

Abstract
Sunitinib is used as standard treatment for metastatic or unresectable clear cell renal cell carcinoma (ccRCC). However, ccRCC eventually develops resistance to sunitinib in most cases, and the mechanisms underlying such resistance have not been fully determined. Nuclear receptors (NRs) are a class of transcription factors that regulate many cellular functions by controlling gene expression, and they also play important roles in tumor development, proliferation and progression in various types of cancers. In the present study, we aimed to explore the mechanisms underlying sunitinib resistance in RCC and the potential role of NRs in sunitinib resistance. The expression profile of NRs was obtained from the Gene Expression Omnibus (GEO) RNAseq database. A total of 138 patients from GSE65615 were examined in this study. From the GEO metadata, we found that the expressions of three genes, encoding peroxisome proliferator activated receptor alpha (PPARα), androgen receptor (AR) and PPARγ, were significantly increased in sunitinib-treated samples compared with control samples. RT-PCR analysis showed that the PPARα expression at the mRNA level was significantly increased in sunitinib-resistant A498, CaKi-1 and 780-O ccRCC lines compared with their sunitinib-sensitive parental cells. Furthermore, knockdown of PPARα significantly inhibited cell proliferation in all three sunitinib-resistant ccRCC lines, successfully overcoming the resistance to sunitinib. Our results also showed that nuclear factor kappa B (NF-κB) signaling pathway was activated in sunitinib-resistant ccRCC lines, indicating that PPARα and NF-κB inhibition could play a synergistic role to modulate sunitinib resistance and suggesting that PPARα could be used as a potential target to overcome sunitinib resistance via the NF-κB pathway.
Keywords: Renal cell carcinoma, PPARα, NF-κB pathway, sunitinib resistance
Introduction
As the 13th most common cancer worldwide, renal cell carcinoma (RCC) is responsible for about 2% of all malignancies in adults, with an incidence that is increasing by 1.5%-5.9% per year [1,2]. In 2017, approximately 63,990 new cases of renal cancer and 14,440 deaths from the disease are expected in the United States [3]. Clear cell RCC (ccRCC) is the most common histologic type (70%-80%) of RCC [4]. Despite developments in the diagnosis and treatment strategies of RCC, one-third of RCC patients present with metastatic disease at diagnosis [5]. Metastatic disease is usually resistant to radiotherapy and chemotherapy, and immunotherapy shows limited response rates of 15%-20% in these cases [6]. Therefore, surgery remains the only effective treatment for RCC. However, approximately 20%-40% of RCC patients subjected to surgical nephrectomy will develop metastasis [7].
Targeted therapies have become increasingly available in recent years, showing considerable promise for the treatment of ccRCC and other malignancies. Sunitinib is a small molecule inhibitor of multiple receptor tyrosine kinases, including vascular endothelial growth factor (VEGF) receptors (VEGFR-1, VEGFR-2 and VEGFR-3), platelet-derived growth factor receptors (PDGFR-α and PDGFR-β), fms-related tyrosine kinase 3 (FLT3) and stem cell growth factor receptors (KIT and RET) [8]. Sunitinib functions by interfering with these cell signaling pathways, exhibiting anti-tumor and anti-angiogenesis activities. Sunitinib is currently considered as a standard first-line treatment for metastatic ccRCC, resulting in longer overall survival (OS) and progression-free survival compared with patients treated with conventional drugs [9-11]. Approximately 38% of patients with metastatic RCC treated with sunitinib show significant tumor control [12]. However, the prognosis of non-responding patients remains poor, with a mean OS of 14.5 months [12]. Furthermore, despite the efficacy of sunitinib, ccRCC often develops resistance to sunitinib, and the majority of patients receiving sunitinib for treatment of advanced ccRCC exhibit progressive disease after 1 year of treatment [7]. Several hypotheses have been proposed regarding the mechanism(s) underlying resistance to sunitinib, but the precise pathways remain largely unexplored [13,14].
PPARα (peroxisome proliferator activated receptor alpha) is a nuclear receptor (NR) that is activated through ligand binding, and it induces the expression of a large number of target genes. PPARα functions in various pathways, including the circadian rhythm pathway and NOTCH1-mediated pathway for nuclear factor kappa B (NF-κB) activity modulation [15]. Diseases associated with the aberrant expression and function of PPARα include fatty liver disease and alcoholic cardiomyopathy, showing altered expressions of target genes involved in cell proliferation and cell differentiation as well as in immune and inflammation responses. The function of PPARα has also been linked to various cancers. Inhibition of PPARα activity induces tumor suppression in melanoma [16] and glioblastoma [17], and increased PPARα activation has also been found to lead to progression of tumor growth in hepatocellular carcinoma [18] and breast cancer [19]. Previous studies have also identified PPARα as a novel therapeutic target for RCC [20,21]. Mouse xenograft metabolomics studies have reported an inhibitory effect of PPARα on tumor growth and demonstrated that PPARα-mediated inhibition of tumor growth is comparable to that of sunitinib, suggesting that PPARα is involved in RCC tumorigenesis through its effects on reprogrammed metabolic pathways [15]. A previous report has found that the expression of PPARα is correlated with RCC progression, and PPARα inhibition attenuates RCC cell viability [20]. Furthermore, the specific PPARα antagonist, GW6471, induces apoptosis and cell cycle arrest at G0/G1 in RCC cell lines, which is associated with alterations in expressions of cell cycle regulatory proteins, with a pronounced dependence on histologic grade of expression and a synergistic effect with glycolysis inhibition. Taken together, these studies have established that PPARα can be used as a potential target in treatment of RCC.
In the present study, we aimed to further clarify the mechanisms underlying resistance to sunitinib and the related signaling pathways using sunitinib-resistant RCC cell lines. In addition, several potential targets to overcome sunitinib resistance were identified.
Materials and methods
Gene expression omnibus (GEO) data
The metadata of a ccRCC patient GEO cohort were downloaded from the GEO. A total of 138 patients were included in the analysis. The expressions of 48 NRs at the mRNA level in mRCC were obtained from GSE65615.
Cell lines and reagents
Three human RCC cell lines (A498, Caki-1 and 786-O) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Grand Island, NY, USA) and 100 units/mL penicillin and streptomycin (Thermo Fisher Scientific) at 37°C in a humidified incubator containing 5% CO2. The sunitinib-resistant A498, Caki-1 and 786-O cell lines were generated in a stepwise manner by exposing parental cells to increasing doses (1, 3, 5 and 10 µg/mL) of sunitinib. The surviving cells were subsequently maintained in the conditioned medium containing 1 µg/mL sunitinib (Sigma-Aldrich Co., St Louis, MO, USA) to retain the drug-resistant phenotype. The parental controls were performed in A498, Caki-1 and 786-O cells with similar passage numbers.
PPARα downregulation and overexpression constructs
The lentiviral vector system was purchased from Clontech (Mountain View, CA, USA). The vector system included the plasmids as follows: pLV-CMV-GFP, pLVX-U6-GFP, psPAX and pMD2G. The sequences for the target siRNAs against human PPARα (GenBank, NM_001001928.2) and control siRNAs were as follows: shPPARα-F, 5’-gatccTATCTGAAGAGTTCCTGCAAGAAATTTCAAGAGAATTTCTTGCAGGAACTCTTCAGATATTTTTTg-3’; shPPARα-R, 5’-aattcAAAAAATATCTGAAGAGTTCCTGCAAGAAATTCTCTTGAAATTTCTTGCAGGAACTCTTCAGATAg-3’; control shRNA-F, 5’-gatccGACTTCATAAGGCGCATGCTTCAAGAGAGCATGCGCCTTATGAAGTCTTTT TTg-3’; and control shRNA-R, 5’-aattcAAAAAAGACTTCATAAGGCGCATGCTCTCTTGAAGCATGCGCCTTATGAAGTCg-3’. The siRNAs were cloned into the lentiviral vector and named as pLV-CMV-shPPARα and pLV-CMV-sh-control, respectively. The lentiviral vector for PPARα overexpression, named pLV-CMV-PPARα, was constructed by Genelily (Shanghai, China). The lentiviral vectors containing the PPARα shRNA or PPARα coding sequence were confirmed by polymerase chain reaction (PCR) and sequencing. The lentiviral vector containing a non-silencing sequence was used as the negative control sequence (Con) for lentiviral vector production and cell infection. Lentiviral vectors were produced by transient transfection of HEK293 cells according to standard protocols. The HEK293 cells was cultured in DMEM supplemented with 10% FBS. When the cells were subconfluent, transfection was performed using 1.8 mL DNA solution containing 10 µg of Lv-shControl, Lv-shPPARα or Lv-PPARα, 10 µg of psPAX and 10 µg of pMD2G. All virus stocks were produced by Lipofectamine®-mediated transfection. After 48 h, the cell supernatants containing viral particles were filtered using the 0.45-µm Steriflip® vacuum filtration system (EMB Millipore, Billerica, MD, USA) and concentrated by ultracentrifugation at 25,000 rpm at 4°C. The titer of the virus was tested based on the expression level of green fluorescent protein (GFP). In experiments, A498, Caki-1 and 786-O cells were seeded at a confluency of 30%-40% on the day before infection. On the following day, cells were infected by packaged lentiviral production. In parallel, non-infected cells were simultaneously observed. Cells were cultured in normal growth medium for 72 h after infection. Subsequently, cells were propagated in selection medium containing puromycin.
Cell viability assay and IC50 determination
Cells were seeded into 96-well plates for 24 h, then 10 µL CCK8 (Sigma, USA) reagent was added into each well, and the mixture was incubated for additional 2 h. The absorbance at a wavelength of 450 nm was recorded using a microplate absorbance reader (Tecan, Safire II, Switzerland). Data were used to generate curves of drug effect or cell proliferation rate.
Western blotting analysis
Cells were lysed in RIPA lysis buffer (Solarbio, Beijing, China) containing 0.1 mM PMSF and a protease inhibitor (Roche) on ice for 30 min. Samples were subjected to 12% SDS-PAGE and then transferred onto nitrocellulose membranes. Blots were incubated with properly diluted primary antibodies at room temperature for 2 h, including anti-androgen receptor (AR, 1:2,000, Abcam, Cambridge, UK), anti-PPARα (1:2,000, Abcam), anti-PPARγ (1:3,000, Abcam), anti-NF-κB p65 (1:1,000, Abcam) and anti-GAPDH (1:5,000, Santa Cruz Biotechnology). After washed with PBST for four times, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies. The signals were detected with Pierce® ECL western blotting substrate (Pierce, Rockford, IL, United States) according to the manufacturer’s instructions and exposed on x-ray films (Kodak, Rochester, NY, USA).
Quantitative real-time PCR
Total RNA was extracted from cells with TRIzol reagent (Invitrogen, Shanghai, China) according to the manufacturer’s instructions. Purified RNA (1 µg) was reversely transcribed into cDNA by Superscript II reverse transcriptase (Invitrogen) and random primer oligonucleotides (Invitrogen). Real-time PCR was performed on the 7900 HT RealTime PCR System (Applied Biosystems), and GAPDH was selected as a housekeeping gene. Table 1 lists the sequences of the primers used in the study.
Table 1.
Primer Sequences
| Genes | Sequences of primers | Product sizes (bp) |
|---|---|---|
| GAPDH | F: 5’-TGACTTCAACAGCGACACCCA-3’ | 121 |
| R: 5’-CACCCTGTTGCTGTAGCCAAA-3’ | ||
| Androgen Receptor | F: 5’-CCCACTTGTGTCAAAAGCGA-3’ | 192 |
| R: 5’-GCAGCTTCCACATGTGAGAG-3’ | ||
| PPARα | F: 5’-ATCGGCGAGGATAGTTCTGG-3’ | 205 |
| R: 5’-CCTTGTCCCCGCAGATTCTA-3’ | ||
| PPARγ | F: 5’-GAGCCTTCCAACTCCCTCAT-3’ | 231 |
| R: 5’-ATGAGACATCCCCACTGCAA-3’ |
NF-κB transcription assay
Cells were seeded into 24-well plates, precultured to 75% confluence and then transfected with an HIV promoter-driven luciferase cDNA plasmid as described previously. Luciferase activity was assessed using a kit from Promega. The firefly luciferase activity was normalized to Renilla luciferase activity. The assay was repeatedthree times in duplicate.
Renal cancer tumor xenograft in female nude mice
Athymic BALB/c nude mice (female, 4-week-old, weight 20 g) were purchased from Shanghai Slac Laboratory Animal Corporation (Shanghai, China). Female nude mice were maintained under specific pathogen-free conditions with 12-h light/12-h dark cycle for 3 days. A xenograft model of human renal cancer was established in female nude mice through subcutaneously injecting PPARα-overexpression or PPARα-knockdown sunitinib-resistant 780-O and parental cells into each mouse. The mice were randomly divided into six groups. Treatment was started when the tumor reached about 150 mm3-200 mm3. Briefly, 20 mg/mL sunitinib was intraperitoneally injected once every 2 days. Tumor diameter was measured using a caliper every 2 days, and the tumor volume was calculated according to the formula as follows: length × width × height × 0.5. After treatment with cisplatin, the tumors were excised.
Immunohistochemistry
Briefly, 4 µm sections of xenografted tumors were blocked with 10% goat serum for 1 h and then incubated with antibodies against PPARα and NF-κB p65 (1:200 each) at room temperature for 2 h. Subsequently, sections were washed with TBST (10 mM Tris pH 7.4, 150 mM NaCl, and 0.1% Tween-20) for three times (10 min for each), processed by the UltraVision Quanto Detection System (Termo Scientific) and counterstained with hematoxylin. Immunohistochemical results of each marker were quantified with Aperio ImageScope and Spectrum Software ver. 10.0.
Statistical analyses
Statistical analyses were conducted using SPSS 21.0. The samples were separated into sunitinib-resistant and sunitinib-sensitive groups, and t-tests were used to compare differences between two groups. All statistical tests were two-sided. One-way analysis of variance (ANOVA) was used to compare differences among more than two groups. P < 0.05 was considered as statistically significant.
Results
The expressions of PPARα, AR and PPARγ are increased in ccRCC patients with resistance to sunitinib
Previous studies have shown an upregulation of NR-related metabolites and genes in RCC [12]. To examine the relevance of NRs in sunitinib resistance in RCC, we examined the expressions of 48 NRs at the mRNA level in sunitinib-resistant and non-resistant ccRCC patients. The expressions of NR family members (IlluminaHiSeq) and metadata of an mRCC patient cohort were downloaded from the GEO. A total of 138 patients were included in the analysis. We used a paired t-test to compare expression data between sunitinib-treated and untreated mRCC patients. Our analyses revealed that the expressions of three genes, PPARα, AR and PPARγ, were significantly increased in sunitinib-treated samples compared with untreated samples (all P < 0.05; Table 2), suggesting that PPARα, AR and PPARγ were up-regulated in sunitinib-treated mRCC patients compared with untreated mRCC patients.
Table 2.
Correlation between expressions of PPARα, AR and PPARγ and sunitinib treatment
| Gene | Untreated Mean | Untreated Std | Resistant Mean | Resistant Std | t value | Raw p | Adjust P |
|---|---|---|---|---|---|---|---|
| AR | 4.941 | 0.509 | 4.606 | 0.502 | 3.5534 | 5.92E- | 5.92E- |
| PPARα | 4.017 | 0.570 | 3.682 | 0.281 | 3.7462 | 4.05E- | 4.05E- |
| PPARγ | 4.321 | 0.72 | 3.971 | 0.514 | 2.8509 | 0.00562 | 0.00562 |
PPARα shows increased expression in sunitinib-resistant RCC cells compared with sunitinib-sensitive RCC cells
To examine the potential involvement of PPARα, AR and PPARγ in sunitinib resistance in RCC, we established three sunitinib-resistant RCC cell lines using 786-O, A498 and Caki-1 cells. We next examined the expressions of PPARα, AR and PPARγ at the protein (Figure 1A) and mRNA (Figure 1B) levels in the three sunitinib-resistant cell lines. The expression of AR at both the protein and mRNA levels was decreased in sunitinib-resistant CaKi-1 and 786-O cells compared with their parental lines, while its expression was increased in sunitinib-resistant A498 cells. The expression of PPARγ was decreased in sunitinib-resistant A498 and Caki-1 cells compared with their parental lines, while its expression was slightly increased in sunitinib-resistant 786-O cells. However, the expression of PPARα at the mRNA and protein levels was increased in all three sunitinib-resistant RCC cell lines compared with their parental lines. Based on these results, we hypothesized that PPARα might be involved in sunitinib resistance of ccRCC.
Figure 1.
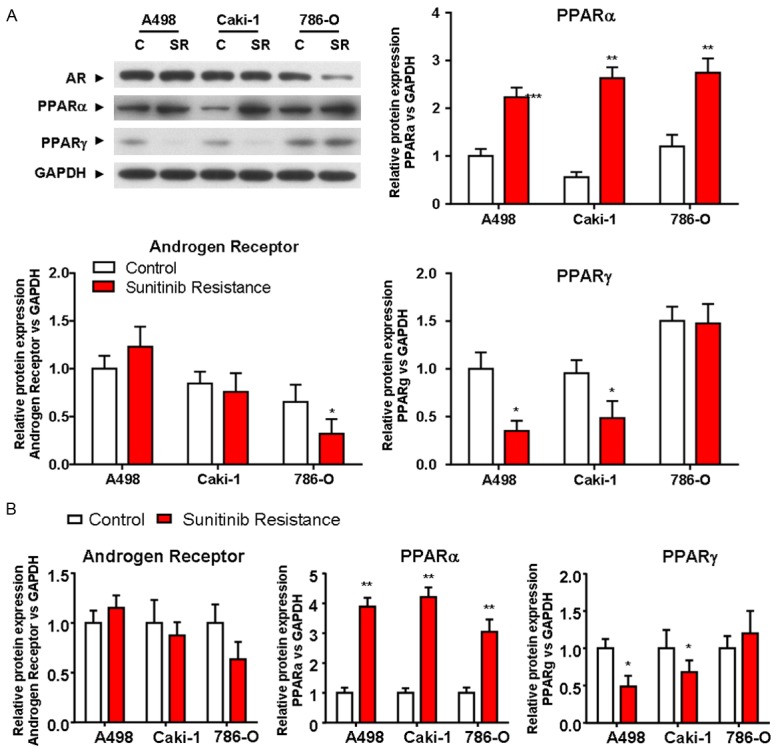
Expressions of AR, PPARα and PPARγ in response to sunitinib resistance in ccRCC lines. Western blotting analysis (A) and RT-PCR (B) were used to examine the expressions of AR, PPARα and PPARγ at the protein and mRNA levels in sunitinib-resistant or parental cells (A498, CaKi-1 and 786-O). GAPDH was used as loading control. Data were expressed as the mean ± S.D. P < 0.05 versus control; Students’ t-test. All experiments were performed in triplicate independently. *P < 0.05, **P < 0.01. C, control; SR, sunitinib resistant.
Effects of sunitinib on sunitinib-resistant ccRCC cell lines with different expression levels of PPARα
We next evaluated the functional role of PPARα in ccRCC cell viability. We overexpressed or downregulated PPARα in A498, CaKi-1 and 786-O sunitinib-resistant or parental cells (Figure 2A). Cells were treated with sunitinib for 72 h, and the cell viability was assessed (Figure 2B). In all three parental cell lines (A498, CaKi-1 and 786-O), the upregulation of PPARα decreased sensitivity to sunitinib compared with PPARα knockdown lines (Figure 2B). However, the downregulation of PPARα in sunitinib-sensitive cells had little or no impact on the cell viability compared with control cells, while the downregulation of PPARα reduced the cell viability in sunitinib-resistant ccRCC lines (Figure 2B). Sunitinib IC50 analysis showed that the upregulation of PPARα decreased the sensitivity to sunitinib in ccRCC (Figure 2B). These results suggested that PPARα played a role in modulating the sensitivity to sunitinib treatment in ccRCC cells.
Figure 2.
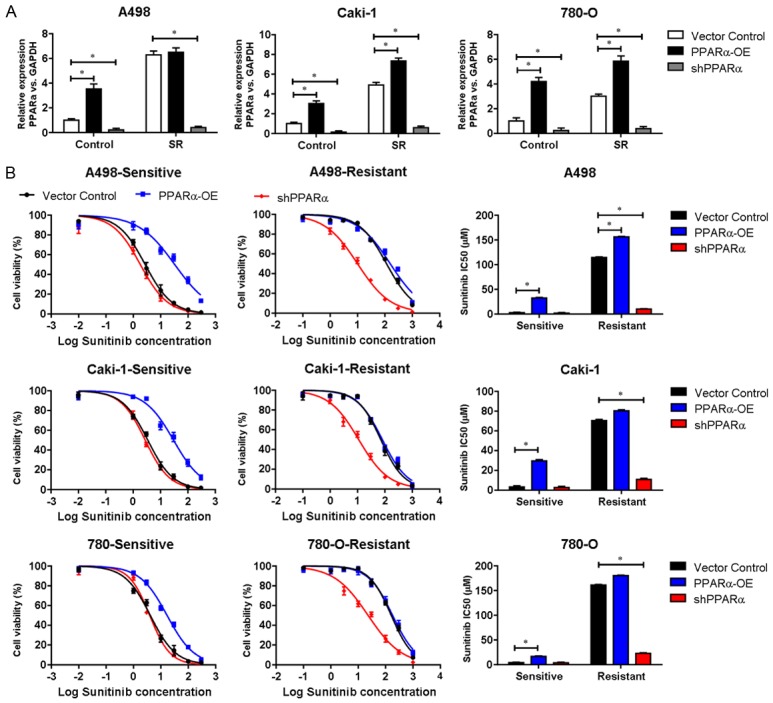
The expression of PPARα modulates sunitinib resistance in cell lines. A. RT-PCR was used to examine the expression of PPARα at the mRNA level in sunitinib-resistant or parental cells (A498, CaKi-1 and 786-O). B. Cell viability of A498, Caki-1 and 780-O sunitinib-resistant or parental cells with PPARα upregulation or shRNA-mediated PPARα knockdown treated with sunitinib for 72 h. Sunitinib IC50 was calculated and presented. Data were presented as means ± S.D. of triplicate experiments. *P < 0.05. PPARα-OE, PPARα overexpression.
Effects of sunitinib treatment in RCC cell lines after treatment with NF-κB pathway inhibitors
Our results suggested that PPARα modulated the sensitivity to sunitinib treatment in ccRCC cells. A previous study has revealed that upregulation of PAK1 kinase activity confers a stem-like phenotype via NF-κB/IL-6 activation in vitro and in vivo, defining a novel potential mechanism underlying tumor metastasis and sunitinib resistance in RCC patients [15]. Therefore, we next examined whether the NF-κB signaling pathway was involved in PPARα-modulated response to sunitinib treatment. We found that both NF-κB p65 expression (Figure 3A) and NF-κB transcription activity (Figure 3B) were significantly increased in sunitinib-resistant cells compared with sunitinib-sensitive cells. Furthermore, the upregulation of PPARα induced the expression (Figure 3A) and activity (Figure 3B) of NF-κB p65, while the downregulation of PPARα led to opposite results. These findings demonstrated that NF-κB p65 signaling was elevated in response to the development of sunitinib resistance in RCC cells, and PPARα was required for induction of NF-κB signaling.
Figure 3.
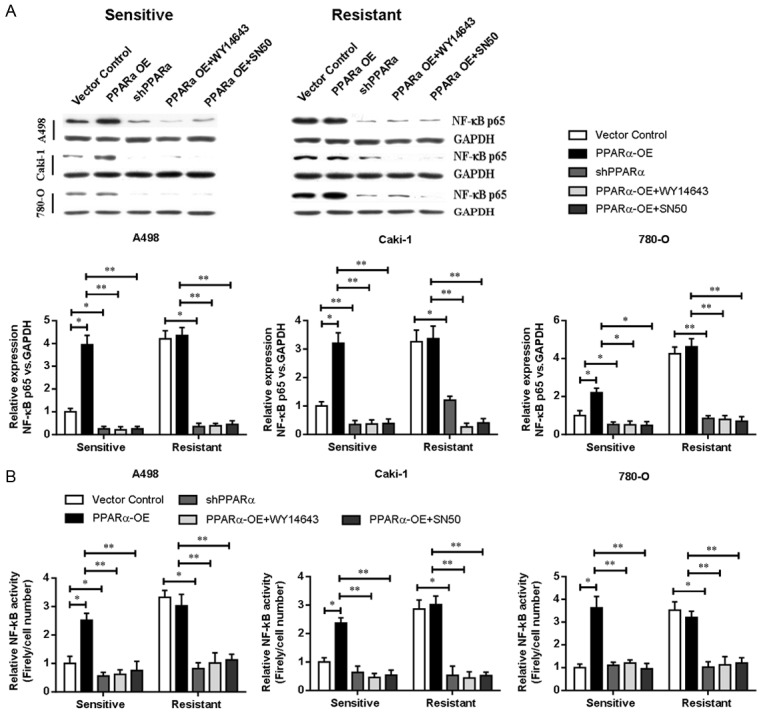
NF-κB p65 is involved in sunitinib resistance. (A) Western blotting analysis of NF-κB p65 expression and (B) NF-κB activity assay in sunitinib-sensitive or resistant RCC cell lines with PPARα overexpression or shRNA-mediated knockdown of PPARα. Cells were cultured with WY14643 or SN50 NF-κB inhibitors at 25 µM for 24 h. GADPH was used as loading control. *P < 0.05, **P < 0.01. PPARα-OE, PPARα overexpression.
Furthermore, PPARα-overexpressing RCC cells were treated with WY14643 or SN50 (NF-κB inhibitors), and we found that the cell viability was decreased compared with untreated cells (Figure 4A), suggesting that the inhibited NF-κB signaling pathway by WY14643 or SN50 in PPARα-overexpressing cells could reduce the cell viability and decrease the sensitivity (Figure 4B) to sunitinib in sunitinib-resistant cell lines. These data indicated that inhibition of the NF-κB signaling pathway resulted in increased efficacy of sunitinib treatment, and PPARα synergistically modulated the sunitinib sensitivity and NF-κB signal inhibition.
Figure 4.
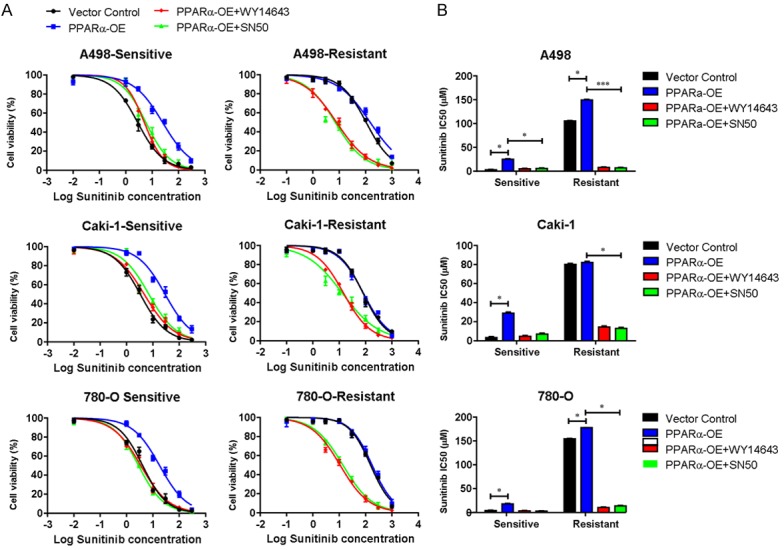
PPARα mediates sunitinib resistance in RCC cell lines via the NF-κB signaling pathway. A. Cell viability of sunitinib-sensitive or resistant RCC cell lines with PPARα overexpression or shRNA-mediated knockdown of PPARα. Cells were cultured with 25 µM WY14643 or SN50 (NF-κB inhibitors). Cell viability was then tested following sunitinib treatment. B. Sunitinib IC50 was calculated and presented. Data were presented as means ± S.D. of triplicate experiments. *P < 0.05, **P < 0.01. PPARα-OE, PPARα overexpression.
PPARα inhibits sunitinib-resistant tumor growth in mice xenograft models
To investigate the potential therapeutic effects of PPARα inhibition in vivo, we established a mice xenograft model using 786-O sunitinib-resistant or parental cells. Various groups of xenografts in mice were treated with sunitinib, and then the tumor volume and weight were examined. Figure 5A shows that in 786-O parental cells, downregulation of PPARα increased the sensitivity to sunitinib compared with control cells, while overexpression of PPARα in sunitinib-sensitive cells had little impact on tumor weight. In sunitinib-resistant cells, knockdown of PPARα enhanced the sunitinib efficacy, and upregulated PPARα expression decreased the sensitivity of sunitinib compared with control cells. Furthermore, the immunochemistry results (Figure 5B) showed that knockdown of PPARα inhibited the expression of NF-κB p65. These data suggested that PPARα-mediated NF-κB signaling could serve as an important therapeutic checkpoint in the sunitinib-resistant RCC.
Figure 5.
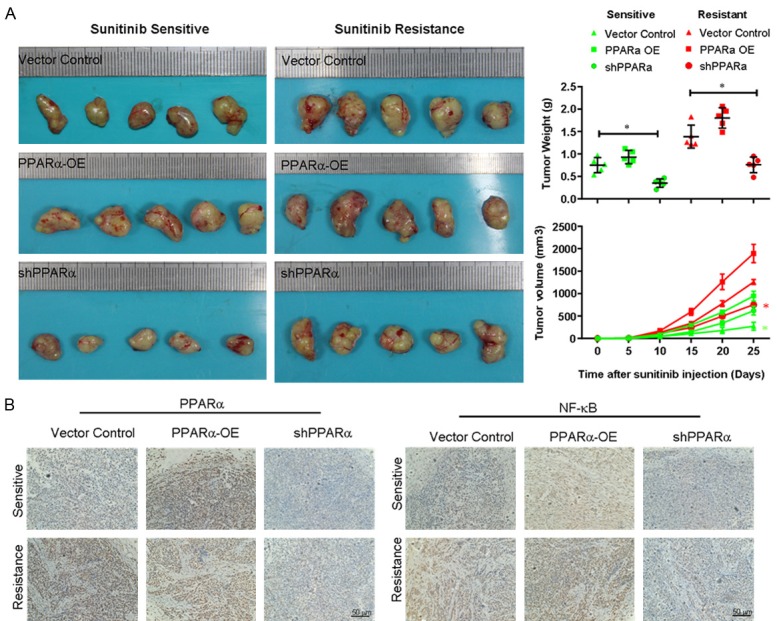
PPARα inhibits sunitinib-resistant tumor growth in mice xenograft models. A. A xenograft model of human renal cancer was established in female nude mice, and various groups of xenografts in mice were treated with sunitinib. Tumor diameter was measured using a caliper every 2 days, and the tumor volume was calculated according to the formula: length × width × height × 0.5. B. Immunochemical analysis of PPARα and NF-κB p65 expression in the xenografted tumors. *P < 0.05. PPARα-OE, PPARα overexpression.
Discussion
The growth of solid neoplasms, such as RCC, is always accompanied by neovascularization [22]. ccRCCs are highly vascularized tumors, which are highly dependent on VEGF-mediated angiogenesis. Indeed, anti-angiogenic therapy targeting VEGF offers significant clinical benefit in metastatic RCC [23]. In addition to sunitinib, a number of anti-angiogenic therapies targeting the VEGF pathway have also shown satisfactory effects in the treatment of ccRCC [13,24]. Despite the significant improvement in metastatic RCC treatment using anti-angiogenic drugs, such as sunitinib and sorafenib, the duration of therapeutic effect is often short [13]. Typically, the development of resistance occurs after a median of 6-15 months of treatment [25]. Resistance to these anti-angiogenic tyrosine kinase inhibitors is a major clinical obstacle in the treatment of metastatic RCC, highlighting that novel therapeutic approaches are urgently required for sunitinib-resistant patients with metastatic RCC [26]. However, the molecular mechanism underlying sunitinib-resistance in RCC remains largely unexplored.
Previous studies have shown that sunitinib exerts its antitumor effect on ccRCC mainly through the suppression of VEGF/VEGFR-mediated tumor angiogenesis in vivo [27]. Sunitinib resistance is considered to develop as a result of changes in the tumor microenvironment or gene expression, which facilitate continuous tumor growth independently of VEGFR [28]. With the development of resistance to sunitinib, new angiogenesis could cause alterations of energy pathways, increase the glycolysis and elevate the production of pyruvate and lactate.
Several studies have demonstrated an upregulation of PPARα-related metabolites [13] and genes [14] in RCC, suggesting that PPARα inhibition can be used as a novel therapeutic approach for advanced RCC. For the first time, we have shown that the expression of PPARα was upregulated in sunitinib-resistant RCC cell lines.
The role of PPARα has been previously restricted to lipid and lipoprotein metabolism, glucose homeostasis and cellular differentiation [15]. PPARα also regulates fatty acid oxidation (FAO) through targeted gene expression [29]. A previous study has indicated that pharmacologically blocked transport of long-chain fatty acids into mitochondria results in hepatic steatosis, severe hypoketonemia, hypoglycemia, and hypertriglyceridemia [30]. In addition, synthetic antidiabetic thiazolidinediones and lipid-lowering fibrates can functionally activate PPARα [31]. PPARα is involved in increasing FAO, and this activity may enhance the dependence of cells on glycolysis due to the attenuation of FAO [10]. Our finding that the expression of PPARα was increased in sunitinib-resistant RCC cell lines compared with sunitinib-sensitive RCC cell lines, suggesting that the increased expression of PPARα at the protein level was associated with sunitinib resistance in RCC.
It is known that VEGF can induce vascular permeability, act as a critical survival factor for endothelial cells, and promote endothelial proliferation and migration [32]. Although only relatively low levels of VEGFR2 (formerly termed KDR/Flk-1) are detected in adult vasculature, it can be markedly upregulated by blood vessels during chronic inflammation, tumor growth, and wound repair [32]. Endothelial expression of VEGFR2 closely parallels VEGF expression in angiogenic responses. As a result, suppression of the VEGF/VEGFR2 signaling pathway has been intensely explored as a therapeutic prospect to interfere with formation of new blood vessels. Another study has identified that VEGFR2 is an additional target of PPARα activation in endothelial cells, indicating that the repressive effects of fibrates on VEGFR2 occur at the transcriptional level via activation of PPARα. Since PPARα activators suppress steady-state VEGFR2 mRNA levels in human endothelial cells, negative control mechanisms will likely help to better define the therapeutic potential and clinical indications of PPAR activators in vascular-dependent diseases [33].
To evaluate whether PPARα is a valuable potential therapeutic target for sunitinib resistance in RCC, we analyzed the efficacy of PPARα in sunitinib-resistant RCC cell lines with PPARα upregulation or downregulation. In our study, we found that upregulation or downregulation of PPARα in RCC cell lines sensitive or resistant to sunitinib modulated the efficacy of sunitinib in RCC. Knockdown of PPARα significantly inhibited the cell proliferation in sunitinib-resistant RCC cell lines. We speculated these effects were associated with PPARα-inhibited endothelial cell proliferation as well as PPARα-dependent downregulation of cytochrome P450, leading to inhibited neoangiogenesis [34,35].
NF-κB is a family of transcription factors that are involved in apoptosis and inflammation [36,37]. The NF-κB family consists of five genes, including NF-κB1 (p59/p105), NF-κB2 (p52/p100), Re1A (p65), c-Rel and RelB. Most prior studies have focused on the p65 subunit when assessing the association between NF-κB and tumor development. A previous study has demonstrated that increased NF-κB activity is associated with the development and progression of RCC [38]. Oya et al. have demonstrated the role of increased NF-κB activation in the development of RCC [18]. Some data have indicated that NF-κB has a dual effect on apoptosis, acting as an inhibitor or an activator depending on the levels of p65 and cRel subunits [7]. However, it is commonly accepted that the activation of NF-κB leads to apoptosis resistance. In our study, treatment by NF-κB inhibitors resulted in increased apoptosis. Our findings suggested that inhibition of NF-κB activity played a significant role in the resistance of sunitinib treatment.
NF-κB mediates the secretion of pro-inflammatory cytokines (IL-6 and IL-8) in various types of cancers [39]. More recently, a study has shown that IL-6 can induce a highly oncogenic, drug-resistant, stem-like phenotype in cancer cells. In our study, the expression of NF-κB p65 was significantly increased in sunitinib-resistant RCC cell lines compared with sunitinib-sensitive RCC cell lines. According to these results, both p65 subunits of NF-κB exhibited an increased activation in RCC, confirming that the p65 subunit was involved in tumor development.
A previous study has revealed that upregulation of PAK1 kinase activity confers a stem-like phenotype via NF-κB/IL-6 activation in vitro and in vivo, defining a novel potential mechanism underlying tumor metastasis and sunitinib resistance in RCC patients. Arai et al. have reported the decrease of factors, such as VEGF, by the inhibition of NF-κB with dexamethasone administration in RCC cases. These studies have demonstrated that inhibition of NF-κB decreases the expression of VEGF [40,41]. Furthermore, our work established that sunitinib-induced upregulation of PPARα activity and NF-κB activation contributed to the development of sunitinib resistance.
In the present study, our findings showed that sunitinib-induced upregulation of PPARα activity and downstream NF-κB activation could be an important mechanism of sunitinib resistance. These results suggested that PPARα could be used as a predictive biomarker for sunitinib response and resistance in RCC treatment, and PPARα-induced overexpression of NF-κB signaling could substantially promote resistance to targeted drugs.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Number: 81760452) and the Natural Science Foundation of Xinjiang Uygur Autonomous Region (Grant Number: 2016D01C334).
Disclosure of conflict of interest
None.
References
- 1.Ljungberg B, Campbell SC, Choi HY, Jacqmin D, Lee JE, Weikert S, Kiemeney LA. Corrigendum to “the epidemiology of renal cell carcinoma” [Eur Urol 2011;60:615-21] . Eur Urol. 2011;60:1317. doi: 10.1016/j.eururo.2011.06.049. [DOI] [PubMed] [Google Scholar]
- 2.Buttner F, Winter S, Rausch S, Reustle A, Kruck S, Junker K, Stenzl A, Agaimy A, Hartmann A, Bedke J, Schwab M, Schaeffeler E. Survival prediction of clear cell renal cell carcinoma based on gene expression similarity to the proximal tubule of the nephron. Eur Urol. 2015;68:1016–20. doi: 10.1016/j.eururo.2015.05.045. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 4.Rosner I, Bratslavsky G, Pinto PA, Linehan WM. The clinical implications of the genetics of renal cell carcinoma. Urol Oncol. 2009;27:131–6. doi: 10.1016/j.urolonc.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motzer RJ, Rini BI, Bukowski RM, Curti BD, George DJ, Hudes GR, Redman BG, Margolin KA, Merchan JR, Wilding G, Ginsberg MS, Bacik J, Kim ST, Baum CM, Michaelson MD. Sunitinib in patients with metastatic renal cell carcinoma. JAMA. 2006;295:2516–24. doi: 10.1001/jama.295.21.2516. [DOI] [PubMed] [Google Scholar]
- 6.Motzer RJ, Russo P. Systemic therapy for renal cell carcinoma. J Urol. 2000;163:408–17. [PubMed] [Google Scholar]
- 7.Liang L, Li L, Zeng J, Gao Y, Chen YL, Wang ZQ, Wang XY, Chang LS, He D. Inhibitory effect of silibinin on EGFR signal-induced renal cell carcinoma progression via suppression of the EGFR/MMP-9 signaling pathway. Oncol Rep. 2012;28:999–1005. doi: 10.3892/or.2012.1874. [DOI] [PubMed] [Google Scholar]
- 8.Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6:734–45. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- 9.Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2006;24:16–24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- 10.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 11.Mena AC, Pulido EG, Guillen-Ponce C. Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: sunitinib. Anticancer Drugs. 2010;21(Suppl 1):S3–11. doi: 10.1097/01.cad.0000361534.44052.c5. [DOI] [PubMed] [Google Scholar]
- 12.Molina AM, Lin X, Korytowsky B, Matczak E, Lechuga MJ, Wiltshire R, Motzer RJ. Sunitinib objective response in metastatic renal cell carcinoma: analysis of 1059 patients treated on clinical trials. Eur J Cancer. 2014;50:351–8. doi: 10.1016/j.ejca.2013.08.021. [DOI] [PubMed] [Google Scholar]
- 13.Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009;10:992–1000. doi: 10.1016/S1470-2045(09)70240-2. [DOI] [PubMed] [Google Scholar]
- 14.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganti S, Taylor SL, Abu Aboud O, Yang J, Evans C, Osier MV, Alexander DC, Kim K, Weiss RH. Kidney tumor biomarkers revealed by simultaneous multiple matrix metabolomics analysis. Cancer Res. 2012;72:3471–9. doi: 10.1158/0008-5472.CAN-11-3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res. 2006;12:3028–36. doi: 10.1158/1078-0432.CCR-05-2556. [DOI] [PubMed] [Google Scholar]
- 17.Liu DC, Zang CB, Liu HY, Possinger K, Fan SG, Elstner E. A novel PPAR alpha/gamma dual agonist inhibits cell growth and induces apoptosis in human glioblastoma T98G cells. Acta Pharmacol Sin. 2004;25:1312–9. [PubMed] [Google Scholar]
- 18.Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? J Mol Med (Berl) 2005;83:774–85. doi: 10.1007/s00109-005-0678-9. [DOI] [PubMed] [Google Scholar]
- 19.Suchanek KM, May FJ, Robinson JA, Lee WJ, Holman NA, Monteith GR, Roberts-Thomson SJ. Peroxisome proliferator-activated receptor alpha in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol Carcinog. 2002;34:165–71. doi: 10.1002/mc.10061. [DOI] [PubMed] [Google Scholar]
- 20.Abu Aboud O, Wettersten HI, Weiss RH. Inhibition of PPARalpha induces cell cycle arrest and apoptosis, and synergizes with glycolysis inhibition in kidney cancer cells. PLoS One. 2013;8:e71115. doi: 10.1371/journal.pone.0071115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abu Aboud O, Donohoe D, Bultman S, Fitch M, Riiff T, Hellerstein M, Weiss RH. PPARalpha inhibition modulates multiple reprogrammed metabolic pathways in kidney cancer and attenuates tumor growth. Am J Physiol Cell Physiol. 2015;308:C890–8. doi: 10.1152/ajpcell.00322.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 23.Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res. 2012;72:1909–14. doi: 10.1158/0008-5472.CAN-11-3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reeves DJ, Liu CY. Treatment of metastatic renal cell carcinoma. Cancer Chemother Pharmacol. 2009;64:11–25. doi: 10.1007/s00280-009-0983-z. [DOI] [PubMed] [Google Scholar]
- 25.Rini BI, Flaherty K. Clinical effect and future considerations for molecularly-targeted therapy in renal cell carcinoma. Urol Oncol. 2008;26:543–9. doi: 10.1016/j.urolonc.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, Kahnoski R, Futreal PA, Furge KA, Teh BT. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010;70:1063–71. doi: 10.1158/0008-5472.CAN-09-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Chen J, Hu X, Huang Y, Li Z, Zhou L, Tian Z, Ma H, Wu Z, Chen M, Han Z, Peng Z, Zhao X, Liang C, Wang Y, Sun L, Chen J, Zhao J, Jiang B, Yang H, Gui Y, Cai Z, Zhang X. Comparative mRNA and microRNA expression profiling of three genitourinary cancers reveals common hallmarks and cancer-specific molecular events. PLoS One. 2011;6:e22570. doi: 10.1371/journal.pone.0022570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J, Vandenbeldt K, Qian CN, Teh BT. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 2010;70:1053–62. doi: 10.1158/0008-5472.CAN-09-3722. [DOI] [PubMed] [Google Scholar]
- 29.Torra IP, Chinetti G, Duval C, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors: from transcriptional control to clinical practice. Curr Opin Lipidol. 2001;12:245–54. doi: 10.1097/00041433-200106000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–32. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Djouadi F, Weinheimer CJ, Saffitz JE, Pitchford C, Bastin J, Gonzalez FJ, Kelly DP. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator- activated receptor alpha- deficient mice. J Clin Invest. 1998;102:1083–91. doi: 10.1172/JCI3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 33.Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 2001;169:453–9. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 34.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–4. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 35.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 36.Meteoglu I, Erdogdu IH, Meydan N, Erkus M, Barutca S. NF-KappaB expression correlates with apoptosis and angiogenesis in clear cell renal cell carcinoma tissues. J Exp Clin Cancer Res. 2008;27:53. doi: 10.1186/1756-9966-27-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pozzi A, Ibanez MR, Gatica AE, Yang S, Wei S, Mei S, Falck JR, Capdevila JH. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J Biol Chem. 2007;282:17685–95. doi: 10.1074/jbc.M701429200. [DOI] [PubMed] [Google Scholar]
- 38.Panigrahy D, Kaipainen A, Huang S, Butterfield CE, Barnés CM, Fannon M, Laforme AM, Chaponis DM, Folkman J, Kieran MW. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci U S A. 2008;105:985–90. doi: 10.1073/pnas.0711281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 40.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Oya M, Takayanagi A, Horiguchi A, Mizuno R, Ohtsubo M, Marumo K, Shimizu N, Murai M. Increased nuclear factor-kappa B activation is related to the tumor development of renal cell carcinoma. Carcinogenesis. 2003;24:377–84. doi: 10.1093/carcin/24.3.377. [DOI] [PubMed] [Google Scholar]