

Abstract.
An individual’s sexual phenotype is usually determined by the presence or absence of the Y chromosome in the embryo’s karyotype, however, due to abnormal X/Y terminal exchange through male meiosis, a few individuals develop male genitalia in the absence of the Y chromosome. This case report presents an adolescent referred to the Pediatric Endocrinology Unit due to bilateral gynecomastia. A diagnosis of hypergonadotropic hypogonadism was established and chromosomal analysis disclosed 46,XX karyotype, with the SRY gene locus found on one of his X chromosomes. A multidisciplinary approach, including psychological support and genetic counseling, is ideal for the management of these patients. Neoplastic transformation of the dysgenetic gonads has been described in several cases, and hence self-examinations and regular ultrasounds are commonly advised.
Keywords: 46, XX male disorder of sexual development, hypergonadotropic hypogonadism, gynecomastia
Introduction
The 46,XX testicular disorder of sexual development is a rare condition, previously known as the Chapelle Syndrome after its first description in 1964 (1).
This disorder is thought to affect one in every 20,000 male newborns and accounts for approximately 2% of male infertility cases. It is characterized by a variable degree of mismatch between the individual’s phenotype and genotype. Furthermore, despite several cases describing phenotypic heterogeneity in this genetic condition, 90% of the affected individuals present a normal male phenotype at birth. Generally, diagnosis occurs after the onset of puberty, upon presentation of hypogonadism and/or gynecomastia, or later due to an assessment of infertility, as a lack of spermatogenesis is observed in all cases (2, 3).
The sexual phenotype is determined by the presence or absence of the Y chromosome in the karyotype of the embryos. However, the vast majority of 46,XX males carry a portion of the Y chromosome, as a result of recombination between the distal portions of the short arms of the X and Y chromosomes during male meiosis. These individuals develop male genitalia in the absence of the Y chromosome. In fact, on the basis of the SRY (sex-determining region of Y) gene, this condition may be divided into two groups: SRY positive (which accounts for 90% of 46,XX males) and SRY negative (2, 4).
Case Report
A previously healthy 13-yr-old male adolescent was referred to the Pediatric Endocrinology outpatient clinic due to progressive bilateral gynecomastia. This feature was a major concern for the adolescent during the previous year. The family medical history was negative for gynecomastia, sexual development disorders, or other endocrinology diseases. We were unable to collect any further information about his parents’ medical history. Unfortunately, we were also unable to access his growth chart or his parents’ height, which would have helped us better characterize his phenotype.
A full physical examination revealed adequate somatometry, with both height and weight between 25 and 50 percentiles for age and sex and gynoid body fat distribution. The vital signs were in the normal range for age. In regard to the features of sexual development, he presented marked bilateral and symmetric gynecomastia, with palpable breast tissue of nearly five centimeters. The male external genitalia appeared normal, with a penis size of 13 centimeters and thick black pubic hair at its base corresponding to stage III on the Tanner scale. The testicles were palpable in the scrotum, both small for his age, with a volume of approximately 2 mm.
The endocrinological study revealed high levels of both gonadotropins, with a luteinizing hormone level of 20.04 mUI/mL (normal range 1.70–8.60 mUI/mL) and follicle-stimulating hormone level of 59.17 mUI/mL (normal range 1.50–12.40 mUI/mL). In addition, a low testosterone level of 2.33 ng/mL (normal range 2.80–8.00 ng/mL) was recorded, with undetectable androstenedione. The sex hormone-binding globulin levels were high, 94.5 nmol/L (normal range 14.50–48.40 nmol/L), and the estradiol levels were 19.8 pg/mL (normal range 7.6–42.6 pg/mL), with 2.38 ng/mL 17-hydroxyprogesterone (normal range 0.42–3.50 ng/mL). These results were consistent with the hypergonadotropic hypogonadism diagnosis.
The bone age was assessed using a hand and wrist X-ray and matched his chronological age.
The chromosomal analysis revealed an apparent 46,XX karyotype. Fluorescence in situ hybridization (FISH) revealed that the SRY gene locus was translocated to the short arm of the X chromosome, reported as: 46,XX.ish der(X)t(X;Y)(p22.3;p11.3)(DXZ1x2, SRY+) – see Fig. 1 for FISH image.
Fig. 1.
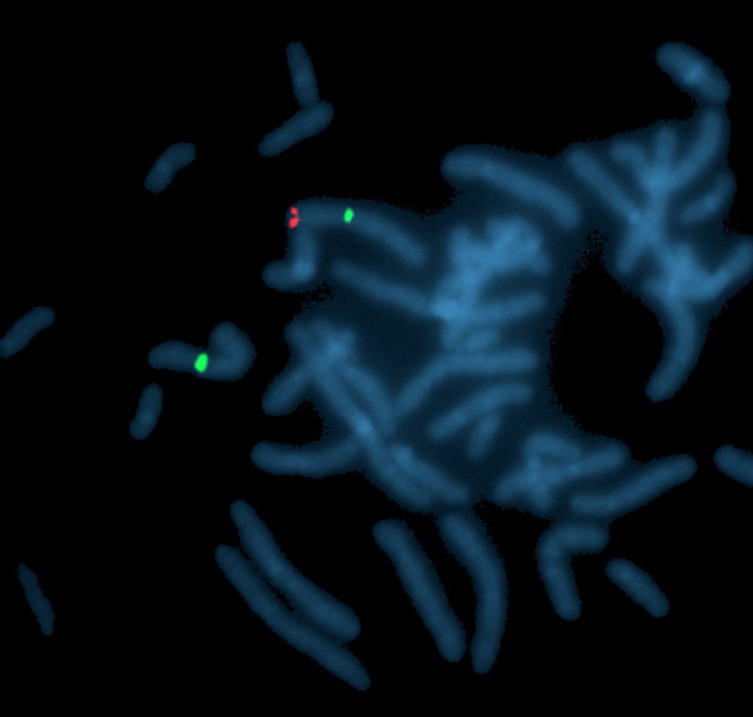
FISH showing SRY gene locus translocated on the short arm of chromosome X; Probes from Vysis LSI SRY Spectrum Orange (for SRY gene – red color). CEP X Spectrum Green Probes (for X chromosomes centromeres).
A diagnosis of congenital hypergonadotropic hypogonadism due to a genetic abnormality of the sex chromosomes was established, and imaging exams were performed. The scrotal ultrasound and pelvic Magnetic Resonance Imaging confirmed the presence of both testicles inside the scrotum, both smaller than for the normal range (maximum diameter of 18 mm), without other abnormalities of male genitalia or remaining Mullerian structures. Both clinical and analytical features were compatible with the 46,XX male disorder of sexual development.
A multidisciplinary approach, including psychological support, assisted in the management of this case. The hypogonadism was managed with testosterone replacement to achieve adequate pubertal development.
In this particular case, due to azoospermia and the risk of gonadoblastoma associated with the 46,XX male disorders of sexual development and very small testicles, surgical removal and testicular prothesis were recommended to the adolescent, which is still under consideration. Meanwhile, self-examinations and regular ultrasound were advised. Furthermore, cosmetic surgery for gynecomastia could be performed, preferably when he reaches stage V on the Tanner scale as his fat distribution may vary due to testosterone therapy.
Genetic counseling was provided to help the patient and his family comprehend the different aspects of this condition and consider future options. With regards to fertility, his options are currently limited to artificial insemination or in vitro fertilization using donor sperm or deciding on adoption.
Discussion
Most 46,XX testicular disorders of sexual development present the translocation of the SRY gene to one of the existing chromosomes, usually the X, and SRY gene appears as one of the determinant genes for the development of the male genitalia (2, 3).
A recent study including 144 males with 46,XX karyotype confirmed that the presentation of 46,XX male disorder of sexual development is hypergonadotropic hypogonadism, regardless of the presence or absence of the SRY gene. As the initiation of spermatogenesis is mainly regulated by this gene, these males present with azoospermia (5, 6).
Neoplastic transformation (gonadoblastoma) of dysgenetic gonads has been described in up to 30% of cases specifically when Y chromosome material is detected (7). This highlights the relevance of gonadal removal in these cases, besides the associated infertility and psychological impact of having smaller testicles and otherwise normal-sized genitalia.
In addition, the hormonal unbalance in hypergonadotropic hypogonadism, due to androgen deficiency, causes a predominance of estrogenic activity over androgen, ultimately resulting in an increase in mammary stroma and growth of ductal tissue and causing gynecomastia (8).
In the present case, upon physical examination, we observed gynecomastia and small testicles. Biochemical features indicated hypergonadotropic hypogonadism and 46,XX karyotype with SRY translocation to the X chromosome. This presentation is a classical form 46,XX testicular disorders of sex development, which is a rare variant of the overall causes of disorders of sexual development.
Conflicts of Interest: The authors declare that they have no conflicts of interest.
References
- 1.Chapelle A, Hortling H, Niemi M, Wennstroem J. XX sex cromossomas in a human male. First Case. Acta Med Scand 1964;175(suppl 412): 25–8. doi: 10.1111/j.0954-6820.1964.tb04630.x [DOI] [PubMed] [Google Scholar]
- 2.Ergun-Longmire B, Vinci G, Alonso L, Matthew S, Tansil S, Lin-Su K, et al. Clinical, hormonal and cytogenetic evaluation of 46,XX males and review of the literature. J Pediatr Endocrinol Metab 2005;18: 739–48. doi: 10.1515/JPEM.2005.18.8.739 [DOI] [PubMed] [Google Scholar]
- 3.Majzoub A, Arafa M, Starks C, Elbardisi H, Al Said S, Sabanegh E., Jr Asian J Androl 2017;19: 168–72. doi: 10.4103/1008-682X.181224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vorona E, Zitzmann M, Gromoll J, Schüring AN, Nieschlag E. Clinical, endocrinological, and epigenetic features of the 46,XX male syndrome, compared with 47,XXY Klinefelter patients. J Clin Endocrinol Metab 2007;92: 3458–65. doi: 10.1210/jc.2007-0447 [DOI] [PubMed] [Google Scholar]
- 5.Chen T, Tian L, Wu F, Xuan X, Ma G, Tang R, et al. Clinical and genetic analysis in males with 46,XX disorders of sex development: A reproductive centre experience of 144 cases. Andrologia 2019;51: e13232. doi: 10.1111/and.13232 [DOI] [PubMed] [Google Scholar]
- 6.Hamada AJ, Esteves SC, Agarwal A. A comprehensive review of genetics and genetic testing in azoospermia. Clinics (Sao Paulo) 2013;68(Suppl 1): 39–60. doi: 10.6061/clinics/2013(Sup01)06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manuel M, Katayama PK, Jones HW., Jr Am J Obstet Gynecol 1976;124: 293–300. doi: 10.1016/0002-9378(76)90160-5 [DOI] [PubMed] [Google Scholar]
- 8.Guerreo-Fdez J, Bonis AB. Endocrinologia pediátrica, chapter 86. In: Manual de Diagnóstico y Terapéutica em Pediatría. 6th edition. Editorial Medica; 2017. p. 805-811. [Google Scholar]