAAACAGAC | 807/887 | |
| R: GTACGACTCACTATAGGGAATACGCCGCACAACTTTG | |||
| ETV6-RUNX1 | F: AGGTGACACTATAGAATACTCATCGGGAAGACCTGGCTTAC | 657/696 | |
| R: GTACGACTCACTATAGGGATTTCTGCCGATGTCTTCGAG | |||
| BCR-ABL | M-BCR | F: AGGTGACACTATAGAATAAGCATTCCGCTGACCATCA | M-BCR: 405/480 (p210) |
| m-BCR | F: AGGTGACACTATAGAATACCACCACCTACCGCATGT | m-BCR: 587 (p190) | |
| Common primer | R: GTACGACTCACTATAGGGAAGATACTCAGCGGCATTGC | ||
| E2A-PBX1 | F: AGGTGACACTATAGAATACTACGACGGGGGTCTCCAC | 222 | |
| R: GTACGACTCACTATAGGGACACGCCTTCCGCTAACAG | |||
| PML-RARα | F: AGGTGACACTATAGAATACTGGACATGCACGGTTTC | 266/740 | |
| R: GTACGACTCACTATAGGGAGCTTGTAGATGCGGGGTAGAG | |||
| AML1-ETO | F: AGGTGACACTATAGAATATCACTCTGACCATCACTGTCTTC | 357 | |
| R: GTACGACTCACTATAGGGAAGAAGAGGAAGGCCCATTGC | |||
| MLL-AF4 | F: AGGTGACACTATAGAATACCGCCCAAGTATCCCTGTA | 538 | |
| R: GTACGACTCACTATAGGGAGTTCCTTGCTGAGAATTTGAGTG | |||
| GAPDH | F: AGGTGACACTATAGAATACAAGGTCATCCATGACAACTTTG | 175 | |
| R: GTACGACTCACTATAGGGATGGTGAAGACGCCAGTGGA | |||
| Universal primers | F: AGGTGACACTATAGAATA | ||
| R: GTACGACTCACTATAGGGA | |||
One step mono RT-PCR assay
GAPDH was used as internal positive control, RNA of Cancer-free control subjects were as negative controls and positive clinical samples with different fusion genes as positive controls. Specificity of each pair of chimeric primers was evaluated in these samples to ascertain the actual amplicon size of each target region. One step mono RT-PCR assay was performed using 2 μL of RNA, 5X one step mono RT-PCR buffer (Mg2+ plus) 5 μL, 10X solution I 2.5 μL, 10 mM dNTPs 2 μL, HotStart Hitaq DNA polymerase (5 U/Μl) 1 μL, M-MLV (200 U/μL) 0.5 μL, 500 nM each of universal primers, and 50 nM each of chimeric primers. RNAse-free water was added to the RT-PCR system to a final volume of 25 μL. The reaction was incubated on a PCR system at 50°C for 25 min, 95°C for 10 min; 95°C for 15 s, 58°C for 30 s, 72°C for 50 s, 10 cycles; 95°C for 15 s, 65°C for 30 s, 72°C for 50 s, 10 cycles; 95°C for 15 s, 48°C for 30 s, 72°C for 50 s, 20 cycles; 72°C for 5 min. After amplification, PCR product separation and detection were performed on the QIAxcel Advanced system (QIAGEN, Germany) based on size using high-resolution capillary electrophoresis. The peak height for each PCR target was reported using the electropherograms and matched to the appropriate genes.
The specificity, sensitivity and cross-reactivity of one step multiplex RT-PCR combined with capillary electrophoresis assay
Plasmids of fusion transcripts served as positive controls to optimize one step multiplex RT-PCR combined with capillary electrophoresis assay by varying single parameters, including annealing temperature and the optimal proportion of primers. To estimate the specificity, sensitivity, and cross-reactivity, 10-fold serial dilutions of plasmids of fusion transcripts, positive and negative samples were used in the one step multiplex RT-PCR assay. GAPDH as internal positive control was added in each action. Serial 10-fold dilution of the plasmids were generated to evaluate the technical sensitivity in the optimized one step multiplex RT-PCR assay system.
Repeatability and reproducibility of one step multiplex RT-PCR combined with capillary electrophoresis assay
Two different dilutions of nine transcribed RNA products (105 and 103 copies/μL) were tested respectively to evaluate the repeatability and reproducibility of one step multiplex RT-PCR assay. Each dilution was tested three times by the same operator for the assessment of repeatability. As for the reproducibility, each dilution was analyzed three times by two different operators on three different days in two different laboratories.
Real-time-PCR for testing fusion genes
The one step multiplex RT-PCR assay was assessed by those clinical positive specimens, which had been tested by real-time RT-PCR kits of the nine fusion transcripts respectively (YuanQi BIO, China). The cycling conditions were based on those provided in the manufacturer’s instructions on 7500 real-time PCR system (Applied Biosystems Corporation, USA): 30 min at 42°C, 5 min at 94°C, and 40 cycles of 15 s at 94°C and 60 s at 60°C. The results were considered positive when Ct values were below 36.
Application of optimized one step multiplex RT-PCR combined with capillary electrophoresis assay in clinical positive specimens
A total of 122 clinical positive specimens from leukemia patients were tested by optimized one step multiplex RT-PCR combined with capillary electrophoresis assay. Multiplex RT-PCR products performed as described previously were added into the QIAxcel Advanced system (QIAGEN, German) for 8 mins at 2 kV with an alignment marker 15 bp to 1000 bp size, or 15 bp to 3000 bp size. Fragment sizing was performed automatically by the QIAxcel software. Sequencing analysis was used on specimens with inconsistent results of one step multiplex RT-PCR and real-time RT-PCR, as the final results.
Results
One step mono RT-PCR in detection leukemia fusion genes
In one step mono RT-PCR assay, each pair of fusion transcript-specific primers could amplify the target region of the corresponding fusion transcripts with no false migration. The amplification strategy was shown in Figure 1. The amplicon size for each fusion transcript was almost identical to the target size in Table 1, and the difference among amplicons did not exceed 3%. The length of each fragment is listed as follows: E2A-PBX1, 222 bp; PML-RARα, 266 bp; CBFβ-MYH11, 309 bp; AML1-ETO, 357 bp; BCR-ABL (p210) (M-BCR): 405/480 bp; BCR-ABL (p190) (m-BCR): 587 bp; MLL-AF4, 538 bp; ETV6-RUNX1, 657/696 bp; SIL-TAL1, 807/887 bp and GAPDH, 175 bp as positive control.
Figure 1.
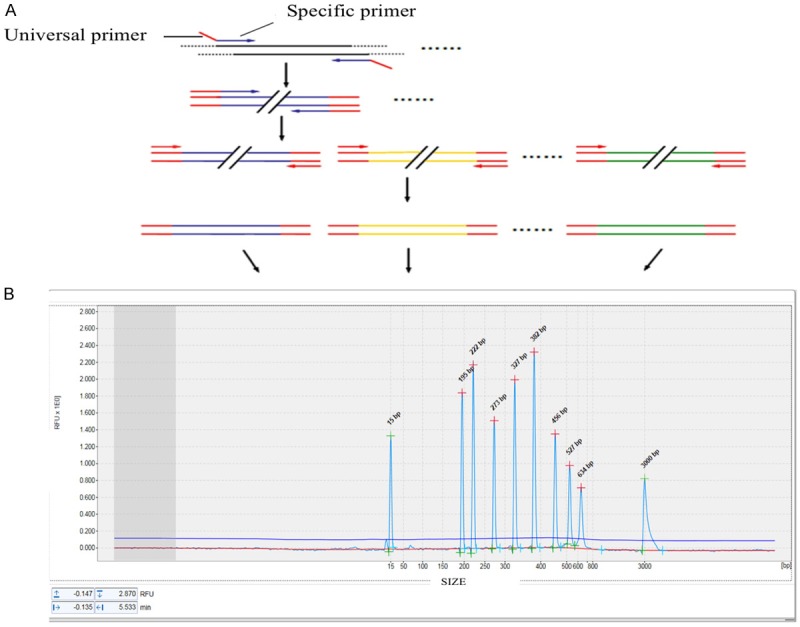
Workflow of one step multiple RT-PCR combining capillary electrophoresis analysis system.
Optimization of one step multiplex RT-PCR combined with capillary electrophoresis assay
According to the results of the orthogonal experiments, the concentration of each forward and reverse chimeric primers in the optimized one step multiplex RT-PCR assay were as follows: the concentration of GAPDH was 45 nM; MLL-AF4, ETV6-RUNX1, SIL-TAL1, CBFβ-MYH11 and E2A-PBX1 were 50 nM; M-BCR, m-BCR, PML-RARα and AML1-ETO were 60 nM; universal primers were 500 nM. The best PCR reaction condition was as follows: 50°C for 25 min, 95°C for 10 min; 95°C for 15 s, 58°C for 30 s, 72°C for 50 s, 10 cycles; 95°C for 15 s, 65°C for 30 s, 72°C for 50 s, 10 cycles; 95°C for 15 s, 48°C for 30 s, 72°C for 50 s, 20 cycles; 72°C for 5 min.
Specificity of one step multiplex RT-PCR combined with capillary electrophoresis assay
In the optimized one step multiplex RT-PCR system, nine chosen positive clinical samples with different fusion genes were tested, whose results were consistent with real-time PCR, shown in Table 2. In Figure 2, only specific amplification peaks and a GAPDH positive control were observed in positive samples with different fusion genes. The observed peaks showed specific amplification products without cross-amplification, while different combinations of fusion transcript plasmids were added in the amplification system (showed in Figure 3).
Table 2.
Positive control samples with different fusion transcripts
| No. | CBFB-MYH11 | SIL-TAL1 | ETV6-RUNX1 | M-bcr | m-bcr | E2A-PBX1 | PML-RARα | AML1-ETO | MLL-AF4 |
|---|---|---|---|---|---|---|---|---|---|
| 5 | + | - | - | - | - | - | - | - | - |
| 13 | - | + | - | - | - | - | - | - | - |
| 28 | - | - | + | - | - | - | - | - | - |
| 42 | - | - | - | + | - | - | - | - | - |
| 69 | - | - | - | - | + | - | - | - | - |
| 88 | - | - | - | - | - | + | - | - | - |
| 95 | - | - | - | - | - | - | + | - | - |
| 101 | - | - | - | - | - | - | - | + | - |
| 107 | - | - | - | - | - | - | - | - | + |
Figure 2.

Specificity results of one step multiplex RT-PCR combined with capillary electrophoresis assay in testing different fusion transcripts respectively. (A~I) showed amplified results of E2A-PBX1 (P1), PML-RARα (P2), CBFB-MYH11 (P3), AML1-ETO (P4), M-BCR p210 (P5), MLL-AF4 (P6), m-BCR p190 (P7), ETV6-RUNX1 (P8), SIL-TAL1 (P9) from different positive samples. RNA from healthy children’s peripheral blood was used as negative control. GAPDH (P10) was used as endogenous positive control in every one step GX-M-RT-PCR assay system. In (A~J), 15 bp and 1000 bp were as align markers.
Figure 3.

Evaluation of cross-reactivity of one step multiplex RT-PCR combined with capillary electrophoresis assay. In one step multiplex RT-PCR assay, different combinations of fusion transcript plasmids were observed showing specific component amplification peaks, without cross-amplification. The fusion transcripts were marked as E2A-PBX1 (P1), PML-RARα (P2), CBFB-MYH11 (P3), AML1-ETO (P4), M-BCR p210 (P5), MLL-AF4 (P6), m-BCR p190 (P7), ETV6-RUNX1 (P8), SIL-TAL1 (P9) and GAPDH (P10) in the Figure.
Sensitivity of one step multiplex RT-PCR combined with capillary electrophoresis assay
Serial 10-fold dilutions of quantitative plasmids of fusion genes were used to evaluate the sensitivity of one step multiplex RT-PCR combined with capillary electrophoresis assay. The results show that AML1-ETO, MLL-AF4 and M-BCR are 102 copies/μL respectively, CBFβ-MYH11, SIL-TAL1, ETV6-RUNX1, m-BCR, E2A-PBX1 and PML-RARα are 103 copies/μL respectively. When nine pre-mixed plasmids were added, the one step multiplex RT-PCR combined with capillary electrophoresis assay could detect as few as 103 copies/μL (showed in Figure 4).
Figure 4.

Sensitivity of one step multiplex RT-PCR combined with capillary electrophoresis assay. The one step multiplex RT-PCR combined with capillary electrophoresis assay was used to detect 10-fold concentration of eight premixed recombinant plasmids with 105 copies/μL (A), 104 copies/μL (B), 103 copies/μL (C) and 102 copies/μL (D) respectively. ALL fusion transcript plasmids were successfully tested at levels of 103 copies/μL. AML1-ETO, MLL-AF4 and M-bcr were tested as low as 102 copies/μL. The fusion transcripts were marked as E2A-PBX1 (P1), PML-RARα (P2), CBFB-MYH11 (P3), AML1-ETO (P4), M-bcr p210 (P5), MLL-AF4 (P6), m-bcr p190 (P7), ETV6-RUNX1 (P8), SIL-TAL1 (P9), GAPDH (P10).
Repeatability and reproducibility of one step multiplex RT-PCR combined with capillary electrophoresis assay
Two different dilutions of nine quantitative plasmids (105 and 103 copies/μL) were tested respectively and each dilution was tested three times by the same operator for the assessment of repeatability. As for the reproducibility, each dilution was analyzed three times by two different operators on three different days in two different laboratories. These results were similar, and the difference between the same fusion transcript sizes did not exceed 5% (results not shown), indicating that one step multiplex RT-PCR combined with capillary electrophoresis assay was satisfactory and with high repeatability and reproducibility.
Interference analysis of one step multiplex RT-PCR combined with capillary electrophoresis assay
Two specific amplification peaks in two different fusion gene templates which are 103 copies/μL and 105 copies/μL, respectively, were observed by using one step multiplex RT-PCR combined with capillary electrophoresis assay. The peak of mixed templates is similar to the peak of single template. For example, one step multiplex RT-PCR combined with capillary electrophoresis assay could detect two specific amplification peaks in AML1-ETO (105 copies/μL) and M-bcr (103 copies/μL) templates. No significant difference was detected in AML1-ETO and M-bcr peaks between single template (AML1-ETO or M-bcr) and mixed template (AML1-ETO and M-bcr) (showed in Figure 5). The results of these experiments show that this interference has the least effect on detecting hybrid templates.
Figure 5.

Interference results of one step multiplex RT-PCR combined with capillary electrophoresis assay determination. One step multiplex RT-PCR combined with capillary electrophoresis assay was performed using three different templates, showing mixed template (AML1-ETO and M-bcr p210) (A), single template AML1-ETO (B), single M-bcr p210 (C). The different peaks on three graphs represent GAPDH (P10), AML1-ETO (P4), M-bcr p210 (P5), respectively.
Application of clinical specimens
A total of 122 clinical specimens, which were positive fusion transcripts from leukemia patients were tested by one step multiplex RT-PCR combined with capillary electrophoresis assay. All the specimens were confirmed by real-time PCR. In comparison with the results of real-time PCR, the sensitivities of one step multiplex RT-PCR combined with capillary electrophoresis assay for nine fusion transcripts, BCR-ABL were 100% (21/21), CBFβ-MYH11 were 100% (13/13), SIL-TAL1 were 100% (13/13), ETV6-RUNX1 were 100% (16/16), E2A-PBX1 were 100% (16/16), PML-RARα were 100% (15/15), AML1-ETO were 93.3% (14/15), MLL-AF4 were 100% (13/13). In particular, 21 cases showed BCR-ABL positive, including 8 cases showed p190 only, 9 cases showed p210 only, 4 cases showed both p190 and p210. A positive clinical sample of AML1-ETO gene was not detected. However, the Ct value of the amplification curve was large by second Real-time PCR analysis, indicating that the proportion of AML1-ETO gene was very low in this sample.
Split out PCR
A rare result showed that three peaks didn’t coincide with the size of nine fusion transcripts when the clinical positive specimen was tested using one step multiplex RT-PCR combined with capillary electrophoresis assay. Therefore, these special specimens were tested again using one step mono RT-PCR assay containing only one pair of chimeric primers (showed in Figure 6). Finally, ETV6-RUNX1 was confirmed positive and showed deletion of 5th exon in ETV6 gene and deletion of 2th exon and/or 3th exon and/or 4th exon in RUNX1 gene by sequencing analysis. After sequencing analysis, the sequence followed 5th exon of ETV6 was found with fracture. Besides, RUNX1 was found be broken in multiple sites then spliced, making the possibility of the missing of RUNX1’s 1 to 4 exons (showed in Figure 7).
Figure 6.
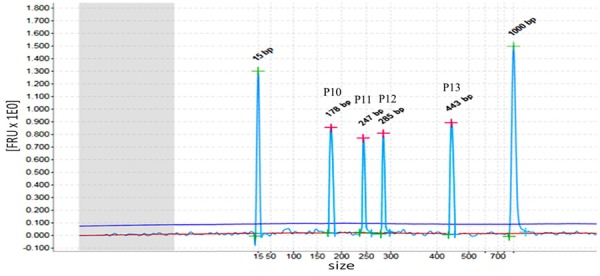
ETV6-RUNX1 positive sample. In this specimen, three different peaks were detected, namely 247 bp (P11), 285 bp (P12), 443bp (P13). GAPDH (P10), as a positive control, was shown at 178 bp.
Figure 7.

Sequencing analysis of one ETV6-RUNX1 positive sample. A. The ETV6 part of the three sequencing fragment results showed all the peaks containing the sequence followed 5th exon of ETV6. B. The RUNX1 part of the first fragment contained the 2th and 5th exon of RUNX1. C. The RUNX1 part of the second fragment contained the 2th, 4th and 5th exon of RUNX1. D. The RUNX1 part of the third fragment contained the 5th exon of RUNX1.
Discussion
The one step multiplex RT-PCR combined with capillary electrophoresis assay was an effective and economical method for detecting fusion genes of leukemia. During the initial 5th to 15th cycles of amplification procedure, all fusion transcripts were amplified by specific chimeric primers leading to the products being labeled with universal primer sequences. Then high concentrations of universal primers played a leading role and amplified all the targets until the amplification ended. This assay made multiplex PCR amplification mediated by nine pair of specific primers change into a mono PCR amplification, which further reduced amplification complexity. PCR products were detected by capillary electrophoresis automatically using QIAxcel Advanced system other than agarose gel electrophoresis with simplified procedure, time saving and less population.
In the present study, one pair of universal primers and ten pairs of gene-specific chimeric primers were designed to develop one-step multiplex RT-PCR assay for simultaneous identification of eight fusion genes, including BCR-ABL (p190), BCR-ABL (p210), SIL-TAL1, ETV6-RUNX1, MLL-AF4, E2A-PBX1, AML1-ETO, PML-RARα, CBFβ-MYH11 and GAPDH (as a positive control). The selection of the eight significant fusion genes associated with childhood leukemia could help clinicians give proper treatment for patients [24,25]. In this assay, the multiplex RT-PCR amplified nine fusion genes in one tube, which covered all possible cases of the nine fusion transcripts. Finally, the products were automatically analyzed by QIAxcel Advanced system to avoid product contamination and save time. The results of 122 positive cases by real-time PCR showed 121 cases were accurately detected by one step multiplex RT-PCR combined with capillary electrophoresis assay, indicating the high accuracy and detection rate of this assay. Besides, one AML1-ETO positive sample failed to be detected by one step multiplex RT-PCR combined with capillary electrophoresis assay because of low concentration.
Clinical specimens might inhibit enzyme-based nucleic acid amplification by containing inhibitors. A few studies have indicated that internal control increased the sensitivity of this assay [26-28]. However, several studies about GeXP analyzer-based multiplex PCR assay didn’t add internal control. In this study, GAPDH, as a co-amplification and co-detection internal endogenous transcript was added in this system which protected the generation of potential false negative results and monitored the entire process of every tested sample, including nucleic acid extraction and presence of residual PCR inhibitors [29]. The quantity of GAPDH was also used as an indicator of the degradation of target RNA and the re-extraction of the specimen. According to results of analysis of 122 samples, GAPDH was one of the best housekeeping genes in our system.
Based on the complication of childhood leukemia fusion transcripts, different length of amplicons might appear in the same fusion transcript and the generation of new fusion transcripts do not have positive control. In this study, a new-found ETV6-RUNX1 translocation was confirmed in one of the 122 cases. So the special specimen was tested by using one step mono RT-PCR assay containing only one pair of chimeric primers. Finally, ETV6-RUNX1 was positively confirmed which showed the deletion of 5th exon in ETV6 gene and 2th exon and/or 3th exon and/or 4th exon in RUNX1 gene by sequencing analysis. The complexity of fracture sites of leukemia fusion genes was compounded by the different sizes of amplified fragments in the same type of translocation or the occurrence of new fractures resulting in missing match with positive control. In order to get reliable results, it was necessary to determine the type of fusion gene by splitting out the PCR, then the cleavage site and DNA information was determined by sequencing.
In conclusion, the newly developed and validated one-step multiplex RT-PCR combined with capillary electrophoresis provides a sensitive and reliable technology platform for detecting nine common fusion transcripts in pediatric leukemia. The assay provides accuracy in detecting eight distinct leukemia fusion genes in four hours including RNA extraction, which saves money, time, and labor compared with previous methods.
Acknowledgements
This work was supported by Nanjing Science and Technique Development Foundation (201402022) and Nanjing Medical Science and Technology Development Foundation (QRX17078).
Disclosure of conflict of interest
None.
References
- 1.Schurch CM, Riether C, Ochsenbein AF. Dendritic cell-based immunotherapy for myeloid leukemias. Front Immunol. 2013;4:496. doi: 10.3389/fimmu.2013.00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basilico S, Gottgens B. Dysregulation of haematopoietic stem cell regulatory programs in acute myeloid leukaemia. J Mol Med (Berl) 2017;95:719–727. doi: 10.1007/s00109-017-1535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ogasawara MA, Liu J, Pelicano H, Hammoudi N, Croce CM, Keating MJ, Huang P. Alterations of mitochondrial biogenesis in chronic lymphocytic leukemia cells with loss of p53. Mitochondrion. 2016;31:33–39. doi: 10.1016/j.mito.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127:29–41. doi: 10.1182/blood-2015-07-604496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 2017;35:934–946. doi: 10.1200/JCO.2016.71.2208. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Mao S, Su S, Wei L, Li T, Liu Y. Detection of concurrent TEL-AML1/TEL-ABL fusion genes in a patient with B-acute lymphoblastic leukemia using a multi-fusion gene qRT-PCR screening method. Pathol Int. 2016;66:475–477. doi: 10.1111/pin.12428. [DOI] [PubMed] [Google Scholar]
- 7.Hu Y, He H, Lu J, Wang Y, Xiao P, Li J, Li J, Sun Y, Lv H, Fan J, Yao Y, Chai Y, Hu S. E2A-PBX1 exhibited a promising prognosis in pediatric acute lymphoblastic leukemia treated with the CCLG-ALL2008 protocol. Onco Targets Ther. 2016;9:7219–7225. doi: 10.2147/OTT.S115257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prange KH, Mandoli A, Kuznetsova T, Wang SY, Sotoca AM, Marneth AE, van der Reijden BA, Stunnenberg HG, Martens JH. MLL-AF9 and MLL-AF4 oncofusion proteins bind a distinct enhancer repertoire and target the RUNX1 program in 11q23 acute myeloid leukemia. Oncogene. 2017;36:3346–3356. doi: 10.1038/onc.2016.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szanto A, Pap Z, Denes L, Benedek Lazar E, Horvath A, Tunyogi AB, Baroti BA, Pavai Z. Real-time quantitative PCR detection of WT1 and M-BCR-ABL expressions in chronic myeloid leukemia. Rom J Morphol Embryol. 2015;56:703–707. [PubMed] [Google Scholar]
- 10.Arora R, Sawney S, Saluja D. Potential therapeutic approaches for the treatment of acute myeloid leukemia with AML1-ETO translocation. Curr Cancer Drug Targets. 2016;16:215–225. doi: 10.2174/1568009616666151113120146. [DOI] [PubMed] [Google Scholar]
- 11.Purkait R, Dolai TK. Identification of PML/RARa fusion gene by RT-PCR acute promyelocytic leukaemia. Indian Pediatr. 2014;51:1020–1021. [PubMed] [Google Scholar]
- 12.Schwind S, Edwards CG, Nicolet D, Mrozek K, Maharry K, Wu YZ, Paschka P, Eisfeld AK, Hoellerbauer P, Becker H, Metzeler KH, Curfman J, Kohlschmidt J, Prior TW, Kolitz JE, Blum W, Pettenati MJ, Dal Cin P, Carroll AJ, Caligiuri MA, Larson RA, Volinia S, Marcucci G, Bloomfield CD Alliance for Clinical Trials in Oncology. inv(16)/t(16;16) acute myeloid leukemia with non-type A CBFB-MYH11 fusions associate with distinct clinical and genetic features and lack KIT mutations. Blood. 2013;121:385–391. doi: 10.1182/blood-2012-07-442772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Bockstaele F, Verhasselt B, Philippe J. Prognostic markers in chronic lymphocytic leukemia: a comprehensive review. Blood Rev. 2009;23:25–47. doi: 10.1016/j.blre.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Muhlbacher V, Haferlach T, Kern W, Zenger M, Schnittger S, Haferlach C. Array-based comparative genomic hybridization detects copy number variations with prognostic relevance in 80% of ALL with normal karyotype or failed chromosome analysis. Leukemia. 2016;30:318–324. doi: 10.1038/leu.2015.276. [DOI] [PubMed] [Google Scholar]
- 15.Akhter A, Mughal MK, Elyamany G, Sinclair G, Azma RZ, Masir N, Shuib S, Rashid-Kolvear F, Shabani-Rad MT, Stewart DA, Mansoor A. Multiplexed automated digital quantification of fusion transcripts: comparative study with fluorescent in-situ hybridization (FISH) technique in acute leukemia patients. Diagn Pathol. 2016;11:89. doi: 10.1186/s13000-016-0541-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arora T, Dhir R. A review of metaphase chromosome image selection techniques for automatic karyotype generation. Med Biol Eng Comput. 2016;54:1147–1157. doi: 10.1007/s11517-015-1419-z. [DOI] [PubMed] [Google Scholar]
- 17.Younis A, Ramzan F, Hwang YJ, Lim KB. FISH and GISH: molecular cytogenetic tools and their applications in ornamental plants. Plant Cell Rep. 2015;34:1477–1488. doi: 10.1007/s00299-015-1828-3. [DOI] [PubMed] [Google Scholar]
- 18.Krauter J, Peter W, Pascheberg U, Heinze B, Bergmann L, Hoelzer D, Lubbert M, Schlimok G, Arnold R, Kirchner H, Port M, Ganser A, Heil G. Detection of karyotypic aberrations in acute myeloblastic leukaemia: a prospective comparison between PCR/FISH and standard cytogenetics in 140 patients with de novo AML. Br J Haematol. 1998;103:72–78. doi: 10.1046/j.1365-2141.1998.00926.x. [DOI] [PubMed] [Google Scholar]
- 19.Pine SR, Yin C, Matloub YH, Sabaawy HE, Sandoval C, Levendoglu-Tugal O, Ozkaynak MF, Jayabose S. Detection of central nervous system leukemia in children with acute lymphoblastic leukemia by real-time polymerase chain reaction. J Mol Diagn. 2005;7:127–132. doi: 10.1016/S1525-1578(10)60018-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jinawath N, Norris-Kirby A, Smith BD, Gocke CD, Batista DA, Griffin CA, Murphy KM. A rare e14a3 (b3a3) BCR-ABL fusion transcript in chronic myeloid leukemia: diagnostic challenges in clinical laboratory practice. J Mol Diagn. 2009;11:359–363. doi: 10.2353/jmoldx.2009.090008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Velden VH, Hochhaus A, Cazzaniga G, Szczepanski T, Gabert J, van Dongen JJ. Detection of minimal residual disease in hematologic malignancies by real-time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia. 2003;17:1013–1034. doi: 10.1038/sj.leu.2402922. [DOI] [PubMed] [Google Scholar]
- 22.Ye F, Laosinchai-Wolf W, Labourier E. An optimized technology platform for the rapid multiplex molecular analysis of genetic alterations associated with leukemia. Cancer Genet. 2012;205:488–500. doi: 10.1016/j.cancergen.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagel MA, Gilden D, Shade T, Gao B, Cohrs RJ. Rapid and sensitive detection of 68 unique varicella zoster virus gene transcripts in five multiplex reverse transcription-polymerase chain reactions. J Virol Methods. 2009;157:62–68. doi: 10.1016/j.jviromet.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pakakasama S, Kajanachumpol S, Kanjanapongkul S, Sirachainan N, Meekaewkunchorn A, Ningsanond V, Hongeng S. Simple multiplex RT-PCR for identifying common fusion transcripts in childhood acute leukemia. Int J Lab Hematol. 2008;30:286–291. doi: 10.1111/j.1751-553X.2007.00954.x. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Mancilla M, Rodriguez-Aguirre I, Tejocote-Romero I, Medina-Sanson A, Ocadiz-Delgado R, Gariglio P. Clinical relevance of the fusion transcripts distribution pattern in mexican children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2013;35:170–173. doi: 10.1097/MPH.0b013e318286d241. [DOI] [PubMed] [Google Scholar]
- 26.Rosenstraus M, Wang Z, Chang SY, DeBonville D, Spadoro JP. An internal control for routine diagnostic PCR: design, properties, and effect on clinical performance. J Clin Microbiol. 1998;36:191–197. doi: 10.1128/jcm.36.1.191-197.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao XL, He YQ, Yu YG, Yang H, Chen G, Li HF, Zhang JW, Liu DM, Li XF, Yang XQ, Wu H. Simultaneous detection of human enterovirus 71 and coxsackievirus A16 in clinical specimens by multiplex real-time PCR with an internal amplification control. Arch Virol. 2009;154:121–125. doi: 10.1007/s00705-008-0266-8. [DOI] [PubMed] [Google Scholar]
- 28.Sidoti F, Bergallo M, Terlizzi ME, Piasentin Alessio E, Astegiano S, Gasparini G, Cavallo R. Development of a quantitative real-time nucleic acid sequence-based amplification assay with an internal control using molecular beacon probes for selective and sensitive detection of human rhinovirus serotypes. Mol Biotechnol. 2012;50:221–228. doi: 10.1007/s12033-011-9432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vandenbussche F, Mathijs E, Lefebvre D, De Clercq K, Van Borm S. A tale of tails: dissecting the enhancing effect of tailed primers in real-time PCR. PLoS One. 2016;11:e0164463. doi: 10.1371/journal.pone.0164463. [DOI] [PMC free article] [PubMed] [Google Scholar]