

Abstract
Aim: We aimed to assess the effect of autophagy and stromal interaction molecule 1 (STIM1) on podocyte epithelial-mesenchymal transition in diabetic nephropathy. Methods: The sera of 8-week-old db/db and C57BL/KsJ rats were used to culture MPC5 cells. The experiment was divided into 4 groups: MPC5 + siRNA-Scr + 10% C57BL/KsJ (Group A), MPC5 + siRNA-STIM1 + 10% C57BL/KsJ (Group B), MPC5 + siRNA-Scr + 10% db/db (Group C), and MPC5 + siRNA-STIM1 + 10% db/db (Group D). Podocyte autophagy was evaluated via immunofluorescence staining for LC3II and P62, and via Western blotting for P62 and LC3 (LC3II/LC3I). Western blotting was also used to assess the expression of TRPC6, Orai1, Beclin-1, Bcl-2, Caspase3, E-cadherin, fibronectin, and α-SMA protein. Furthermore, podocyte apoptosis was assessed via flow cytometry. Results: We found that, in podocytes cultured in the serum of diabetic nephrotic rats, the autophagy level decreased, whereas the apoptosis level increased, and EMT can be advanced. However, after silencing STIM1 with siRNA, a converse outcome was noted. Furthermore, in diabetic nephropathy rats, the up-regulated expression of podocyte STIM1 can activate TRPC6 and Orai1 channels, which results in Ca2+ entry. Conclusions: We found that, in podocytes cultured in the serum of diabetic nephrotic rats, the autophagy level increased, whereas the apoptosis level decreased, and EMT can be inhibited by silencing STIM1 with siRNA.
Keywords: Autophagy, diabetic nephropathy, epithelial-mesenchymal transition, podocyte, STIM1
Introduction
The prevalence of diabetes mellitus has markedly increased in China in recent decades [1]. Diabetic nephropathy (DN) is a serious complication of diabetes mellitus, which itself is the major cause of end stage renal disease (ESRD) [2]. Albuminuria is a common feature of early-stage DN, which is associated with glomerular hypertrophy, thickening of the glomerular basement membrane (GBM), and expansion of the mesangial extracellular matrix. Moreover, advanced DN is characterised by glomerulosclerosis, vascular and capillary rarefaction, tubulointerstitial degeneration, and fibrosis associated with glomerular filtration rate (GFR) decline and substantial proteinuria [3].
Podocytes are one of 2 cell types (partial mesenchymal and partial epithelial cell) that contribute to the formation of the glomerular filtration barrier (GFB). Indeed, podocytes clearly undergo a set of phenotypic and morphological changes during DN including foot process effacement, podocyte loss, and mesangial extracellular matrix expansion, and the process has been reported to be similar to epithelia-mesenchymal transition (EMT). Besides, although podocytes are terminally differentiated cells, they show high basal levels of autophagy in order to maintain intracellular homeostasis and cell integrity. Therefore, podocyte injury and autophagy levels are believed to be crucial in DN pathogenesis [4].
Autophagy is a highly regulated lysosomal protein degradation pathway that removes protein aggregates as well as damaged or excess organelles in order to maintain intracellular homeostasis and cell integrity [5]. A growing body of evidence suggests that dysregulation of the autophagic pathway is involved in the pathogenesis of kidney aging and in DN [6]. The high basal levels of autophagy enable podocytes to eliminate protein aggregates and damaged organelles and thus maintain intracellular homeostasis and cell integrity.
The EMT process is highly regulated and consists of 4 key steps: loss of epithelial cell adhesion, de novo-SMA (smooth muscle actin) expression and actin reorganisation, disruption of the tubular basement membrane, and enhanced cell migration and invasion [7]. Evidence for EMT in vivo has been observed in various animal models of chronic kidney diseases (CKD), including diabetic nephropathy [8]. When podocytes are exposed to various stresses, the initial response is cell hypertrophy, which is an adaptive change to compensate for lost function. However, if the injury is progressive, podocytes will undergo EMT and escape from apoptosis [9]. Podocytes become motile after EMT, which eventually results in the disruption of the delicate three-dimensional architecture of podocytes, thereby impairing GFB function and leading to podocyte dysfunction, proteinuria, and glomerular sclerosis [10].
STIM1 is an endoplasmic reticulum (ER) transmembrane protein, which is a sensor of endoplasmic reticulum (ER) calcium levels; it is activated by a decrease in ER calcium levels. Once activated, STIM1 interacts with a plasma membrane protein, Orai1, to activate Orai1-containing calcium-selective plasma membrane channels [11]. Both STIM1 and Orai1 are expressed in podocytes. TRPC6 is also found in the podocyte foot processes and along the slit diaphragm, which is vital for normal renal function [12]. STIM1 has been implicated in the development of diabetes. In particular, the abnormal expression of STIM1 induces podocyte injury by causing an imbalance in Ca2+ homeostasis, changing the expression of podocyte-associated molecules, inducing oxidative stress, and leading to mitochondrial dysfunction.
In the present study, we assess the effects of autophagy and STIM1 on podocyte EMT in DN rats. Our findings indicate that silencing STIM1 not only promotes podocyte autophagy, but inhibits apoptosis and EMT in DN rats. Moreover, STIM1 upregulation leads to the activation of TRPC6 and Orai1 channels, which results in Ca2+ entry. However, the mechanisms of autophagy and STIM1 in podocyte dysfunction under diabetic conditions remain unclear. Here, we aimed to determine the significance of autophagy and STIM1 in podocyte injury and EMT, as well as its involvement in the pathogenesis of diabetic nephropathy.
Materials and methods
Male db/db and C57BL/KsJ rats (8-week-old) were purchased from Cavens Lab Animal Co. Ltd., Changzhou, China. MPC5 cells were acquired from the America type culture collection (ATCC).
Cell culture and RNA interference
Cells were cultured at 33°C in RPMI-1640 (Hyclone, Shanghai, China) medium supplemented with 10% PBS (JRDUN Biotechnology Co. Ltd, Shanghai, China) and recombinant interferon-γ (PeproTech China, Suzhou, China), with a transmission ratio of 1:4. To induce differentiation, podocytes were grown under non-permissive conditions at 37°C for 11 days, with a transmission ratio of 1:2. When had matured, MPC5 cells were cultured without RPMI-1640 for 24 h. The experiment was divided into 4 groups: MPC5 + siRNA-Scr + 10% C57BL/KsJ (Group A), MPC5 + siRNA-STIM1 + 10% C57BL/KsJ (Group B), MPC5 + siRNA-Scr + 10% db/db (Group C), and MPC5 + siRNA-STIM1 + 10% db/db (Group D). After each group was transfected with siRNA for 48 h, the MPC5 cells were collected and the interference effect was tested after 24 h.
Cell transfection
Podocytes were plated at a density of 5×105 on glass cover slips in 6-well plates and cultured at 37°C in a 5% CO2 cell incubator (Thermo, Shanghai, China) for 24 h. Transient transfections with siRNA were conducted using Lipofectamine 2000. In brief, 250 μL of Opti-MEM was added to an EP tub (A) and was diluted with 5 μL of Lipofectamine 2000. The mixture was then incubated for 5 min at 37°C. Another EP tub (B) was filled with 250 μL of Opti-MEM to dilute 10 μL siRNA. Mixing of both solutions A and B were done and then the mixture was incubated for 20 min at 37°C. The siRNA-Lipofectamine 2000 mixture was then applied to 6-well plates containing approximately 1.5 mL of complete medium.
Immunofluorescence staining
Indirect immunofluorescence staining was performed according to a previously described procedure. In brief, cells cultured on coverslips were washed once with cold PBS (JRDUN Biotechnology Co. Ltd, Shanghai, China) and fixed with 3% methyl aldehyde (1:1) for 10-15 min at room temperature. The cells were washed with PBS 3 times, then treated with 1% Triton X-100 for 5-10 min and blocked with 3% Bull Serum Albumin (BSA) for 30 min at room temperature. The cells were then incubated with specific primary antibodies, including rabbit anti-mouse polyclonal antibodies against LC3-II (1:200) and antibodies against P62 (1:200), followed by staining with a secondary antibody (1:500). Cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) (Beyotime Company, Shanghai, China) hydrochloride to visualise the nuclei. Slides were viewed using an epifluorescence microscope (Leica Company, Shanghai, China).
Measurements of intracellular Ca2+ concentrations
Podocytes were washed once with PBS, and the cellular deposits following trypsin digestion were collected. These cellular deposits were incubated with a buffer containing 10 μmol/L fluo-3, AM (Life Technologies, New York, USA) for 1 h, after which MPC5 cells were washed in calcium-free buffer, and the fluorescence levels were monitored using a FluorChen E ascent single channel fluorimeter (Protein Simple Company, California, USA). The findings were recorded in F. The cells were incubated with 1% Triton X-100 for 30 min at room temperature, and then with CaCl2 for 1-10 min and the cellular fluorescence was recorded as Fmax. In addition, cells were also added to 10 mM EDTA (Solarbio, Beijing, China) and incubated for 1-10 min. Fluorescence measurements were performed at the single cell level using 488 nm excitation wavelengths and the recorded values were considered as Fmin. Ca2+ concentrations were estimated using the following formula: [Ca2+] free = Kd (F-Fmin)/(Fmax-F). (Kd = 390 nM).
Flow cytometry
The cell culture solution was extracted into a centrifuge tube. The cells were washed once with PBS, and added EDTA solution, then the mixture was incubated at room temperature. The collected cell culture medium in the previous step was added, and the solution was mixed, transferred to a centrifuge tube, and centrifuged at 1000 rpm for 5 min. The supernatant was discarded and the cells treated with PBS were counted. Following another centrifugation, the cells were resuspended in 195 μL Annexin V-APC at 5-10×104 and incubated at 4°C for 15 min. Propidium iodide (PI) was then added and the mixture was incubated at 4°C for 5 min. Moreover, an Annexin V-APC-free and PI-free mixture was used as the negative control. Flow cytometry was subsequently performed using BD Annexin V Apoptosis Detection Kit APC (eBioscience, Shanghai, China) according to the manufacturer’s instructions. For BD flow cytometry, the Annexin V-APC corresponded with the FL4 channel and the PI corresponded with the FL2 channel.
Western blot analysis
Total protein extracts were collected from each group in RIPA lysis buffer (JRDUN Biotechnology Co. Ltd, Shanghai, China). Protein concentrations were determined using a BCA protein assay kit (Thermos, Shanghai, China) according to the manufacturer’s protocol. Proteins were separated via sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) using 10% gels and were transferred onto nitrocellulose (NC) membranes (Millipore, Shanghai, China). Non-specific sites were blocked using 5% powdered milk diluted in TBS (13.7 mM NaCl, 0.27 mM KCl, and 1.0 M Tris) containing 0.05% Tween 20 (TBST) for 1.5-2 h. The membranes were incubated with primary antibodies, including rabbit anti-mouse polyclonal antibodies against STIM1, antibodies against LC3-II, antibodies against P62, antibodies against TRPC6, antibodies against Orai1, antibodies against Beclin-1, antibodies against Bcl-2, antibodies against Caspase3, antibodies against E-cadherin, antibodies against fibronectin, and antibodies against α-SMA (1:1,000) overnight at 4°C. After washing the membranes with TBST 3 times, incubation with secondary antibodies, including those of donkey anti-goat, goat anti-mouse, or goat anti-rabbit (1:5,000), was performed at 37°C for 1-2 h. The signals were finally detected using the ECL advanced system. β-actin was used as a control. Furthermore, blots were analysed using FluorChen E Image software (Protein Simple Company, California, USA).
Statistical analysis
Data are expressed as means ± standard error. Comparisons between groups were made using Student’s t-test (= 2) or one-way analysis of variance (>2). Throughout the text, Figures, and Figure legends, * and ** indicate P values of <0.05 and <0.01, respectively. A P value of <0.05 was considered to indicate statistical significance.
Results
Effect of siRNA-mediated silencing of STIM1 with Western blots
The effect of siRNA1 was optimal, therefore, siRNA1 was selected for subsequent experiments (Figure 1A and 1B).
Figure 1.
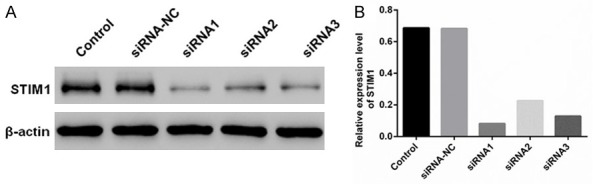
Western blot indicates the effect of siRNA-mediated silencing of STIM1. A. STIM1 expression was standardized using β-actin, and STIM1 was treated with siRNA in MPC5 cells. B. Western blotting was used to detect the relative expression levels of STIM1 after the MPC5 cells were treated with STIM1 siRNA. Control, negative control-siRNA, and complex STIM1-siRNA (siRNA1, siRNA2, and siRNA3).
Silencing STIM1 reverses DN-induced autophagy
Compared with that in Group A, a higher fluorescence intensity for LC3II (Figure 2A and 2B; P<0.01) and a lower fluorescence intensity for P62 (Figure 2C and 2D; P<0.01) were observed in Group B. These findings suggest that podocyte autophagy can be enhanced by silencing STIM1. In contrast, the fluorescence intensity for LC3II was lower (Figure 2A and 2B; P<0.01), and the P62 was greater (Figure 2C and 2D; P<0.05) in Group D, which indicates that podocyte autophagy can be decreased by culturing in DN rats serum. The LC3II levels (Figure 2A and 2B) in Group D were significantly greater (P<0.01) than those in Group C, although the P62 levels (Figure 2C and 2D) were lower (P<0.05). These observations show that the podocyte autophagy levels were stronger in cultured with serum of DN rats by silencing STIM1. Furthermore, on Western blotting, expression of LC3II/LC3I (Figure 2E and 2G) in Groups C and D (cultured in DN rats serum) was significantly lower as compared to that in Groups A and B (cultured in normal rats serum), whereas expression of P62 (Figure 2E and 2F) was markedly greater. These results indicate that autophagy in podocytes cultured in the serum of DN rats is significantly reduced, but can be enhanced by silencing STIM1.
Figure 2.

Silencing of STIM1 with siRNA reverses DN-induced autophagy. A-D. Immunofluorescence staining and Western blotting were performed to detect LC3II and P62 proteins in MPC5 cells cultured in different sera. E-G. Expression and quantification of P62, LC3I, and LC3II proteins were standardised using Western blots in MPC5 cells cultured in different sera. (**P<0.01, *P<0.05).
Upregulation of STIM1 can contribute to Ca2+ entry
To assess the potential role of STIM1 activity in Ca2+ level regulation, we evaluated the Ca2+ concentrations in 4 groups (Figure 3A-C). We observed that the Ca2+ levels were decreased (P>0.05; Figure 3C), whereas TRPC6 and Orai1 protein levels were significantly decreased in Group B, but the result was completely opposite in Group C (P<0.01; Figure 3C), compared to that in Group A (Figure 3D-F). Compared to those noted in Group B, the Ca2+ levels were markedly increased (P<0.01; Figure 3C), whereas the TRPC6 and Orai1 protein levels were also increased in Group C (Figure 3D-F), but converse findings were noted in Group D (P<0.01; Figure 3C). In conclusion, the upregulation of STIM1 can contribute to Ca2+ entry by activating the TRPC6 and Orai1 channels.
Figure 3.

Upregulation of STIM1 facilitates Ca2+ entry by activating the TRPC6 and Orai1 channels. A-C. Intracellular Ca2+ concentrations were measured in the different treatment groups. D. Expression of TRPC6 and Orai1 proteins was standardized using β-actin. E, F. Western blotting was performed to detect TRPC6 and Orai1 protein expression levels in different treatment groups. (**P<0.01).
Upregulation of STIM1 can induce podocytes apoptosis in vitro
Compared to that noted in Group A, the apoptosis rate was slightly lower (P<0.01) in Group B, but was significantly greater in Group C (P<0.01; Figure 4A and 4B). Compared to that noted in Group B, the apoptosis rate was markedly greater in Group D (P<0.01). However, compared with that in Group C, the apoptosis rate was markedly lower in Group D (P<0.01). Furthermore, upregulation of STIM1 can lead to podocyte apoptosis in DN. Western blot analysis (Figure 4C-F) indicated that compared with those in Group A, the Beclin-1 and Bcl-2 protein levels were markedly increased, but the Caspase3 protein levels were significantly decreased in Group B. Converse findings were noted in Group C. Compared with those in Group C, the Beclin-1 and Bcl-2 protein levels were markedly increased, whereas the Caspase3 protein levels were markedly decreased in Group D. These results suggest that autophagy was inhibited and apoptosis was enhanced in podocytes cultured in the serum of DN rats. However, with silencing of STIM1, a converse outcome was noted.
Figure 4.

Upregulation of STIM1 can induce podocyte apoptosis. A, B. Cellular apoptosis and the apoptosis rate were detected using flow cytometry in the different treatment groups. C. The expressions of Beclin-1, Bcl-2, and Caspase-3 proteins were standardized using β-actin. D-F. Western blotting was performed to detect the expression levels of Beclin-1, Bcl-2, and Caspase-3 proteins in MPC5 cells cultured in different sera. (**P<0.01).
Silencing of STIM1 inhibits podocyte EMT in vitro
Compared with those in Group A, the E-cadherin levels were greater (Figure 5A and 5B), but the α-SMA (Figure 5A and 5C) and Fibronectin (Figure 5A and 5D) protein levels were lower in Group B. However, converse findings were noted in Group C. Moreover, compared with those in Group B, E-cadherin levels were lower, but α-SMA and fibronectin protein levels were greater in Group D. Compared with those in Group C, E-cadherin levels were greater, but α-SMA and fibronectin protein levels were lower in Group D. These results suggest that silencing STIM1 can inhibit podocyte EMT in DN rats.
Figure 5.
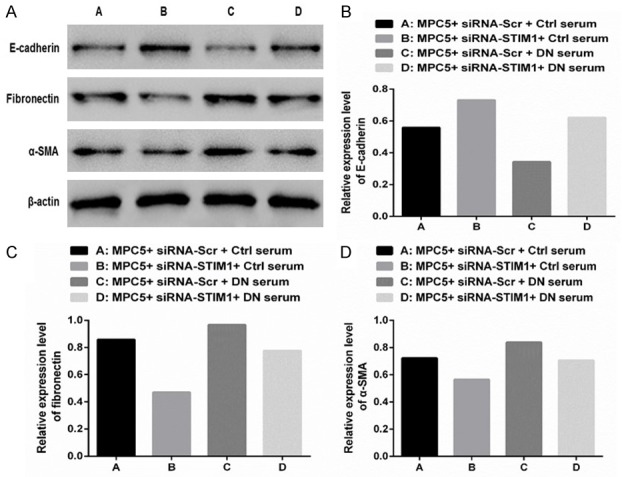
Silencing STIM1 with siRNA could significantly inhibit podocyte epithelial-mesenchymal transition. A. The expression of E-cadherin, fibronectin, and α-SMA proteins were standardized using β-actin. B-D. Western blotting was performed to detect the expression levels of E-cadherin, fibronectin, and α-SMA proteins in different treatment groups.
Discussion
Podocytes are a crucial part of the GFB, and their loss and injury can lead to proteinuria and EMT, as well as glomerulosclerosis [13]. Crucial evidence suggests that podocytes exhibit a high level of constitutive autophagy, which attenuates diabetic glomerular damage by protecting against hyperglycemia-induced podocyte injury [14]. Autophagy acts as a survival mechanism for maintaining cellular integrity, particularly under unfavourable circumstances [15]. Tagawa et al. [16] showed that insufficient podocyte autophagy in patients with diabetes and rats with massive proteinuria was accompanied by podocyte loss and stimulation of cultured podocytes. Furthermore, sera from patients with diabetes or rats with massive proteinuria-impaired autophagy resulted in apoptosis. In our study, compare with cultured podocytes with normal serum of rats, the podocytes autophagy increased, and apoptosis decreased (Figures 2 and 4). Preservation of lysosome- and autophagy-mediated proteostasis may be critical for podocytes to cope with increased amounts of damaged proteins produced during diabetes [17]. Moreover, Hartleben et al. [18] indicated that the podocyte-specific deletion of autophagy-related proteins, such as Atg5, leads to an increased susceptibility to glomerulopathy in aging rats, along with the accumulation of oxidised and ubiquitinated proteins, ER stress, and proteinuria. In particular, the ER play a vital role in regulating autophagic flux, autophagy may be impaired following unmitigated ER induced by high glucose levels and Mammalian target of rapamycin complex 1 (mTORC1) activation, such as resulting from defective autophagy might accelerate the irreparable progression of DN [14]. These changes eventually led to podocyte loss and late-onset glomerulosclerosis. However, the recently identified potential pathophysiologic mechanism of podocyte autophagy may represent only a minimal part of the entire process.
EMT is a dynamic development process, wherein epithelial cells acquire features of mesenchymal cells such as myofibroblasts; this leads to the loss of epithelial cell markers such as E-cadherin, acquisition of mesenchymal markers such as α-SMA and fibronectin, disruption of the tubular basement membrane, and enhanced cell migration and invasion [7]. Moreover, EMT may be important in later stages of renal disease progression, which may lead to interstitial fibrosis and tubular atrophy due to the disappearance of epithelial cells [19]. However, several established intracellular signal transduction pathways such as TGFβ/Smad, integrin-linked kinase (ILK), and Wnt/β-catenin signalling are essential for controlling the EMT process in DN. Furthermore, we suggest that silencing STIM1 also can inhibit EMT.
The term store-operated Ca2+ entry (SOCE) was first used to describe the process whereby the depletion of intracellular Ca2+ stores results in the movement of extracellular Ca2+ into cells [20]. This Ca2+ entry pathway plays an essential role in a wide variety of physiological functions, including exocytosis, enzymatic activity, gene transcription, cell proliferation, apoptosis, and it can activate by depletion of ER [21]. Previous studies have identified STIM1 and Orai1 as the essential components of the SOCE channels [22], and have indicated the overexpression of STIM1 and/or Orai1 in various types of cells, for instance, in glomerular mesangial cells, high glucose, and diabetes enhanced SOCE and increased expression of STIM1/Orai1 [23]. A recent study suggests a beneficial effect of SOC in glomerular mesangial cells (MCs) by inhibiting extracellular matrix (ECM) protein expression, which may protect the kidney from diabetic injury at early stages of DN [24]. However, the effect of STIM1 in podocytes in DN remains unclear. The transient receptor potential canonical 1 (TRPC1) was shown to mediate SOCE, contribute to the calcium-permeable currents generated by store depletion, and regulate physiological function in many cell types [25]. Recent studies demonstrate that STIM1 activates TRPC and via a different domain. The initial observation showed that knockdown of TRPC reduced SOCE by about 60%, while loss of Orai1 or STIM1 induced complete elimination of SOCE [26]. We also show that upregulation STIM1 can activate TRPC6 and Orai1 channels, which results in Ca2+ entry.
STIM1 enhances migration by promoting EMT following the activation of the Snail, TGF-β, and Wnt/β-Catenin signal pathways in human prostate cancer cell lines [27]; however, blocking SOCE activity by using a specific blocker or by applying siRNAs that target STIM1 and Orai1 can inhibit the formation of focal adhesions, and thus reduce the migration and invasion of tumor cells [28]. Further studies have shown that that SOCE functionally interacts with the pro-apoptotic protein during apoptosis [29], and that overexpression of STIM1 increases SOCE activity and can also accelerate apoptosis [30]. Similarly, our study suggests that silencing STIM1 not only decrease apoptosis and EMT, but also increase autophagy.
In conclusion, our study suggests that STIM1 can play an important role in podocytes injury and EMT in DN rats. Silencing STIM1 may enhance podocytes, which is cultured with serum of DN rats. It can also induce autophagy and inhibit apoptosis and EMT. Thus, we consider that STIM1 may be associated with modulation of calcium ion concentration by blocking TRPC6 and Orai1 channels in podocytes. The underlying mechanisms need further investigation.
Acknowledgements
We would like to thank the native English speaking scientists of Elixigen Company (Huntington Beach, California) for editing our manuscript. This work was supported by the National Natural Science Foundation of China [grant number 81641026], the Major Projects of Science and Technology Department of Zhejiang Province [grant number 2014C03047-2], the Natural Science Foundation of Zhejiang Province [grant numbers LY16H050005, LZ17H050001], the Project of Scientific Research Foundation of Chinese Medicine [grant number 2017ZA010], and the General Project of the Medical and Health of Zhejiang Province [grant number 2016KYB007].
Disclosure of conflict of interest
None.
References
- 1.Xu Y, Wang L, He J, Bi Y, Li M, Wang T, Wang L, Jiang Y, Dai M, Lu J, Xu M, Li Y, Hu N, Li J, Mi S, Chen CS, Li G, Mu Y, Zhao J, Kong L, Chen J, Lai S, Wang W, Zhao W, Ning G 2010 China Noncommunicable Disease Surveillance Group. Prevalence and control of diabetes in Chinese adults. JAMA. 2013;310:948–959. doi: 10.1001/jama.2013.168118. [DOI] [PubMed] [Google Scholar]
- 2.Lasaridis AN, Sarafidis PA. Diabetic nephropathy and antihypertensive treatment: what are the lessons from clinical trials? Am J Hypertens. 2003;16:689–697. doi: 10.1016/s0895-7061(03)00864-1. [DOI] [PubMed] [Google Scholar]
- 3.Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int. 1999;56:1627–1637. doi: 10.1046/j.1523-1755.1999.00721.x. [DOI] [PubMed] [Google Scholar]
- 4.May CJ, Saleem M, Welsh GI. Podocyte dedifferentiation: a specialized process for a specialized cell. Front Endocrinol (Lausanne) 2014;5:148. doi: 10.3389/fendo.2014.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 6.Huber TB, Edelstein CL, Hartleben B, Inoki K, Jiang M, Koya D, Kume S, Lieberthal W, Pallet N, Quiroga A, Ravichandran K, Susztak K, Yoshida S, Dong Z. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8:1009–1031. doi: 10.4161/auto.19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- 8.Holian J, Qi W, Kelly DJ, Zhang Y, Mreich E, Pollock CA, Chen XM. Role of Kruppel-like factor 6 in transforming growth factor-beta1-induced epithelial-mesenchymal transition of proximal tubule cells. Am J Physiol Renal Physiol. 2008;295:F1388–1396. doi: 10.1152/ajprenal.00055.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moeller MJ, Soofi A, Hartmann I, Le Hir M, Wiggins R, Kriz W, Holzman LB. Podocytes populate cellular crescents in a murine model of inflammatory glomerulonephritis. J Am Soc Nephrol. 2004;15:61–67. doi: 10.1097/01.asn.0000102468.37809.c6. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mukherjee S, Brooks WH. Stromal interaction molecules as important therapeutic targets in diseases with dysregulated calcium flux. Biochim Biophys Acta. 2014;1843:2307–2314. doi: 10.1016/j.bbamcr.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 13.Stitt-Cavanagh E, MacLeod L, Kennedy C. The podocyte in diabetic kidney disease. ScientificWorldJournal. 2009;9:1127–1139. doi: 10.1100/tsw.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W, Dai C, Yang J. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS One. 2013;8:e60546. doi: 10.1371/journal.pone.0060546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Asanuma K, Kim EH, Haneda M, Kajiwara N, Hayashi K, Ohashi H, Ugi S, Maegawa H, Uzu T. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes. 2016;65:755–767. doi: 10.2337/db15-0473. [DOI] [PubMed] [Google Scholar]
- 17.Newsholme P, Rebelato E, Abdulkader F, Krause M, Carpinelli A, Curi R. Reactive oxygen and nitrogen species generation, antioxidant defenses, and beta-cell function: a critical role for amino acids. J Endocrinol. 2012;214:11–20. doi: 10.1530/JOE-12-0072. [DOI] [PubMed] [Google Scholar]
- 18.Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, Cohen CD, Pavenstadt H, Kerjaschki D, Mizushima N, Shaw AS, Walz G, Huber TB. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ziyadeh F, Wolf G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev. 2008;4:39–45. doi: 10.2174/157339908783502370. [DOI] [PubMed] [Google Scholar]
- 20.Putney JW Jr. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 21.Parekh AB, Putney JW Jr. Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 22.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 23.Chaudhari S, Wu P, Wang Y, Ding Y, Yuan J, Begg M, Ma R. High glucose and diabetes enhanced store-operated Ca(2+) entry and increased expression of its signaling proteins in mesangial cells. Am J Physiol Renal Physiol. 2014;306:F1069–1080. doi: 10.1152/ajprenal.00463.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu P, Wang Y, Davis ME, Zuckerman JE, Chaudhari S, Begg M, Ma R. Store-operated Ca2+ channels in mesangial cells inhibit matrix protein expression. J Am Soc Nephrol. 2015;26:2691–2702. doi: 10.1681/ASN.2014090853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ong HL, de Souza LB, Cheng KT, Ambudkar IS. Physiological functions and regulation of TRPC channels. Handb Exp Pharmacol. 2014;223:1005–1034. doi: 10.1007/978-3-319-05161-1_12. [DOI] [PubMed] [Google Scholar]
- 26.Ong HL, Ambudkar IS. STIM-TRP pathways and microdomain organization: contribution of trpc1 in store-operated Ca(2+) entry: impact on Ca(2+) signaling and cell function. Adv Exp Med Biol. 2017;993:159–188. doi: 10.1007/978-3-319-57732-6_9. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y, Zhang S, Niu H, Ye Y, Hu F, Chen S, Li X, Luo X, Jiang S, Liu Y, Chen Y, Li J, Xiang R, Li N. STIM1 accelerates cell senescence in a remodeled microenvironment but enhances the epithelial-to-mesenchymal transition in prostate cancer. Sci Rep. 2015;5:11754. doi: 10.1038/srep11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang N, Tang Y, Wang F, Zhang H, Xu D, Shen Y, Sun S, Yang G. Blockade of store-operated Ca2+ entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013;330:163–169. doi: 10.1016/j.canlet.2012.11.040. [DOI] [PubMed] [Google Scholar]
- 29.Li N, Zheng L, Lin P, Danielpour D, Pan Z, Ma J. Overexpression of Bax induces down-regulation of store-operated calcium entry in prostate cancer cells. J Cell Physiol. 2008;216:172–179. doi: 10.1002/jcp.21385. [DOI] [PubMed] [Google Scholar]
- 30.Chiu WT, Tang MJ, Jao HC, Shen MR. Soft substrate up-regulates the interaction of STIM1 with store-operated Ca2+ channels that lead to normal epithelial cell apoptosis. Mol Biol Cell. 2008;19:2220–2230. doi: 10.1091/mbc.E07-11-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]