

Abstract
Objective:
Generalized convulsive status epilepticus is associated with high mortality. We tested whether α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor plasticity plays a role in sustaining seizures, seizure generalization, and mortality observed during focal onset status epilepticus. We also determined whether modified AMPA receptors generated during status epilepticus could be targeted with a drug.
Methods:
Electrically induced status epilepticus was characterized by electroencephalogram and behavior in GluA1 knockout mice and in transgenic mice with selective knockdown of the GluA1 subunit in hippocampal principal neurons. Excitatory and inhibitory synaptic transmission in CA1 neurons was studied using patch clamp electrophysiology. The dose response of N,N,H,-trimethyl-5-([tricyclo(3.3.1.13,7)dec-1-ylmethyl]amino)-1-pentanaminiumbromide hydrobromide (IEM-1460), a calcium-permeable AMPA receptor antagonist, was determined.
Results:
Global removal of the GluA1 subunit did not affect seizure susceptibility; however, it reduced susceptibility to status epilepticus. GluA1 subunit knockout also reduced mortality, severity, and duration of status epilepticus. Absence of the GluA1 subunit prevented enhancement of glutamatergic synaptic transmission associated with status epilepticus; however, γ-aminobutyric acidergic synaptic inhibition was compromised. Selective removal of the GluA1 subunit from hippocampal principal neurons also reduced mortality, severity, and duration of status epilepticus. IEM-1460 rapidly terminated status epilepticus in a dose-dependent manner.
Interpretation:
AMPA receptor plasticity mediated by the GluA1 subunit plays a critical role in sustaining and amplifying seizure activity and contributes to mortality. Calcium-permeable AMPA receptors modified during status epilepticus can be inhibited to terminate status epilepticus.
Generalized convulsive status epilepticus (SE) is associated with high mortality; however, the mechanisms leading to generalized convulsions and associated mortality have not been elucidated.1,2 One factor contributing to mortality is cardiorespiratory compromise associated with generalized tonic–clonic seizures. In one prospective randomized study, 22.5% of patients in generalized convulsive SE who were treated with placebo had cardiac instability and respiratory compromise requiring intubation prior to arrival at the emergency department.3 A detailed analysis of patients suffering from sudden unexplained death in epilepsy (SUDEP) also revealed that generalized tonic–clonic seizures pose a particular risk of death.4,5 Mice undergoing convulsive SE have a high mortality rate that occurs during or soon after generalized tonic–clonic seizures.6,7
Neuronal plasticity mechanisms that facilitate the spread of seizures from the hippocampus to the motor cortex during generalized convulsive SE are not understood. Generalized convulsive seizures likely arise from the activation of neurons in the motor cortex.8,9 The hippocampus is a common site of origin of focal seizures, and it is injured during generalized convulsive SE in patients with complex febrile SE.10,11
We hypothesized that α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor plasticity mediated by the GluA1 subunit is critical for secondary generalization of seizures and thus death during SE. We determined the role of GluA1 subunit trafficking in SE using a combination of knockout (KO) and transgenic (TG) mice and electrophysiological analysis in an electrical stimulation–based mouse model of SE, which is associated with recurrent generalized tonic–clonic seizures and mortality.7 Finally, we tested whether calcium-permeable AMPA receptors, which are generated by AMPA receptor plasticity during SE, can be targeted to terminate SE.
Materials and Methods
All of the common chemicals were obtained from Sigma-Aldrich (St Louis, MO). N,N,H,-Trimethyl-5-([tricyclo (3.3.1.13,7)dec-1-ylmethyl]amino)-1-pentanaminiumbromide hydrobromide (IEM-1460), amantadine hydrochloride, atropine, DL-2-amino-5-phosphonopentanoic acid, 6,7-dinitroquinoxaline-2,3-dione, and picrotoxin were obtained from Tocris (Minneapolis, MN), and the anti-GluA1 subunit antibody (clone RH95) was from Millipore (Burlington, MA).
These studies were performed according to protocols approved by the animal care and use committee of the University of Virginia. Adult male and female mice (22–25g, 40–50 days old) were used in these studies; the mice were kept 4 per cage, had ad libitum access to food and water, and were maintained in a 12 hours light/12 hours dark cycle. The results were similar between male and female mice, and hence the data were pooled.
The GluA1 global KO mice were obtained from Seeburg (Max-Plank Institute for Medical Research, Heidelberg, Germany) and have been characterized in detail previously.12,13 GluA1fl mice with floxed exon 11 (GluR-A2lox, B6N.129-Gria1tm2Rsp/J) were obtained from Jackson Laboratory (Bar Harbor, ME). TG mice were originally generated on the 129X1/SvJ background; they were subsequently crossed with C57BL/6NCrl mice for 6 generations.
Virus Injection
To generate mice with local deletion of Gria1, which encodes the GluA1 subunit of the AMPA receptor, in the CA1 and CA3 pyramidal neurons, subiculum, and entorhinal cortex, a virus specific to excitatory glutamatergic neurons expressing Cre recombinase under the CaM kinase II promotor, AAV9.CamKII. HI.GFP-Cre.WPRE.SV40, was used. Transduction of cells in the Gria1-floxed animals resulted in deletion of GluA1 and expression of the green fluorescent protein (GFP) marker protein in transduced neurons. The control virus used in the study, AAV9.CamKII0.4.eGFP.WPRE.rBG, marked transduced neurons with GFP without gene deletion. Both viruses were procured from the University of Pennsylvania viral vector core.
GluA1-floxed animals were stereotaxically (Stoelting, Wood Dale, IL) injected with Cre or control virus bilaterally in the ventral and dorsal CA1 region of the hippocampus (coordinates used: anteroposterior [AP], −2.54; mediolateral [ML], −/+2.00; dorsoventral [DV], −1.20 for ventral hippocampus and AP, −3.40; ML, −/+3.35; DV, −2.25 for dorsal hippocampus). A Hamilton Company (Reno, NV) syringe (Hamilton 7000 Glass, 5μl, 0.3302mm) was loaded with virus solution that was previously kept on dry ice and mounted in the peristaltic pump holder (Harvard Apparatus, Holliston, MA; P-1500). The following settings were used to infuse the virus: 0.1μl/min constant flow rate and 0.25μl per spot. Prior to starting infusion, the needle was left in the tissue for 1 minute to decrease the pressure generated; the same procedure was repeated after infusion before withdrawal of the needle. Animals recovered for 2 weeks, and then electrodes were implanted.
Electrode Implantation and Induction of SE
Animals were stereotaxically implanted with a bipolar insulated stainless-steel electrode in the left hippocampus, bilateral cortical electrodes, and a cerebellar reference electrode. One week later, the hippocampus was stimulated by a continuous stimulation protocol as described previously.7 A 0.75-millisecond biphasic square wave pulse at 50Hz was applied for 10 seconds to determine the afterdischarge threshold (ADT). The initial stimulation was set at 40μA, and the pulse amplitude was increased by 20μA in successive stimulation trains separated by at least 60 seconds until a seizure with a duration of 5 seconds or longer was observed on the electroencephalogram (EEG). If a seizure was not triggered by a stimulation intensity of 200μA, the animal was excluded from the study. The current intensity was set to twice the magnitude of ADT with a minimum at 100μA, and continuous stimulation was performed by cyclic application of 10-second, 50Hz stimulation trains followed by a 5-second off period. The cycle was repeated for 30 minutes or for 60 minutes. Induction of SE was marked by rhythmic and evolving spike-wave discharges for ≥5 minutes at the end of the stimulation period. During hippocampal stimulation, EEG was recorded from cortical electrodes only and from hippocampal and cortical electrodes following stimulation. Animals were monitored by continuous-video EEG until the end of SE, which was determined based on the decline of spike-wave discharge frequency to <1Hz, the disappearance of thigmotactic behavior, and the resumption of feeding and grooming.
Behavioral seizures were scored using a modified Racine scale.14 The baseline behavior of animals during SE, which involved continuous exploration of the cage (thigmotactic behavior), was classified as stage 1. Facial twitching, head bobbing, or behavioral arrest was classified as stage 2, unilateral forelimb clonus as stage 3, bilateral forelimb clonus as stage 4, rearing and falling as stage 5, and running fits and involuntary jumping in the cage as stage 6.
For the mortality study, we discriminated between stage 5 and 6, because death was mainly correlated with stage 6. For the severity study, we pooled stages 5 and 6 together and considered this group stage 5. When behavioral seizures were scored independent of EEG characteristics, the animals were observed for 1 minute, and the highest behavioral score during the epoch was scored. Behavior was also scored in conjunction with EEG characteristics during fast discharges on EEGs; animals were observed for the entire duration of the event, and the highest observed seizure score was assigned.
Treatment of Animals with IEM-1460, Amantadine, and Atropine
SE was induced in adult male and female C57BL/6 mice using continuous hippocampal stimulation as described above. Animals in SE were treated with increasing doses of IEM-1460 ranging from 10mg/kg to 100mg/kg, or with 45mg/kg amantadine hydrochloride or 1mg/kg atropine (intraperitoneal [i.p.]) 15 minutes after the end of hippocampal stimulation. Animals in SE treated with saline at 15 minutes were used as controls.
Western Blotting
Animals were injected with adeno-associated virus (AAV)-CamKII-Cre in the left hemisphere, and hippocampal slices were prepared 2 weeks later. The GFP-expressing hippocampal tissue from the left hemisphere was collected as the transduced tissue, and the hippocampal tissue from the right hemisphere was collected as the control. Tissue lysis and Western blotting to evaluate GluA1 subunit expression in the transduced and control tissue was performed as described previously.15
Electrophysiology
EEG data were exported to LabChart 7 (ADInstruments, Colorado Springs, Colorado). One hippocampal and 1 cortical EEG channel were selected to generate a power spectrogram. The analysis consisted of a fast Fourier transform using a cosine bell data window with a window size of 1,024 data points (2.56 seconds). This method resulted in a frequency resolution of 0.375Hz. A window overlap of 87.5% was used to help smooth the x-axis of the spectrogram. The power was expressed as square microvolts.
AMPA receptor–mediated synaptic currents were recorded from CA1 pyramidal neurons as described previously using acutely prepared slices from animals in SE or control animals.15 The plasticity of AMPA receptors during SE is rapid and transient. Hence, all recordings were completed within 2 hours of slicing. γ-Aminobutyric acid type A (GABAA) receptor–mediated synaptic currents were also recorded as described before.16
Statistics
GraphPad (San Diego, CA) Prism 7 software was used for statistical tests and generation of heat maps of behavioral seizures. The proportions of animals surviving SE were tested using Fisher exact test. The behavioral seizure scores were described by median values and compared using the Mann–Whitney test. The cumulative frequency distributions of high-frequency discharges were compared with the distribution-free Kolmogorov–Smirnov test. Normally distributed data (median synaptic current amplitudes, frequency, Western blot of GluA1 expression) were compared using t tests. The duration of SE was described with Kaplan–Meier survival analysis, and difference between curves was determined by the Gehan–Breslow–Wilcoxon test. Normalized SE duration was plotted against log–IEM-1460 doses and fitted to a 4-parameter equation for sigmoidal curve to determine the median effective dose (ED50). Sex was considered as a biological variable in a post hoc analysis of seizure latency, duration, ADT, and excitatory postsynaptic current (EPSC) amplitude. No sex-dependent effects were evident; however, the study was not powered to detect subtle differences.
Results
SE in GluA1-KO Mice
Global deletion of the GluA1 subunit (GluA1-KO) reduced mortality during SE from 30% to 0% (Fig 1). GluA1-KO and GluA1 wild-type (GluA1-WT) littermates received hippocampal stimulation for 60 minutes, and the survival of animals was compared. The GluA1-WT animals were more prone to death during the stimulation or self-sustaining phase of SE. Because generalized tonic–clonic seizures increase the risk of death, we scored behavioral seizures every 10 minutes; severe behavioral seizures (grade 4 or 5) were less common in the KO animals compared to WT controls. The EEG power spectrum during the first 2 hours of the self-sustaining phase of SE was quite distinct between the GluA1-KO and GluA1-WT animals. The power spectrum in the WT animals contained several high-power streaks associated with fast spike-wave discharges on EEGs. Such events were infrequent in the GluA1-KO animals. Visual examination of the EEGs revealed rhythmic high-amplitude, theta-frequency, spike-wave discharges in both WT and KO animals. In WT animals, both hippocampal and cortical electrodes recorded rhythmic theta-frequency discharges that were intermittently interrupted by faster, lower-amplitude, evolving discharges in the beta and gamma frequency range; these were observed as power spikes on the spectrogram. The high-frequency discharges were often associated with severe behavioral seizures, as described in detail previously.7,17 In the KO animals, low-amplitude, high-frequency discharges were not observed in the cortex, and these events were seen less frequently in the hippocampus. We counted the high-frequency discharges recorded in the hippocampus from 7 representative animals from each group; the cumulative number of these events was substantially higher in the WT animals than in the KO animals. Finally, the SE duration in KO and WT animals was compared; a Kaplan–Meier survival curve revealed that SE was shorter in the KO animals compared to the WT animals.
FIGURE 1:
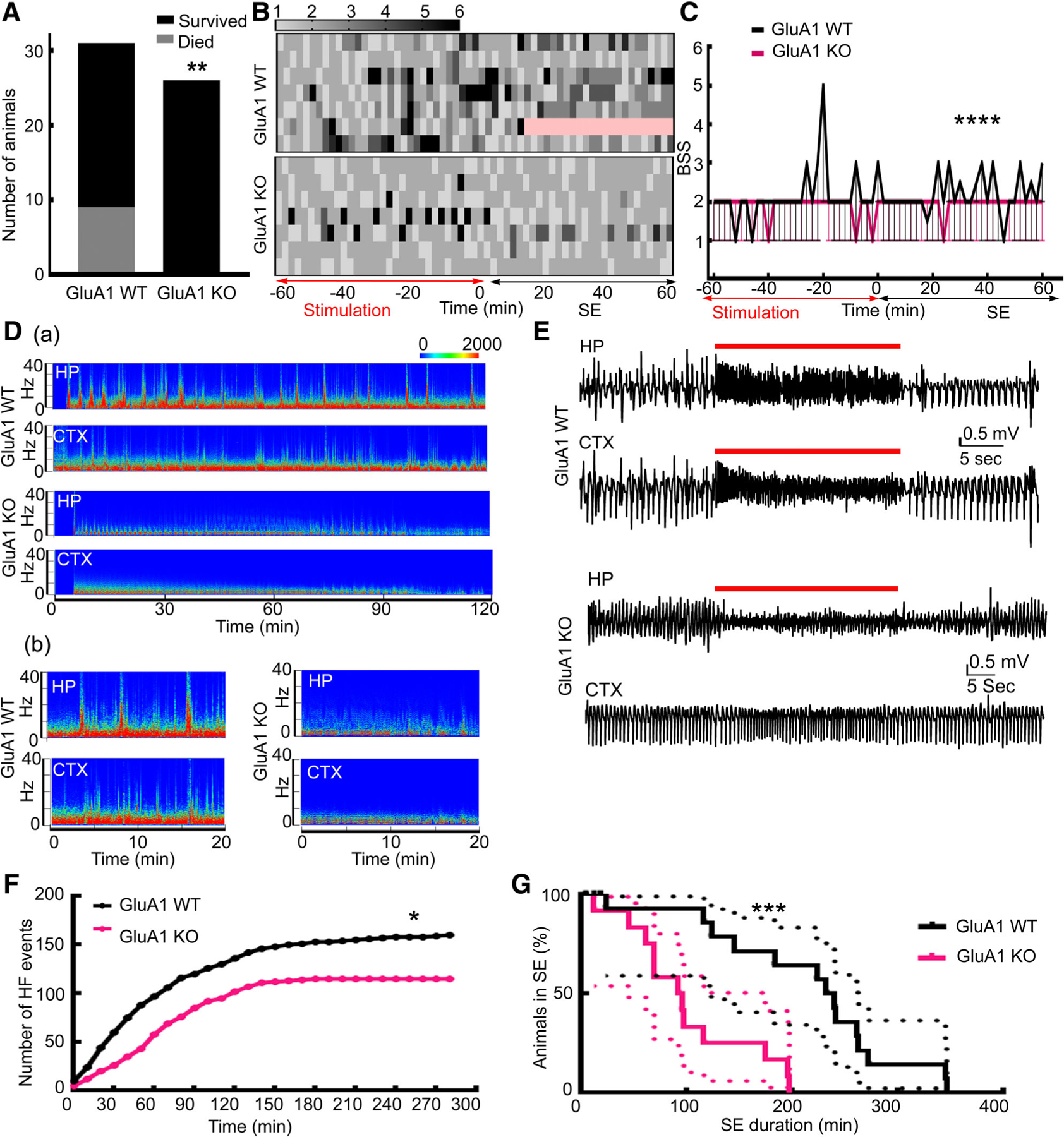
Status epilepticus (SE) was not fatal, was less intense, and had shorter duration in GluA1 knockout (KO) mice. (A) Survival during SE. All 26 GluA1-KO animals survived SE; 9 GluA1 wild-type (WT) animals died during SE, and 22 survived (**p = 0.0022, 1-sided Fisher exact test). (B) Heat maps illustrating the severity of behavior seizure scores in GluA1-WT and GluA1-KO animals during SE. Death is marked in pink. Rows signify individual animals, and 7 animals were randomly chosen to represent each group. The seizures were scored every alternate minute during stimulation and for the first hour of the self-sustained SE seizures. The red and black arrows mark the end of stimulation and the first hour of self-sustaining SE. (C) The median behavioral seizure scores (BSSs) along with 95% CI in the GluA1-WT and GluA1-KO animals illustrated in B, n = 7 each; ****p < 0.0001, 2-tailed Mann–Whitney test. (D, a) Spectrograms illustrating the power of electroencephalograms (EEGs) recorded from the hippocampus (HP) and cortex (CTX) during the initial 120 minutes of the self-sustaining phase of SE from a representative GluA1-WT and GluA1-KO animal. The scale bar represents power in square microvolts. Please note that SE duration was shorter in the GluA1-KO animal, and there was a substantial reduction in the power of EEG by 120 minutes. (D, b) A part of the spectrogram is magnified to illustrate the high-frequency spike-wave discharges marked by streaks of elevated power prominent in the GluA1-WT animal but not in the GluA1-KO animal. These events were generally associated with severe behavioral seizures (4–6). (E) Hippocampal and cortical EEGs illustrating a representative high-frequency spike-wave discharge, marked by red lines, in the GluA1-WT animal. Please note that in the GluA1-KO animals, the amplitude of discharges was reduced in the hippocampal recording during an event, and the event was not prominent in cortical EEGs. (F) The cumulative frequency distribution of the high-frequency (HF) spike-wave discharges in 12 randomly selected GluA1-WT and GluA1-KO animals. The data were binned every 10 minutes. *Approximate p = 0.027, Kolmogorov–Smirnov test. (G) Kaplan–Meier survival curves illustrating the duration of SE, which occurred earlier in the GluA1-KO animals than in GluA1-WT animals, n = 17 GluA1-WT and n = 12 GluA1-KO; *** p = 0.0006 Gehan–Breslow–Wilcoxon test.
To determine whether global deletion of the GluA1 subunit reduces seizure susceptibility, we compared the effect of a single dose of pentylenetetrazol (PTZ; 45mg/kg) in KO and WT animals. PTZ induced seizures in all the WT (n = 10) and KO animals (n = 9); the latency, duration, and severity of seizures were also similar (Fig 2A, B).
FIGURE 2:
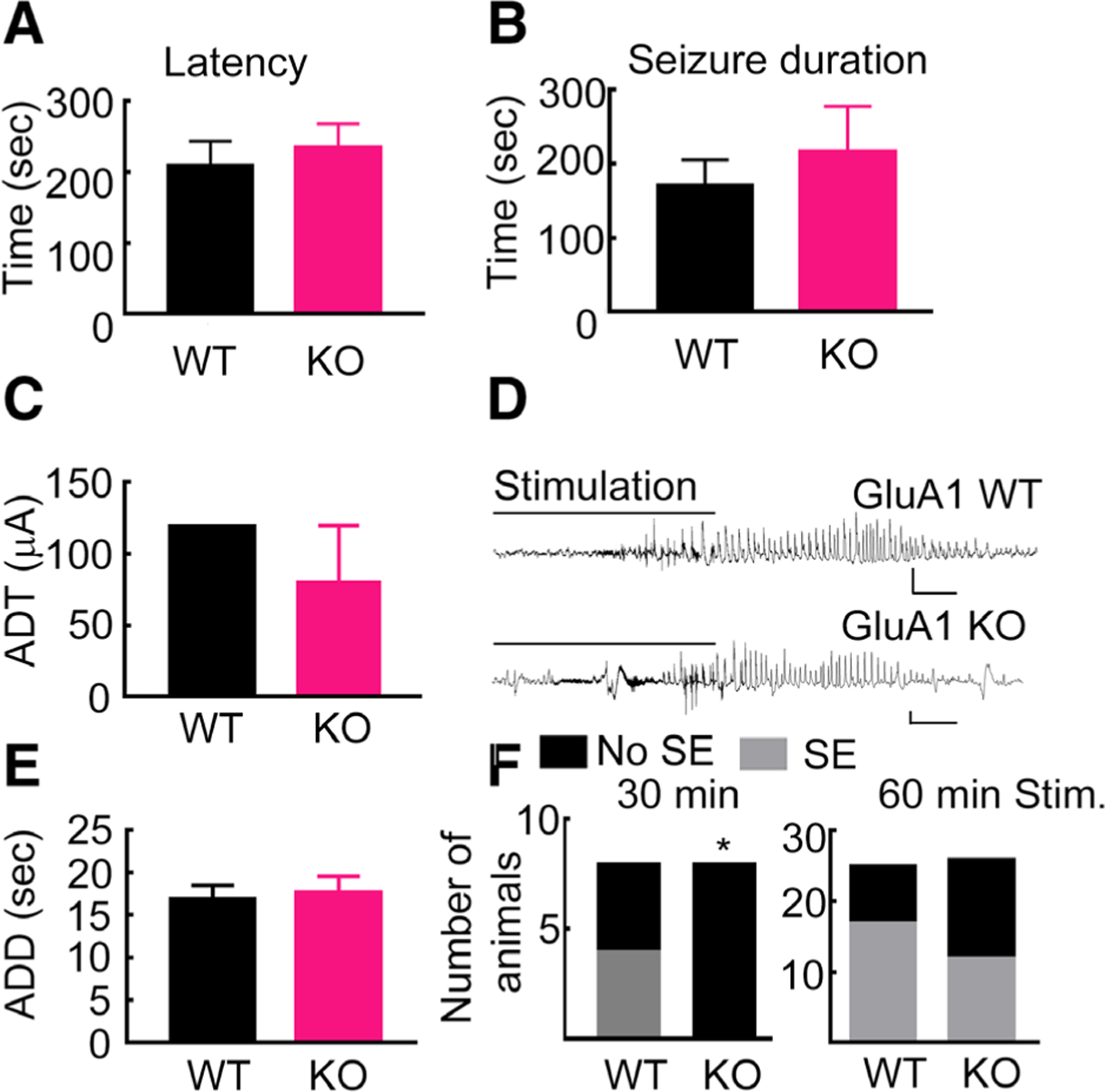
Seizure susceptibility in GluA1 knockout (KO) and wild-type (WT) animals was similar. (A) For the pentylenetetrazol (PTZ; 45mg/kg)-evoked seizure, latency was 210 ± 31 seconds in WT animals and 236 ± 32 seconds in KO animals (n = 10 WT and n = 9 KO animals, p = 0.59 unpaired t test). (B) PTZ-evoked seizure duration in the WT and KO animals; WT, 170 ± 35 seconds; KO, 217 ± 61 seconds; p = 0.55, Mann–Whitney test. The median behavioral seizure score (Racine scale) was 5 in WT animals and 4.5 in KO animals, p = 0.54, Mann–Whitney test (data not shown). (C) The intensity of current necessary to evoke a hippocampal seizure (after discharge threshold [ADT]) in WT (range = 40–200μA, median = 120, n = 19) and in KO animals (range = 20–160μA, median = 80, n = 20; p = 0.23, Mann–Whitney test). (D) A representative seizure recorded from an ipsilateral cortical electrode in response to 10 seconds of hippocampal stimulation in the WT and KO animals. The x and y scale bars represent 2 seconds and 1mV, respectively. (E) The duration of afterdischarges evoked in the WT and KO animals. The afterdischarge duration (ADD) in the WT animals was 7.0 to 37 seconds, median = 17 seconds, n = 19 and that in the KO animals was 6.7 to 49 seconds, median = 16.6 seconds, n = 20; p = 0.98, Mann–Whitney test. (F) The number of animals that experienced status epilepticus (SE; self-sustaining seizures lasting >5 minutes) following hippocampal stimulation in the 2 genotypes. SE was induced in 50% of WT (n = 8) and 0% of KO (n = 8) animals after 30 minutes of stimulation (p = 0.038, Fisher exact test) and in 68% of WT (n = 25) and 46% of KO (n = 26) animals after 60 minutes of stimulation (p = 0.098, Fisher exact test).
We then tested the susceptibility to seizures and SE following hippocampal stimulation. Seizures evoked by 10 seconds of electrical stimulation of the hippocampus had similar current thresholds and comparable durations in the WT and KO animals (see Fig 2). Thirty minutes of stimulation did not cause self-sustained seizures lasting 5 minutes or longer (SE) in any of the KO animals, whereas it caused SE in 50% of the WT animals. However, a similar proportion of WT and KO animals experienced SE following 60 minutes of hippocampal stimulation. In summary, seizure susceptibility of GluA1-KO animals was not altered, but global deletion of the GluA1 subunit reduced susceptibility to SE, decreased the severity, shortened the duration, and lowered the mortality rate associated with SE. We then tested the mechanism behind this protection.
SE-Associated Synaptic Plasticity in KO Animals
SE in rats is associated with AMPA receptor–mediated plasticity of excitatory transmission of CA1 pyramidal neurons.15,18 We tested whether this plasticity is observed in electrical stimulation–induced SE in WT mice and prevented in the KO mice. Hippocampal slices were prepared from WT animals in self-sustaining SE or WT controls, and spontaneous EPSCs (sEPSCs) were recorded from CA1 pyramidal neurons (Fig 3A). The sEPSCs recorded from animals in SE had larger amplitudes than in the control animals (see Fig 3C).
FIGURE 3:
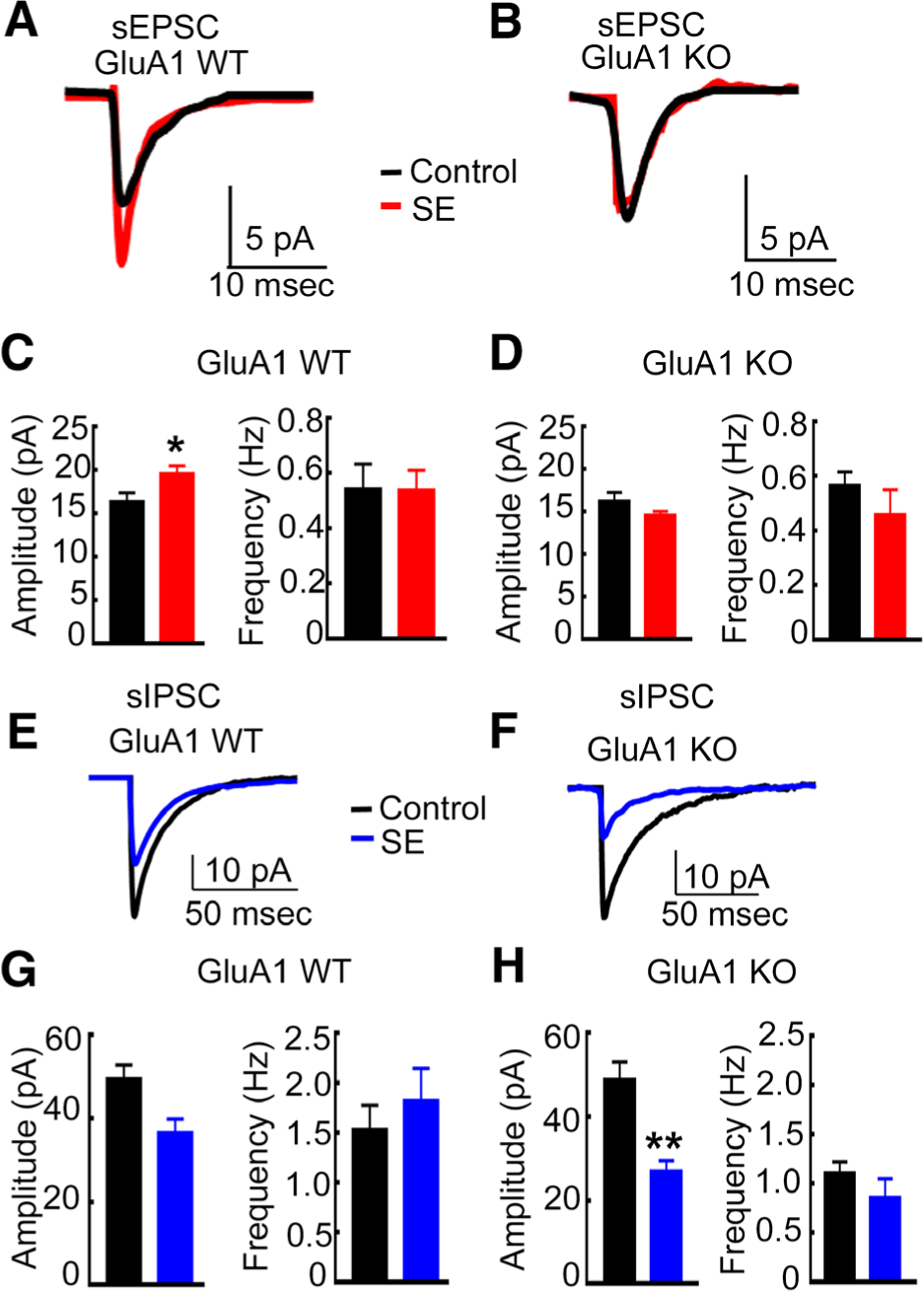
The enhancement of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor–mediated synaptic transmission during status epilepticus (SE) was blocked, but the diminution in γ-aminobutyric acid receptor–mediated synaptic inhibition during SE occurred in GluA1 knockout (KO) animals. (A) Averaged spontaneous excitatory postsynaptic current (sEPSC) traces recorded from CA1 pyramidal neurons of a representative control GluA1 wild-type (WT) animal (black) and a WT animal in SE (red). (B) Averaged sEPSC traces from a control GluA1-KO animal and in a KO animal in SE. (C) The mean of the median amplitude and the average frequency of sEPSCs recorded from control and SE WT animals. The amplitude in WT controls was 16.4 ± 1.0pA (n = 8 cells, n = 4 animals), and that in WT SE animals was 19.7 ± 0.9pA (n = 8 cells, 5 animals; *p = 0.028, unpaired t test). The frequency in control WT animals was 0.54 ± 0.9Hz, and that in WT SE animals was 0.54 ± 0.07Hz (n is the same as for amplitude measurements; p = 0.98, unpaired t test). (D) The mean of the median amplitude and the average frequency of sEPSCs recorded from control and SE KO animals. The amplitude in KO control and KO SE animals was 16.2 ± 1.0pA (5 cells from 3 animals) and 14.6 ± 0.43pA (8 cells from 5 animals), respectively (p = 0.132, unpaired t test). The frequency in control and SE animals was 0.57 ± 0.05Hz and 0.46 ± 0.09Hz, respectively (n is the same as for amplitude measurements; p = 0.4, unpaired t test). (E) Averaged spontaneous inhibitory postsynaptic current (sIPSC) recorded from the CA1 pyramidal neurons of a control GluA1-WT animal (black) and a WT animal in SE (blue). (F) Averaged sIPSCs recorded from CA1 pyramidal neurons of control and SE GluA1-KO animals. (F) Mean of the median sIPSC amplitude in WT control (49.5 ± 3.3pA, 8 cells from 5 animals) and WT SE animals (36.6 ± 3.3pA, 7 cells from 5 animals; p = 0.017, unpaired t test) and the mean frequency of events in the control and SE animals (1.5 ± 0.25Hz and 1.8 0.33Hz, respectively; p = 0.49, unpaired t test). (G) The mean of the median amplitude and the mean frequency of sIPSCs recorded from GluA1-KO control and SE animals. (H) The mean sIPSC amplitude in the control and SE animals was 48.9 ± 4.3pA (8 cells from 5 animals) and 26.9 ± 2.7pA (6 cells from 5 animals), respectively (**p = 0.0018, unpaired t test). Mean sIPSC frequency in control and SE animals was 1.1 ± 0.12Hz and 0.86 ± 0.19Hz, respectively (p = 0.27, unpaired t test).
SE-associated enhancement of EPSCs did not occur in the KO animals (see Fig 3). In agreement with previous reports, the properties of AMPA receptor–mediated, action potential–dependent sEPSCs recorded from KO animals were similar to those of WT animals.12,13 We then compared sEPSCs recorded from KO animals in SE with those of control KO animals. The amplitude of sEPSCs recorded from KO-SE animals was similar to that recorded from control KO animals. Thus, the SE-induced enhancement of EPSCs in CA1 neurons did not occur in GluA1-KO animals. SE did not affect the frequency of sEPSCs in the WT or KO animals. Furthermore, the 10 to 90% rise time and weighted decay of the events were also similar between the 4 groups of animals (data not shown). In summary, SE-associated plasticity of AMPA receptor–mediated neurotransmission did not occur in the KO animals.
SE was shorter and less intense in the GluA1-KO animals, and the excitatory transmission of CA1 pyramidal neurons was also not enhanced. This raised the possibility that seizures in the KO animals were too short or weak to provoke synaptic plasticity; we addressed that possibility by studying GABAergic synaptic transmission. We first confirmed that SE in WT mice resulted in reduced GABAA receptor–mediated synaptic inhibition. We recorded spontaneous inhibitory postsynaptic currents (sIPSCs) from CA1 pyramidal neurons (see Fig 3). The sIPSCs recorded from WT animals in SE were of smaller amplitude than those recorded from control WT mice. A similar diminution of GABAergic transmission was also observed in the KO mice in SE compared to KO controls. The frequency, 10 to 90% rise time, and decay of sIPSCs were not different between WT and KO animals in SE or controls (data not shown). Taken together, these studies revealed that in the global-KO animals, the enhancement in AMPA receptor–mediated glutamatergic transmission was blocked, but the diminution in GABAA receptor (GABAR)-mediated inhibitory neurotransmission remained unaffected.
SE in TG Mice with GluA1 Knockdown in Excitatory Neurons in the Hippocampus
GluA1 subunit is also expressed in extrahippocampal regions, including frontal, entorhinal, and perirhinal cortices, olfactory bulbs, and anterior olfactory nucleus, and in lateral septum,19 and seizures propagate through these regions.20,21 Global KO removes GluA1 subunit from inhibitory and excitatory neurons of all these regions and hippocampus, whereas SE-induced plasticity has been described in the principal, excitatory neurons of the hippocampus.15,18 Therefore, we tested whether knocking down the GluA1 subunit specifically in the excitatory neurons of the hippocampus also impacts SE. We used the GluR-A2lox mice in which exon 11 of Gria1 is flanked by loxP sites, which allows deletion of the sequences encoding the glutamate receptor transmembrane domain lining the ion channel pore in cells and tissues expressing Cre-recombinase. The GluAfl mice were injected with either AAV9-CamKII-eGFP (virus control) or AAV9-CamKII-eGFP-Cre virus (CamKII-Cre). AAV9 viruses have high tropism and transduction efficiency in the hippocampus.22 We used the Cam kinase promoter to restrict GluA1 deletion to the CA1 pyramidal neurons and other excitatory neurons in the hippocampus.23 TG mice without viral injection (uninjected controls) were also studied.
We confirmed that the injected virus was expressed in hippocampal excitatory neurons and that it reduced GluA1 expression. Two weeks after viral injection, GFP was expressed in the CA1 pyramidal neurons, subiculum, superficial entorhinal cortex, and dentate granule cells (Fig 4A). A Western blot detecting the GluA1 subunit confirmed that its expression was reduced in the virus-injected hippocampus compared to the contralateral uninjected hippocampus (see Fig 4B).
FIGURE 4:
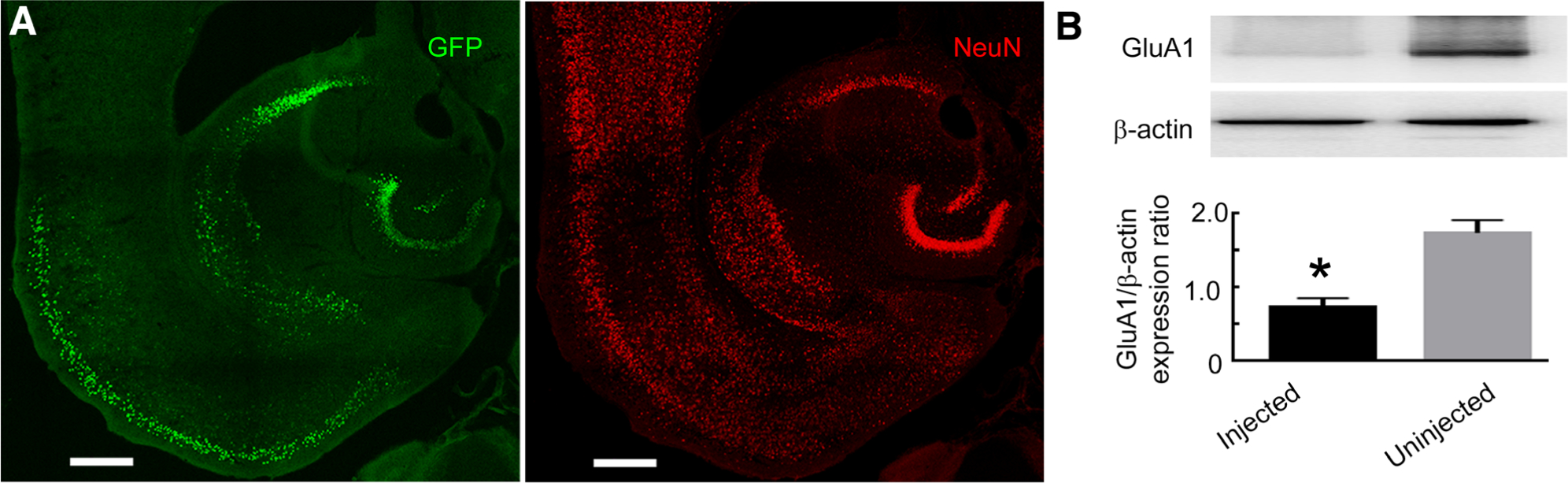
Conditional deletion of the GluA1 subunit using adeno-associated virus (AAV)-expressing green fluorescent protein (GFP)-tagged CamKII-Cre. (A) Images illustrating GFP expression in the hippocampus of a GluA1-floxed animal injected with AAV serotype 9 expressing a GFP-tagged Cre recombinase under the control of the CamKII promoter. Immunoreactivity of NeuN (red) illustrates the neurons. Colocalization of GFP fluorescence corresponding to Cre expression with NeuN immunoreactivity labeled transduced neurons. The scale bars correspond to 400μm. (B) A Western blot illustrating the expression of the GluA1 subunit in the hippocampal tissue injected with AAV-expressing GFP-tagged CamKII-Cre. The GluA1 subunit expression in the contralateral hippocampus, which was not injected with the AAV, was used as a control. The expression of β-actin was used as a loading control. The graph shows GluA1 to β-actin expression ratio in the injected and uninjected hippocampi. The GluA1 subunit expression in the virus-injected hippocampus was 45 ± 11% of that in the contralateral hemisphere without virus injection (n = 3, *p = 0.0079, t test).
The properties of SE were similar in the control virus-injected animals and in the uninjected animals; hence, the data were pooled, and the animals were referred to as TG controls. Death occurred during stimulation or subsequent SE in many of the TG control animals but rarely in the CamKII-Cre virus–injected animals (Fig 5A). Furthermore, death occurred during or shortly after a severe grade 5/6 seizure (see Fig 5B).
FIGURE 5:
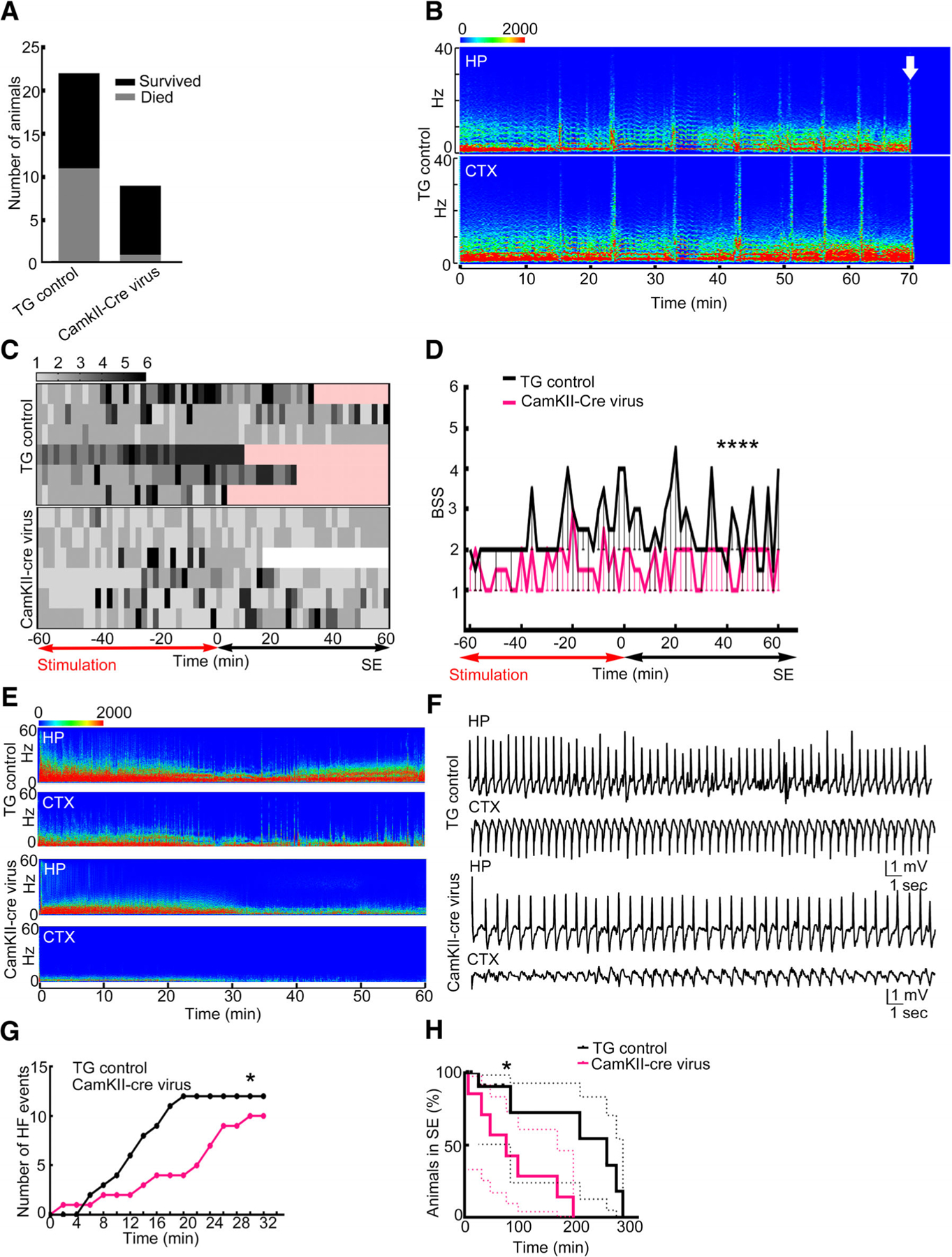
Conditional deletion of the GluA1 subunit using adeno-associated virus (AAV)-expressing green fluorescent protein (GFP)-tagged CamKII-Cre was protective. (A) A comparison of floxed GluA1 animals injected with AAV9-expressing CamKII-GFP (transgenic [TG] control control) and CamKII-Cre-GFP (CamKII-Cre) that died during status epilepticus (SE); death occurred in 11 of the 22 TG control animals and in 1 of the 9 CamKII-Cre virus animals (p = 0.0496, 1-sided Fisher exact test). (B) The electroencephalographic (EEG) power spectrum shows death in an animal marked by an arrow. High-frequency discharge immediately preceded death. CTX = cortex; HP = hippocampus. (C) Heat maps illustrating behavioral seizure scores in the TG control and CamKII-Cre virus–injected floxed GluA1 animals. Each row represents 1 animal; 7 animals were randomly scored in each group. The time subsequent to the death of the animal is marked in pink and after the end of SE is marked in white. (D) Median and 95% confidence interval of the behavioral seizure score (BSS) in the animals illustrated in A (****p < 0.0001, 2-tailed Mann–Whitney test). (E) Spectrogram illustrating the power of EEGs recorded from the hippocampus and cortex during the first hour of SE from a TG control and CamKII-Cre virus–injected animal. (F) Hippocampal and cortical EEGs during SE from a TG control and a CamKII-Cre virus–injected animal. (G) Cumulative frequency distribution of high-frequency spike-wave discharges recorded from 7 representative TG control and CamKII-Cre virus–injected animals. The data are binned every 10 minutes (*p = 0.0207, Kolmogorov–Smirnov test). (H) A Kaplan–Meyer curve illustrating duration of SE in TG control (n = 14) and CamKII-Cre virus–injected (n = 7) animals (*p = 0.0405 Gehan–Breslow–Wilcoxon test).
SE was less severe in the CamKII-Cre virus–injected animals compared to control TG mice. Analysis of the behavioral seizures during and after stimulation revealed that the control TG animals had severe seizures during stimulation, which continued past the end of stimulation into the self-sustaining phase (see Fig 5C, D). In contrast, the CamKII-Cre virus–injected animals had milder seizures during stimulation and during SE. The median behavioral seizure scores were higher in the TG controls compared to the CamKII-Cre–injected animals (see Fig 5D).
The EEGs recorded from the cortex of animals injected with CamKII-Cre were quite distinct from the TG controls (see Fig 5). In these animals, the spike-wave discharges recorded from the cortex were smaller in amplitude than the hippocampal discharges, with poorly formed sharp waves and few high-frequency discharges; in TG controls, there were high-amplitude spike-wave discharges and fast discharges present. There were robust rhythmic spike-wave discharges in the hippocampus of the CamKII-Cre–injected animals, but there were fewer high-frequency discharges compared to the TG controls. The compressed spectral display of EEGs recorded from the TG controls demonstrated persistently high power in the 1 to 10Hz range, with intermittent peaks at higher (20–60Hz) frequencies, and similar patterns were observed in the EEGs recorded from the hippocampal and cortical leads. In contrast, the compressed spectral displays of EEGs recorded from animals injected with CamKII-Cre were distinct; recording from the hippocampus displayed high power in the 1 to 10Hz range, but cortical recordings were devoid of these high-power activities. We compared the occurrence of high-frequency discharges in the first 30 minutes following the end of stimulation; there were infrequent high-frequency discharges in the CamKII-Cre–injected animals compared to the TG controls. Finally, SE was shorter in the CamKII-Cre virus–injected animals than in the TG controls. Thus, deletion of the GluA1 subunit in the hippocampal excitatory neurons had a similar effect on SE severity to global deletion of this subunit.
SE Was Treated with an Antagonist for Calcium-Permeable AMPA Receptors
A key impact of GluA1-mediated AMPA plasticity during SE is that calcium-permeable AMPA receptors are expressed in CA1 pyramidal neurons.18 We tested whether inhibiting calcium-permeable AMPA receptors reduced the severity, shortened the duration of SE, and lessened mortality. C57BL/6 animals in SE were treated with either saline or IEM-1460 15 minutes after stimulation ended. A small dose of IEM-1460 (10mg/kg) did not affect the seizures; in contrast, 18mg/kg and 30mg/kg shortened the duration of SE (Fig 6A). Higher doses of IEM-1460 were toxic and resulted in the death of animals (5 of 7 at 58mg/kg; 6 of 8 at 100mg/kg). Fitting of the electrographic SE duration–IEM-1460 dose data to a 4-parametric equation for sigmoidal curve yielded an ED50 of 17.8mg/kg. Sedation was not observed in animals treated with IEM-1460 doses of 10, 18, and 30mg/kg; however, these animals displayed a mild ataxic gait following the drug administration.
FIGURE 6:
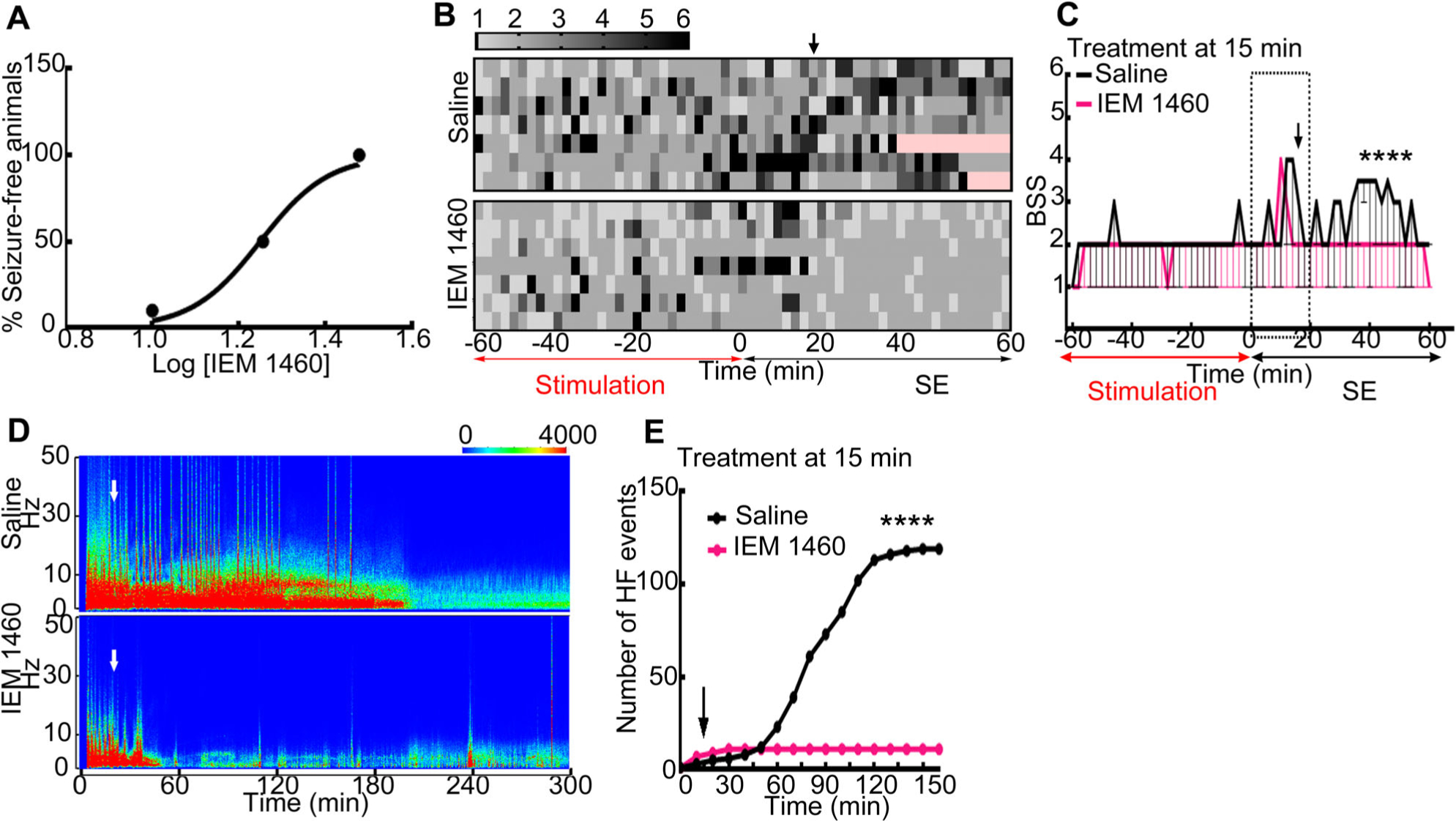
Blockade of calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors with IEM-1460 terminated status epilepticus (SE). (A) Effect of IEM-1460 administered at 15 minutes after the end of hippocampal stimulation on the duration of SE. Log dose of IEM-1460 was plotted against the percentage of animals free of electrographic seizure activity within 60 minutes of the drug administration (n = 11 treated with 10mg/kg, n = 8 treated with 18mg/kg, and n = 12 treated with 30mg/kg IEM-1460). The line represents the best fit of the data to a 4-parametric equation for sigmoidal curve. These studies were performed in a blinded manner; the investigator reading the electroencephalogram (EEG) was blinded to the drug administered to the animals. (B) A heat map illustrating the behavioral seizure scores (BSSs) in C57BL/6 animals in continuous hippocampal stimulation (CHS)-induced SE treated with saline or IEM-1460 (30mg/kg, intraperitoneal) at 15 minutes after the end of hippocampal stimulation. (C) The seizure score and 95% confidence interval from animals depicted in B (****p < 0.0001, 2-tailed Mann–Whitney test). (D) Spectrograms illustrating EEG power during SE recorded from hippocampi of representative saline-treated and IEM-1460–treated animals. The white arrows mark the time of injection of saline or IEM-1460. The EEG power dropped in IEM-1460–treated animal. (E) Cumulative frequency distribution of high-frequency (HF) spike-wave discharges in the saline-treated and IEM-1460–treated animals. The treatments were performed at 15 minutes from the end of CHS (n = 7 each, ****p < 0.0001, Kolmogorov–Smirnov test).
The increased expression of calcium-permeable AMPA receptors in the CA1 neurons of animals in SE could have contributed to the observed toxicity of 58 and 100mg/kg doses of IEM-1460, which is a competitive antagonist of these receptors.24 Therefore, we also evaluated the effect of these doses on naive animals. Both doses of IEM-1460 caused the death of naive animals within minutes (3 of 5 at 58mg/kg; 4 of 4 at 100mg/kg).
In addition to shortening the duration of SE, IEM-1460 also reduced the severity of seizures (see Fig 6). Saline-treated animals had frequent grade 5 or 6 behavioral seizures, whereas there was a dramatic reduction in the behavioral seizure score after 30mg/kg IEM-1460 administration. The median behavioral seizure score was also reduced following administration of IEM-1460. The power of EEG also reduced substantially following administration of IEM-1460. High-frequency discharges became infrequent after IEM-1460 injection compared to saline treatment.
To evaluate whether the above effects were specific to the blockade of calcium-permeable AMPA receptors or inhibition of any excitatory neurotransmitter receptors would exert similar effects, we determined the effect of amantadine hydrochloride, a structural analog of IEM-1460 that is ineffective against AMPA receptors but blocks N-methyl-D-aspartate (NMDA) receptors.25 We treated animals with amantadine hydrochloride (45mg/kg, i.p.) at 15 minutes after the end of hippocampal stimulation. One of 7 amantadine-treated animals died in SE, and it did not reduce seizure duration in the remaining animals (254 ± 39 minutes, n = 6), which was comparable to that in animals treated with saline (180 ± 18 minutes, n = 13, p = 0.067).
Cholinergic stimulation is excitatory, and muscarinic receptor activation causes SE, so we determined whether blocking these receptors would reduce SE duration and prevent death. Thirty-three percent (3 of 9) of atropine (1mg/kg)-treated animals died in SE, and seizure duration was shortened in the remaining animals (98 ± 10 minutes, n = 6, p = 0.01). Thus, muscarinic receptor antagonist did not prevent mortality despite reducing SE duration in some animals.
Discussion
SE results from either the failure of seizure termination mechanisms or the initiation of mechanisms that prolong seizures.26 A large body of evidence suggests that failure of GABAergic inhibition contributes to abnormally prolonged seizures.27,28 The current study suggests that GluA1-mediated plasticity, which is initiated by seizures, plays a critical role in sustaining intense, fatal SE. Previous studies have demonstrated the expression of calcium-permeable AMPA receptors and enhancement of AMPA receptor–mediated neurotransmission in CA1 pyramidal neurons during SE.15,18 Furthermore, multiple studies have demonstrated that AMPA receptor antagonist drugs terminate SE.18,29,30 This study suggests that multiple plasticity mechanisms are critical for the occurrence of severe, fatal SE.
This study points to a specific role of GluA1 plasticity in generating calcium-permeable AMPA receptors during SE. Endogenous and exogenously applied polyamines such as philanthotoxin and IEM-1460 block calcium-permeable AMPA receptors by use-dependent open channel block mechanisms.24,31–33 Prior studies have revealed that philanthotoxin reduced the amplitude of evoked EPSCs of CA1 neurons and that IEM-1460 reduced the amplitude of evoked EPSCs of medial prefrontal cortical neurons of animals in SE.18,34 Furthermore, IEM-1460 also blocked the increased calcium influx in an in vitro model of SE.18 These findings, together with increased surface expression of GluA1 subunit and reduced surface expression of GluA2 subunit in the hippocampi of animals in SE, indicated expression of calcium-permeable AMPA receptors.15,18 However, it was unclear whether calcium-permeable AMPA receptors were generated by GluA1 or GluA2 trafficking; the current study demonstrates a specific role for GluA1 subunit.
Blocking calcium-permeable AMPA receptors reduced mortality by reducing severity and duration of SE. In contrast, a prior study has found that IEM-1460 does not affect seizure threshold.35 Thus, the increased expression of GluA1 subunit–containing receptors during SE is likely to underlie the observed efficacy of this drug in rapidly terminating SE. The severity and duration of SE influence its mortality. In experimental studies, cardiorespiratory arrest occurs during or shortly after a generalized tonic–clonic seizure, suggesting that the two are related.7 Cardiorespiratory compromise is a common feature of generalized convulsive SE in human clinical trials.3,36,37 Treatment with benzodiazepines controls seizures and reduces cardiovascular compromise, suggesting a close causal relationship between seizures and cardiorespiratory compromise.3 Studies on SUDEP also emphasize the risk posed by generalized tonic–clonic seizures.4,5
Antagonists of certain excitatory neurotransmitter receptors alter the course of SE but do not reduce severity, duration, and mortality as observed with AMPA receptor antagonists. In cholinergic animal models of SE, NMDA receptor antagonists do not shorten the duration of SE as measured by EEG.38–40 We further confirmed this finding in the current study. Muscarinic agonists cause seizures, which indicates that blockade of muscarinic receptors would reduce severity of SE. Although atropine reduced the duration of SE, it did not protect against mortality. However, a single dose of the drug was tested and higher doses may offer greater protection. The observed protective effect of calcium-permeable AMPA receptor blockade may be specific to these receptors, because blocking NMDA receptors or muscarinic receptors did not have the same effect.
These studies point to a novel therapeutic target for treating SE: calcium-permeable AMPA receptors. Numerous studies have demonstrated that AMPA receptors can be targeted to terminate SE28,29; however, the current study for the first time demonstrates that a subset of receptors, the calcium-permeable AMPA receptors, can be targeted to terminate SE. Previous studies demonstrated that SE-induced modification of AMPA receptors renders them calcium permeable15,18; IEM-1460 is available to block these receptors. This drug crosses the blood–brain barrier, rapidly blocks calcium-permeable AMPA receptors, and has been tested against seizures.41 However, administration of higher doses of IEM-1460 caused respiratory depression in animals in SE and in naive animals. Thus, detailed toxicology and safety studies are needed to further develop this compound for the treatment of SE. Other drugs targeting calcium-permeable AMPA receptors may be developed to terminate SE.
Enhancement of AMPA receptor–mediated transmission can amplify seizures exiting the hippocampus. The CA1 pyramidal neurons serve as hippocampal output neurons, with projections to the subiculum, septum, contralateral hippocampus, entorhinal cortex, retrosplenial cortex, midline thalamus, hypothalamus, and mammillary body.42 The GluA1 subunit mRNA and protein are widely distributed in the hippocampus, cortex, and striatum; thus, GluA1 subunit–containing AMPA receptors could participate in enhancing synaptic transmission and seizure activity at multiple points in the seizure pathway.19 Previous studies have demonstrated that the entorhinal cortex and piriform cortex can generate and amplify seizures.43,44 Blocking AMPA receptors in the piriform cortex shortens the duration of SE.45
Genetic knockdown of the GluA1 subunit leads to compensatory changes in expression of calcium channels and NMDA receptors.46 AMPA receptor–mediated excitatory synaptic transmission is preserved in GluA1-KO mice, which further suggests that other AMPA receptor subunits are compensating for this subunit. Thus, the reduction in severity and mortality observed in KO and CamKII-Cre–injected animals could in part be due to these changes. However, seizure-induced plasticity of glutamatergic EPSCs was prevented in KO animals, suggesting that GluA1-mediated receptor plasticity contributes to SE. Furthermore, studies with IEM-1460 further support the notion that receptor plasticity plays a role in sustaining seizures.
In contrast to the prevention of potentiation of AMPA receptor–mediated excitatory synaptic transmission of CA1 pyramidal neurons of GluA1-KO animals in SE, the diminution of GABAergic synaptic transmission persisted. The reduced amplitude of mIPSCs of dentate granule cells and CA1 pyramidal neurons of animals in SE is accompanied by accelerated internalization of γ2 subunit–containing GABARs.47–50 Protein phosphatase–mediated dephosphorylation of GABARs during SE increases their interaction with endocytotic proteins.49,51 However, calcium influx during SE is the main factor triggering protein phosphatase activation; preventing the expression of calcium-permeable AMPA receptors during SE was not sufficient to block this signaling. Calcium influx through activation of NMDA receptors during SE in the GluA1-KO animals may be sufficient to accelerate internalization of synaptic GABARs.
The GluA1 subunit is critical for certain forms of learning and memory and has been proposed to play a role in epileptogenesis. AMPA receptors in the hippocampus are heteromers primarily composed of GluA1/2 subunits, with minor contribution of GluA2/3 heteromers.52 In the absence of the GluA1 subunit (GluA1-KO animals), the EPSCs recorded from CA1 pyramidal neurons were similar to those in the WT animals, as seen in past studies.12 This subunit is essential for the expression of certain forms of long-term potentiation in the CA1 region of the hippocampus12; however, heterosynaptic theta-burst potentiation is preserved.53 The absence of the subunit interferes with functioning spatial working memory, but spatial reference memory is preserved.54,55 Activity-induced trafficking of the GluA1 subunit is believed to be mediated by post-translational modification of the intracellular C-terminal domain of the protein.56,57 Two sites, S831 and S845, contribute to lowering the threshold for long-term potentiation.58 The S845 site is proposed to stabilize calcium-permeable AMPA receptors at the synapse.59 SE modifies the phosphorylation of the GluA1 subunit; however, diminished phosphorylation of S831 and S845 sites was observed.15 Previous studies have also implicated GluA1 subunit plasticity in hypoxia-induced hyperexcitability.60
In summary, the current study demonstrated an important role for GluA1 subunit–mediated plasticity in sustaining severe SE, which can lead to cardiorespiratory compromise. It suggests a novel therapeutic target for the treatment of SE.
Acknowledgment
This work was supported by NIH (NINDS) grants R01 NS 040337 and R01 NS 044370 to J.K. and R01 NS110863 to S.J.
Footnotes
Potential Conflicts of Interest
Nothing to report.
References
- 1.DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46:1029–1035. [DOI] [PubMed] [Google Scholar]
- 2.Neligan A, Shorvon SD. Frequency and prognosis of convulsive status epilepticus of different causes: a systematic review. Arch Neurol 2010;67:931–940. [DOI] [PubMed] [Google Scholar]
- 3.Alldredge BK, Gelb AM, Isaacs SM, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med 2001;345:631–637. [DOI] [PubMed] [Google Scholar]
- 4.Hesdorffer D, Tomson T, Benn E, et al. Combined analysis of risk factors for SUDEP. Epilepsia 2011;52:1150–1159. [DOI] [PubMed] [Google Scholar]
- 5.Devinsky O, Hesdorffer DC, Thurman DJ, et al. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 2016;15:1075–1088. [DOI] [PubMed] [Google Scholar]
- 6.Buckmaster PS, Abrams E, Wen X. Seizure frequency correlates with loss of dentate gyrus GABAergic neurons in a mouse model of temporal lobe epilepsy. J Comp Neurol 2017;525:2592–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewczuk E, Joshi S, Williamson J, et al. Electroencephalography and behavior patterns during experimental status epilepticus. Epilepsia 2017;59:369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dabrowska N, Joshi S, Williamson J, et al. Parallel pathways of seizure generalization. Brain 2019;142(8):2336–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.York GK III, Steinberg DA. Hughlings Jackson’s neurological ideas. Brain 2011;134:3106–3113. [DOI] [PubMed] [Google Scholar]
- 10.Shinnar S, Bello JA, Chan S, et al. MRI abnormalities following febrile status epilepticus in children: the FEBSTAT study. Neurology 2012; 79:871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nordli DR Jr, Moshe SL, Shinnar S, et al. Acute EEG findings in children with febrile status epilepticus: results of the FEBSTAT study. Neurology 2012;79:2180–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zamanillo D, Sprengel R, Hvalby O, et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 1999;284:1805–1811. [DOI] [PubMed] [Google Scholar]
- 13.Mack V, Burnashev N, Kaiser KM, et al. Conditional restoration of hippocampal synaptic potentiation in Glur-A-deficient mice. Science 2001;292:2501–2504. [DOI] [PubMed] [Google Scholar]
- 14.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972;32: 281–294. [DOI] [PubMed] [Google Scholar]
- 15.Joshi S, Rajasekaran K, Sun H, et al. Enhanced AMPA receptor-mediated neurotransmission on CA1 pyramidal neurons during status epilepticus. Neurobiol Dis 2017;103:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun C, Mtchedlishvili Z, Erisir A, Kapur J. Diminished neurosteroid sensitivity of synaptic inhibition and altered location of the alpha4 subunit of GABA(A) receptors in an animal model of epilepsy. J Neurosci 2007;27:12641–12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandt C, Glien M, Potschka H, et al. Epileptogenesis and neuropathology after different types of status epilepticus induced by prolonged electrical stimulation of the basolateral amygdala in rats. Epilepsy Res 2003;55:83–103. [DOI] [PubMed] [Google Scholar]
- 18.Rajasekaran K, Todorovic M, Kapur J. Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann Neurol 2012;72:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin LJ, Blackstone CD, Levey AI, et al. AMPA glutamate receptor subunits are differentially distributed in rat brain. Neuroscience 1993;53:327–358. [DOI] [PubMed] [Google Scholar]
- 20.Collins RC, Tearse RG, Lothman EW. Functional anatomy of limbic seizures: focal discharges from medial entorhinal cortex in rat. Brain Res 1983;280:25–40. [DOI] [PubMed] [Google Scholar]
- 21.VanLandingham KE, Lothman EW. Self-sustaining limbic status epilepticus. I. Acute and chronic cerebral metabolic studies: limbic hypermetabolism and neocortical hypometabolism. Neurology 1991; 41:1942–1949. [DOI] [PubMed] [Google Scholar]
- 22.Aschauer DF, Kreuz S, Rumpel S. Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS One 2013;8:e76310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McHugh TJ, Blum KI, Tsien JZ, et al. Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout mice. Cell 1996;87:1339–1349. [DOI] [PubMed] [Google Scholar]
- 24.Twomey EC, Yelshanskaya MV, Vassilevski AA, Sobolevsky AI. Mechanisms of channel block in calcium-permeable AMPA receptors. Neuron 2018;99:956–968.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammad H, Sekar S, Wei Z, et al. Perampanel but not amantadine prevents behavioral alterations and epileptogenesis in pilocarpine rat model of status epilepticus. Mol Neurobiol 2019;56:2508–2523. [DOI] [PubMed] [Google Scholar]
- 26.Trinka E, Cock H, Hesdorffer D, et al. A definition and classification of status epilepticus—report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015;56:1515–1523. [DOI] [PubMed] [Google Scholar]
- 27.Chen JW, Naylor DE, Wasterlain CG. Advances in the pathophysiology of status epilepticus. Acta Neurol Scand Suppl 2007;186:7–15. [PubMed] [Google Scholar]
- 28.Joshi S, Kapur J. GABAA receptor plasticity during status epilepticus In: Noebels JL, Avoli M, Rogawski MA, et al. , eds. Jasper’s basic mechanisms of the epilepsies. Bethesda, MD, 2012:545–554. [Google Scholar]
- 29.Fritsch B, Stott JJ, Joelle DJ, Rogawski MA. Treatment of early and late kainic acid-induced status epilepticus with the noncompetitive AMPA receptor antagonist GYKI 52466. Epilepsia 2010;51:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pitkanen A, Mathiesen C, Ronn LC, et al. Effect of novel AMPA antagonist, NS1209, on status epilepticus. An experimental study in rat. Epilepsy Res 2007;74:45–54. [DOI] [PubMed] [Google Scholar]
- 31.Donevan SD, Rogawski MA. Intracellular polyamines mediate inward rectification of Ca(2+)-permeable AMPA receptors. Proc Natl Acad Sci U S A 1995;92:9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samoilova MV, Buldakova SL, Vorobjev VS, et al. The open channel blocking drug, IEM-1460, reveals functionally distinct α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors in rat brain neurons. Neuroscience 1999;94:261–268. [DOI] [PubMed] [Google Scholar]
- 33.Magazanik LG, Buldakova SL, Samoilova MV, et al. Block of open channels of recombinant AMPA receptors and native AMPA/kainate receptors by adamantane derivatives. J Physiol 1997;505:655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malkin SL, Amakhin DV, Veniaminova EA, et al. Changes of AMPA receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience 2016; 327:146–155. [DOI] [PubMed] [Google Scholar]
- 35.Borowicz K, Banach M. Effect of IEM 1460—a selective antagonist of GluR2-lacking AMPA receptors—on the action of conventional anti-epileptic drugs against maximal electroshock in mice. J Preclin Clin Res 2007;1:39–40. [Google Scholar]
- 36.Silbergleit R, Durkalski V, Lowenstein D, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med 2012;366:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chamberlain JM, Okada P, Holsti M, et al. Lorazepam vs diazepam for pediatric status epilepticus: a randomized clinical trial. JAMA 2014;311:1652–1660. [DOI] [PubMed] [Google Scholar]
- 38.Rice AC, DeLorenzo RJ. NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res 1998;782:240–247. [DOI] [PubMed] [Google Scholar]
- 39.Martin BS, Kapur J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia 2008;49:248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niquet J, Baldwin R, Suchomelova L, et al. Treatment of experimental status epilepticus with synergistic drug combinations. Epilepsia 2017;58:e49–e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szczurowska E, Mares P. An antagonist of calcium permeable AMPA receptors, IEM1460: anticonvulsant action in immature rats? Epilepsy Res 2015;109:106–113. [DOI] [PubMed] [Google Scholar]
- 42.Oh SW, Harris JA, Ng L, et al. A mesoscale connectome of the mouse brain. Nature 2014;508:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laura U, Federica T, Giovanni C, et al. Seizure-like discharges induced by 4-aminopyridine in the olfactory system of the in vitro isolated guinea pig brain. Epilepsia 2013;54:605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Guzman P, D’Antuono M, Avoli M. Initiation of electrographic seizures by neuronal networks in entorhinal and perirhinal cortices in vitro. Neuroscience 2004;123:875–886. [DOI] [PubMed] [Google Scholar]
- 45.Tortorella A, Halonen T, Sahibzada N, Gale K. A crucial role of the AMPA subtype of glutamate receptors in piriform and perirhinal cortex for the initiation and propagation of limbic motor seizures. J Pharmacol Exp Ther 1997;280:1401–1405. [PubMed] [Google Scholar]
- 46.Rulun Z, Andrew H, Jing D, et al. Genome-wide gene expression profiling in GluR1 knockout mice: key role of the calcium signaling pathway in glutamatergically mediated hippocampal transmission. Eur J Neurosci 2009;30:2318–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodkin HP, Joshi S, Mtchedlishvili Z, et al. Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J Neurosci 2008; 28:2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodkin HP, Yeh JL, Kapur J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci 2005;25: 5511–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terunuma M, Xu J, Vithlani M, et al. Deficits in phosphorylation of GABA(A) receptors by intimately associated protein kinase C activity underlie compromised synaptic inhibition during status epilepticus. J Neurosci 2008;28:376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naylor DE, Liu H, Wasterlain CG. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci 2005;25:7724–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joshi S, Rajasekaran K, Hawk KM, et al. Phosphatase inhibition prevents the activity-dependent trafficking of GABAA receptors during status epilepticus in the young animal. Epilepsia 2015;56: 1355–1365. [DOI] [PubMed] [Google Scholar]
- 52.Lu W, Shi Y, Jackson AC, et al. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 2009;62:254–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoffman DA, Sprengel R, Sakmann B. Molecular dissection of hippocampal theta-burst pairing potentiation. Proc Natl Acad Sci U S A 2002;99:7740–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reisel D, Bannerman DM, Schmitt WB, et al. Spatial memory dissociations in mice lacking GluR1. Nat Neurosci 2002;5:868–873. [DOI] [PubMed] [Google Scholar]
- 55.Schmitt WB, Sprengel R, Mack V, et al. Restoration of spatial working memory by genetic rescue of GluR-A-deficient mice. Nat Neurosci 2005;8:270–272. [DOI] [PubMed] [Google Scholar]
- 56.Lu W, Roche KW. Posttranslational regulation of AMPA receptor trafficking and function. Curr Opin Neurobiol 2012;22:470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron 2003;40:361–379. [DOI] [PubMed] [Google Scholar]
- 58.Makino Y, Johnson RC, Yu Y, et al. Enhanced synaptic plasticity in mice with phosphomimetic mutation of the GluA1 AMPA receptor. Proc Natl Acad Sci U S A 2011;108:8450–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He K, Song L, Cummings LW, et al. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc Natl Acad Sci U S A 2009;106:20033–20038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rakhade SN, Zhou C, Aujla PK, et al. Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J Neurosci 2008;28:7979–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]