

Abstract
Autonomic nervous system control of the heart is a dynamic process in both health and disease. A multilevel neural network is responsible for control of chronotropy, lusitropy, dromotropy, and inotropy. Intrinsic autonomic dysfunction arises from diseases that directly affect the autonomic nerves, such as diabetes mellitus and the syndromes of primary autonomic failure. Extrinsic autonomic dysfunction reflects the changes in autonomic function that are secondarily induced by cardiac or other disease. An array of tests interrogate various aspects of cardiac autonomic control in either resting conditions or with physiological perturbations from resting conditions. The prognostic significance of these assessments have been well established. Clinical usefulness has not been established, and the precise mechanistic link to mortality is less well established. Further efforts are required to develop optimal approaches to delineate cardiac autonomic dysfunction and its adverse effects to develop tools that can be used to guide clinical decision-making.
Keywords: arrhythmia, autonomic, heart failure, myocardial infarction
Central Illustration
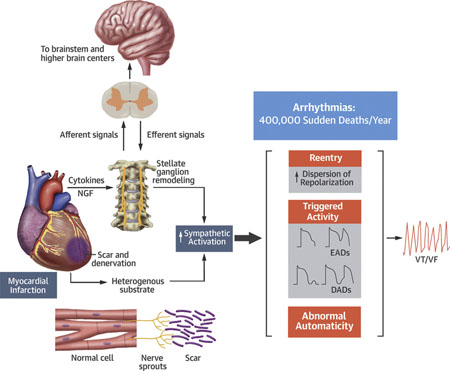
The complex autonomic changes that may occur secondary to acute myocardial infarction. There is regional denervation in the ischemic zones. Nerve sprouting may then appear at the border zone. Nerve growth factors and cytokines may also induce remodeling at the stellate ganglion. Afferent and efferent feedback loops to the brainstem and higher brain centers may modulate the effects of these changes. Stimuli, such as heart failure, will enhance sympathoexcitation and act upon this altered autonomic substrate. These effects may serve to further enhance heart failure or promote arrhythmias that may be responsible for sudden cardiac death. DADs = delayed afterdepolarizations; EADs = early afterdepolarizations; NE = norepinephrine; NGF = nerve growth factor; VT/VF = ventricular tachycardia/ventricular fibrillation.
Autonomic nervous system (ANS) control of the heart is a dynamic process in both health and disease. ANS dysfunction may result from primary disorders of the autonomic nerves or secondarily in response to cardiac (or other systemic) disease. Cardiac disease may promote both anatomic (primary) and functional (secondary) changes in cardiac autonomic function. These changes may, in turn, contribute to the progression of disease and/or be involved in arrhythmogenesis. Beta-adrenergic blockers are the most established autonomic intervention associated with improved outcomes. Other interventions (e.g., cardiac sympathetic denervation) have shown promise for the management of refractory ventricular arrhythmias (1). Much has been learned about the complex interactions along the neuroaxis and their role in cardiac control. What has been most challenging is the development of a simple method of assessment of the autonomic effects on the heart and/or autonomic dysfunction. Consequently, there have been a plethora of methods that assess some aspect of autonomic function. Although many measures have been shown to have some prognostic significance, none have been adapted or adopted into clinical practice. This review highlights some of the background and newer concepts in autonomic control of the heart.
NORMAL AUTONOMIC FUNCTION
A brief description of the advances in our understanding of the physiology of cardiac autonomic control is in order to place autonomic testing and emerging therapies in context. Autonomic control of the heart is achieved by afferent neural impulses that are transmitted from the heart to the intrinsic neurons of the heart, to extracardiac intrathoracic ganglia (e.g., stellate ganglion), to the spinal cord, and to the brain stem. These afferent neural signals are processed by various parts of the nervous system to regulate the cardiomotor neural output to the heart via the sympathetic and parasympathetic nerves. It is important to emphasize that the anatomical nerve trunks that reach the heart, which are traditionally described as the sympathetic and parasympathetic trunks, have both afferent and efferent nerve fibers.
ORGANIZATION OF CARDIAC NEURAL CONTROL.
This neuroaxis is organized as multiple levels of integrative centers. At the level of the heart, the intrinsic cardiac nervous system (ICNS) is a distributed network system located in the cardiac ganglia that are ganglionated plexi (GPs) that exist in the fat pads around the heart, predominantly in the posterior and superior aspects of the atria (2). These connect with the intrathoracic extracardiac ganglia (the sympathetic paravertebral ganglia), the extrathoracic cardiac ganglia (the nodose, dorsal root ganglia), and the central nervous system (3,4). At each level, the system has the ability to modulate cardiac activity with efferent feedback loops (Figure 1).
FIGURE 1. Autonomic Neural Control of the Heart.

Autonomic neural control of the heart. Modified with permission from Shivkumar et al. (4). DRG= dorsal root ganglion; ICNS = intrinsic nervous system; SG = stellate ganglion.
Afferent neural signals are transmitted to the ICNS, the stellate ganglia, and via the dorsal root ganglia to the spinal cord and to the nodose ganglia (Figure 2) (5,6). The efferent projections to the heart occur via short and long feedback loops that occur at the central nervous system, the spinal cord (intermediolateral columns), the intrathoracic ganglia, and the ICNS. Sensory neurites associated with the ICNS are found in areas of the atrioventricular junction and the adventitia of major vessels (e.g., coronary arteries). It was previously believed that the different levels within the ANS were just relay stations, but it is now well-established that each level processes neural information and coordinates a response by communicating with other levels by these feedback loops (Figure 2).
FIGURE 2. Functional Organization of Cardiac Neural Control.

The shaded areas (yellow = sympathetic fibers, grey = parasympathetic fibers [vagal trunk]) roughly correspond to the anatomical sympathetic and parasympathetic nerve fibers that are seen macroscopically. The afferent neural fibers run along with nerves anatomically referred to as sympathetic and parasympathetic trunks. Modified with permission from Shivkumar et al. (4). Aff = afferent; LCN = local circuit neuron; other abbreviation as Figure 1.
The ICNS consists of ganglia composed of afferent, efferent, and interconnecting neurons to other cardiac ganglia. These ganglia coordinate the sympathetic and parasympathetic inputs received from the rest of the cardiac ANS. The ICNS has regional control over different cardiac functions, such as sinus node electrical activation and propagation, as well as atrioventricular nodal conduction (7,8). Emerging therapies that target these structures have shown the importance of emerging research in this area.
Sympathetic afferents and efferents.
The sympathetic neurons that regulate cardiac function are located in the stellate ganglion. It receives pre-ganglionic sympathetic input from the intermediolateral column (and other spinal neurons) and coordinates efferent neural responses either directly or via the ansa subclavia to the middle cervical ganglia to the heart (9–12). Efferent post-ganglionic fibers travel alongside the coronary vasculature to penetrate the epicardial regions and travel toward the endocardium. Stimulation of the stellate ganglion results in an increase in dromotropy, chronotropy, lusitropy, and inotropy (13). Cardiac afferent neurons are mechanosensory, chemosensory, or multimodal in nature (4). They transduce a variety of chemicals, including various neuropeptides, such as substance P, bradykinin, and calcitonin gene–related peptide. These cardiac afferents are also involved in initiating local inflammatory and vascular reactions that may play an important role in cardiac remodeling (14).
The sympathetic nerve fibers are located in the atria and the ventricles. There is a regional response to the right sympathetic paravertebral ganglia versus the left sympathetic paravertebral ganglia, with predominant effects on the anterior and posterior ventricular walls, respectively (15). In addition to increased sinoatrial node firing and enhancement of atrioventricular nodal conduction, sympathetic output results in ventricular action potential duration shortening (16).
Parasympathetic afferents and efferents.
The parasympathetic nervous system also has important afferent and efferent components (Figures 1 and 2). The cardiomotor function of this system helps slow the heart rate, reduce blood pressure, and balances the system to ensure there is a counterbalance to sympathoexcitation. The parasympathetic effects are coordinated via the cervical vagus nerve, which divides into the superior and inferior cardiac nerves to finally enter the heart via the cardiac plexus. The parasympathetic nerve fibers are much more heterogeneously distributed, with significant innervation of the sinoatrial node, atrioventricular node (7), and the ventricles (17).
The parasympathetic cardiomotor response is coordinated by neurons in the ICNS that receive pre-ganglionic parasympathetic input from the cervical vagus and provide a homogeneous or coordinated response to the atria and ventricles (18,19). Cervical vagus stimulation can produce different responses depending on what aspect of the neuroaxis is engaged. In the intact state, stimulation can result in both direct and reactive, or reflex initiated responses on the intrinsic cardiac nervous system as a result of central and peripheral interactions initiated throughout the cardiac neuronal hierarchy. Low-level stimulation delivered to the cervical vagus can result in tachycardia, probably as a result of engaging cardiac afferent fibers, whereas higher level stimulation results in bradycardia (20). In the resting state, the cardiac ANS is an intricate balance between sympathetic and parasympathetic inputs. Once the cervical vagus is transected from the central nervous system, there is a significant increase in heart rate that suggests that the central cholinergic neuronal drive plays a considerable role in controlling the basal heart rate (20).
Integration of cardiac and vascular afferents.
Another important afferent control system includes the baroreceptors, which are stretch receptors embedded in the adventitia of the aortic wall and carotid sinus that transduce pressure fluctuations to the cardiovascular neural reflex pathways responsible for heart rate and blood pressure. There is a continuous strip of mechanosensory neurites along the inner aortic arch (21). They are believed to transduce combined mechanosensory information that measures distortion from pulsatile waves in the aorta in space and time. The arterial chemoreceptors are in the carotid arteries, aortic bodies, and medulla, and respond to hypoxemia and hypercapnia, respectively. The aortic arch mechanoreceptors can be found in the nodose ganglion, whereas some of the carotid sinus afferents are found in the petrosal ganglia. These receptors then transmit information to the nucleus of the tractus solitarius, which modulates sympathetic and parasympathetic output to the cardiovascular system.
Finally, the regulation of the cardiac ANS is also under central nervous system control. The balance of activation and inhibitory neurons is finely tuned and is designed to provide dynamic control of the heart under a range of physiological conditions ranging from rest to severe exertion. This exquisite multilevel neural network is profoundly altered in the presence of cardiac dysfunction and leads to progression of heart disease (22).
AUTONOMIC DYSFUNCTION AND ITS ROLE IN CARDIOVASCULAR DISEASE.
Autonomic dysfunction may arise from 2 mechanisms—intrinsic or extrinsic. Intrinsic autonomic dysfunction arises from diseases that directly affect the autonomic nerves, such as diabetes mellitus and the various syndromes of primary autonomic failure. Extrinsic autonomic dysfunction reflects the changes in autonomic function that are secondarily induced by cardiac or other disease. Cardiac diseases, such as myocardial infarction (MI), can also primarily disrupt the autonomic nerves.
PRIMARY AUTONOMIC DYSFUNCTION.
Primary autonomic dysfunction resulting from diabetic neuropathy.
Diabetes mellitus is associated with the development of both peripheral and autonomic neuropathy. Because of its widespread prevalence, diabetes is the leading cause of primary autonomic dysfunction. Pathological studies have noted loss of and/or damage to myelinated vagus nerve axons (23– 25). Although findings in the sympathetic nerves and ganglia have been disputed (26), they include giant nerve cells, vacuolization, and neuroaxonal dystrophy (24,26,27). Appenzeller and Richardson (27) noted abnormalities in 4 of 5 patients with diabetes with neuropathy and in 0 of 4 patients without neuropathy. These studies showed significant pathological changes in the autonomic nerves of patients with diabetes who predominantly had advanced disease and peripheral neuropathy. The pathological basis of early diabetic autonomic neuropathy has not been elucidated. Numerous biochemical mechanisms for hyperglycemic injury have been implicated, including the production of advanced glycosylation end products, hyperglycemic activation of the polyol pathway, as well as protein kinase C, immunological processes, and neurovascular insufficiency that leads to local ischemia. The proposed mechanisms come largely from research on somatic nerves in experimental diabetic animal models. Although biochemically diverse, the various degenerative mechanisms have a common predisposition for distal axon terminals. The pathogenesis of diabetic cardiac autonomic neuropathy in vivo is likely to be heterogeneous and results from the interaction of multiple pathways (26,28–30). Because the vagus nerve is the longest autonomic nerve in the body, abnormalities in parasympathetic innervation of the heart are typically the earliest manifestation of cardiac autonomic neuropathy (31).
The prevalence of cardiac autonomic neuropathy in patients with diabetes varies widely depending on the cohort studied and the tests and/or criteria used for assessment, but approximately 15% to 20% of asymptomatic people with diabetes appear to have abnormal cardiovascular autonomic function (28,32–34). Only later stages of the disease are associated with symptoms. Autonomic abnormalities may even be detected before the onset of diabetes (35–37). Early evidence of cardiac autonomic neuropathy can be detected in children with type 1 diabetes (38). Many methods are used to diagnose cardiac autonomic neuropathy. Because the heart rate is easily measured and responds to autonomic stimuli (39), noninvasive studies to assess cardiac autonomic neuropathy focus primarily on heart rate or heart rate responses to physiological manipulations. Cardiac autonomic neuropathy has been diagnosed based on abnormalities in heart rate variability (HRV), baroreflex sensitivity (BRS), Ewing’s tests (heart rate and/or blood pressure responses to deep breathing, standing, Valsalva maneuver, and handgrip), and heart rate recovery (HRR) (40–44). These parameters are not highly correlated with each other (45,46), which presents a unique challenge in the diagnostic approach to this entity. Although cardiac autonomic reflex tests (Ewing’s tests) are generally recommended for diagnosis (47), it is unclear which method, if any, is preferred. It is also unclear how often any assessment for cardiac autonomic neuropathy is performed.
The presence of cardiac autonomic neuropathy confers an adverse prognosis (48). In a meta-analysis of 15 studies among subjects with diabetes mellitus (49), the pooled relative risk for mortality related to cardiac autonomic neuropathy was 3.45 (95% confidence interval: 2.66 to 4.47; p < 0.001). The mechanism for the increased mortality has not been clarified, but various possibilities have been proposed, including QT interval prolongation (50–54), renal disease (55–57), and asymptomatic myocardial ischemia (28,56,58). Some of these potential mechanisms may arise directly from the primary disturbance of cardiac autonomic function.
Other primary disorders of autonomic dysfunction.
Other primary disorders of autonomic dysfunction and/or failure include a range of disorders—pure autonomic failure, idiopathic orthostatic hypotension, Parkinson’s disease with autonomic failure, and multiple system atrophy (59). These disorders are characterized by orthostatic hypotension in combination with other manifestations. Postural orthostatic tachycardia syndrome is a complex syndrome with a major autonomic component. An autoimmune etiology that targets ganglionic receptors (nicotinic acetylcholine receptors) may be the underlying etiology for some of these disorders (60). Extensive pathological studies have not been reported, but in autonomic failure, Lewy bodies (abnormal protein aggregates) have been identified in the sympathetic ganglia (61,62). These disorders are discussed in further detail in other studies (63,64).
AUTONOMIC DYSFUNCTION SECONDARY TO MI, CARDIOMYOPATHY, AND HEART FAILURE, AND ITS ROLE IN THE GENESIS OF VENTRICULAR ARRHYTHMIAS.
In contrast to disorders that primarily affect the autonomic nerves, a variety of cardiac pathologies, such as MI, heart failure, and cardiomyopathy result in secondary acute and chronic changes within the ANS (65). Direct ischemic damage to cardiac autonomic nerves may result from acute MI (66). In addition, the cardiac ANS responds acutely to preserve homeostasis. This may involve increased sympathetic and decreased parasympathetic activity to maintain cardiac contractility and cardiac output in the setting of a major insult. Hemodynamic changes that occur as a consequence of these conditions cause a cascade of changes in neurohumoral activity in the cardiovascular, peripheral vascular, and renal vascular systems. The problem arises when the immediate threat has abated, but the imbalance in the ANS, driven by altered afferent signaling, persists and is further modulated by cytokines generated as a response to the disease state (65). This can lead to a vicious cycle, with MI, cardiomyopathy, and heart failure leading to an outpouring of catecholamines (and inflammatory cytokines), and the catecholamines, in turn, lead to worsening of cardiomyopathy. This cycle of adrenergic activation and cardiac dysfunction has been well established in congestive heart failure with reduced ejection fraction. The marked benefit of beta-adrenergic blockers in this entity, which improve both ejection fraction and outcomes, is a testament to the critical role of the secondary changes in the ANS in this disease state and reversibility of congestive heart failure with treatment.
Persistent maladaptations within the ANS as a result of cardiac dysfunction have been linked to atrial and ventricular arrhythmias (4). These changes result in a functional reorganization of the ANS and alterations in neural processing at each level of the cardiac neural hierarchy, which leads to a conflict between the central (central nervous system) and peripheral (intrathoracic cardiac ganglia) control. Cardiac dysfunction can cause short- and long-term changes in the neural networks, which results in exaggerated reflex responses. Central sensitization of neurons can occur secondarily to constant afferent signaling from the site of injury, such as the infarct zone, which can elicit a cellular process that changes the excitability of cell membranes and reduces the inhibitory mechanisms within the nervous system (67).
Morphological changes seen in the stellate ganglion secondarily to ischemic and nonischemic cardiomyopathy include neuronal enlargement (68), as well as alterations in neurochemical expression patterns in the stellate (69). Other changes such as nerve sprouting around the border zones or periphery of an ischemic territory also occur (70). Regional adrenergic overexpression with functional denervation may be seen in the border zone after MI in animal models (71) and humans with ischemic cardiomyopathy (72). Clinically, it is well appreciated that excess adrenergic activity is a negative prognostic indicator post-MI and in congestive cardiac failure. This forms the basis for the success of beta-blocker therapy in these conditions.
There is neuronal enlargement within the stellate ganglion in both the right and left stellate ganglion, regardless of the territory of the MI (73). This is in contrast to the ICNS, where there are regional changes as a result of MI (74). The afferent IC neuronal activity is attenuated in the territory of the MI, whereas those IC neurons in remote regions are preserved. As such, this creates a heterogenous neural response within the myocardium (74). This suggests that the ICNS receives and coordinates the information that is transmitted to the intrathoracic and extrathoracic ganglia. In relation to other ganglionic changes seen as a result of MI, the dorsal root ganglia appear not to change morphologically, but they do have a significant change in neurochemical properties of the neurons present (increased neuronal nitric oxide synthase and calcitonin gene-related peptide) (69). Alterations in neurohumoral control, circulating catecholamines, and the renin-angiotensin-aldosterone system also occur as a result of cardiac injury.
Ventricular arrhythmias that occur in the setting of ischemic and nonischemic cardiomyopathy may be precipitated as a result of sympathetic overactivity. These occur as a result of an excessive sympatho-neurohumoral response and a reduced parasympathetic input that results in increased dispersion of electrical activation and repolarization in the ventricles from the endocardium to epicardium. In recent years, neuromodulation, with the goal of increasing parasympathetic tone and suppressing sympathetic tone, has become an emerging therapeutic strategy for the treatment of ventricular arrhythmias. Emerging therapeutic approaches include left cardiac sympathetic denervation, cervical vagal stimulation, and spinal cord stimulation (75). Removal of sympathetic efferent input to the heart by surgical resection or thoracic epidural anesthesia of the stellate ganglion to T4 can significantly reduce ventricular arrhythmias in the setting of a severe life-threatening ventricular electrical storm. Although animal studies have clearly shown that vagal nerve stimulation can suppress ventricular arrhythmias in heart failure, vagal stimulation has only been used in patients with heart failure in an attempt to improve cardiac remodeling and heart failure (76,75). Similarly, spinal cord stimulation has been shown to be improve symptoms, functional status, and left ventricular function and remodeling in patients with severe symptomatic heart failure (77); however, spinal cord stimulation has not been systematically evaluated for its antiarrhythmic benefits in clinical studies.
Despite the potential success of neuromodulation in pre-clinical or clinical studies in preventing ventricular arrhythmias, it is important to remember that cardiac dysfunction results in adverse adaptions of the afferent and efferent inputs at various levels throughout the cardiac neuroaxis. These adaptions may perpetuate cardiac dysfunction and are generally pro-arrhythmic. Further in-depth mechanistic understanding is required to improve the sophistication of these therapies.
ROLE OF AUTONOMIC DYSFUNCTION IN CREATING A VULNERABLE SUBSTRATE FOR ATRIAL FIBRILLATION.
Atrial fibrillation (AF) is the most common sustained arrhythmia disturbance and is associated with significant morbidity and mortality. The morbidity and mortality associated with AF are especially increased in the setting of congestive heart failure, with up to one-half of all patients with heart failure having concomitant AF (78). Several mechanisms contribute to the electrophysiological and structural substrate for AF, including fibrosis, stretch, oxidative stress, and altered calcium handling characteristics (79). The ANS has been hypothesized to have a likely role in creation of a vulnerable AF substrate (80), with both sympathetic and parasympathetic nerves believed to be involved in the genesis of AF (81,82). Armour et al. (2) demonstrated an intricate pattern of autonomic innervation in the heart with the atria being innervated by at least 5 major atrial fad pads or GPs (80). Direct nerve recordings from the stellate ganglia, vagus nerve, and the cardiac GPs also demonstrated a dynamic interaction between the sympathetic and parasympathetic nervous system in creating AF (83–86). Hou et al. (87,88) suggested the presence of an intricate, interconnecting neural network in the left atrium that could contribute to a substrate for AF.
Over the last several years, the pulmonary veins (PVs) and adjoining posterior left atrium have been shown to play a significant role in the genesis of AF, with this region demonstrating unique structural, molecular, and electrophysiological characteristics that appear to contribute to the AF substrate. Anatomical and physiological studies of the autonomic innervation of the atria indicate that the pulmonary veins and posterior left atria have a unique autonomic profile (86,89–91). Chevalier et al. (92) described several gradients of innervation in and around the PVs (92). In a canine study, Arora et al. (93) showed that the posterior left atrium was more richly innervated than the rest of the left atrium, with nerve bundles containing both parasympathetic and sympathetic fibers, with parasympathetic fibers predominating over sympathetic fibers. These canine studies were in agreement with human studies that demonstrated co-localization of sympathetic and parasympathetic nerve fibers in the human left atrium (80,94). Deneke et al. (95) also demonstrated co-localization of sympathetic and parasympathetic nerves, with patients in persistent AF that demonstrated a shift toward a lower density of cholinergic nerves and a higher density of adrenergic nerves. There was evidence of sympathetic hyperinnervation in patients with persistent AF (96).
Using direct nerve recordings from the stellate ganglia and the vagus nerve in a canine study, Ogawa et al. (86) showed increased sympathetic and vagal nerve discharge before the onset of atrial arrhythmias in pacing-induced heart failure [these atrial arrhythmias were preventable by prophylactic ablation of the stellate ganglion and the T2 to T4 thoracic sympathetic ganglia (85)]. In a canine HF model, Ng et al. (78) demonstrated a profound increase in parasympathetic—and to a lesser extent sympathetic—nerves in the left atrium (78), with nerve growth being most pronounced in the PVs and posterior left atrium. The increase in vagal innervation noted in this model was believed to contribute to the AF substrate by affecting conduction characteristics of the PVs and posterior left atrium.
Taken together, the previous studies indicated an important role for the ANS in the genesis of AF not only in normal hearts, but also in structural heart disease. These data underscore the potential importance of the ANS as a suitable therapeutic target in AF. A variety of strategies targeting ≥1 GPs either surgically (97,98) or through a transvenous, endocardial approach (alone or together with PV isolation) have been used in patients with both paroxysmal and persistent AF with variable success (99–101). Scanavacca et al. (102) demonstrated the feasibility of selective atrial vagal denervation, as guided by evoked vagal reflexes, to treat patients with paroxysmal AF. Pokushalov et al. (103) reported that regional ablation at the anatomic sites of the left atrial GP could be safely performed and enabled maintenance of sinus rhythm in 71% of patients with paroxysmal AF. Karitsis et al (104) and others (105) demonstrated that when GP ablation was combined with PV isolation, it yielded better results than PV isolation alone, with success rates approaching 80% (104). Recent surgical studies also attempted to add GP ablation and/or excision to PV isolation with varying efficacy (97,98,106,107). The efficacy and durability of this approach has not been established.
Renal denervation, which is known to lead to a reduction of renal norepinephrine spillover (108) and a reduction in firing of single sympathetic vasoconstrictor fibers (a measure of central sympathetic nerve outflow) (109), has been shown to reduce atrial sympathetic nerve sprouting, structural alterations, and AF complexity in goats with persistent AF, independent of changes in blood pressure (110). Early stage clinical data suggests that renal denervation may improve the results of pulmonary vein isolation in patients with persistent AF and/or severe resistant hypertension (111). However, the Symplicity HTN-3 trial did not demonstrate efficacy of renal denervation in reducing blood pressure in patients with resistant hypertension (112). Its role as a potential therapeutic tool in AF has not been established.
Recent approaches attempted to disrupt autonomic signaling by using novel pharmacological and biological methods. Pokushalov et al. (113,114) reported that injection of botulinum toxin in the GPs at the time of open-heart surgery led to a decrease in the incidence of post-operative AF, as well as a decrease of AF burden at 1 year of follow-up. The clinical usefulness of this approach was not established. In animal studies, Aistrup et al. (115) showed that parasympathetic signaling in the atrium could be selectively disrupted by using G-protein inhibitory peptides targeting the C-terminus of the Gαi/o subunits, with the peptides injected either directly (115) or as plasmid expression vectors to obtain constitutive administration of Gαi2ctp and Gαo1ctp (116). In ongoing preclinical studies, the same group of investigators is actively exploring the efficacy and duration of expression of genes targeting the ANS in canine models of chronic AF.
ASSESSMENT OF CARDIAC AUTONOMIC FUNCTION
Assessment of normal and abnormal autonomic function is challenging because of the proliferation of techniques to assess cardiac autonomic function. Furthermore, most of the techniques currently available to assess autonomic function—whether direct nerve recordings or indirect assessments of autonomic activity by HRV, heart rate turbulence (HRT), and so on—are difficult to use and reliably interpret in day-to-day clinical practice. Nonetheless, a brief review is presented to provide a historical perspective on how autonomic testing in cardiovascular disease has evolved over the years. This review largely focuses on the most widely used heart rate–based tests (HRV, HRT, BRS, HRR, and QT-RR slope), with a secondary emphasis on direct nerve recording techniques. Less widely used imaging-based techniques (sympathetic imaging with meta-iodobenzylguanidine, C-meta-hydroxyephedrine) will not be discussed here. There are many heart rate–based tests, but the core of these involve evaluation of heart rate or heart rate changes. It is critical to note that these heart rate evaluations largely rely on how autonomic input affects the sinus node. However, regional autonomic effects on the sinus node, atrium, atrioventricular node, and ventricles differ in normal subjects and may be even more accentuated in the presence of cardiac disease with regional abnormalities in innervation and ganglionic function. How closely abnormalities in sinus node autonomic function parallel abnormalities in other areas of the heart may depend on regional differences in innervation, interruption of feedback loops, and the underlying disease state. This conundrum extends to the prognostic significance of the heart rate-based measures. For example, although sympathoexcitation has been considered to be an important component in the pathogenesis of life-threatening ventricular arrhythmias, a meta-analysis of predictors of sudden cardiac death and arrhythmic events in patients with nonischemic dilated cardiomyopathy noted that HRV, BRS, or HRT, which are all tests that focus on autonomic effects on the sinus node, were not significant predictors of ventricular arrhythmic events (117).
Heart rate can be evaluated under resting and steady state, conditions in which parasympathetic effects normally predominate. This manifests as respiratory sinus arrhythmia, the “high-frequency” oscillations of the heart rate that are noted at the respiratory frequency due to inspiratory suppression of vagal nerve discharges. Next, the responsiveness of the sinus node to small perturbations from the steady state and resting hemodynamic status can be assessed. Tests such as BRS and HRT evaluate the heart rate response to an acute increase in blood pressure and premature ventricular beats, respectively. Finally, larger perturbations in autonomic activity can be provoked with exercise. Both the acceleration of heart rate with exercise and the HRR after cessation of exercise have been studied. The spectrum of evaluation from steady state to mild perturbations from steady state to the large changes noted with exercise provides different assessments of autonomic function, likely related to the different multilevel feedback loops that may be engaged in each condition.
HEART RATE VARIABILITY.
HRV represents a measure of the oscillation in the intervals between consecutive heart beats. The most common methods for measuring the variation in heart rate can be broadly categorized into either time- or frequency-domain analyses. Various nonlinear analyses have also been proposed. Time-domain measures of HRV, such as the SD of normal RR intervals, the root mean square of successive RR interval differences, and the percentage of normal RR intervals that differ by >50 ms, are calculated based on statistical and/or mathematical operations on RR intervals (118). Frequency-domain measures use spectral analysis of a sequence of RR intervals and provide information on how power (variance) is distributed as a function of frequency (39), using either short- (2 to 5 min) or long-term (24 h) recordings.
Respiratory sinus arrhythmia is a reflection of the oscillatory parasympathetic effects on the sinus node related to the respiratory cycle (119,120), which yields the high-frequency component of HRV. The low-frequency and very–low-frequency components of HRV have a more complex physiology that integrates both sympathetic and parasympathetic activities. More specifically, the absolute and normalized low-frequency powers are influenced by sympathetic modulation of the heart rate (121). Upright tilt-table testing increases sympathetic tone (39); the low frequency and the low-frequency/high-frequency ratio (122) increase with upright tilt, and beta-blockade blunts these changes. Although the low-frequency/high-frequency ratio is often considered an index of sympathovagal balance (123,124), the RR interval may actually be a more precise index of the net effect of sympathetic and parasympathetic effects on the sinus node (125) than the time- or frequency-domain HRV parameters. Fluctuations in the renin-angiotensin-aldosterone system and variations in thermoregulatory mechanisms have been speculated to underlie the very–low-frequency component. In long-term recordings, the physiological basis for ultralow frequency oscillations has not been well defined (126).
Nonlinear analysis techniques have been applied to further characterize HRV (127,128). A variety of analysis techniques and parameters have been used to measure nonlinear properties of HRV. Analysis of 1/f characteristics (i.e., the inverse power–law slope), which describes the slope of the spectral powers in the ultra-low and very–low-frequency areas has provided prognostic information beyond the traditional HRV measures (129). None of these techniques have become widely used. Importantly, autonomic blockade markedly attenuates all HRV, regardless of the measure, providing further evidence of the complexity of the various different approaches to assess autonomic effects on the sinus node.
Diminished HRV has been associated with both sudden cardiac death and nonsudden death in MI and in chronic left ventricular dysfunction, independent of the left ventricular ejection fraction (130–132). Large epidemiological studies have also demonstrated the prognostic significance of diminished HRV in the general population (133–135). At present, however, measures of HRV by themselves do not provide adequate refinement of risk of sudden cardiac death due to ventricular tachyarrhythmias. Furthermore, the pathophysiological link between reduced HRV and increased mortality is unclear. Although HRV provides a measure of autonomic modulation, it has not entered the realm of clinical evaluation even after many decades of study.
BAROREFLEX SENSITIVITY.
BRS refers to the reflex bradycardia that accompanies a transient increase in systemic blood pressure, and, as such, reflects ventricular fibrillation. intrinsic properties of arterial baroreceptors, but is also attenuated by afferent cardiac sympathetic stimulation (136). BRS is typically measured after a change in blood pressure has been provoked. This is classically done by acute administration of phenylephrine (137). From the simultaneous record of RR intervals and blood pressure, BRS is calculated as the slope of the regression line (to assess the dependency of RR intervals on systolic blood pressure values). As a result, the greater the slope of the regression line, the stronger the baroreflex.
BRS can also be determined by using neck chamber devices that help activate and/or deactivate carotid baroreceptors by applying positive pressure or suction to the neck (121). An increase in the pressure around the neck is perceived by the baroreceptors as a decrease in the arterial pressure, whereas neck suction stimulates an increase in blood pressure. Neck suction is typically better tolerated and is therefore more commonly used. Using this method, BRS is typically quantified by the maximum RR interval lengthening taken from repeated applications.
Spontaneous fluctuations in arterial pressure and RR interval may also be assessed to determine BRS. This is most often done by assessing the coherence of the power spectra of simultaneously recorded RR and blood pressure modulations. The coherence between the power spectra is usually estimated in the low-frequency band and less often in the high-frequency band (121).
Animal studies suggest that diminished BRS after MI denotes an increased risk of ventricular fibrillation (138). Depressed BRS assessed by the phenylephrine method was found to be a significant predictor of 5-year mortality in survivors of acute MI with preserved ventricular function (139). However, data in humans after MI are more conflicting (140,141). In the Marburg Cardiomyopathy Study, BRS did not appear to be helpful for arrhythmia risk stratification for patients with idiopathic cardiomyopathy (142).
HEART RATE TURBULENCE.
In 1999, Schmidt et al. described HRT that was manifested by short-term heart rate changes induced by a premature ventricular beat (143). In healthy subjects, a ventricular premature beat provokes an early acceleration followed by a late deceleration of heart rate, whereas in subjects with cardiac dysfunction, such a reaction is diminished or even completely nonexistent. Based on data from experimental and clinical studies, HRT is most likely mediated via the baroreceptor reflex; however, other mechanisms such as post-extrasystolic potentiation have been proposed (144–146). A premature ventricular ectopic beat results in a transient drop in blood pressure that results in baroreceptor activation and immediate vagal inhibition, and therefore, an increase in heart rate. Augmented myocardial contractility following a ventricular premature beat and the subsequent increase in blood pressure lead to an opposite reaction with a subsequent decrease in sinus node activity; thus, the biphasic HRT curve of acceleration and deceleration is created (144).
Several large population studies have shown that decreased (or abnormal) HRT identifies patients at high risk of mortality, including sudden death (147). HRT is represented by 2 numeric descriptors (144): turbulence onset, which reflects the initial acceleration of heart rate after a ventricular premature beat; and turbulence slope, which describes subsequent deceleration of heart rate following a ventricular premature beat. La Rovere et al. (148) studied the relationship between HRT and BRS in 157 heart failure patients in whom Holter-derived HRT and phenylephrine-induced BRS were evaluated. Both turbulence onset and turbulence slope significantly correlated with phenylephrine-derived BRS. These findings strongly support the concept that HRT is mediated by the baroreflex response. HRT was evaluated in 577 survivors of acute MI in the Multicenter Postinfarction Program and in 614 post-MI patients randomized to the placebo arm in the EMIAT (European Myocardial Infarction Amiodarone Trial). Diminished HRT was a significant predictor of all-cause mortality (143). In contrast, HRT was not associated with a significant reduction in all-cause mortality in the MADIT-II (Multicenter Automatic Defibrillator Implantation Trial II) (149).
HEART RATE RECOVERY.
During exercise, there is a well-described increase in sympathetic activity and withdrawal of parasympathetic activity (150,151). During recovery from exercise, there is an initially rapid and then gradual return of heart rate to its previous resting level. The dynamic range of changes in sympathetic and parasympathetic activity related to exercise are greater than those assessed with HRV, BRS, or HRT. HRR after exercise reflects the sympathetic withdrawal and parasympathetic reactivation that occurs. Although some earlier studies (152) suggested that sympathetic withdrawal is the major autonomic limb contributing to HRR soon after peak exercise cessation (with parasympathetic activation contributing later in recovery), more recent studies indicated that parasympathetic reactivation was the key factor in early HRR (153). Kannankeril et al. (154) studied heart rate and HRR in healthy individuals during peak exercise and recovery under normal physiological conditions, as well as during selective parasympathetic blockade with atropine. They noted that even during peak exercise, parasympathetic withdrawal was not complete. In recovery, parasympathetic effects on heart rate appeared rapidly within the first minute, increased steadily until 4 min into recovery, and then plateaued.
Another important factor in the analysis of HRR is the parameter to use: heart rate or the RR interval. These variables are inversely related and cannot be used interchangeably. In an analysis of heart rate and the RR interval after submaximal exercise in 33 healthy subjects at baseline conditions and during selective beta-adrenergic blockade and/or parasympathetic blockade (155), it was shown that the heart rate changes provided more physiological information and should therefore be the preferred variable.
Abnormal HRR is also associated with an adverse prognosis. Jouven et al. (156) reported a relative risk of 2.2 for sudden cardiac death in those with 1-min HRR <25 beats/min versus those with HRR >40 beats/min. Cole et al. (157) demonstrated a 4-fold risk of death in those with abnormal HRR after peak exercise in a cohort of 2,428 subjects without a history of heart failure, coronary revascularization, coronary angiography, or exercise testing. After adjustment for age, sex, medications, perfusion defects on thallium scintigraphy, standard cardiac risk factors, resting heart rate, change in heart rate with exercise, and workload achieved, there was still a 2-fold risk of death in those with an abnormal HRR.
QT-RR SLOPE.
Ventricular repolarization can be assessed on the electrocardiogram (the QT interval) and is subject to autonomic effects. Because the QT interval is also strongly influenced by heart rate, determining the independent autonomic effects on the QT interval is challenging. A variety of approaches have been proposed. Although rate correction formulas are widely used to adjust the QT interval for the underlying heart rate, these formulas are inadequate to assess autonomic effects on the QT interval. One approach, assessing the QT-RR slope, has been shown to provide reliable and reproducible delineation of cardiac repolarization (158,159). The autonomic effects on the QT-RR relationship in the first 5 min of recovery has been defined (160) with selective autonomic blockade. Figure 3 provides a diagram of the autonomic effects on the QT-RR relationship, showing a strong parasympathetic effect because atropine steepens the slope dramatically, and a smaller beta-adrenergic effect as propranolol blunts the slope. Thus, there are characteristic autonomic effects on the QT-RR slope.
FIGURE 3. Diagram of the QT-RR Relationship.

The QT-RR relationship in the early post-exercise recovery period without autonomic blockade (baseline [orange]), with beta-blockade (green), with parasympathetic blockade (atropine [grey]), and double blockade with propranolol and atropine (blue). Original data from Sundaram et al. (160).
DIRECT NERVE RECORDINGS.
Sympathetic microneurography.
Although heart rate–based tests are simple noninvasive tools, they do not directly measure autonomic activity. The development of microneurography, in which nerve activity can be recorded directly from intraneural microelectrodes inserted percutaneously in a peripheral nerve in awake patients, has provided a wealth of information on the control of sympathetic outflow to muscle and skin. Recordings of muscle sympathetic nerve activity and skin sympathetic nerve activity in different disease states have increased our understanding of sympathetic function, both in physiological and pathological settings. Several studies have suggested that muscle sympathetic nerve activity is a reliable marker of sympathetic response in some internal organs (161). Compared with healthy individuals, muscle sympathetic nerve activity is altered in the setting of orthostatic hypotension and syncope, in neurological disorders such as Parkinson’s disease, multiple system atrophy, familial dysautonomia, Guillain-Barre syndrome, and in cardiovascular disease states such as hypertension and heart failure (162). Increased muscle sympathetic nerve activity in patients with heart failure is associated with reduced exercise capacity (163) and also helps predict mortality in this patient population (164). Importantly, interventions, such as exercise training, appear to significantly decrease sympathetic activity, as assessed by muscle sympathetic nerve activity, as well as by noninvasive parameters such as HRV and HRR (165).
Direct recordings from cardiac autonomic nerves.
Recently, direct recordings of autonomic nerve activity have been made from the stellate ganglia, vagus nerve, and from the cardiac GPs. These studies, in large animal models of AF, suggest that sympathetic and parasympathetic nerve discharges frequently precede the onset of atrial arrhythmias, both in the setting of heart failure and in a model of rapid atrial pacing-induced AF (166). Recent data suggest that it may be possible to record autonomic nerve activity by electrodes implanted in the subcutaneous space of the left thorax, and that this subcutaneous nerve activity correlates with the stellage ganglia nerve activity (167). Of great interest is the possibility of making these types of recordings from skin sympathetic activity in humans (168). Future studies are needed to assess the applicability of these techniques in humans, to better understand the role of the ANS in the genesis of atrial and ventricular arrhythmias.
SUMMARY
The link between ANS dysfunction and various cardiovascular disease states has been clearly established (Table 1). The role of exercise and other conditions associated with sympathoexcitation as a major precipitant for MI (169,170) and ventricular arrhythmias and/or sudden cardiac death (171,172), and the proven usefulness of beta-adrenergic blockers to improve survival after MI and in heart failure with reduced ejection fraction highlight the prominent role autonomics play in clinical heart disease (Central Illustration).
TABLE 1.
Common Cardiovascular Conditions Linked to the Autonomic Nervous System
| Conditions | Link | Strength of Evidence |
|---|---|---|
| Myocardial infarction | Imaging: sympathetic denervation Beta-blockers improve survival |
++++ |
| Congestive heart failure | Imaging: diminished NE reuptake Elevated plasma catecholamines Downregulation of beta-adrenergic receptors Beta-blockers improve survival |
++++ |
| Hypertension | Sympathetic activation | +++ |
| Atrial fibrillation | Experimental models: changes in innervation; induction of AF by vagal stimulation, vagal stimulation plus sympathetic activation Clinical precipitants: vagal AF, sympathetic AF |
+++ |
| Long QT syndrome | Beta-blockers prevent events | +++ |
| Neurocardiogenic syncope | Bezold-Jarisch reflex | ++++ |
| Postural orthostatic tachycardia syndrome | Sympathetic activation | + |
| Diabetic cardiac autonomic neuropathy | Abnormalities in autonomic function tests involving parasympathetic and sympathetic reflexes | ++++ |
AF = atrial fibrillation.
The appreciation of the importance of autonomics led to the development of many noninvasive diagnostics that interrogate the ANS, but have limited to no clinical applicability at this time (Table 2). This may be due to the fact that the tests are focused on autonomic effects on the sinus node, whereas the adverse cardiac effects are generated by autonomic effects on the ventricle. In addition, simple tests of autonomic reflexes may not adequately interrogate the complex interplay of a multilevel system with multiple levels of feedback and excitatory and inhibitory control. Because of the prominent role the ANS plays in cardiac disease, other diagnostic approaches are needed. Potentially useful approaches include direct nerve recordings and cardiac sympathetic imaging.
TABLE 2.
Noninvasive Tests of Cardiac Autonomic Function
| Test | Physiological Information | Clinical Usefulness |
|---|---|---|
| Heart rate | Net autonomic effect on the sinus node | ++ |
| Heart rate variability | Autonomic modulation of sinus node | — |
| Heart rate recovery | Parasympathetic reactivation after cessation of exercise | — |
| Baroreflex sensitivity | Sinus node response to baroreceptor activation | — |
| Heart rate turbulence | Sinus node response to hemodynamic perturbation by a PVC | — |
| Autonomic reflex testing (Ewing’s maneuvers) | Sinus node response to breathing maneuvers, Valsalva, tilt, handgrip | — |
| Sympathetic nerve recordings | Quantify regional sympathetic output | — |
| Plasma/urinary catecholamines/turnover rates | Total body spillover to blood/urine | +++ (pheochromocytoma) |
| Cardiac sympathetic imaging | Sympathetic nerve distribution and function | — |
PVC = premature ventricular complex.
Therapeutically, the key intervention that has demonstrated benefit is beta-blocker therapy. Only limited data are available for other medical therapies, such as scopolamine, which can improve HRV and BRS (173) but may not affect survival (174). Other interventions that have been tested include autonomic nerve stimulation and autonomic nerve disruption, with mixed results. It is possible that further efforts to develop optimal approaches to interrogate this complex system will enable targeted stimulation and disruption interventions that can be more specifically targeted to abnormalities that can be ameliorated with these approaches.
Acknowledgments
Dr. Goldeberger is supported by the National Heart, Lung, and Blood Institute (NHLBI) (HL70179). Dr. Arora is supported by the NHLBI (HL093490). Dr. Shivkumar is supported by the NHLBI (HL084261), by NHLBI grant R01HL084261, and by National Institutes of Health grant NIHOT2OD023848. Dr. Arora holds ownership interest in Rhythm Therapeutics, Inc.
ABBREVIATIONS AND ACRONYMS
- AF
atrial fibrillation
- ANS
autonomic nervous system
- BRS
baroreflex sensitivity
- GP
ganglionated plexus
- HRR
heart rate recovery
- HRT
heart rate turbulence
- HRV
heart rate variability
- ICNS
intrinsic cardiac nervous system
- MI
myocardial infarction
- PV
pulmonary vein
Footnotes
All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
REFERENCES
- 1.Vaseghi M, Barwad P, Malavassi Corrales FJ, et al. Cardiac sympathetic denervation for refractory ventricular arrhythmias. J Am Coll Cardiol 2017;69:3070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armour JA, Murphy DA, Yuan BX, Macdonald S, Hopkins DA. Gross and microscopic anatomy of the human intrinsic cardiac nervous system. Anat Rec 1997;247:289–98. [DOI] [PubMed] [Google Scholar]
- 3.Buckley U, Shivkumar K, Ardell JL. Autonomic regulation therapy in heart failure. Curr Heart Fail Rep 2015;12:284–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shivkumar K, Ajijola OA, Anand I, et al. Clinical neurocardiology defining the value of neuroscience-based cardiovascular therapeutics. J Physiol 2016;594:3911–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armour JA. Cardiac neuronal hierarchy in health and disease. Am J Physiol Regul Integr Comp Physiol 2004;287:R262–71. [DOI] [PubMed] [Google Scholar]
- 6.Hoover DB, Shepherd AV, Southerland EM, Armour JA, Ardell JL. Neurochemical diversity of afferent neurons that transduce sensory signals from dog ventricular myocardium. Auton Neurosci 2008;141:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Randall DC, Brown DR, McGuirt AS, Thompson GW, Armour JA, Ardell JL. Interactions within the intrinsic cardiac nervous system contribute to chronotropic regulation. Am J Physiol Regul Integr Comp Physiol 2003;285:R1066–75. [DOI] [PubMed] [Google Scholar]
- 8.Csepe TA, Zhao J, Hansen BJ, et al. Human sinoatrial node structure: 3D microanatomy of sinoatrial conduction pathways. Prog Biophys Mol Biol 2016;120:164–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irie T, Yamakawa K, Hamon D, Nakamura K, Shivkumar K, Vaseghi M. Cardiac sympathetic innervation via middle cervical and stellate ganglia and antiarrhythmic mechanism of bilateral stellectomy. Am J Physiol Heart Circ Physiol 2017;312: H392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buckley U, Yamakawa K, Takamiya T, Andrew Armour J, Shivkumar K, Ardell JL. Targeted stellate decentralization: implications for sympathetic control of ventricular electrophysiology. Heart Rhythm 2016;13:282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellison JP, Williams TH. Sympathetic nerve pathways to the human heart, and their variations. Am J Anat 1969;124:149–62. [DOI] [PubMed] [Google Scholar]
- 12.Mizeres NJ. The cardiac plexus in man. Am J Anat 1963;112:141–51. [Google Scholar]
- 13.Ajijola OA, Vaseghi M, Zhou W, et al. Functional differences between junctional and extra-junctional adrenergic receptor activation in mammalian ventricle. Am J Physiol Heart Circ Physiol 2013;304:H579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang HJ, Wang W, Cornish KG, Rozanski GJ, Zucker IH. Cardiac sympathetic afferent denervation attenuates cardiac remodeling and improves cardiovascular dysfunction in rats with heart failure. Hypertension 2014;64:745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaseghi M, Zhou W, Shi J, et al. Sympathetic innervation of the anterior left ventricular wall by the right and left stellate ganglia. Heart Rhythm 2012;9:1303–9. [DOI] [PubMed] [Google Scholar]
- 16.Vaseghi M, Yamakawa K, Sinha A, et al. Modulation of regional dispersion of repolarization and T-peak to T-end interval by the right and left stellate ganglia. Am J Physiol Heart Circ Physiol 2013;305:H1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ulphani JS, Cain JH, Inderyas F, et al. Quantitative analysis of parasympathetic innervation of the porcine heart. Heart Rhythm 2010;7:1113–9. [DOI] [PubMed] [Google Scholar]
- 18.Yamakawa K, Rajendran PS, Takamiya T, et al. Vagal nerve stimulation activates vagal afferent fibers that reduce cardiac efferent parasympathetic effects. Am J Physiol Heart Circ Physiol 2015;309:H1579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamakawa K, So EL, Rajendran PS, et al. Electrophysiological effects of right and left vagal nerve stimulation on the ventricular myocardium. Am J Physiol Heart Circ Physiol 2014;307:H722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ardell JL, Rajendran PS, Nier HA, KenKnight BH, Armour JA. Central-peripheral neural network interactions evoked by vagus nerve stimulation: functional consequences on control of cardiac function. Am J Physiol Heart Circ Physiol 2015;309:H1740–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kember GC, Armour JA, Zamir M. Mechanism of smart baroreception in the aortic arch. Phys Rev E Stat Nonlin Soft Matter Phys 2006;74:031914. [DOI] [PubMed] [Google Scholar]
- 22.Kember G, Ardell JL, Shivkumar K, Armour JA. Recurrent myocardial infarction: mechanisms of free-floating adaptation and autonomic derangement in networked cardiac neural control. PLoS One 2017;12:e0180194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo YP, McLeod JG, Baverstock J. Pathological changes in the vagus nerve in diabetes and chronic alcoholism. J Neurol Neurosurg Psychiatry 1987; 50:1449–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duchen LW, Anjorin A, Watkins PJ, Mackay JD. Pathology of autonomic neuropathy in diabetes mellitus. Ann Intern Med 1980;92:301–3. [DOI] [PubMed] [Google Scholar]
- 25.Kristensson K, Nordborg C, Olsson Y, Sourander P. Changes in the vagus nerve in diabetes mellitus. Acta Pathol Microbiol Scand A 1971;79:684–5. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt RE. Neuropathology and pathogenesis of diabetic autonomic neuropathy. Int Rev Neurobiol 2002;50:257–92. [DOI] [PubMed] [Google Scholar]
- 27.Appenzeller O, Richardson EP Jr. The sympathetic chain in patients with diabetic and alcoholic polyneuropathy. Neurology 1966;16:1205–9. [DOI] [PubMed] [Google Scholar]
- 28.Vinik AI, Maser RE, Mitchell BD, Freeman R. Diabetic autonomic neuropathy. Diabetes Care 2003;26:1553–79. [DOI] [PubMed] [Google Scholar]
- 29.Cameron NE, Cotter MA. Metabolic and vascular factors in the pathogenesis of diabetic neuropathy. Diabetes 1997;46 Suppl 2:S31–7. [DOI] [PubMed] [Google Scholar]
- 30.Schonauer M, Thomas A, Morbach S, Niebauer J, Schonauer U, Thiele H. Cardiac autonomic diabetic neuropathy. Diab Vasc Dis Res 2008;5:336–44. [DOI] [PubMed] [Google Scholar]
- 31.Pop-Busui R Cardiac autonomic neuropathy in diabetes: a clinical perspective. Diabetes Care 2010;33:434–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Brien IA, O’Hare JP, Lewin IG, Corrall RJ. The prevalence of autonomic neuropathy in insulindependent diabetes mellitus: a controlled study based on heart rate variability. Q J Med 1986;61: 957–67. [PubMed] [Google Scholar]
- 33.Neil HA, Thompson AV, John S, McCarthy ST, Mann JI. Diabetic autonomic neuropathy: the prevalence of impaired heart rate variability in a geographically defined population. Diabet Med 1989;6:20–4. [DOI] [PubMed] [Google Scholar]
- 34.Ziegler D, Gries FA, Spuler M, Lessmann F. The epidemiology of diabetic neuropathy. Diabetic Cardiovascular Autonomic Neuropathy Multicenter Study Group. J Diab Comp 1992;6:49–57. [DOI] [PubMed] [Google Scholar]
- 35.Carnethon MR, Prineas RJ, Temprosa M, Zhang ZM, Uwaifo G, Molitch ME. The association among autonomic nervous system function, incident diabetes, and intervention arm in the Diabetes Prevention Program. Diabetes Care 2006; 29:914–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carnethon MR, Jacobs DR Jr., Sidney S, Liu K. Influence of autonomic nervous system dysfunction on the development of type 2 diabetes: the CARDIA study. Diabetes Care 2003;26:3035–41. [DOI] [PubMed] [Google Scholar]
- 37.Carnethon MR, Golden SH, Folsom AR, Haskell W, Liao D. Prospective investigation of autonomic nervous system function and the development of type 2 diabetes: the Atherosclerosis Risk In Communities study, 1987–1998. Circulation 2003;107:2190–5. [DOI] [PubMed] [Google Scholar]
- 38.Lucini D, Zuccotti G, Malacarne M, et al. Early progression of the autonomic dysfunction observed in pediatric type 1 diabetes mellitus. Hypertension 2009;54:987–94. [DOI] [PubMed] [Google Scholar]
- 39.Lahiri MK, Kannankeril PJ, Goldberger JJ. Assessment of autonomic function in cardiovascular disease: physiological basis and prognostic implications. J Am Coll Cardiol 2008;51:1725–33. [DOI] [PubMed] [Google Scholar]
- 40.Vinik AI, Ziegler D. Diabetic cardiovascular autonomic neuropathy. Circulation 2007;115: 387–97. [DOI] [PubMed] [Google Scholar]
- 41.Spallone V, Menzinger G. Diagnosis of cardiovascular autonomic neuropathy in diabetes. Diabetes 1997;46 Suppl 2:S67–76. [DOI] [PubMed] [Google Scholar]
- 42.Ng F, Wong S, Cruz AL, Hernandez MI, Gomis P, Passariello G. Heart rate recovery in the diagnosis of diabetic cardiovascular autonomic neuropathy. Comput Cardiol 2007:681–4. [Google Scholar]
- 43.Tesfaye S, Boulton AJ, Dyck PJ, et al. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 2010;33:2285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ewing DJ, Martyn CN, Young RJ, Clarke BF. The value of cardiovascular autonomic function tests: 10 years experience in diabetes. Diabetes Care 1985;8:491–8. [DOI] [PubMed] [Google Scholar]
- 45.Mestivier D, Chau NP, Chanudet X, Bauduceau B, Larroque P. Relationship between diabetic autonomic dysfunction and heart rate variability assessed by recurrence plot. Am J Physiol 1997;272:H1094–9. [DOI] [PubMed] [Google Scholar]
- 46.Khandoker AH, Jelinek HF, Palaniswami M. Identifying diabetic patients with cardiac autonomic neuropathy by heart rate complexity analysis. Biomed Eng Online 2009;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.American Diabetes Association. Standards of medical care in diabetes–2013. Diabetes Care 2013;36 Suppl 1:S11–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balcıoğlu AS, Müderrisoğlu H. Diabetes an d cardiac autonomic neuropathy: clinical manifestations, cardiovascular consequences, diagnosis and treatment. World J Diabetes 2015;6:80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maser RE, Mitchell BD, Vinik AI, Freeman R. The association between cardiovascular autonomic neuropathy and mortality in individuals with diabetes: a meta-analysis. Diabetes Care 2003;26: 1895–901. [DOI] [PubMed] [Google Scholar]
- 50.Naas AA, Davidson NC, Thompson C, et al. QT and QTc dispersion are accurate predictors of cardiac death in newly diagnosed non-insulin dependent diabetes: cohort study. BMJ 1998; 316:745–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossing P, Breum L, Major-Pedersen A, et al. Prolonged QTc interval predicts mortality in patients with type 1 diabetes mellitus. Diabet Med 2001;18:199–205. [DOI] [PubMed] [Google Scholar]
- 52.Sawicki PT, Dahne R, Bender R, Berger M. Prolonged QT interval as a predictor of mortality in diabetic nephropathy. Diabetologia 1996;39: 77–81. [DOI] [PubMed] [Google Scholar]
- 53.Sawicki PT, Kiwitt S, Bender R, Berger M. The value of QT interval dispersion for identification of total mortality risk in non-insulin-dependent diabetes mellitus. J Intern Med 1998;243:49–56. [DOI] [PubMed] [Google Scholar]
- 54.Veglio M, Sivieri R, Chinaglia A, Scaglione L, Cavallo-Perin P. QT interval prolongation and mortality in type 1 diabetic patients: a 5-year cohort prospective study. Neuropathy Study Group of the Italian Society of the Study of Diabetes, Piemonte Affiliate. Diabetes Care 2000;23: 1381–3. [DOI] [PubMed] [Google Scholar]
- 55.Ewing DJ, Campbell IW, Clarke BF. Mortality in diabetic autonomic neuropathy. Lancet 1976;1: 601–3. [DOI] [PubMed] [Google Scholar]
- 56.Orchard TJ, Lloyd CE, Maser RE, Kuller LH. Why does diabetic autonomic neuropathy predict IDDM mortality? An analysis from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Res Clin Pract 1996;34 Suppl 1:S165–71. [DOI] [PubMed] [Google Scholar]
- 57.Suarez GA, Clark VM, Norell JE, et al. Sudden cardiac death in diabetes mellitus: risk factors in the Rochester Diabetic Neuropathy Study. J Neurol Neurosurg Psychiatry 2005;76:240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wackers FJ, Young LH, Inzucchi SE, et al. Detection of silent myocardial ischemia in asymptomatic diabetic subjects: the DIAD study. Diabetes Care 2004;27:1954–61. [DOI] [PubMed] [Google Scholar]
- 59.The Consensus Committee of the American Autonomic Society, the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology 1996;46:1470. [DOI] [PubMed] [Google Scholar]
- 60.Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med 2000;343: 847–55. [DOI] [PubMed] [Google Scholar]
- 61.Hague K, Lento P, Morgello S, Caro S, Kaufmann H. The distribution of Lewy bodies in pure autonomic failure: autopsy findings and review of the literature. Acta Neuropathol (Berl) 1997;94:192–6. [DOI] [PubMed] [Google Scholar]
- 62.Hishikawa N, Hashizume Y, Hirayama M, et al. Brainstem-type Lewy body disease presenting with progressive autonomic failure and lethargy. Clin Auton Res 2000;10:139–43. [DOI] [PubMed] [Google Scholar]
- 63.Freeman R, Abuzinadah AR, Gibbons C, Jones P, Miglis MG, Sinn DI. Orthostatic hypotension: JACC state-of-the-art review. J Am Coll Cardiol 2018;72:1294–309. [DOI] [PubMed] [Google Scholar]
- 64.Bryarly M, Phillips LT, Fu Q, Vernino S, Levine BD. Postural orthostatic tachycardia syndrome: JACC focus seminar. J Am Coll Cardiol 2019;73:1207–28. [DOI] [PubMed] [Google Scholar]
- 65.Fukuda K, Kanazawa H, Aizawa Y, Ardell JL, Shivkumar K. Cardiac innervation and sudden cardiac death. Circ Res 2015;116:2005–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Inoue H, Zipes DP. Time course of denervation of efferent sympathetic and vagal nerves after occlusion of the coronary artery in the canine heart. Circ Res 1988;62:1111–20. [DOI] [PubMed] [Google Scholar]
- 67.Costigan M, Moss A, Latremoliere A, et al. Tcell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci 2009;29: 14415–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ajijola OA, Wisco JJ, Lambert HW, et al. Extracardiac neural remodeling in humans with cardiomyopathy. Circ Arrhythm Electrophysiol 2012;5:1010–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakamura K, Ajijola OA, Aliotta E, Armour JA, Ardell JL, Shivkumar K. Pathological effects of chronic myocardial infarction on peripheral neurons mediating cardiac neurotransmission. Auton Neurosci 2016;197:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li C-Y, Li Y- G. Cardiac sympathetic nerve sprouting and susceptibility to ventricular arrhythmias after myocardial infarction. Cardiol Res Pract 2015;2015:698368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ajijola OA, Yagishita D, Patel KJ, et al. Focal myocardial infarction induces global remodeling of cardiac sympathetic innervation: neural remodeling in a spatial context. Am J Physiol Heart Circ Physiol 2013;305:H1031–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vaseghi M, Lux RL, Mahajan A, Shivkumar K. Sympathetic stimulation increases dispersion of repolarization in humans with myocardial infarction. Am J Physiol Heart Circ Physiol 2012;302: H1838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ajijola OA, Yagishita D, Reddy NK, et al. Remodeling of stellate ganglion neurons after spatially targeted myocardial infarction: neuropeptide and morphologic changes. Heart Rhythm 2015;12:1027–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rajendran PS, Nakamura K, Ajijola OA, et al. Myocardial infarction induces structural and functional remodelling of the intrinsic cardiac nervous system. J Physiol 2016;594:321–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hou Y, Zhou Q, Po SS. Neuromodulation for cardiac arrhythmia. Heart Rhythm 2016;13: 584–92. [DOI] [PubMed] [Google Scholar]
- 76.Premchand RK, Sharma K, Mittal S, et al. Autonomic regulation therapy via left or right cervical vagus nerve stimulation in patients with chronic heart failure: results of the ANTHEM-HF trial. J Card Fail 2014;20:808–16. [DOI] [PubMed] [Google Scholar]
- 77.Tse HF, Turner S, Sanders P, et al. Thoracic Spinal Cord Stimulation for Heart Failure as a Restorative Treatment (SCS HEART study): firstin-man experience. Heart Rhythm 2015;12: 588–95. [DOI] [PubMed] [Google Scholar]
- 78.Ng J, Villuendas R, Cokic I, et al. Autonomic remodeling in the left atrium and pulmonary veins in heart failure: creation of a dynamic substrate for atrial fibrillation. Circ Arrhythm Electrophysiol 2011;4:388–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Iwasaki YK, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology: implications for management. Circulation 2011;124:2264–74. [DOI] [PubMed] [Google Scholar]
- 80.Shen MJ, Choi EK, Tan AY, et al. Neural mechanisms of atrial arrhythmias. Nat Rev Cardiol 2011;9:30–9. [DOI] [PubMed] [Google Scholar]
- 81.Amar D, Zhang H, Miodownik S, Kadish AH. Competing autonomic mechanisms precede the onset of postoperative atrial fibrillation. J Am Coll Cardiol 2003;42:1262–8. [DOI] [PubMed] [Google Scholar]
- 82.Patterson E, Po SS, Scherlag BJ, Lazzara R. Triggered firing in pulmonary veins initiated by in vitro autonomic nerve stimulation.[see comment]. Heart Rhythm 2005;2:624–31. [DOI] [PubMed] [Google Scholar]
- 83.Tan AY, Zhou S, Ogawa M, et al. Neural mechanisms of paroxysmal atrial fibrillation and paroxysmal atrial tachycardia in ambulatory canines. Circulation 2008;118:916–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi EK, Shen MJ, Han S, et al. Intrinsic cardiac nerve activity and paroxysmal atrial tachyarrhythmia in ambulatory dogs. Circulation 2010; 121:2615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ogawa M, Tan AY, Song J, et al. Cryoablation of stellate ganglia and atrial arrhythmia in ambulatory dogs with pacing-induced heart failure. Heart Rhythm 2009;6:1772–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ogawa M, Zhou S, Tan AY, et al. Left stellate ganglion and vagal nerve activity and cardiac arrhythmias in ambulatory dogs with pacing-induced congestive heart failure. J Am Coll Cardiol 2007; 50:335–43. [DOI] [PubMed] [Google Scholar]
- 87.Hou Y, Scherlag BJ, Lin J, et al. Ganglionated plexi modulate extrinsic cardiac autonomic nerve input: effects on sinus rate, atrioventricular conduction, refractoriness, and inducibility of atrial fibrillation. J Am Coll Cardiol 2007;50:61–8. [DOI] [PubMed] [Google Scholar]
- 88.Hou Y, Scherlag BJ, Lin J, et al. Interactive atrial neural network: determining the connections between ganglionated plexi. Heart Rhythm 2007; 4:56–63. [DOI] [PubMed] [Google Scholar]
- 89.Arora R, Ng J, Ulphani J, et al. Unique autonomic profile of the pulmonary veins and posterior left atrium. J Am Coll Cardiol 2007;49:1340–8. [DOI] [PubMed] [Google Scholar]
- 90.Arora R, Ulphani JS, Villuendas R, et al. Neural substrate for atrial fibrillation: implications for targeted parasympathetic blockade in the posterior left atrium. Am J Physiol 2008;294:H134–44. [DOI] [PubMed] [Google Scholar]
- 91.Arora R Recent insights into the role of the autonomic nervous system in the creation of substrate for atrial fibrillation: implications for therapies targeting the atrial autonomic nervous system. Circ Arrhythm Electrophysiol 2012;5: 850–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chevalier P, Tabib A, Meyronnet D, et al. Quantitative study of nerves of the human left atrium. Heart Rhythm 2005;2:518–22. [DOI] [PubMed] [Google Scholar]
- 93.Arora R, Ulphani JS, Villuendas R, et al. Neural substrate for atrial fibrillation: implications for targeted parasympathetic blockade in the posterior left atrium. Am J Physiol Heart Circ Physiol 2008;294:H134–44. [DOI] [PubMed] [Google Scholar]
- 94.Tan AY, Li H, Wachsmann-Hogiu S, Chen LS, Chen PS, Fishbein MC. Autonomic innervation and segmental muscular disconnections at the human pulmonary vein-atrial junction: implications for catheter ablation of atrial-pulmonary vein junction. J Am Coll Cardiol 2006;48:132–43. [DOI] [PubMed] [Google Scholar]
- 95.Deneke T, Chaar H, de Groot JR, et al. Shift in the pattern of autonomic atrial innervation in subjects with persistent atrial fibrillation. Heart Rhythm 2011;8:1357–63. [DOI] [PubMed] [Google Scholar]
- 96.Gould PA, Yii M, McLean C, et al. Evidence for increased atrial sympathetic innervation in persistent human atrial fibrillation. Pacing Clin Electrophysiol 2006;29:821–9. [DOI] [PubMed] [Google Scholar]
- 97.Edgerton JR, Brinkman WT, Weaver T, et al. Pulmonary vein isolation and autonomic denervation for the management of paroxysmal atrial fibrillation by a minimally invasive surgical approach. J Thorac Cardiovasc Surg 2010;140: 823–8. [DOI] [PubMed] [Google Scholar]
- 98.Edgerton JR, Edgerton ZJ, Weaver T, et al. Minimally invasive pulmonary vein isolation and partial autonomic denervation for surgical treatment of atrial fibrillation. Ann Thorac Surg 2008; 86:35–8; discussion 39. [DOI] [PubMed] [Google Scholar]
- 99.Pokushalov E, Romanov A, Artyomenko S, Turov A, Shirokova N, Katritsis DG. Left atrial ablation at the anatomic areas of ganglionated plexi for paroxysmal atrial fibrillation. Pacing Clin Electrophysiol 2010;33:1231–8. [DOI] [PubMed] [Google Scholar]
- 100.Pokushalov E, Romanov A, Artyomenko S, et al. Ganglionated plexi ablation for longstanding persistent atrial fibrillation. Europace 2010;12: 342–6. [DOI] [PubMed] [Google Scholar]
- 101.Pokushalov E, Romanov A, Shugayev P, et al. Selective ganglionated plexi ablation for paroxysmal atrial fibrillation. Heart Rhythm 2009;6: 1257–64. [DOI] [PubMed] [Google Scholar]
- 102.Scanavacca M, Pisani CF, Hachul D, et al. Selective atrial vagal denervation guided by evoked vagal reflex to treat patients with paroxysmal atrial fibrillation. Circulation 2006;114: 876–85. [DOI] [PubMed] [Google Scholar]
- 103.Pokushalov E, Romanov A, Artyomenko S, et al. Ganglionated plexi ablation directed by high-frequency stimulation and complex fractionated atrial electrograms for paroxysmal atrial fibrillation. Pacing Clin Electrophysiol 2012;35: 776–84. [DOI] [PubMed] [Google Scholar]
- 104.Katritsis DG, Giazitzoglou E, Zografos T, Pokushalov E, Po SS, Camm AJ. Rapid pulmonary vein isolation combined with autonomic ganglia modification: a randomized study. Heart Rhythm 2011;8:672–8. [DOI] [PubMed] [Google Scholar]
- 105.Zhou Q, Hou Y, Yang S. A meta-analysis of the comparative efficacy of ablation for atrial fibrillation with and without ablation of the ganglionated plexi. Pacing Clin Electrophysiol 2011;34:1687–94. [DOI] [PubMed] [Google Scholar]
- 106.Nitta T, Ishii Y, Sakamoto S. Surgery for atrial fibrillation: recent progress and future perspective. Gen Thorac Cardiovasc Surg 2012;60:13–20. [DOI] [PubMed] [Google Scholar]
- 107.Bagge L, Blomstrom P, Nilsson L, Einarsson GM, Jideus L, Blomstrom-Lundqvist C. Epicardial off-pump pulmonary vein isolation and vagal denervation improve long-term outcome and quality of life in patients with atrial fibrillation. J Thorac Cardiovasc Surg 2009;137:1265–71. [DOI] [PubMed] [Google Scholar]
- 108.Krum H, Schlaich M, Whitbourn R, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet 2009;373: 1275–81. [DOI] [PubMed] [Google Scholar]
- 109.Hering D, Lambert EA, Marusic P, et al. Substantial reduction in single sympathetic nerve firing after renal denervation in patients with resistant hypertension. Hypertension 2013;61: 457–64. [DOI] [PubMed] [Google Scholar]
- 110.Linz D, van Hunnik A, Hohl M, et al. Catheterbased renal denervation reduces atrial nerve sprouting and complexity of atrial fibrillation in goats. Circ Arrhythm Electrophysiol 2015;8: 466–74. [DOI] [PubMed] [Google Scholar]
- 111.Pokushalov E, Romanov A, Katritsis DG, et al. Renal denervation for improving outcomes of catheter ablation in patients with atrial fibrillation and hypertension: early experience. Heart Rhythm 2014;11:1131–8. [DOI] [PubMed] [Google Scholar]
- 112.Bhatt DL, Kandzari DE, O’Neill WW, et al. A controlled trial of renal denervation for resistant hypertension. N Engl J Med 2014;370:1393–401. [DOI] [PubMed] [Google Scholar]
- 113.Pokushalov E, Kozlov B, Romanov A, et al. Long-term suppression of atrial fibrillation by botulinum toxin injection into epicardial fat pads in patients undergoing cardiac surgery: one-year follow-up of a randomized pilot study. Circ Arrhythm Electrophysiol 2015;8:1334–41. [DOI] [PubMed] [Google Scholar]
- 114.Pokushalov E, Kozlov B, Romanov A, et al. Botulinum toxin injection in epicardial fat pads can prevent recurrences of atrial fibrillation after cardiac surgery: results of a randomized pilot study. J Am Coll Cardiol 2014;64:628–9. [DOI] [PubMed] [Google Scholar]
- 115.Aistrup GL, Villuendas R, Ng J, et al. Targeted G-protein inhibition as a novel approach to decrease vagal atrial fibrillation by selective parasympathetic attenuation. Cardiovasc Res 2009;83:481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aistrup GL, Cokic I, Ng J, et al. Targeted nonviral gene-based inhibition of Gai/o-mediated vagal signaling in the posterior left atrium decreases vagal induced AF. Heart Rhythm 2011: 1722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goldberger JJ, Subacius H, Patel T, Cunnane R, Kadish AH. Sudden cardiac death risk stratification in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol 2014;63: 1879–89. [DOI] [PubMed] [Google Scholar]
- 118.Kleiger RE, Stein PK, Bosner MS, Rottman JN. Time domain measurements of heart rate variability. Cardiol Clin 1992;10:487–98. [PubMed] [Google Scholar]
- 119.Laude D, Weise F, Girard A, Elghozi JL. Spectral analysis of systolic blood pressure and heart rate oscillations related to respiration. Clin Exp Pharmacol Physiol 1995;22:352–7. [DOI] [PubMed] [Google Scholar]
- 120.Elghozi JL, Laude D, Girard A. Effects of respiration on blood pressure and heart rate variability in humans. Clin Exp Pharmacol Physiol 1991;18:735–42. [DOI] [PubMed] [Google Scholar]
- 121.Malik MSG. Autonomic testing and cardiac risk In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology: From Cell to Bedside. 6th ed Philadelphia: Elsevier, 2014:649–56. [Google Scholar]
- 122.Vybiral T, Bryg RJ, Maddens ME, Boden WE. Effect of passive tilt on sympathetic and parasympathetic components of heart rate variability in normal subjects. Am J Cardiol 1989;63:1117–20. [DOI] [PubMed] [Google Scholar]
- 123.Pagani M, Lombardi F, Guzzetti S, et al. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res 1986; 59:178–93. [DOI] [PubMed] [Google Scholar]
- 124.Chiou CW, Zipes DP. Selective vagal denervation of the atria eliminates heart rate variability and baroreflex sensitivity while preserving ventricular innervation. Circulation 1998;98:360–8. [DOI] [PubMed] [Google Scholar]
- 125.Goldberger JJ. Sympathovagal balance: how should we measure it? Am J Physiol 1999;276: H1273–80. [DOI] [PubMed] [Google Scholar]
- 126.Fox K, Borer JS, Camm AJ, et al. Resting heart rate in cardiovascular disease. J Am Coll Cardiol 2007;50:823–30. [DOI] [PubMed] [Google Scholar]
- 127.Huikuri HV, Stein PK. Heart rate variability in risk stratification of cardiac patients. Prog Cardiovasc Dis 2013;56:153–9. [DOI] [PubMed] [Google Scholar]
- 128.Huikuri HV, Stein PK. Clinical application of heart rate variability after acute myocardial infarction. Front Physiol 2012;3:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huikuri HV, Makikallio TH, Peng CK, Goldberger AL, Hintze U, Moller M. Fractal correlation properties of R-R interval dynamics and mortality in patients with depressed left ventricular function after an acute myocardial infarction. Circulation 2000;101:47–53. [DOI] [PubMed] [Google Scholar]
- 130.Kleiger RE, Miller JP, Bigger JT Jr., Moss AJ. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol 1987;59:256–62. [DOI] [PubMed] [Google Scholar]
- 131.La Rovere MT, Pinna GD, Maestri R, et al. Short-term heart rate variability strongly predicts sudden cardiac death in chronic heart failure patients. Circulation 2003;107:565–70. [DOI] [PubMed] [Google Scholar]
- 132.Deyell MW, Krahn AD, Goldberger JJ. Sudden cardiac death risk stratification. Circ Res 2015;116: 1907–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dekker JM, Crow RS, Folsom AR, et al. Low heart rate variability in a 2-minute rhythm strip predicts risk of coronary heart disease and mortality from several causes: the ARIC Study. Atherosclerosis Risk In Communities. Circulation 2000;102:1239–44. [DOI] [PubMed] [Google Scholar]
- 134.de Bruyne MC, Kors JA, Hoes AW, et al. Both decreased and increased heart rate variability on the standard 10-second electrocardiogram predict cardiac mortality in the elderly: the Rotterdam Study. Am J Epidemiol 1999;150:1282–8. [DOI] [PubMed] [Google Scholar]
- 135.Tsuji H, Larson MG, Venditti FJ Jr., et al. Impact of reduced heart rate variability on risk for cardiac events. The Framingham Heart Study. Circulation 1996;94:2850–5. [DOI] [PubMed] [Google Scholar]
- 136.Gnecchi Ruscone T, Lombardi F, Malfatto G, Malliani A. Attenuation of baroreceptive mechanisms by cardiovascular sympathetic afferent fibers. Am J Physiol 1987;253:H787–91. [DOI] [PubMed] [Google Scholar]
- 137.Stein KM. Noninvasive risk stratification for sudden death: signal-averaged electrocardiography, nonsustained ventricular tachycardia, heart rate variability, baroreflex sensitivity, and QRS duration. Prog Cardiovasc Dis 2008;51:106–17. [DOI] [PubMed] [Google Scholar]
- 138.Billman GE, Schwartz PJ, Stone HL. Baroreceptor reflex control of heart rate: a predictor of sudden cardiac death. Circulation 1982;66: 874–80. [DOI] [PubMed] [Google Scholar]
- 139.De Ferrari GM, Sanzo A, Bertoletti A, Specchia G, Vanoli E, Schwartz PJ. Baroreflex sensitivity predicts long-term cardiovascular mortality after myocardial infarction even in patients with preserved left ventricular function. J Am Coll Cardiol 2007;50:2285–90. [DOI] [PubMed] [Google Scholar]
- 140.Huikuri HV, Tapanainen JM, Lindgren K, et al. Prediction of sudden cardiac death after myocardial infarction in the beta-blocking era. J Am Coll Cardiol 2003;42:652–8. [DOI] [PubMed] [Google Scholar]
- 141.La Rovere MT, Pinna GD, Hohnloser SH, et al. Baroreflex sensitivity and heart rate variability in the identification of patients at risk for lifethreatening arrhythmias: implications for clinical trials. Circulation 2001;103:2072–7. [DOI] [PubMed] [Google Scholar]
- 142.Grimm W, Christ M, Bach J, Muller HH, Maisch B. Noninvasive arrhythmia risk stratification in idiopathic dilated cardiomyopathy: results of the Marburg Cardiomyopathy Study. Circulation 2003;108:2883–91. [DOI] [PubMed] [Google Scholar]
- 143.Schmidt G, Malik M, Barthel P, et al. Heartrate turbulence after ventricular premature beats as a predictor of mortality after acute myocardial infarction. Lancet 1999;353:1390–6. [DOI] [PubMed] [Google Scholar]
- 144.Cygankiewicz I Heart rate turbulence. Prog Cardiovasc Dis 2013;56:160–71. [DOI] [PubMed] [Google Scholar]
- 145.Mrowka R, Persson PB, Theres H, Patzak A. Blunted arterial baroreflex causes “pathological” heart rate turbulence. Am J Physiol Regul Integr Comp Physiol 2000;279:R1171–5. [DOI] [PubMed] [Google Scholar]
- 146.Davies LC, Francis DP, Ponikowski P, Piepoli MF, Coats AJ. Relation of heart rate and blood pressure turbulence following premature ventricular complexes to baroreflex sensitivity in chronic congestive heart failure. Am J Cardiol 2001;87:737–42. [DOI] [PubMed] [Google Scholar]
- 147.Bauer A, Malik M, Schmidt G, et al. Heart rate turbulence: standards of measurement, physiological interpretation, and clinical use: International Society for Holter and Noninvasive Electrophysiology Consensus. J Am Coll Cardiol 2008;52:1353–65. [DOI] [PubMed] [Google Scholar]
- 148.La Rovere MT, Maestri R, Pinna GD, Sleight P, Febo O. Clinical and haemodynamic correlates of heart rate turbulence as a non-invasive index of baroreflex sensitivity in chronic heart failure. Clin Sci (Lond) 2011;121:279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Berkowitsch A, Zareba W, Neumann T, et al. Risk stratification using heart rate turbulence and ventricular arrhythmia in MADIT II: usefulness and limitations of a 10-minute Holter recording. Ann Noninvasive Electrocardiol 2004;9:270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Robinson BF, Epstein SE, Beiser GD, Braunwald E. Control of heart rate by the autonomic nervous system. Studies in man on the interrelation between baroreceptor mechanisms and exercise. Circ Res 1966;19:400–11. [DOI] [PubMed] [Google Scholar]
- 151.Ekblom B, Goldbarg AN, Kilbom A, Astrand PO. Effects of atropine and propranolol on the oxygen transport system during exercise in man. Scand J Clin Lab Invest 1972;30:35–42. [DOI] [PubMed] [Google Scholar]
- 152.Savin WM, Davidson DM, Haskell WL. Autonomic contribution to heart rate recovery from exercise in humans. J Appl Physiol Respir Environ Exerc Physiol 1982;53:1572–5. [DOI] [PubMed] [Google Scholar]
- 153.Imai K, Sato H, Hori M, et al. Vagally mediated heart rate recovery after exercise is accelerated in athletes but blunted in patients with chronic heart failure. J Am Coll Cardiol 1994;24:1529–35. [DOI] [PubMed] [Google Scholar]
- 154.Kannankeril PJ, Le FK, Kadish AH, Goldberger JJ. Parasympathetic effects on heart rate recovery after exercise. J Investig Med 2004; 52:394–401. [DOI] [PubMed] [Google Scholar]
- 155.Goldberger JJ, Johnson NP, Subacius H, Ng J, Greenland P. Comparison of the physiologic and prognostic implications of the heart rate versus the RR interval. Heart Rhythm 2014;11:1925–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jouven X, Empana JP, Schwartz PJ, Desnos M, Courbon D, Ducimetiere P. Heart-rate profile during exercise as a predictor of sudden death. N Engl J Med 2005;352:1951–8. [DOI] [PubMed] [Google Scholar]
- 157.Cole CR, Blackstone EH, Pashkow FJ, Snader CE, Lauer MS. Heart-rate recovery immediately after exercise as a predictor of mortality. N Engl J Med 1999;341:1351–7. [DOI] [PubMed] [Google Scholar]
- 158.Batchvarov VN, Ghuran A, Smetana P, et al. QT-RR relationship in healthy subjects exhibits substantial intersubject variability and high intrasubject stability. Am J Physiol Heart Circ Physiol 2002;282:H2356–63. [DOI] [PubMed] [Google Scholar]
- 159.Fenichel RR, Malik M, Antzelevitch C, et al. Drug-induced torsades de pointes and implications for drug development. J Cardiovasc Electrophysiol 2004;15:475–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sundaram S, Carnethon M, Polito K, Kadish AH, Goldberger JJ. Autonomic effects on QT-RR interval dynamics after exercise. Am J Physiol Heart Circ Physiol 2008;294: H490–7. [DOI] [PubMed] [Google Scholar]
- 161.Carter JR, Ray CA. Sympathetic neural adaptations to exercise training in humans. Auton Neurosci 2015;188:36–43. [DOI] [PubMed] [Google Scholar]
- 162.Macefield VG. Sympathetic microneurography. Handb Clin Neurol 2013;117:353–64. [DOI] [PubMed] [Google Scholar]
- 163.Notarius CF, Millar PJ, Floras JS. Muscle sympathetic activity in resting and exercising humans with and without heart failure. Appl Physiol Nutr Metab 2015;40:1107–15. [DOI] [PubMed] [Google Scholar]
- 164.Barretto AC, Santos AC, Munhoz R, et al. Increased muscle sympathetic nerve activity predicts mortality in heart failure patients. Int J Cardiol 2009;135:302–7. [DOI] [PubMed] [Google Scholar]
- 165.Pearson MJ, Smart NA. Exercise therapy and autonomic function in heart failure patients: a systematic review and meta-analysis. Heart Fail Rev 2018;23:91–108. [DOI] [PubMed] [Google Scholar]
- 166.Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res 2014;114:1500–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Robinson EA, Rhee KS, Doytchinova A, et al. Estimating sympathetic tone by recording subcutaneous nerve activity in ambulatory dogs. J Cardiovasc Electrophysiol 2015;26: 70–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Doytchinova A, Hassel JL, Yuan Y, et al. Simultaneous noninvasive recording of skin sympathetic nerve activity and electrocardiogram. Heart Rhythm 2017;14:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mittleman MA, Maclure M, Tofler GH, Sherwood JB, Goldberg RJ, Muller JE. Triggering of acute myocardial infarction by heavy physical exertion: protection against triggering by regular exertion. N Engl J Med 1993;329: 1677–83. [DOI] [PubMed] [Google Scholar]
- 170.Mittleman MA, Maclure M, Sherwood JB, et al. Triggering of acute myocardial infarction onset by episodes of anger. Determinants of Myocardial Infarction Onset Study Investigators. Circulation 1995;92:1720–5. [DOI] [PubMed] [Google Scholar]
- 171.Albert CM, Mittleman MA, Chae CU, Lee IM, Hennekens CH, Manson JE. Triggering of sudden death from cardiac causes by vigorous exertion. N Engl J Med 2000;343:1355–61. [DOI] [PubMed] [Google Scholar]
- 172.Lampert R, Joska T, Burg MM, Batsdord WP, McPherson CA, Jain D. Emotional and physical precipitants of ventricular arrhythmia. Circulation 2002;106:1800–5. [DOI] [PubMed] [Google Scholar]
- 173.De Ferrari GM, Mantica M, Vanoli E, Hull SS Jr., Schwartz PJ. Scopolamine increases vagal tone and vagal reflexes in patients after myocardial infarction. J Am Coll Cardiol 1993;22:1327–34. [DOI] [PubMed] [Google Scholar]
- 174.Hull SS Jr., Vanoli E, Adamson PB, De Ferrari GM, Foreman RD, Schwartz PJ. Do increases in markers of vagal activity imply protection from sudden death? The case of scopolamine. Circulation 1995;91:2516–9. [DOI] [PubMed] [Google Scholar]