

Abstract
Objective: To report newly identified mutations in two families in China with cystic renal disease. Case presentations: Two fetuses were found by prenatal ultrasound to have symmetrically enlarged kidneys with increased echogenicity and cystic changes. We isolated fetal and parental genomic DNAs from umbilical cord blood and circulating leukocytes, performed next generation sequencing for mutations, followed by Sanger sequencing for confirmation. We discovered two new heterozygous mutations in PKHD1: c.2507_2515delTGAAGGAGG (p.Val836_Glu838del) in exon 24 among the fetus and father, as well as c.6840G>A (p.Trp2280*) in exon 42 among the fetus and mother. A mutation of c.2507_2515delTGAAGGAGG caused deletion of three amino acids. Two heterozygous mutations in AHI1, c.1304G>A (p.Arg435Gln), and c.3257A>G (p.Glu1086Gly) were identified in the second fetus, while the former was also found in the mother. The mutated locus in AHI1 is highly conserved among humans, dogs, mice, and monkeys. Conclusions: We report two newly identified mutations in PKHD1 and AHI1. An accurate genetic diagnosis is crucial for genetic counseling of parents with offspring carrying cystic renal disease.
Keywords: Cystic renal disease, PKHD1 gene, AHI1 gene, autosomal recessive polycystic kidney disease, Joubert syndrome
Introduction
Cystic renal disease is classified into four types: autosomal recessive polycystic kidney disease (ARPKD)/infantile polycystic kidney disease, renal cystic dysplasia, autosomal dominant polycystic kidney disease (ADPKD), and cystic/hydronephrotic kidneys caused by prolonged obstruction [1]. However, cystic renal disease may be an important manifestation of other inherited disorders with renal involvement, such as ciliopathy syndromes, or tuberous sclerosis, and von Hippel-Lindau disease.
When cases with cystic renal disease are discovered, it is crucial that we make an accurate genetic diagnosis in order to facilitate subsequent prenatal diagnosis, genetic counseling, and to administer appropriate treatment. In this case series, we sequenced two probands with cystic renal disease and their parents from our institute, and report novel mutation loci.
Case presentation
All procedures performed in this study were approved by the Ethics Committee of Shengjing Hospital of China Medical University (2016PS30J), and informed written consent from the probands’ parents was obtained.
A 25-year-old pregnant woman with one prior unsuccessful pregnancy due to early embryonic death at 80 days was found to have a fetus with bilateral enlarged hyperechoic kidneys at the gestational age of 27 weeks. Fetal ultrasonography showed a second-trimester pregnancy with oligohydramnios, enlargement of fetal kidneys, and hyperechoic renal parenchyma. Infantile polycystic kidney disease was considered (Figure 1A). Renal biopsy found fusiform dilation of the renal collecting ducts and distal renal tubules with low-flat epithelium, and epithelial cell exfoliation of partial tubules (Figure 1D). A decision was made to terminate the pregnancy immediately.
Figure 1.

(A) Fetal ultrasonography of the first proband showed bilateral kidney enlargement and enhanced echogenicity of the renal parenchyma (arrow). (B) Fetal ultrasonography of the second proband showed similar findings. (C) Renal biopsy of the second proband’s older brother showed massive cystic dilation of the proximal tubules with low-flat epithelium, epithelial cell exfoliation of partial tubules, as well as massive interstitial fibrosis (A: PAS 200 ×; B: Masson 200 ×). (D) Renal pathology of the first proband showed fusiform dilation of collecting ducts and distal renal tubules (light microscope, HE 100 ×). (E) Fetal magnetic resonance imaging of the second proband showed enlargement of both kidneys and mild dilation and deformation of the renal pelvis and calices (A: T2-weighted cross-sectional view, B: T2-weighted coronal view). (F) Gross examination of the fetal kidneys from the second proband, showing cystic enlargement.
Another woman had one prior successful pregnancy, and her first male baby developed end-stage renal disease at the age of 11 months, had obvious developmental retardation, and eventually died. Ultrasonography of his urinary system showed a 7.6 cm × 4.1 cm left kidney, a 7.3 cm × 3.9 cm right kidney, and with generally hyperechoic parenchyma and poor corticomedullary differentiation.
The first male baby received a renal biopsy, which showed subacute interstitial nephritis and cystic dilations of the proximal tubules (Figure 1C), raising suspicion of congenital cystic renal diseases. He received no genetic diagnosis at that time due to limited resources. Her second fetus was found to have bilateral enlarged echogenic kidneys at the gestational age of 29 weeks, accompanied by situs inversus viscerum, mirror-image dextrocardia, and bilateral renal enlargement with enhanced echogenicity and infantile polycystic kidney disease was suspected (Figure 1B).
Magnetic resonance imaging (MRI) indicated an enlargement of the fetal kidneys and mild dilation and deformation of the renal pelvis and calices (Figure 1E). According to the clinical presentations of the second proband and his older brother, ARPKD was proven clinically, and the fetus was terminated soon after diagnosis. Gross examination of the kidney showed cystic enlargement (Figure 1F). The babies and their parents were referred for genetic diagnoses.
A detailed evaluation of both fetuses’ parents found no clinical signs of renal abnormality (Figure 2). We collected umbilical cord blood from both probands (at the time of induction) as well as their parental blood, sending the samples to the pediatric laboratory of Peking University First Hospital for genetic analysis of any cystic renal diseases. Mutation screening was performed using next generation sequencing (NGS) technologies, with results confirmed by Sanger sequencing.
Figure 2.
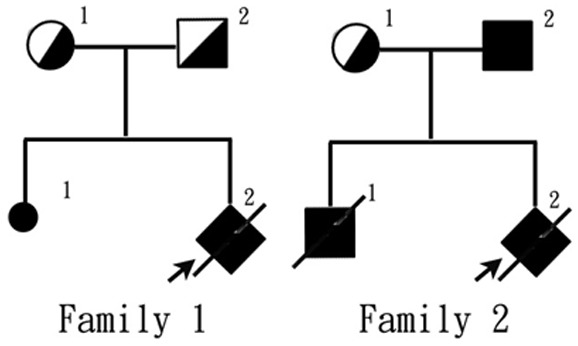
The genealogical tree of the two probands in this case series.
The sequencing results of the first proband showed a heterozygous deletion mutation in the PKHD1 gene (NM_138694.3)-c.2507_2515delTGAAGGAGG (p.Val836_Glu838del) located in exon 24 (Figure 3A)-causing deletion of three amino acids. This mutation was thus considered pathogenic. Another heterozygous missense mutation-c.6840G>A (p.Trp2280*) located in exon 42 (Figure 3B)-was also discovered causing protein truncation and the production of a peptide consisting of 2,279 amino acids only (normally 4,074 amino acids). This mutation was also considered pathogenic.
Figure 3.

A: Sequencing results of PKHD1 exon 24 in the first family. B: Sequencing results of PKHD1 exon 42 in the first family. C: Sequencing results of AHI1 exon 9 in the second family. D: Sequencing results in AHI1 exon 24 in the second family. (1) Reference sequence; (2) Sequence of the proband; (3) Sequence of the proband’s mother; (4) Sequence of the proband’s father.
A database search (http://www.humgen.rwth-aachen.de) did not find these two pathogenic mutations in PKHD1. A heterozygous mutation in exon 42-c.6840G>A (p.Trp2280*)-was found in the first proband’s mother (Figure 3B), and a heterozygous mutation in exon 24-c.2507_2515delTGAAGGAGG (p.Val836_Glu838del)-was found in the first proband’s father (Figure 3A). Since his parents each had a heterozygous mutation in PKHD1 without phenotypes, while the proband had a compound heterozygous mutation with phenotypes, we believed that the inheritance pattern of this case was likely autosomal recessive, suggesting the diagnosis of ARPKD.
On the other hand, the sequencing results of the second proband showed two heterozygous missense mutations of AHI1 gene (NM_001134830.1): c.1304G>A (p.Arg435Gln) in exon 9 (Figure 3C) and c.3257A>G (p.Glu1086Gly) in exon 24 (Figure 3D). The heterozygous mutation of c.1304G>A (p.Arg435Gln) was also found in the second proband’s mother (Figure 3C), and this mutation could cause substitution of the 435th arginine by glutamine. This locus is highly conserved among humans, dogs, mice, and monkeys, and this mutation has not been reported in the existing genomic sequencing databases including 1000 Genomes Database, dbSNP database, ESP6500 database, and BGI internal database of the general population. This mutation was predicted to be neutral according to software (EnsCondelPred) analysis. In addition, a homozygous mutation of c.3257A>G (p.Glu1086Gly) was found in the second proband’s father (Figure 3D) which has been reported to be pathogenic (HGMD, 2008) by causing the substitution of the 1,086th glutamine by glycine.
Discussion
ARPKD is a severe, early-onset form of cystic renal disease, and affected fetuses often have oligohydramnios and massively enlarged kidneys. Renal findings in ARPKD patients include bilateral kidney enlargement, hyperechogenecity with poor corticomedullary differentiation, and multiple tiny cysts originating from distal renal tubules and collecting ducts [2]. The clinical diagnosis relies upon imaging results and pathological examinations, but the molecular diagnosis (i.e. PKHD1 mutation) is also of great importance for the clinical management, genetic counseling, and prenatal diagnosis of ARPKD patients and their families.
PKHD1 is located at 6p12 and has a length of 472 kb [3]. Its longest open reading frame consists of 67 exons that encodes a type I transmembrane protein consisting of 4,074 amino acids, called fibrocystin/polyductin (FPC) [4]. Evidence indicates that FPC has multiple alternative splicing variants [5].
At present, 750 mutations have been catalogued in the ARPKD mutation database (http://www.humgen.rwth-aachen.de), but PKHD1 does not contain mutational hotspots [6]. Moreover, most patients may have compound heterozygous PKHD1 mutation. A genotype-phenotype correlation study found that (in terms of collecting duct dilatation without portal fibrosis) the phenotypes of fetal and neonatal ARPKD were significantly associated with the presence of two truncating mutations [7]. However, exceptions do exist, such as a case of homozygous PKHD1 mutation due to a large deletion that reportedly survived the neonatal period [8]. In addition, the phenotype may be highly variable among patients with the same genotype, probably due to gene modification [9].
The first proband was considered to have infantile polycystic kidneys, based on ultrasonographic findings. The pathological examination identified fusiform dilation of distal renal tubules and collecting ducts. In addition, we found compound heterozygous changes of PKHD1 including a deletion mutation (c.2507_2515delTGAAGGAGG) and a truncating mutation (c.6840G>A), which were also found separately in his mother and father. This is consistent with the autosomal recessive inheritance pattern seen in ARPKD.
For the second proband, using renal pathology results from his older brother showing cystic dilations of proximal tubules, the primary clinical diagnosis of ARPKD was highly likely. However, gene sequencing in this proband did not find any mutation in PKHD1 but instead two heterozygous missense mutations (c.1304G>A and c.3257A>G in AHI1), one of pathogenic genes of Joubert syndrome (JBTS) [10] type 3 and located on chromosome 6q23.3 [11]. JBTS is a rare autosomal recessive ciliopathy, characterized by developmental delay and oculomotor abnormalities, with renal manifestations similar to those of ARPKD [12].
We believe that the proband and his older brother might have JBTS3. The missense mutation of c.3257A>G in the second proband was transmitted from his father who was homozygous for the mutation but had no phenotypes of renal cystic disease. This mutation site has been reported by Kroes in 2008, and it is located in exon 24 of AHI1 and encodes SH3 protein binding domain [13]. The father in this family similarly did not have renal involvement, consistent with the above report. Homozygosity of mutation in his father, in combination with the heterozygous mutation in his mother and a compound heterozygous mutation in the proband, is compatible with autosomal recessive inheritance pattern of JBTS.
In conclusion, we found that autosomal recessive ciliopathy JBTS is difficult to distinguish from ARPKD in term of renal presentations only. Therefore, a genetic analysis provides a more accurate diagnosis of JBTS. Newly discovered mutations of ARPKD expand the mutation spectrum of this gene, assisting subsequent prenatal diagnosis and genetic counseling of these two families.
Acknowledgements
We thank Dr Jie Ding, Fang Wang, and Yanqin Zhang for analyzing the data in this study. This work was partially supported by grants from the Natural Science Foundation of Liaoning Province, China (2013225086, 2013021099, 2015020492), Science and Technology Planning Project of Shenyang City, China (F13221-9-59).
Disclosure of conflict of interest
None.
References
- 1.Osathanondh V, Potter EL. Pathogenesis of polycystic kidneys. Survey of results of microdissection. Arch Pathol. 1964;77:510–512. [PubMed] [Google Scholar]
- 2.Bergmann C. ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol. 2015;30:15–30. doi: 10.1007/s00467-013-2706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 4.Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schoneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, Germino GG. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boddu R, Yang C, O’Connor AK, Hendrickson RC, Boone B, Cui X, Garcia-Gonzalez M, Igarashi P, Onuchic LF, Germino GG, Guay-Woodford LM. Intragenic motifs regulate the transcriptional complexity of Pkhd1/PKHD1. J Mol Med (Berl) 2014;92:1045–1056. doi: 10.1007/s00109-014-1185-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossetti S, Harris PC. Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2007;18:1374–1380. doi: 10.1681/ASN.2007010125. [DOI] [PubMed] [Google Scholar]
- 7.Denamur E, Delezoide AL, Alberti C, Bourillon A, Gubler MC, Bouvier R, Pascaud O, Elion J, Grandchamp B, Michel-Calemard L, Missy P, Zaccaria I, Le Nagard H, Gerard B, Loirat C, Societe Francaise de F, Barbet J, Beaufrere AM, Berchel C, Bessieres B, Boudjemaa S, Buenerd A, Carles D, Clemenson A, Dechelotte P, Devisme L, Dijoud F, Esperandieu O, Fallet C, Gonzales M, Hillion Y, Jacob B, Joubert M, Kermanach P, Lallemand A, Laquerriere A, Laurent N, Liprandi A, Loeuillet L, Loget P, Martinovic J, Menez F, Narcy F, Roux JJ, Rouleau-Dubois C, Sinico M, Tantau J, Wann AR. Genotype-phenotype correlations in fetuses and neonates with autosomal recessive polycystic kidney disease. Kidney Int. 2010;77:350–358. doi: 10.1038/ki.2009.440. [DOI] [PubMed] [Google Scholar]
- 8.Zvereff V, Yao S, Ramsey J, Mikhail FM, Vijzelaar R, Messiaen L. Identification of PKHD1 multiexon deletions using multiplex ligation-dependent probe amplification and quantitative polymerase chain reaction. Genet Test Mol Biomarkers. 2010;14:505–510. doi: 10.1089/gtmb.2009.0188. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann C, Senderek J, Windelen E, Kupper F, Middeldorf I, Schneider F, Dornia C, Rudnik-Schoneborn S, Konrad M, Schmitt CP, Seeman T, Neuhaus TJ, Vester U, Kirfel J, Buttner R, Zerres K APN (Arbeitsgemeinschaft für Pädiatrische Nephrologie) Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD) Kidney Int. 2005;67:829–848. doi: 10.1111/j.1523-1755.2005.00148.x. [DOI] [PubMed] [Google Scholar]
- 10.Tuz K, Hsiao YC, Juarez O, Shi B, Harmon EY, Phelps IG, Lennartz MR, Glass IA, Doherty D, Ferland RJ. The Joubert syndrome-associated missense mutation (V443D) in the Abelson-helper integration site 1 (AHI1) protein alters its localization and protein-protein interactions. J Biol Chem. 2013;288:13676–13694. doi: 10.1074/jbc.M112.420786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagier-Tourenne C, Boltshauser E, Breivik N, Gribaa M, Betard C, Barbot C, Koenig M. Homozygosity mapping of a third Joubert syndrome locus to 6q23. J Med Genet. 2004;41:273–277. doi: 10.1136/jmg.2003.014787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guay-Woodford LM. Autosomal recessive polycystic kidney disease: the prototype of the hepato-renal fibrocystic diseases. J Pediatr Genet. 2014;3:89–101. doi: 10.3233/PGE-14092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kroes HY, van Zon PH, Fransen van de Putte D, Nelen MR, Nievelstein RJ, Wittebol-Post D, van Nieuwenhuizen O, Mancini GM, van der Knaap MS, Kwee ML, Maas SM, Cobben JM, De Nef JE, Lindhout D, Sinke RJ. DNA analysis of AHI1, NPHP1 and CYCLIN D1 in Joubert syndrome patients from the Netherlands. Eur J Med Genet. 2008;51:24–34. doi: 10.1016/j.ejmg.2007.10.001. [DOI] [PubMed] [Google Scholar]