

Abstract
Objectives: Lung adenocarcinoma (LUAD) accounts for a majority of cancer-related deaths worldwide annually. The identification of prognostic biomarkers and prediction of prognosis for LUAD patients is necessary.
Materials and Methods: In this study, LUAD RNA-Seq data and clinical data from the Cancer Genome Atlas (TCGA) were divided into TCGA cohort I (n = 338) and II (n = 168). The cohort I was used for model construction, and the cohort II and data from Gene Expression Omnibus (GSE72094 cohort, n = 393; GSE11969 cohort, n = 149) were utilized for validation. First, the survival-related seed genes were selected from the cohort I using the machine learning model (random survival forest, RSF), and then in order to improve prediction accuracy, the forward selection model was utilized to identify the prognosis-related key genes among the seed genes using the clinically-integrated RNA-Seq data. Second, the survival risk score system was constructed by using these key genes in the cohort II, the GSE72094 cohort and the GSE11969 cohort, and the evaluation metrics such as HR, p value and C-index were calculated to validate the proposed method. Third, the developed approach was compared with the previous five prediction models. Finally, bioinformatics analyses (pathway, heatmap, protein-gene interaction network) have been applied to the identified seed genes and key genes.
Results and Conclusion: Based on the RSF model and clinically-integrated RNA-Seq data, we identified sixteen key genes that formed the prognostic gene expression signature. These sixteen key genes could achieve a strong power for prognostic prediction of LUAD patients in cohort II (HR = 3.80, p = 1.63e-06, C-index = 0.656), and were further validated in the GSE72094 cohort (HR = 4.12, p = 1.34e-10, C-index = 0.672) and GSE11969 cohort (HR = 3.87, p = 6.81e-07, C-index = 0.670). The experimental results of three independent validation cohorts showed that compared with the traditional Cox model and the use of standalone RNA-Seq data, the machine-learning-based method effectively improved the prediction accuracy of LUAD prognosis, and the derived model was also superior to the other five existing prediction models. KEGG pathway analysis found eleven of the sixteen genes were associated with Nicotine addiction. Thirteen of the sixteen genes were reported for the first time as the LUAD prognosis-related key genes. In conclusion, we developed a sixteen-gene prognostic marker for LUAD, which may provide a powerful prognostic tool for precision oncology.
Keywords: Lung adenocarcinoma, Prognosis prediction, RNA-Seq data, Random survival forest, Forward selection model
Introduction
Lung cancer is the leading cause of cancer death worldwide, and approximately 1 million people die of this disease each year 1-3. Lung adenocarcinoma (LUAD) is the most common histological subtype of lung cancer, and the incidence and mortality are increasing 4-6. The average 5-year survival rate for LUAD patients is only 15%, although progress has been made in treatment, such as combination of chemotherapy and chemoradiation, survival rate has been improved very little over the past a few decades 7-9. Therefore, it is particularly important to find prognostic markers for LUAD and provide precise targeted therapy.
With the development of high-throughput sequencing techniques to interrogate genome-wide genetic variations, RNA-Seq based markers have recently been studied as prognostic markers for lung cancer 10, 11. Sudbanshu Shukla et al. undertook the first prognostic analysis of LUAD RNA-Seq data and generated the prognostic feature using the Cox model, which can provide a powerful prognostic tool for precision oncology as part of an integrated RNA-Seq clinical sequencing program 5. Subsequently, Li B et al. used the RNA-Seq dataset to develop a robust, individualized clinical immune signature that can assess the prognosis and overall survival in patients with early-stage nonsquamous non-small cell lung cancer 12. As a subset of RNA-Seq, certain long non-coding RNAs (lncRNAs) also encode proteins and play crucial roles in gene transcription and regulation 13, 14. Zheng S et al. used the Cox model to develop an 8-lncRNA prognostic signature in LUAD patients, which provided an effective independent prognostic prediction model for LUAD patients 15. These studies were based solely on RNA-Seq data. Yuan Yuan et al. used molecular data in combination with clinical variables to predict the survival of four cancer types, the results showed that the comprehensive model had little predictive power for lung squamous cell carcinoma (LUSC), while the predictive power of the other three kinds of cancers (kidney renal clear cell carcinoma, glioblastoma multiforme, ovarian serous cystadenocarcinoma) were significantly improved 16.
Broadly defined, machine learning is a branch of computer science that deals with making predictions from complex data through statistical models and is widely applied in the biomedical field 17, 18. As an efficient machine learning algorithm, RSF is considered as a more powerful method for survival analysis 19, 20. In the present study, the high-throughput RNA-Seq data and clinical data were downloaded from the TCGA database, the Cox model and the RSF model were used to identify the survival-related seed genes from the TCGA cohort I, and then the forward selection model was generated and the prognosis-related key genes were identified. Finally, based on the RSF model and the clinically-integrated RNA-Seq data, we obtained a subset of sixteen genes that formed the prognostic gene expression signature and improved the power for predicting the prognosis of LUAD patients.
Materials and Methods
Data collection and preprocessing
The RNA-Seq data and the corresponding clinical information for LUAD were downloaded from the publicly available TCGA database. After filtering out the missing data, a total of 506 LUAD patients were kept as our study samples. The 506 LUAD patients were further randomly assigned to a training cohort (TCGA cohort I, n = 338) and an internal validation cohort (TCGA cohort II, n = 168). Moreover, the other two external validation cohorts consisting of 393 and 149 LUAD patients were downloaded from the GEO database (GSE72094 cohort, GSE11969 cohort). In the RNA-Seq dataset for the above four cohorts, the numerical distribution of Reads per Kilo-base per Million mapped (RPKM) reads are too wide to be used in model analysis, thus we formulated each RPKM value in 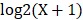 , where X is the RPKM value 21.
, where X is the RPKM value 21.
Machine learning model: random survival forest
Random survival forest (RSF) is an adaptation of random forests (RF) designed to be used for survival data 22. The mathematical principle of the RSF model is introduced as follows: bootstrap methods are used to randomly extract the ntree bootstrap samples from the raw data and create a binary recursive survival tree for each sample. In the experiment, the good division was determined by the log-rank splitting rule to maximize the survival difference between the daughter nodes 23. The Cumulative Hazard Function (CHF) gives the values of the terminal nodes associated with time. For a terminal node h of a survival tree n at time t, this is given by the Nelson-Aalen estimator:
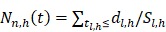 |
Among them, 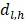 represents the number of deaths,
represents the number of deaths, 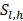 indicates individuals at risk,
indicates individuals at risk, 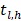 indicates distinct time events. All cases of
indicates distinct time events. All cases of 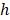 are assigned the same CHF. In order to calculate the ensemble CHF of the survival forest of ntree trees with a given d-dimensional case
are assigned the same CHF. In order to calculate the ensemble CHF of the survival forest of ntree trees with a given d-dimensional case 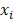 ,
,
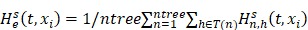 |
Where 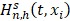 is equal to the Nelson-Aalen estimator, if
is equal to the Nelson-Aalen estimator, if 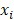 ends with the survival tree falling to h
24,
ends with the survival tree falling to h
24,
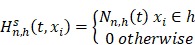 |
The RSF model provides a unified treatment of Breiman's RF 25 for a variety of data settings, such as survival, regression, classification and so on. Survival is grown for right-censored survival data. Survival settings require a time and censoring variable which should be identified as the response in the Surv function for the “randomForestSRC” R package (http://www.r-project.org) 26. The censoring variable must be coded as a non-negative integer with 0 reserved for censor and 1 reserved for death. Furthermore, this model generates a random forest using the training data, and the function var.select implements random forest variable selection using tree minimal depth methodology 25, 27.
Survival-related seed genes generation
The association between genes and patients' overall survival was analyzed in the TCGA cohort I. The survival-related seed genes were screened and identified from all the genes using the following two models.
All the genes among the TCGA cohort I were included in the Cox univariate survival analysis by the “survival” R package, the genes with expressing significance p values less than 0.05 were extracted as the first group of survival-related seed genes. The Cox model is a traditional method in the biostatistical field. In addition, the RSF model in machine learning field was used to select the second group of survival-related seed genes from all the genes among the TCGA cohort I again, which was built by the “randomForestSRC” R package 26.
To search the key pathways that were associated with LUAD survival, we next performed the KEGG pathway enrichment analysis for the two sets of survival-related seed genes by using the Database for Annotation, Visualization and Integrated Discovery (DAVID) 28, 29.
Prognosis-related key genes selection
The seed genes in primary selection were not suitable for clinical diagnosis 21. To increase the feasibility and reliability of clinical prognosis based on gene, the forward selection model was used to select the prognosis-related key genes from seed genes. We implemented prognosis-related key genes selection by the “rbsurv” R package 30-32. The procedure was as followings:
(I) All the samples of the TCGA cohort I were randomly divided into 2/3 training set and 1/3 validation set. A gene was fitted into the training set and the parameter estimate  of this gene was obtained. Then the parameter estimate
of this gene was obtained. Then the parameter estimate 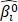 and the validation set were used to calculate the log-likelihood. This evaluation was carried out for each gene.
and the validation set were used to calculate the log-likelihood. This evaluation was carried out for each gene.
(II) The above procedure was repeated n times, and each gene obtained n log-likelihoods. The gene with the largest mean log-likelihood was selected as the top gene and labeled g (1). Subsequently, we searched the next top two genes by evaluating every two-gene model and selected an optimal one with the largest mean log-likelihood.
(III) We continued this forward gene selection procedure and eventually generated a series of models. In order to prevent overfitting, the akaike information criterion (AIC) was used to evaluate these modes, rather than log-likelihood. Finally, the best model with minimum AIC was selected.
(IV) Risk factors were included in the model. Then steps II-IV were repeated.
Verification and comparison of key genes
In order to comprehensively investigate the association between these key genes and the prognosis of LUAD, we developed the survival risk score systems using the TCGA cohort II, the GSE72094 cohort and the GSE11969 cohort. The risk scores were calculated by taking into account both the expression data of these prognosis-related key genes and correlation coefficients. Then the patients of the TCGA cohort II, the GSE72094 cohort and the GSE11969 cohort were divided into two groups separately at high- and low-risk using the 50th percentile of risk score 33. The KM survival curves were used to assess the efficacy of key genes in the three groups. We then calculated the concordance index (C-index) using function rcorr.cens in the R “Hmisc” package 34. In addition, previous studies used various methods (such as decision trees, risk scoring, semi-supervised, and various online tools) to obtain prognostic genetic features of lung cancer or non-small cell lung cancer 5, 35-38. We compared the sixteen-gene prognostic signatures to the five published lung cancer prognostic signatures by rederiving a multivariable Cox model using the gene list from each signature.
Results
The overall flowchart of this work was summarized in Fig. 1A. Firstly, the Cox model and the RSF model were used to screen each one group of survival-related seed genes from all the genes in the TCGA cohort I, and then the forward selection model employed the seed genes or the clinically-integrated seed genes to select four sets of prognosis-related key genes. Finally, the TCGA cohort II, the GSE72094 cohort and the GSE11969 cohort were used to verify the performance of the four sets of prognosis-related key genes.
Figure 1.

Identification of prognostic gene signature. A) Flowchart of RNA-Seq analysis and signature generation. Briefly, survival-related seed genes of the 506 TCGA LUAD patients were first identified by the Cox model and the machine learning model (random survival forest, RSF) from the TCGA cohort I. Next the forward selection model was used to select four sets of key genes for prognosis prediction. The survival risk score systems were built based on the expression data of gene signatures in the TCGA cohort II and the GSE72094 cohort and the GSE11969 cohort, which divided patients into high- and low-risk groups. B) KEGG enrichment pathway analysis of 5376 survival-related seed genes obtained by the Cox model. C) KEGG enrichment pathway analysis of 1113 survival-related seed genes obtained by the RSF model. D) The venn diagram showed that the common key genes obtained from RNA-Seq data and clinically-integrated RNA-Seq data using both the Cox model and the RSF model.
Identification of seed genes associated with survival
The full TCGA cohort included 24991 genes and 506 samples, each sample had survival time and survival status. The Cox univariate survival analysis model and the RSF model were used respectively to select the survival-related seed genes from all the 24 991 genes in TCGA cohort I. The Cox model showed that 5376 genes were statistically significantly correlated with overall survival at the p value less than 0.05. Besides, in the RSF model, 1113 genes were screened using the minimum depth selection method. Pathway analysis showed that the 5376 genes were statistically significantly enriched for Metabolic pathways (p = 3.28e-13) and Focal adhesion (p = 6.14e-09) (Fig. 1B), and the 1113 genes were statistically significantly enriched for Metabolic pathways (p = 1.85e-06) (Fig. 1C). These two sets of survival-related seed genes were used for further analysis.
Identification of key genes associated with prognosis
We selected the prognosis-related key genes from the two groups of seed genes based on the forward selection model. We firstly performed the Cox survival analysis for clinical data of the TCGA cohort I, and selected the risk factors with p value less than 0.05 for construction of clinically-integrated RNA-Seq data (Supplementary Table S1). In the TCGA cohort I, a series of gene models were generated, and then the best model was selected by the minimum AIC. The best model identified by the Cox model and RNA-Seq data, the Cox model and clinically-integrated RNA-Seq data, the RSF model and RNA-Seq data, the RSF model and clinically-integrated RNA-Seq data respectively contained 13, 13, 15 and 16 genes (Supplementary Table S2, S3, S4 and S5). In addition, the parameters in the forward selection model were shown in Supplementary Table S6. The venn diagram showed that four genes were both selected from the Cox model and the RSF model (Fig. 1D).
Development of survival risk score system
We employed the four sets of prognosis-related key genes to construct four survival risk score systems in the TCGA cohort II. The risk scores were calculated by taking into account the expression data of these prognosis-related key genes and correlation coefficients (Supplementary Table S2, S3, S4 and S5), and a threshold was set at the 50th percentile. The higher the risk score, the larger mortality risk for LUAD patients. Then the patients of the TCGA cohort II were divided into high- and low-risk groups based on the threshold. High-risk patients, as defined by the four groups of prognosis-related key genes based on the risk score, had statistically significantly worse prognosis in TCGA cohort II (Fig. 2A, B, C and D). Then through the assessment of the four survival risk score systems (Table 1), the RSF model showed higher predictive power than the Cox model, and the predictive power of clinically-integrated RNA-Seq data was higher than RNA-Seq data. In short, the RSF model and clinically-integrated RNA-Seq data could predict the prognosis of LUAD patients well (HR = 3.80, 95% CI: 2.20-6.55, p = 1.63e-06) (Fig. 2D).
Figure 2.

Validation of the prognosis-related key genes in the TCGA cohort II. A) The KM survival curve was generated in the TCGA cohort II by the Cox model and RNA-Seq data. Patients of the TCGA cohort II were divided into high- and low-risk groups based on the 50th percentile of risk score. B) The KM survival curve was generated in the TCGA cohort II by the Cox model and clinically-integrated RNA-Seq data. Patients of the TCGA cohort II were divided into high- and low-risk groups based on the 50th percentile of risk score. C) The KM survival curve was generated in the TCGA cohort II by the RSF model and RNA-Seq data. Patients of the TCGA cohort II were divided into high- and low-risk groups based on the 50th percentile of risk score. D) The KM survival curve was generated in the TCGA cohort II by the RSF model and clinically-integrated RNA-Seq data. Patients of the TCGA cohort II were divided into high- and low-risk groups based on the 50th percentile of risk score.
Table 1.
Survival analysis based on the TCGA cohort II and GSE72094 cohort
| TCGA cohort II | GSE72094 cohort | |||||
|---|---|---|---|---|---|---|
| model | HRa (95% CIb) | p | C-indexc | HR (95% CI) | p | C-index |
| model 1d | 2.07 (1.25-3.41) | 4.38e-03 | 0.610 | 2.22 (1.50-3.28) | 6.30e-05 | 0.615 |
| model 2e | 2.72 (1.64-4.50) | 1.02e-04 | 0.645 | 2.77 (1.85-4.15) | 8.28e-07 | 0.630 |
| model 3f | 2.53 (1.53-4.17) | 2.88e-04 | 0.643 | 2.94 (1.95-4.43) | 2.94e-07 | 0.623 |
| model 4g | 3.80 (2.20-6.55) | 1.63e-06 | 0.656 | 4.12 (2.68-6.35) | 1.34e-10 | 0.672 |
aHR = hazard ratio; bCI = confidence interval; cC-index = concordance index; dmodel 1: the Cox model and RNA-Seq data; emodel 2: the Cox model and clinically-integrated RNA-Seq data; fmodel 3: the RSF model and RNA-Seq data; gmodel 4: the RSF model and clinically-integrated RNA-Seq data.
The C-index
The C-index for the four sets of key genes in the TCGA cohort II was showed in Table 1. The C-index was mainly used to calculate the degree of discrimination between the predicted value of the Cox model and the reality in the survival analysis 39. We calculated the C-index for both the Cox model and the RSF model, the C-index increased for clinically-integrated RNA-Seq data in comparison to the RNA-Seq data. In addition, the C-index of the RSF model was larger than that of the Cox model.
External validation of key genes in the GSE72094 cohort
The KM curves
The above-mentioned models were further validated by using the GSE72094 cohort data (n=393). We performed the Cox survival analysis for LUAD patients from the GSE72094 cohort (Supplementary Table S7), the p value for stage was much less than 0.05. The risk score of each patient was calculated based on the expression data of the four sets of prognosis-related key genes. We divided 393 LUAD patients of the GSE72094 cohort into high- and low-risk groups, based on the 50th percentile of risk score. The KM curves of the four sets of prognosis-related key genes all indicated that high-risk patients had statistically significantly worse prognosis (Fig. 3A, B, C and D). Furthermore, we compared the four KM survival curves obtained by the four groups of key genes (Table 1), the key genes obtained by the RSF model and clinically-integrated RNA-Seq data can predict the prognosis of LUAD patients well (HR = 4.12, 95% CI: 2.68-6.35, p = 1.34e-09) (Fig. 3D).
Figure 3.

Validation of the prognosis-related key genes in the GSE72094 cohort. A) The KM survival curve was generated in the GSE72094 cohort by the Cox model and RNA-Seq data. Patients of the GSE72094 cohort were divided into high- and low-risk groups based on the 50th percentile of risk score. B) The KM survival curve was generated in the GSE72094 cohort by the Cox model and clinically-integrated RNA-Seq data. Patients of the GSE72094 cohort were divided into high- and low-risk groups based on the 50th percentile of risk score. C) The KM survival curve was generated in the GSE72094 cohort by the RSF model and RNA-Seq data. Patients of the GSE72094 cohort were divided into high- and low-risk groups based on the 50th percentile of risk score. D) The KM survival curve was generated in the GSE72094 cohort by the RSF model and clinically-integrated RNA-Seq data. Patients of the GSE72094 cohort were divided into high- and low-risk groups based on the 50th percentile of risk score.
The C-index
The C-index for the four sets of key genes in the GSE72094 cohort was showed in Table 1. We calculated the C-index for both the Cox model and the RSF model, the C-index increased for clinically-integrated RNA-Seq data in comparison to the RNA-Seq data. In addition, the C-index of the RSF model was larger than that of the Cox model. This result is similar to the TCGA cohort II.
Heat map for the sixteen key genes
We verified the sixteen key genes obtained by the RSF model and clinically-integrated RNA-Seq data based on the GSE72094 cohort. Due to the difference of data preprocessing between GEO and TCGA database, GSE72094 cohort only contains eleven common genes among the sixteen key genes. To comprehensively investigate the expression of eleven genes in high- and low-risk groups, we plotted a heat map using the RNA-Seq data of eleven genes, with red color indicating higher expression and green color indicating lower expression (Fig. 4A). The heat map roughly showed that the higher the patient's risk score, the greater the gene expression value. We next performed a KEGG pathway enrichment analysis for the eleven genes by using the DAVID. These eleven genes were statistically significantly enriched for Nicotine addiction (Fig. 4B).
Figure 4.

The analysis of the sixteen prognosis-related key genes. A) Heat map for the key genes obtained by the RSF model and clinically-integrated RNA-Seq data. The abscissa indicates genes, and the ordinate indicates 393 samples from GSE72094. The rightmost column is the patient's risk score, sorted by ascending order from top to bottom. The low-risk group is above the red dashed line, the high-risk group is under the red dashed line. B) KEGG enrichment pathway analysis of the key genes obtained by the RSF model and clinically-integrated RNA-Seq data. C) Protein-gene network. The yellow hexagons indicate the genes obtained from the RSF model and clinically-integrated RNA-Seq data, and the orange-red ovals indicate the associated proteins. D) The KM survival curve of cross-tumor model.
External validation of key genes in the GSE11969 cohort
These models were further validated by using the GSE11969 cohort data (n = 149). We performed the Cox survival analysis for LUAD patients from the GSE11969 cohort (Supplementary Table S8), the p values for stage and age were much less than 0.05. The 149 LUAD patients were divided into high- and low-risk groups by using the same method as the GSE72094 cohort. The KM curves of the four sets of prognosis-related key genes all indicated that high-risk patients had statistically significantly worse prognosis (Supplementary Figure S1A, B, C and D). The C-index for the four sets of key genes in the GSE11969 cohort were showed in the Supplementary Table S9. Furthermore, we compared the four KM survival curves and the C-index derived from the four groups of key genes (Supplementary Table S9), the key genes obtained by the RSF model and clinically-integrated RNA-Seq data can predict the prognosis of LUAD patients well (HR = 3.87, 95% CI: 2.27-6.61, p = 6.81e-07) (Supplementary Figure S1D). In addition, by comparing the sixteen genes found in this study with the prognostic features of five published lung cancers (Table 2), we found that sixteen gene prognostic features were statistically significant in the three validation cohorts.
Table 2.
Comparison of the sixteen-gene prognostic signatures to the five published lung cancer prognostic signatures
| studies | TCGA cohort II | GSE72094 cohort | GSE11969 cohort | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | p | C-index | HR (95% CI) | p | C-index | HR (95% CI) | p | C-index | |
| Present study 16-gene signature |
3.80 (2.20-6.55) | 1.63e-06 | 0.656 | 4.12 (2.68-6.35) | 1.34e-10 | 0.672 | 3.87 (2.27-6.61) | 6.81e-07 | 0.670 |
| Shukla et al. 4-gene signature |
2.24 (1.36-3.68) | 1.48e-03 | 0.613 | 3.01 (2.00-4.51) | 1.07e-07 | 0.639 | 3.02 (1.85-4.92) | 9.42e-06 | 0.641 |
| Boutros et al. 6-gene signature |
1.70 (1.04-2.77) | 3.49e-02 | 0.561 | 3.17 (2.10-4.79) | 3.84e-08 | 0.656 | 3.09 (1.89-5.03) | 6.10e-06 | 0.646 |
| Chen et al. 5-gene signature |
2.20 (1.34-3.61) | 1.93e-03 | 0.625 | 2.83 (1.88-4.27) | 7.07e-07 | 0.631 | 3.09 (1.89-5.03) | 6.27e-06 | 0.644 |
| Lau et al. 3-gene signature |
1.80 (1.10-2.94) | 1.88e-02 | 0.588 | 2.81 (1.88-4.22) | 5.55e-07 | 0.625 | 2.62 (1.62-4.23) | 7.90e-05 | 0.628 |
| Bianchi et al. 10-gene signature |
3.13 (1.84-5.34) | 2.78e-05 | 0.619 | 2.92 (1.94-4.39) | 2.53e-07 | 0.640 | 3.41 (2.08-5.59) | 1.23e-06 | 0.655 |
Interdependency of the sixteen key prognostic genes
The sixteen key genes were LINC00908, PITX3, GJB3, CRCT1, MELTF, BAIAP2L2, RHOV, GABRA2, ARF3, TRIM7, KRT18, ZNF710.AS1, LOC105370802, LOC100996732, SFTPB and DKK1. Proteins interacting with these sixteen key genes were searched in starbase2.0 (http://starbase.sysu.edu.cn/starbase2/index.php). The results were exported as the nodes and edges of a protein-gene network and were visualized by Cytoscape-v3.6.1. In the protein-gene network, the key genes were mainly associated with nine proteins (Fig. 4C).
Patient prognosis prediction using cross-tumor models
In order to test whether the LUAD patients data could identify commonalities across LUSC, we used the sixteen key gene-trained models obtained from the RSF models and clinically-integrated RNA-Seq data to predict the prognostic characteristics of LUSC patients (n = 486) from the TCGA database. The LUSC patients were divided into high- and low-risk groups according to the 50th percentile of risk score. The KM survival curve showed that the high-risk patients had statistically significantly worse prognosis (HR = 1.58, 95% CI: 1.20-2.07, p = 1.21e-03) (Fig. 4D).
Discussion
Lung cancer is the leading cause of cancer-related mortality, non-small cell lung cancer (NSCLC) accounts for about 80% of the total number of lung cancer patients, while LUAD accounts for more than 40% of NSCLC 40-43. In an effort to bolster clinical tools and biological understanding in LUAD, we presented the RNA-Seq prognostic signatures. We found 1,113 survival-related seed genes from 24,991 genes in the TCGA cohort I using the RSF model, and screened sixteen prognosis-related key genes from the 1,113 clinically-integrated seed genes in TCGA cohort I using the forward selection model. Subsequently the sixteen gene signatures were validated in the TCGA cohort II and the GSE72094 cohort. The sixteen gene signatures can improve the power for predicting the prognosis of LUAD patients.
Previous studies had also found genes associated with lung cancer using methods such as decision trees and Cox models. The sixteen gene prognostic markers obtained in this study had the highest HR and C-index and the lowest p of likelihood ratio compared to the prognostic genes identified from these existing studies. This further validated the effectiveness of using the RSF model and clinically-integrated RNA-Seq data to predict prognosis for LUAD patients.
This study proposed a method for predicting LUAD prognosis markers based on machine learning. The RSF is computed from a set of binary decision trees and can be used to select the most important variables that are linked with time to event 44. The Cox model is a traditional regression model which is not based on any assumptions about the nature or shape of the underlying survival distribution 45, 46. The advantage of RSF is that it is more suitable for the automation of survival analysis than the Cox model, because it requires less input from the user in the highly relevant data settings for covariates 45. In addition, by means of the forward selection model, we can identify multiple sets of genes rather than one large set of genes while adjusting for risk factors.
The expressions of the sixteen key genes were closely associated with the LUAD prognosis. Pubmed was searched for articles on these sixteen key genes related to LUAD. Up to Nov 16, 2018, except for RHOV, SFTPB and DKK1, all other genes among the sixteen key genes were the first time to be identified in LUAD samples. In previous study, RHOV is an atypical RHO GTPase that has been nominated as upregulated in non-small cell lung cancer in a minor study 47. Pro-SFTPB is over expressed in non-small cell lung cancer, especially in LUAD 48. In a multivariate analysis of patients with LUAD, DKK1 was independently associated with poor survival 49. This result may play a guiding role in prognosis prediction and targeted therapy in patients with LUAD.
The RNA-Seq data for individualized therapy intensification has the advantages of high throughput, high sensitivity and high efficiency 12, 50. In addition, the sixteen gene signatures may provide clues for targeted therapy. However, this study also had some limitations. Due to data limitation of the public databases, we only considered three clinical variables (age, gender and tumor stage) in the forward selection model, and future studies may include more clinical variables into the model. The derived prognostic model also needs be validated using more independent cohorts and biological experiments.
In this study, first of all, a machine-learning-based method (RSF) is proposed to identify the seed genes for LUAD prognosis. Second, the clinical data (such as stage, age) are integrated with RNA-Seq data to improve prediction accuracy. Third, three independent cohorts (the TCGA cohort II, GSE72094 cohort and GSE11969 cohort) are utilized to validate the proposed method and the results show that the derived sixteen-gene signature is also superior to the previous five prediction models. Fourth, bioinformatics analyses have been used to the identified seed genes and key genes, and thirteen of the sixteen genes are reported as the key genes related to LUAD prognosis for the first time, which could contribute to the new findings of our study distinct with the previous studies. Finally, we also discussed some limitations of this study. To the best of our knowledge, the average 5-year survival rate for LUAD patients is only 15%, and the sixteen gene markers found in this study are especially vital for improving the prognosis prediction of LUAD patients. In the future, we could consider the application of advanced machine learning methods, such as deep learning for the prognostic prediction of cancer, and provide more powerful tools for improving targeted therapy.
Supplementary Material
Supplementary figure and tables.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (61471078, 81974439); the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry; the visiting scholar grant of the China Scholarship Council (201806575028); the Program for Liaoning Excellent Talents in University (LJQ2015011); the Fundamental Research Funds for the Central Universities (3132014306, 3132015213, 3132017075).
Abbreviations
- AIC
akaike information criterion
- CHF
Cumulative Hazard Function
- C-index
concordance index
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- lncRNAs
long non-coding RNAs
- LUAD
Lung adenocarcinoma
- LUSC
lung squamous cell carcinoma
- NSCLC
non-small cell lung cancer
- RF
random forests
- RPKM
Reads per Kilo-base per Million mapped
- RSF
random survival forest
- TCGA
the Cancer Genome Atlas
References
- 1.Li L, Feng T, Qu J, Feng N, Wang Y, Ma RN. et al. LncRNA Expression Signature in Prediction of the Prognosis of Lung Adenocarcinoma. Genet Test Mol Biomarkers. 2018;22(1):20–28. doi: 10.1089/gtmb.2017.0194. [DOI] [PubMed] [Google Scholar]
- 2.Desantis CE, Siegel RL, Sauer AG, Miller KD, Fedewa SA, Alcaraz KI. et al. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA Cancer J Clin. 2016;66(4):290–308. doi: 10.3322/caac.21340. [DOI] [PubMed] [Google Scholar]
- 3.Torre L, Bray F, Siegel R, Ferlay J, Lortettieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 4.Galvan A, Frullanti E, Anderlini M, Manenti G, Noci S, Dugo M. et al. Gene expression signature of non-involved lung tissue associated with survival in lung adenocarcinoma patients. Carcinogenesis. 2013;34(12):2767–73. doi: 10.1093/carcin/bgt294. [DOI] [PubMed] [Google Scholar]
- 5.Shukla S, Evans JR, Malik R, Feng FY, Dhanasekaran SM, Cao X. et al. Development of a RNA-Seq Based Prognostic Signature in Lung Adenocarcinoma. J Natl Cancer Inst. 2016;109(1):djw200. doi: 10.1093/jnci/djw200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB. et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol. 2015;10(9):1243–1260. doi: 10.1097/JTO.0000000000000630. [DOI] [PubMed] [Google Scholar]
- 7.Yang ZH, Zheng R, Gao Y, Zhang Q, Zhang H. Abnormal gene expression and gene fusion in lung adenocarcinoma with high-throughput RNA sequencing. Cancer Gene Ther. 2014;21(2):74–82. doi: 10.1038/cgt.2013.86. [DOI] [PubMed] [Google Scholar]
- 8.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH. et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–89. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 9.Barrett CL, Christopher DB, Kristen J, Saenz CC, Carson DA, Frazer KA. Systematic transcriptome analysis reveals tumor-specific isoforms for ovarian cancer diagnosis and therapy. Proc Natl Acad Sci U S A. 2015;112(23):E3050–7. doi: 10.1073/pnas.1508057112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu CQ, Tsao MS. Prognostic markers in lung cancer: is it ready for prime time? Transl Lung Cancer Res. 2014;3(3):149–58. doi: 10.3978/j.issn.2218-6751.2014.06.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park YY, Park ES, Sang BK, Sang CK, Bo HS, Chu IS. et al. Development and Validation of a Prognostic Gene-Expression Signature for Lung Adenocarcinoma. Plos One. 2012;7(9):e44225. doi: 10.1371/journal.pone.0044225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B, Cui Y, Diehn M, Li R. Development and Validation of an Individualized Immune Prognostic Signature in Early-Stage Nonsquamous Non-Small Cell Lung Cancer. Jama Oncol. 2017;3(11):1529–1537. doi: 10.1001/jamaoncol.2017.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15(6):423–37. doi: 10.1038/nrg3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–41. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 15.Zheng S, Zheng D, Dong C, Jiang J, Xie J, Sun Y. et al. Development of a novel prognostic signature of long non-coding RNAs in lung adenocarcinoma. J Cancer Res Clin Oncol. 2017;143(9):1649–1657. doi: 10.1007/s00432-017-2411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan Y, Allen EMV, Omberg L, Wagle N, Aminmansour A, Sokolov A. et al. Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat Biotechnol. 2014;32(7):644–52. doi: 10.1038/nbt.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deo RC. Machine Learning in Medicine. Circulation. 2015;132(20):1920–30. doi: 10.1161/CIRCULATIONAHA.115.001593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erickson BJ, Korfiatis P, Akkus Z, Kline TL. Machine Learning for Medical Imaging. Radiographics. 2017;37(2):505–515. doi: 10.1148/rg.2017160130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishwaran H, Kogalur U B, Blackstone E H. et al. Random survival forests. The Annals of Applied Statistics. 2008;2(3):841–860. [Google Scholar]
- 20.Nasejje JB, Mwambi H, Dheda K, Lesosky M. A comparison of the conditional inference survival forest model to random survival forests based on a simulation study as well as on two applications with time-to-event data. BMC Med Res Methodol. 2017;17(1):115. doi: 10.1186/s12874-017-0383-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo D, Deng B, Weng M, Luo Z, Nie X. A prognostic 4-lncRNA expression signature for lung squamous cell carcinoma. Artif Cells Nanomed Biotechnol. 2018;46(6):1207–1214. doi: 10.1080/21691401.2017.1366334. [DOI] [PubMed] [Google Scholar]
- 22.Taylor JMG. Random Survival Forests. J Thorac Oncol. 2011;6(12):1974–5. doi: 10.1097/JTO.0b013e318233d835. [DOI] [PubMed] [Google Scholar]
- 23.Leblanc M, Crowley J. Survival Trees by Goodness of Split. Journal of the American Statistical Association. 1993;88(422):457–67. [Google Scholar]
- 24.Ruyssinck J, van der Herten J, Houthooft R. et al. Random Survival Forests for Predicting the Bed Occupancy in the Intensive Care Unit. Comput Math Methods Med. 2016;2016:7087053. doi: 10.1155/2016/7087053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breiman L. Random Forests. Machine Learning. 2001;45(1):5–32. [Google Scholar]
- 26.Ishwaran H, Gerds TA, Kogalur UB, Moore RD, Gange SJ, Lau BM. Random survival forests for competing risks. Biostatistics. 2014;15(4):757–73. doi: 10.1093/biostatistics/kxu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishwaran H, Kogalur UB, Gorodeski EZ, Minn AJ, Lauer MS. High-Dimensional Variable Selection for Survival Data. Journal of the American Statistical Association. 2010;105(489):205–17. [Google Scholar]
- 28.Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC. et al. DAVID: Database for Annotation, visualization, and Integrated Discovery. Genome Biol. 2003;4(5):P3. [PubMed] [Google Scholar]
- 29.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 30.Wang JY, Tai JJ. Robust Quantitative Trait Association Tests in the Parent-Offspring Triad Design: Conditional Likelihood-Based Approaches. Ann Hum Genet. 2009;73(2):231–44. doi: 10.1111/j.1469-1809.2008.00502.x. [DOI] [PubMed] [Google Scholar]
- 31.Renaud G, Stenzel U, Maricic T, Wiebe V, Kelso J. deML: robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics. 2015;31(5):770–2. doi: 10.1093/bioinformatics/btu719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kendall WL, Brownie C, Pollock KH. A Likelihood-based Approach to Capture Recapture Estimation of Demographic Parameters Under the Robust Design. Biometrics. 1995;51(1):293–308. [PubMed] [Google Scholar]
- 33.Chen H, Sun X, Ge W, Qian Y, Bai R, Zheng S. A seven-gene signature predicts overall survival of patients with colorectal cancer. Oncotarget. 2016;8(56):95054–95065. doi: 10.18632/oncotarget.10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schroder MS, Culhane AC, Quackenbush J, Haibe-Kains B. survcomp: an R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics. 2011;27(22):3206–8. doi: 10.1093/bioinformatics/btr511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bianchi F, Nuciforo P, Vecchi M, Bernard L, Tizzoni L, Marchetti A. et al. Survival prediction of stage I lung adenocarcinomas by expression of 10 genes. J Clin Invest. 2007;117(11):3436–44. doi: 10.1172/JCI32007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boutros PC, Lau SK, Pintilie M, Liu N, Shepherd FA, Der SD. et al. Prognostic gene signatures for non-small-cell lung cancer. Proc Natl Acad Sci U S A. 2009;106(8):2824–8. doi: 10.1073/pnas.0809444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A. et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med. 2007;356(1):11–20. doi: 10.1056/NEJMoa060096. [DOI] [PubMed] [Google Scholar]
- 38.Lau SK, Boutros PC, Pintilie M, Blackhall FH, Zhu CQ, Strumpf D. et al. Three-gene prognostic classifier for early-stage non small-cell lung cancer. J Clin Oncol. 2007;25(35):5562–9. doi: 10.1200/JCO.2007.12.0352. [DOI] [PubMed] [Google Scholar]
- 39.Brentnall AR, Cuzick J. Use of the concordance index for predictors of censored survival data. Stat Methods Med Res. 2018;27(8):2359–2373. doi: 10.1177/0962280216680245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giordano M, Boldrini L, Servadio A, Niccoli C, Melfi F, Lucchi M. et al. Differential microRNA expression profiles between young and old lung adenocarcinoma patients. Am J Transl Res. 2018;10(3):892–900. [PMC free article] [PubMed] [Google Scholar]
- 41.Thunnissen E, Oord KVD, Bakker MD. Prognostic and predictive biomarkers in lung cancer. A review. Virchows Arch. 2014;464(3):347–58. doi: 10.1007/s00428-014-1535-4. [DOI] [PubMed] [Google Scholar]
- 42.Vescovo VD, Denti MA. microRNA and Lung Cancer. Adv Exp Med Biol. 2015;889:153–77. doi: 10.1007/978-3-319-23730-5_9. [DOI] [PubMed] [Google Scholar]
- 43.Ma L, Wen Z. Risk factors and prognosis of pulmonary embolism in patients with lung cancer. Medicine. 2017;96(16):e6638. doi: 10.1097/MD.0000000000006638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dietrich S, Floegel A, Troll M, Kühn T, Rathmann W, Peters A. et al. Random Survival Forest in practice: a method for modelling complex metabolomics data in time to event analysis. Int J Epidemiol. 2016;45(5):1406–1420. doi: 10.1093/ije/dyw145. [DOI] [PubMed] [Google Scholar]
- 45.Datema FR, Moya A, Krause P, Bäck T, Willmes L, Langeveld T. et al. Novel head and neck cancer survival analysis approach: Random survival forests versus cox proportional hazards regression. Head Neck. 2015;34(1):50–8. doi: 10.1002/hed.21698. [DOI] [PubMed] [Google Scholar]
- 46.Liu C, Wang X, Genchev GZ, Lu H. Multi-omics facilitated variable selection in Cox-regression model for cancer prognosis prediction. Methods. 2017;124:100–107. doi: 10.1016/j.ymeth.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 47.Shepelev MV, Korobko IV. The RHOV gene is overexpressed in human non-small cell lung cancer. Cancer Genet. 2013;206(11):393–7. doi: 10.1016/j.cancergen.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 48.He Y, Jiang Z, Tong F, Li M, Yin X, Hu S. et al. Experimental study of peripheral-blood pro-surfactant protein B for screening non-small cell lung cancer. Acta Cir Bras. 2017;32(7):568–575. doi: 10.1590/s0102-865020170070000008. [DOI] [PubMed] [Google Scholar]
- 49.Chambard L, Girard N, Ollier E, Rousseau JC, Duboeuf F, Carlier MC. et al. Bone, muscle, and metabolic parameters predict survival in patients with synchronous bone metastases from lung cancers. Bone. 2018;108:202–209. doi: 10.1016/j.bone.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 50.Han Y, Gao S, Muegge K, Zhang W, Zhou B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 2015;9(Suppl 1):29–46. doi: 10.4137/BBI.S28991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure and tables.