

Abstract
Acute encephalopathy with biphasic seizure and late reduced diffusion (AESD) is a clinico-radiologic syndrome of acute encephalopathy characterized by biphasic seizure and altered consciousness in acute phase followed by restricted diffusion in bilateral cerebral parenchyma on magnetic resonance imaging (MRI) in the subacute stage. Here, we present the MRI and magnetic resonance spectroscopy (MRS) findings in a case of AESD presenting in 4-year child and diagnosed based on clinico-radiological correlation.
Keywords: Acute encephalopathy, biphasic seizure, diffusion-weighted, magnetic resonance imaging, magnetic resonance spectroscopy
Introduction
An acute encephalopathy syndrome characterized by biphasic seizures and late reduced diffusion (AESD) was initially diagnosed in children of East Asian origin (mainly Japanese) during the influenza epidemic. The etiology of AESD has been attributed to a viral infection such as influenza A and human herpesvirus 6.[1] The exact pathogenesis of AESD is uncertain, however, excitotoxic injury with a delayed (or apoptotic) neuronal damage is hypothesized as a possible mechanism.[1] In this case report, we present a case of AESD in the pediatric age group with MRI and MRS findings in subacute stage and at follow-up.
Case History
A 4-year old boy, first born of non consanguineous marriage presented with generalized tonic-clonic seizure, lasting for 30 min after trivial fall. He was also diagnosed as a case of nephrotic syndrome about 5 months previous to this episode. He was also suffering from high-grade fever and upper respiratory tract infection since 24 h for which he has received the first dose of syrup cephalexin from a general practitioner. CT of the brain was done which was unremarkable. He remained in the postictal drowsy state from which he recovered in few hours.
On day 4, he had a cluster of complex partial seizures mainly involving the right side of the body lasting for few minutes. CT of the brain was again done which was unremarkable. Subsequently, he became drowsy and on examination had brisk reflexes. The other investigations such as complete blood count, urine examination, cerebrospinal fluid (CSF) analysis, liver and renal function tests were normal. Electroencephalography revealed diffuse high voltage slow waves. Fundoscopy revealed temporal pallor. The general condition of the patient deteriorated with Glasgow Coma Scale (GCS) of E2 V2 M2 and therefore, patient was put on a ventilator.
MRI of the brain was done on day 7, which revealed extensive symmetrical areas of restricted diffusion involving bilateral cerebral white matter with relative sparing along Sylvian fissure [Figures 1 and 2]. Also, there was sparing around perirolandic region mainly on the right side with partial involvement on the left side [Figures 1 and 2]. The areas of restricted diffusion showed mild T2 and fluid-attenuated inversion recovery (FLAIR) hyperintense signal with minimal swelling of adjacent gyri [Figures 3 and 4]. There was also the involvement of corpus callosum. The deep nuclei and posterior fossa structures (brain stem and cerebellar white matter) were spared. There was no leptomeningeal enhancement on post-contrast T1-weighted images [Figure 5]. MR spectroscopy from frontal white matter revealed reduced N-acetylaspartate (NAA) and peak at 2.1 to 2.5 and 3.8 PPM suggestive of glutamine/glutamate peak [Figures 6 and 7].
Figure 1.
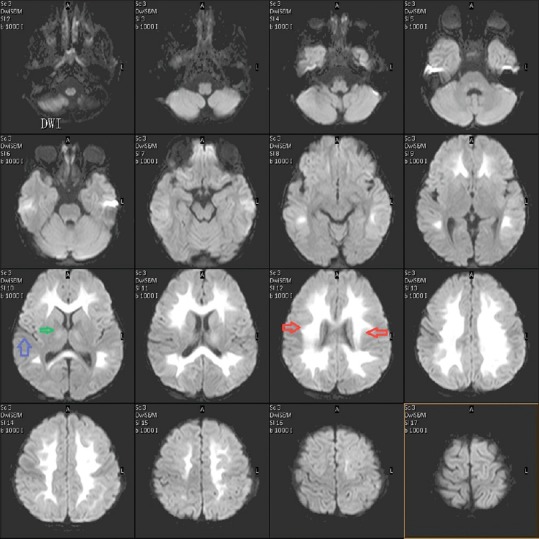
Diffusion-weighted image (DWI) showing restricted diffusion (hyperintense signal) in white matter in bilateral cerebral hemisphere symmetrically (red arrow). There is a sparing of the perisylvian region (blue arrow). A minimal hyperintense signal is also seen in internal capsule (green arrow)
Figure 2.
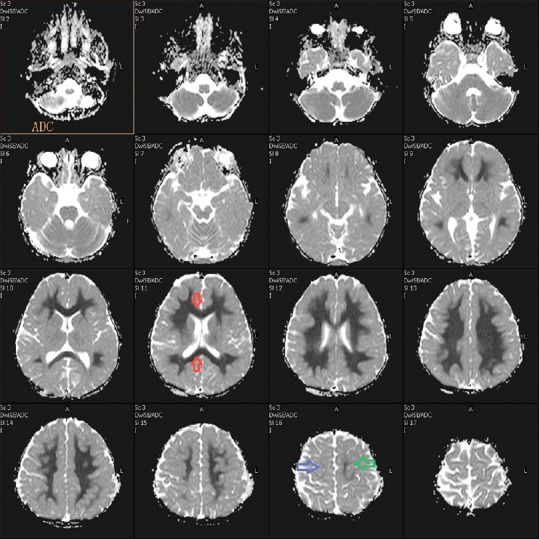
Apparent diffusion coefficient (ADC) image showing hypointense signal (restricted diffusion) in white matter in bilateral cerebral hemisphere symmetrically with the involvement of corpus callosum (red arrow). There is sparing of peri rolandic region on the right side (blue arrow) and minimal involvement on the left side (green arrow)
Figure 3.
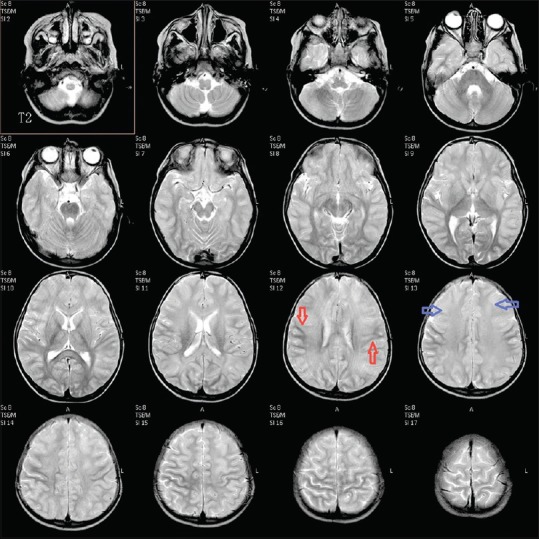
Axial T2-weighted image showing hyperintense signal in bilateral cerebral white matter (blue arrow) with sparing of the central portion (red arrow) showing hypointense signal suggestive of central sparing type
Figure 4.
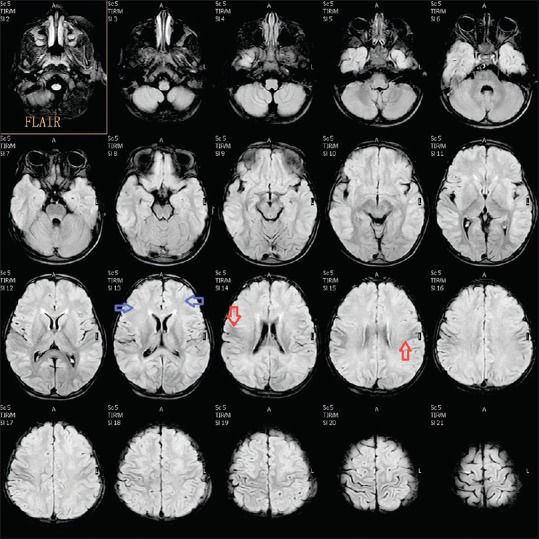
Axial fluid-attenuated inversion recovery (FLAIR) image showing hyperintense signal in bilateral cerebral white matter (blue arrow) with sparing of the central portion (red arrow) showing hypointense signal suggestive of central sparing type
Figure 5.

Axial pre- and post-contrast T1-weighted images showing no abnormal leptomeningeal or parenchymal enhancement
Figure 6.
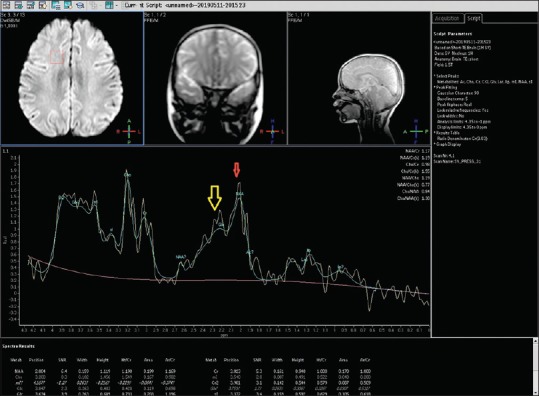
MR spectroscopy from the frontal white matter at low TE (35 ms) showing a reduction in N-acetylaspartate (NAA) peak (red arrow) and prominent glutamine/glutamate peak at 2.1–2.5 PPM (yellow arrow) and at 3.8 PPM
Figure 7.
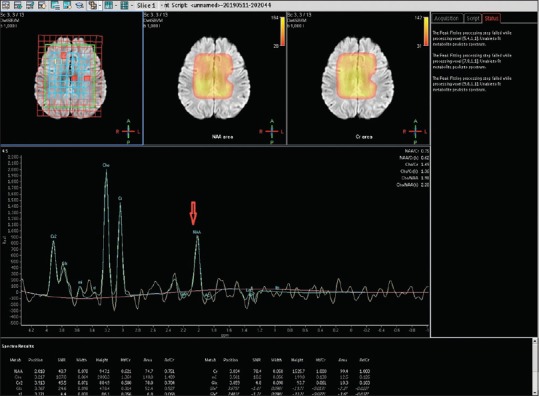
MR spectroscopy from the frontal white matter at intermediate TE (144 ms) showing a reduction in NAA peak (red arrow)
Therefore, the diagnosis of acute encephalopathy with biphasic seizures and late reduced diffusion was made.
The other differentials of metachromatic leukodystrophy and canavans disease which may show a similar pattern of restricted diffusion were ruled out in absence of a history of milestone regression. The possibility of drug-induced toxic leukoencephalopathy was ruled out as cephalexin has not been reported in the literature to be associated with acute leukoencephalopathy. The possibility of hypoxic-ischemic encephalopathy was ruled out as basal ganglia and thalami were spared.
The patient was put on steroid therapy. Although he has not returned to normal mental status, there was a considerable clinical improvement and the patient was off the ventilator on day 10. The GCS on day 12 was E4 V4 M5 and there was residual mild weakness in the right-sided limbs.
Follow-up MRI was done on day 12 which revealed the complete resolution of areas of restricted diffusion in cerebral white matter [Figure 8] with persistent mild T2 and FLAIR hyperintense signal.
Figure 8.

Follow-up DWI and ADC image at 12th day showing resolution of restricted diffusion in cerebral white matter with persistent mild T2 hyperintense signal
Discussion
Acute encephalopathy in association with infectious disease is a well-known entity to pediatricians and pediatric neurologists since the outbreak of influenza-associated encephalopathy during the 1997/1998 winter season in Japan.[2]
Takanashi et al.[1] recently described a spectrum of acute encephalopathy in pediatric age group known as AESD which is diagnosed both by its clinical manifestations and imaging findings. The initial presentation is a prolonged febrile seizure followed by a cluster of subsequent seizures several days later (biphasic seizures). An infective etiology such as influenza A and human herpesvirus 6 has been identified in over half of the cases.[2,3] Few cases of bacterial etiology (Streptococcus pneumoniae associated) have also been reported.[4] CSF examination is mostly unremarkable except few cases showing a mild elevation in CSF tau protein.[5]
Recently okumura[2] described two distinct patterns of brain lesions on the diffusion-weighted image (DWI) in cases of AESD: diffuse lesions and central-sparing lesions. Diffuse lesions are defined as reduced diffusion in the whole cortex and/or subcortical white matter in the bilateral hemisphere during the clinical course, mainly during the subacute stage. In some patients, reduced diffusion in the frontal and occipital areas may precede diffuse lesions. Central-sparing lesions are characterized by lack of reduced diffusion in the areas around the bilateral Sylvian fissures and perirolandic region. In these patients with central sparing lesions, pre- and postcentral areas are clearly spared. In both types of AESD, no restricted diffusion is seen in the basal ganglia and thalami throughout the clinical course. However, T2-weighted images may show signal intensities in the bilateral caudate nuclei in few cases of central-sparing lesions during the subacute period. Patients with central-sparing lesions appear to represent a relatively mild phenotype of acute encephalopathy. Coma is uncommon and laboratory abnormalities in the form of elevated liver enzymes are mild if present. Death is uncommon in central sparing lesions, though various degrees of cognitive impairment are observed as neurologic sequelae. A biphasic clinical course is a characteristic of this group of patients.
Onset is often marked by a prolonged seizure followed by improved consciousness. However, clustered seizures, signs of frontal lobe dysfunction, and worsening of consciousness become apparent at 3–4 days after onset. These features were observed in our patients with central-sparing lesions. It is postulated that pathogenesis of acute encephalopathy with central-sparing lesions may be different from that of acute encephalopathy with diffuse lesions.
Some authors have suggested that this subtype of acute encephalopathy is caused by excitotoxicity[6] because prolonged seizures are often observed at the onset of AESD. MR spectroscopy shows increased glutamine/glutamate concentrations and decreased NAA levels in patients with AESD.[6] Most excitatory neurons in the human cerebral cortex release glutamate (Glu), an excitatory neurotransmitter that is taken up from the synaptic cleft by surrounding astrocytes and is metabolized into a relatively harmless compound, glutamine (Gln). If Glu is released in quantities that cannot be processed by astrocytes or if the astrocytes are not functioning properly, the excessive Glu binding to N-methyl-D-aspartate (NMDA) receptors allows entry of calcium into the postsynaptic neuron, causing necrotic cell death or apoptosis; this condition is referred to as excitotoxicity.[7]
The persistently decreased NAA on follow-up MRI suggests permanent neuronal damage, which is likely related to the neurologic sequelae and the cerebral atrophy observed on follow-up MRI. In contrast, a much smaller decrease in NAA improved to a nearly normal level during follow-up in the patients accompanied by clinical recovery and no persistent abnormality on MRI. Thus, the concentration of NAA on the follow-up MR spectroscopy may be helpful to predict clinical outcome in AESD.[6]
The DWI patterns of our patients were characteristic for AESD, though reduced diffusion in the bilateral hemispheres may be observed with other causes of brain injury, such as hypoxic-ischemic encephalopathy and battered baby syndrome. It is possible that encephalopathy due to substance abuse or intoxication may exhibit similar DWI abnormalities. Thus, the distinction between acute encephalopathy and brain injuries due to other causes may be problematic solely on the basis of imaging findings. However, noninvolvement of deep nuclei favors the possibility of AESD.
Conclusion
Though AESD cases were initially reported from children of Japanese origin, this entity should be kept in mind in the differential diagnosis of acute encephalopathy in pediatric age group especially in the regions where influenza infection has become epidemic. The diagnosis can be established by clinic-radiological correlation and MRI (especially DWI images) with MR spectroscopy done in the subacute stage will help in clinching the diagnosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We are thankful to Mr. Mohan Chandra, Medical Transcriptionist, Rama Medical College in helping to prepare the Manuscript.
References
- 1.Takanashi J, Oba H, Barkovich AJ, Tada H, Tanabe Y, Yamanouchi H, et al. Diffusion MRI abnormalities after prolonged febrile seizures with encephalopathy. Neurology. 2006;66:1304–9. doi: 10.1212/01.wnl.0000210487.36667.a5. [DOI] [PubMed] [Google Scholar]
- 2.Okumura A, Kidokoro H, Tsuji T, Suzuki M, Kubota T, Kato T, et al. Differences of clinical manifestations according to the patterns of brain lesions in acute encephalopathy with reduced diffusion in the bilateral hemispheres. AJNR Am J Neuroradiol. 2009;30:825–30. doi: 10.3174/ajnr.A1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tada H, Takanashi JI, Terada H, Tajima K. Severe form of acute influenza encephalopathy with biphasic seizures and late reduced diffusion. Neuropediatrics. 2008;39:134–6. doi: 10.1055/s-2008-1081459. [DOI] [PubMed] [Google Scholar]
- 4.Kuwata S, Senzaki H, Urushibara Y, Toriyama M, Kobayashi S, Hoshino K, et al. A case of acute encephalopathy with biphasic seizures and late reduced diffusion associated with Streptococcus pneumonia meningoencephalitis. Brain Dev. 2012;34:529–32. doi: 10.1016/j.braindev.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Tanuma N, Miyata R, Kumada S, Kubota M, Takanashi J, Okumura A, et al. The axonal damage marker tau protein in the cerebrospinal fluid is increased in patients with acute encephalopathy with biphasic seizures and late reduced diffusion. Brain Dev. 2010;32:435–9. doi: 10.1016/j.braindev.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Takanashi J, Tada H, Terada H, Barkovich AJ. Excitotoxicity in acute encephalopathy with biphasic seizures and late reduced diffusion. AJNR Am J Neuroradiol. 2009;30:132–5. doi: 10.3174/ajnr.A1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moritani T, Smoker WR, Sato Y, Numaguchi Y, Westesson PL. Diffusion-weighted imaging of acute excitotoxic brain injury. AJNR Am J Neuroradiol. 2005;26:216–28. [PMC free article] [PubMed] [Google Scholar]