

SUMMARY
DAPK1 binding to GluN2B was prominently reported to mediate ischemic cell death in vivo. DAPK1 and CaMKII bind to the same GluN2B region, and their binding is mutually exclusive. Here, we show that mutating the binding region on GluN2B (L1298A/ R1300Q) protected against neuronal cell death induced by cardiac arrest followed by resuscitation. Importantly, the GluN2B mutation selectively abolished only CaMKII, but not DAPK1, binding. During ischemic or excitotoxic insults, CaMKII further accumulated at excitatory synapses, and this accumulation was mediated by GluN2B binding. Interestingly, extra-synaptic GluN2B decreased after ischemia, but its relative association with DAPK1 increased. Thus, ischemic neuronal death requires CaMKII binding to synaptic GluN2B, whereas any potential role for DAPK1 binding is restricted to a different, likely extra-synaptic population of GluN2B.
In Brief
Ischemic insults cause excitotoxic neuronal cell death via NMDA receptor overstimulation. Buonarati et al. find that excitotoxic insults cause DAPK1 movement to extra-synaptic NMDA receptors and CaMKII movement to synaptic NMDA receptors; importantly, preventing this CaMKII movement protects neurons from ischemic death.
Graphical Abstract
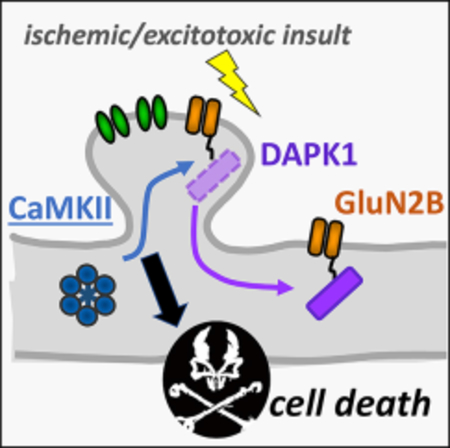
INTRODUCTION
The Ca2+/calmodulin-dependent protein kinase II (CaMKII) and the death-associated protein kinase 1 (DAPK1) are both members of the CaM kinase family (reviewed in Bayer and Schulman, 2019). Both bind to the NMDA-type glutamate receptor (NMDAR) subunit GluN2B but with opposing physiological consequences: although CaMKII binding is required for normal long-term potentiation (LTP) (Barria and Malinow, 2005; Halt et al., 2012), DAPK1 instead mediates the opposing form of synaptic plasticity, long-term depression (LTD) (Goodell et al., 2017). Indeed, the DAPK1 function in LTD is the suppression of CaMKII/GluN2B binding, and binding of DAPK1 and CaMKII to GluN2B is competitive (Goodell et al., 2017), consistent with overlapping binding sites on GluN2B (see Figure 1A). However, this mutually exclusive competitive binding also raises an apparent conundrum, because GluN2B binding of each kinase has been suggested to mediate excitotoxic neuronal cell death: binding of DAPK1 in an in vivo mouse model of stroke (Tu et al., 2010) and binding of CaMKII in neuronal culture models (Vieira et al., 2016). Overlapping binding sites on GluN2B also raised the possibility that the neuroprotective GluN2B-derived peptide that was designed to displace DAPK1 from GluN2B (tatN2Bc; Tu et al., 2010) could additionally interfere with CaMKII binding. Vice versa, the neuroprotective mutations designed to disrupt the CaMKII/GluN2B binding (Strack et al., 2000; Vieira et al., 2016) could potentially additionally disrupt DAPK1 binding. Thus, although there is ample indication for the general involvement of both CaMKII and DAPK1 in ischemic neuronal cell death (Coultrap et al., 2011; Deng et al., 2017; Pei et al., 2014; Shamloo et al., 2005; Velentza et al., 2003; Vest et al., 2010), it is currently not clear which one of these two CaM kinases really needs to specifically bind to GluN2B during the process. Additionally, a recent study questioned any involvement of DAPK1 in ischemic cell death at all (McQueen et al., 2017). Here, we set out to clarify the roles of CaMKII and DAPK1 and their respective binding to GluN2B in ischemic neuronal cell death.
Figure 1. CaMKII Binding to GluN2B Mediates Neuronal Cell Death after Resuscitation from Cardiac Arrest.

Error bars indicate SEM in all panels.
(A) Schematic representation of GluN2B, including the sequence of the overlapping binding region for DAPK1 and CaMKII.
(B) Experimental timeline for the CA/CPR model of global cerebral ischemia.
(C) Representative micrographs of the hippocampal CA1 region, with cell death visualized by H&E staining.
(D) Quantification of neuronal cell death in the CA1 region after CA/CPR. GluN2BDCaMKII mutant mice showed significantly less cell death compared to WT (*p < 0.05, unpaired two-tailed t test).
(E–L) The GluN2BΔCaMKII mutation (L1298A/R1300Q) prevents binding of CaMKII, but not DAPK1.
(E) The GluN2BΔCaMKII mutation blocks the Ca2+/CaM-induced binding of CaMKII in vitro, as assessed by western analysis of GluN2B-bound CaMKII (***p < 0.001, unpaired two-tailed t test; WT, n = 5; mutant, n = 4).
(F) HEK cells co-expressing labeled CaMKII and GluN2Bc WT or mutant, imaged before and after 5 min Ca2+ stimuli induced with ionomycin.
(G) Quantification showed no significant difference in CaMKII/GluN2B colocalization (assessed by Pearson’s correlation) compared to mCherry/GluN2B control(not significant [NS], p > 0.05, one-way ANOVA; WT, n = 42, mutant, n = 38, mCh control, n = 36).
(H) Quantification after ionomycin treatment showed a dramatic increase (two-way ANOVA followed by Bonferroni test) of CaMKII colocalization with GluN2B WT(***p < 0.001, compared to 0 min), but not with the GluN2BΔCaMKII mutant (#p < 0.05, compared to 0 min; ###p < 0.001, compared to WT).
(I) The GluN2BΔCaMKII mutation allows normal DAPK1 binding (in absence of Ca2+/CaM) in vitro, as assessed by western analysis of GluN2B-bound DAPK1 (NS, p > 0.05, unpaired two-tailed t test; n = 4).
(J) HEK cells co-expressing labeled DAPK1 and GluN2Bc WT or mutant, imaged before and after 5 min Ca2+ stimuli induced with ionomycin.
(K) Quantification showed indistinguishable DAPK1 colocalization with GluN2B WT and GluN2BDCaMKII mutant (NS, p > 0.05, Kruskal-Wallis followed by Bonferroni test; WT, n = 34, mutant, n = 41), compared to mCherry control (*p < 0.05; n = 36).
(L) Quantification after ionomycin treatment showed the same dispersal of DAPK1 from GluN2B, with no differences detected between WT and GluN2BΔCaMKII mutant.
Our in vivo mouse model for global cerebral ischemia closely resembles the most prevalent human condition: the ischemic cell death that occurs after cardiac arrest followed by cardiopulmonary resuscitation (CA/CPR). As with focal cerebral ischemia during stroke, neuronal cell death after global cerebral ischemia is thought to be induced largely by excitotoxicity, i.e., by NMDAR overstimulation caused by the massive release of the excitatory neurotransmitter glutamate during anoxic depolarization (Kostandy, 2012; Rossi et al., 2000; Takata et al., 2005).
Here, we show that the GluN2BΔCaMKII mutation (L1298A/R1300Q; Barcomb et al., 2016; Halt et al., 2012) dramatically reduced hippocampal neuronal cell death after global cerebral ischemia induced in vivo by CA/CPR. Importantly, the GluN2BΔCaMKII mutation abolished GluN2B binding for CaMKII but did not affect DAPK1. Additionally, we tested the effect of excitotoxic or ischemic insults on synaptic trafficking and GluN2B binding of CaMKII versus DAPK1. Together, our results demonstrate that CaMKII binding to synaptic GluN2B mediates ischemic neuronal cell death and suggest that any involvement of DAPK1 binding is restricted to extra-synaptic GluN2B.
RESULTS
The GluN2BΔCaMKII Mutation Provides Neuroprotection after Cardiac Arrest
The GluN2BΔCaMKII mutation combines two point mutations (L1298A and R1300Q) within the shared binding region for DAPK1 and CaMKII (Figure 1A). CaMKII inhibition or T286A mutation strongly protects neurons from the ischemic cell death that is induced by CA/CPR in vivo (Deng et al., 2017). Here, we tested the effect of the GluN2BΔCaMKII mutation. Cardiac arrest was induced in wild-type (WT) and homozygous mutant mice by KCl injection; after 6 min, mice were resuscitated with epinephrine injection, oxygen supply, and chest compressions (Figure 1B). Neuronal cell death was assessed by H&E staining 3 days after CA/CPR, specifically in hippocampal CA1 neurons (Figure 1C), one of the most vulnerable neuron populations during global cerebral ischemia (Horn and Schlote, 1992; Ng et al., 1989; Schmidt-Kastner and Freund, 1991). Compared to their WT littermates, GluN2BDCaMKII mutant mice were significantly protected from neuronal cell death (Figure 1D).
In cortex and cerebellum, only minimal damage was observed here, even in WT mice of our cohort that showed significant hippocampal damage in CA1 (Figure S1). Although consistent with the special vulnerability of CA1, it should be noted that prolonged global cerebral ischemia can cause neuronal cell death also in other brain areas (Pulsinelli, 1985), including in the cerebellum after prolonged CA/CPR (Quillinan et al., 2015).
The GluN2BΔCaMKII Mutation Abolishes Binding of CaMKII
Each of the two point mutations that are combined in the GluN2BΔCaMKII mutant individually reduce CaMKII/GluN2B binding (Strack et al., 2000). Although the combined mutation was likely to be additive, the effects of this combined mutation have not been directly tested. Binding was tested here first using an established in vitro micro-titer plate binding assay using purified recombinant proteins (Barcomb et al., 2014; Bayer et al., 2001). As expected, this double mutation abolished CaMKII/GluN2B binding almost completely (Figure 1E). Next, we tested how the GluN2BΔCaMKII mutation affects CaMKII binding to GluN2B within a cellular environment; this utilized an established colocalization assay in HEK293 cells after co-expression of mCherry-CaMKII and EGFP fused to a membrane-localization sequence and the GluN2B C-terminal tail (GluN2Bc; residues 1,120–1,482; note that this GluN2B fusion protein localizes also to intra-cellular membranes of organelles, such as the endoplasmic reticulum; Goodell et al., 2017). CaMKII binding to GluN2B requires CaMKII activation (either by Ca2+/CaM or by T286 phosphorylation; Bayer et al., 2001; O’Leary et al., 2011; Strack et al., 2000); thus, as expected, CaMKII did not colocalize with either WT or mutant GluN2Bc under basal conditions (Figures 1F and 1G). After imaging baseline localization, cells were stimulated with 10 μM ionomycin to induce Ca2+ influx and were imaged again at 2.5 and 5 min. After ionomycin stimulation, a robust translocation of CaMKII to GluN2Bc was observed for WT, but not for the GluN2BΔCaMKII mutation (Figures 1F and 1H). Thus, as expected, the GluN2BΔCaMKII mutation indeed effectively disrupts CaMKII/ GluN2B binding, both in vitro and within cells.
The GluN2BΔCaMKII Mutation Does Not Affect Binding of DAPK1
In contrast to CaMKII/GluN2B binding, the DAPK1/GluN2B binding was unaffected by the GluN2BΔCaMKII mutation (Figures 1I–1L). The binding was first tested in an in vitro pull-down assay using DAPK1 from HEK293 cell extracts after DAPK1 overexpression (Figure 1I). Note that Ca2+/CaM disrupts the DAPK1/ GluN2B binding (Goodell et al., 2017); thus, Ca2+/CaM was not included in the in vitro binding assays with DAPK1. Additionally, we assessed DAPK1/GluN2B binding within HEK293 cells after co-expression of mCherry-DAPK1 and EGFP-GluN2Bc. As reported previously (Goodell et al., 2017), DAPK1 basally localized with GluN2Bc; consistent with the in vitro binding assay, this basal localization was not affected by the GluN2BΔCaMKII mutation (Figures 1J and 1K). Upon ionomycin treatment to induce a Ca2+ stimulus, DAPK1 dispersed to equal extent from GluN2Bc WT and the GluN2BΔCaMKII mutant (Figures 1J and 1L). Thus, the GluN2BΔCaMKII mutation does not affect the DAPK1/GluN2B binding or its disruption by Ca2+ stimuli.
Neuroprotective Inhibitors of CaMKII and DAPK1 Do Not Cross-React
Although our results directly demonstrate that CaMKII/GluN2B binding is involved in ischemic cell death, they do not rule out a potential additional involvement of DAPK1. Notably, DAPK1 involvement has been controversial: although one study described a specific requirement for DAPK1/GluN2B binding in ischemic cell death (Tu et al., 2010), a more recent study questioned any DAPK1 involvement whatsoever (McQueen et al., 2017). Thus, we decided to determine the selectivity of demonstrated neuroprotective peptides in preventing CaMKII versus DAPK1 binding to GluN2B, using our in vitro binding assays (Figure 2A). The CaMKII inhibitor peptide tatCN21 (which binds to CaMKII at the same site as GluN2B; Vest et al., 2007) specifically interfered with GluN2B binding of CaMKII, but not DAPK1. More importantly, the tat2B peptide (which is derived from the binding site on GluN2B and shown to disrupt DAPK1/GluN2B binding in vivo; Tu et al., 2010) instead specifically interfered with GluN2B binding of DAPK1, but not CaMKII (Figure 2A). Thus, neuroprotection by tat2B is not mediated by off-target effects on CaMKII (although it may instead be due to off-target effects on NMDAR function; McQueen et al., 2017).
Figure 2. Selectivity of Neuroprotective Agents Targeting CaMKII versus DAPK1.

(A) CaMKII versus DAPK1 binding to GluN2B in vitro was reduced only by neuroprotective peptides designed to target the respective kinase, i.e., by 5 μM tatCN21 for CaMKII and by 50 μM tat2Bc for DAPK1. The neuroprotective DAPK1 small molecule inhibitor DAPK1-TC6 (TC; 1 μM) did not interfere with GluN2B binding for either kinase, as expected (*p < 0.05, one-way ANOVA followed by Dunnett’s test; n = 4). Error bars indicate SEM in these panels. (B) CaMKII activity (measured by ATP depletion in vitro) was inhibited by tatCN21, but not by the DAPK1 inhibitor TC6 (each at 1 μM; p < 0.05, Kruskal-Wallis followed by Dunn’s test).
TC-DAPK-6 (TC-6) is a small molecule inhibitor of DAPK1 that is ATP competitive and thus expected to interfere only with enzymatic DAPK1 activity, not GluN2B binding. Indeed, TC-6 did not interfere with GluN2B binding of either kinase (Figure 2A) and did not interfere with activity of CaMKII (Figure 2B).
Excitotoxic CaMKII Movement to Dendritic Spines Is Mediated by GluN2B Binding
Biochemical fractionation indicated that CaMKII further accumulates in synaptosomes after GCI induced by CA/CPR (Deng et al., 2017). Here, we first used expression of YFP-CaMKII in order to live-monitor its accumulation in hippocampal neurons exposed to excitotoxic glutamate stimuli (Figures 3A and S2A). As expected, YFP-CaMKII quickly accumulated at excitatory dendritic spine synapses (identified by morphology and by the marker protein PSD95), but not at inhibitory synapses (identified by the marker protein gephyrin). In order to monitor the movement also for endogenous CaMKII, we used our recently described FingR intrabody-based method for simultaneous live imaging of three endogenous proteins, specifically for CaMKII and the two synaptic marker proteins PSD95 and gephyrin (Cook et al., 2019). As expected, endogenous CaMKII also accumulated at excitatory dendritic spine synapses (Figures 3B and S2B). Here, the live-imaging method was adapted for use in mouse neurons, in order to monitor endogenous CaMKII movement also in transgenic neurons. Importantly, the CaMKII movement to excitatory synapses in response to excitotoxic insults was abolished in neurons from the GluN2BΔCaMKII mutant mice (Figures 3C and S2C). Thus, like the CA/CPR-induced neuronal cell death, CaMKII movement to excitatory synapses during excitotoxic insults requires CaMKII/GluN2B binding.
Figure 3. CaMKII Accumulates at Synaptic GluN2B after Excitotoxic Glutamate.

Error bars indicate SEM in all panels. Shown is the movement of CaMKII to excitatory versus inhibitory synapses (marked by PSD95, red, or gephyrin, blue) that is induced by excitotoxic glutamate stimuli. Scale bars indicate 10 μm. Statistical analysis was performed by paired t test (***p < 0.001; NS, p > 0.05).
(A and B) Overexpressed YFP-CaMKII (A) expressed in rat hippocampal neurons and endogenous CaMKII (B) in hippocampal neurons from WT mice (detected by an intrabody) moves to excitatory synapses.
(C) This movement of endogenous CaMKII is blocked in neurons prepared from the GluN2BΔCaMKII mice.
DAPK1 Dispersal from Dendritic Spines May Promote Binding to Extra-synaptic GluN2B
In contrast to CaMKII, mCherry-DAPK1 was highly basally enriched at excitatory dendritic spine synapses but then quickly dispersed in response to excitotoxic glutamate stimuli (Figures 4A and S2D). As no intrabody is available for DAPK1, movement of endogenous DAPK1 was instead monitored by biochemical fractionation (Figures 4B and S3A). Consistent with our imaging results, CaMKII accumulated in a crude synaptic fraction after excitotoxic glutamate simulation although DAPK1 instead moved out (Figure 4B); however, although DAPK1 dispersal from synapses was apparent immediately, it only became statistically significant at later time points (Figure 4B). These results strongly suggest that excitotoxic stimuli do enhance only CaMKII, but not DAPK1, binding to synaptic GluN2B.
Figure 4. DAPK1 Disperses from Dendritic Spines and Synaptic Fractions after In Vitro and In Vivo Insults.

Error bars indicate SEM in all panels.
(A) Excitotoxic glutamate stimuli disperse mCh-DAPK1 from excitatory synapses (identified by PSD95 in red; ***p < 0.001; NS, p > 0.05; paired two-tailed t test). No change was observed at inhibitory synapses (identified by gephyrin in blue). Scale bar indicates 10 μm.
(B–D) All quantifications of western blot analysis were normalized to actin staining, and fractions were verified by enrichment of PSD95 staining as synaptic marker (as shown in Figure S3A). Synaptic and extra-synaptic fractions were prepared by Triton X-100 extraction followed by ultracentrifugation, with the pellet containing synaptic protein. In order to highlight changes induced by the insults, values in all fractions were normalized to the levels detected before the insults.
(B) Endogenous protein in a crude synaptic fraction from cultured hippocampal neurons before and after excitotoxic glutamate stimuli, analyzed by western blot. The amount of DAPK1 was significantly decreased 2 h after 5-min excitotoxic stimuli, compared to control (***p < 0.001, one-way ANOVA followed by Dunnett’s test). The amount of CaMKII increased at each time point compared to control (*p < 0.05, **p < 0.005).
(C) Endogenous protein in a crude synaptic versus extra-synaptic hippocampal fractions, analyzed by western blot. CA/CPR induced a decrease in synapticDAPK1 and an increase in extra-synaptic DAPK1, compared to sham (**p < 0.005, *p < 0.05, unpaired two-tailed t test). Ischemic injury also resulted in increased synaptic CaMKII and decreased extra-synaptic CaMKII, compared to sham (**p < 0.005, unpaired two-tailed t test).
(D) Extra-synaptic GluN2B decreased and synaptic GluN2B increased following CA/CPR (**p < 0.005, ***p < 0.001, unpaired two-tailed t test).
Next, we decided to examine the effect of our CA/CPR in vivo injury model on the movement of CaMKII versus DAPK1 among the hippocampal synaptic versus extra-synaptic fractions (Figures 4C and S3A). Consistent with our cell culture model, the crude synaptic fraction showed an increase in CaMKII and decrease in DAPK1; corresponding opposite changes were detected in the extra-synaptic fractions (Figures 4C and S3A). Interestingly, GluN2B was found basally mainly in the synaptic fraction, and CA/CPR caused a further sharp decrease in extra-synaptic GluN2B (Figure 4D); a similar effect was also seen after excitotoxic glutamate treatments in vitro (Figure S3B). However, consistent with a previous report (Tu et al., 2010), co-immunoprecipitation experiments indicated an increased relative association of DAPK1 with this overall decreased extra-synaptic pool of GluN2B (Figures S3C and S3D).
Together, our data indicate that CaMKII binding to synaptic GluN2B is required for neuronal cell death after ischemia, whereas a potential role of DAPK1 binding is restricted to a decreasing extra-synaptic GluN2B population.
DISCUSSION
DAPK1 binding to the NMDA-receptor subunit GluN2B has been prominently indicated to mediate ischemic neuronal cell death in vivo (Tu et al., 2010). However, here, we provide direct genetic evidence showing instead a requirement for CaMKII binding. This posed a conundrum, as DAPK1 and CaMKII binding occurs at overlapping sites on GluN2B (see Figure 1A) and is mutually exclusive (Goodell et al., 2017). Clearly, only one of the two kinases can bind to a single GluN2B molecule at a time. After the genetic demonstration of CaMKII involvement here, this left several possible explanations: (1) neuroprotective DAPK1 inhibitors may show non-selective interference also with the related CaMKII. However, such cross reactions were not detected here. (2) Binding of both CaMKII and DAPK1 to GluN2B could increase, just on different GluN2B molecules. However, this would require that the binding sites do not get saturated, which appears highly unlikely given the stoichiometric excess of the kinases over the receptor (Feng et al., 2011; Kennedy, 2000). (3) Finally, the kinases may bind to principally distinct GluN2B populations. Indeed, our imaging studies showed that excitotoxic insults cause CaMKII accumulation in dendritic spines but dispersal of DAPK1 away from these sites of excitatory synapses. Furthermore, the CaMKII accumulation required GluN2B binding (as it was abolished by the binding-incompetent GluN2BΔCaMKII mutant). Consistent with a previous report (Tu et al., 2010), there may also be an increased association of DAPK1 with GluN2B, but this is restricted to the extra-synaptic fraction. A synaptic PSD fraction has not been tested for either DAPK1 or CaMKII association with GluN2B, as this would not be very informative: the PSD forms a large protein complex, and co-precipitation would not indicate direct interaction. However, our imaging studies clearly show that DAPK1 does not accumulate at the PSD but instead disperses from its basal colocalization with the marker protein PSD95. Taken together, our data show that CaMKII and DAPK1 associate with different pools of GluN2B after excitotoxic stimuli. Thus, although the competitive mutually exclusive binding of either CaMKII or DAPK1 to synaptic GluN2B participates in determining the direction of physiological synaptic plasticity (i.e., LTP versus LTD; Goodell et al., 2017), excitotoxic insults instead involve increased CaMKII binding to synaptic GluN2B and DAPK1 binding to extra-synaptic GluN2B.
Notably, it has been suggested that extra-synaptic NMDARs mediate neuronal cell death, whereas synaptic NMDARs instead promote neuronal survival (Hardingham and Bading, 2010; Hardingham et al., 2002). This has also been coupled with the notion of differential NMDAR subunit localization, with largely extra-synaptic GluN2B versus largely synaptic GluN2A. However, most NMDARs in the adult hippocampus are now recognized to be triheteromeric (containing GluN1, GluN2B, and GluN2A; Hansen et al., 2014; Rauner and Kö hr, 2011; Tovar et al., 2013), and our results clearly demonstrate a role for synaptic GluN2B in ischemia, at least as synaptic targeting protein for CaMKII, if not as the required Ca2+ source. Additionally, the neuroprotection provided by synaptic SK channels (Allen et al., 2011) is most easily explained by a negative feedback on synaptic NMDARs. Thus, although our results do not rule out a function of extra-synaptic NMDARs in ischemia, they do demonstrate a function of synaptic NMDARs.
Although our findings indicate increased extra-synaptic DAPK1/GluN2B interaction, this does not necessitate functional requirement, and a recent study has seriously questioned any involvement of DAPK1 in ischemic cell death at all (McQueen et al., 2017). This is in contrast to other previous studies that do support a role for DAPK1, even if most of them did not specifically investigate the DAPK1 binding to GluN2B (Pei et al., 2014; Shamloo et al., 2005; Tu et al., 2010; Velentza et al., 2003). Notably, although the involvement of DAPK1 in ischemia may be subject to further debate, the results and claims of our current study would remain consistent with either conclusion. By contrast, for the CaMKII/GluN2B interaction, our data directly indicate a functional role in ischemic cell death.
No efficient pharmacological treatment is clinically available for any excitotoxic conditions (including stroke and global cerebral ischemia), emphasizing the importance of understanding the underlying signal transduction mechanisms (Hall and Traystman, 2009; Quillinan et al., 2016). Tremendous but unsuccessful efforts have been made in the development of a stroke therapy, even though global cerebral ischemia may provide a more amenable condition for therapeutic intervention, in part due to a more homogeneous patient population with full reperfusion of the injured brain regions within a well-defined time period. Our previous studies have indicated the potential for pharmacological success of CaMKII inhibition, at least in mouse models of stroke (Vest et al., 2010), and of the potentially more promising global cerebral ischemia (Deng et al., 2017). The current study provides further target validation by genetic means. Notably, other previous genetic studies appeared conflicting: although CaMKII T286A mutation was protective in global cerebral ischemia (Deng et al., 2017), complete knockout of the brain-specific CaMKIIα isoform instead enhanced injury in a stroke model (Waxham et al., 1996). This could be due to fundamental difference in the injury models or in the effects of specific kinase functions. Indeed, complete loss of CaMKII may sensitize to cell death, although block of over-activated “autonomous” CaMKII after ischemia may be protective (Ashpole and Hudmon, 2011; Coultrap et al., 2011). Notably, autonomous CaMKII activity can be generated by both T286 phosphorylation and GluN2B binding (Bayer et al., 2001; Bayer and Schulman, 2019; Miller and Kennedy, 1986). But why are both forms of autonomy required for cell death yet with protection achieved by abolishing just one? A similar situation actually appears to be the case for the physiological induction of normal LTP, which also requires both T286 phosphorylation and GluN2B binding (Barria and Malinow, 2005; Giese et al., 1998; Halt et al., 2012). Then, a more important question may be this: if ischemic injury is sensitive to so many different individual manipulations in mice, why are we still lacking a therapy in humans? Perhaps the answer will emerge as we shift the focus of clinical efforts from stroke to global cerebral ischemia, with the latter holding more promise for successfully measuring outcome improvements in patients.
STAR⋆METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, K. Ulrich Bayer (ulli.bayer@ucdenver.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Animals
All animal treatment was approved by the University of Colorado Institutional Animal Care and Use Committee, in accordance with NIH guidelines. Animals are housed at the Animal Resource Center at the University of Colorado Anschutz Medical Campus (Aurora, CO) and are regularly monitored with respect to general health, cage changes, and overcrowding. Pregnant Sprague-Dawley rats were supplied by Charles River Labs. Male C57BL/6 adult mice, 8 to 12 weeks old, were also supplied by Charles River Labs. The GluN2BΔCaMKII mice (L1298A/R1300Q) are described previously (Halt et al., 2012) and were kindly provided by Dr. Johannes Hell, University of California Davis. Mouse breeding is done in pairs on a C57BL/6 background, either homozygous (for culture experiments) or heterozygous (for use of littermates in CA/CPR and slice experiments).
Primary hippocampal cultures
Primary hippocampal neurons were imaged after 14–17 days in vitro (DIV14–17). Mouse cultures were prepared from P1-P2 neonatal pups of both sexes. Following decapitation and dissection, mouse hippocampi were incubated in dissociation solution (7 mL HBSS buffered saline, 150 μL 100mM CaCl2, 10 μL 1M NaOH, 10 μL 500mM EDTA, 200 units Papain [Worthington]) at room temperature for 15–30 min. After washing hippocampi in plating media (DMEM, 10% FBS, 50 units/mL pen/strep, 2 mM L-glutamine), cells were dissociated by trituration and counted. Neurons were plated on poly-D-lysine (0.1mg/mL in 1M Borate Buffer: 3.1g boric acid, 4.75 g Borax, in 1 L deionized H20) and laminin (0.01mg/mL in PBS)-coated 18 mm glass coverslips in 12 well plates at a density of 150,000–200,000 cells per mL and maintained at 37°C with 5% CO2. After 1 day in vitro (DIV1), plating media was switched to feeding media (Neurobasal-A, B27 supplement, 2 mM L-glutamine). At DIV4, neurons were treated with anti-mitotic FDU (70 μM 5-fluoro-2’-deoxyuridine/140 μM uridine) to suppress glial growth. Rat cultures were prepared from P0 neonatal pups of both sexes and prepared as described previously (Cook et al., 2019); in contrast to mouse culture preparation, rat hippocampi were dissociated for 1 hour and cells were plated at 100,000 cells per mL for imaging studies and 200,000 cells per mL for biochemical studies.
Human embryonic kidney (HEK) 293 cells
HEK293 cells were purchased from the American Type Culture Collection (ATCC) and certified by the University of Colorado Cancer Center. Cells were maintained in DMEM supplemented with 10% FBS and penicillin/streptomycin. Cells were split and grown to 50% prior to transfection.
METHOD DETAILS
Material and DNA constructs
Material was obtained from Sigma, unless noted otherwise. Expression vectors for YFP-CaMKIIα, mCh-DAPK1 (based on pEGFP) and of the GluN2B C terminus (based on pDisplay) and were described previously (Bayer et al., 2006; Goodell et al., 2017). The expression vectors for the GFP-labeled FingR intrabodies targeting CaMKIIα, PSD-95, and gephyrin were kindly provided by Dr. Donald Arnold (University of Southern California, Los Angeles, CA, USA) as described previously (Gross et al., 2013; Mora et al., 2013). The FingRs against PSD95 and gephyrin contained the CCR5TC repressor (Gross et al., 2013); the FingR against CaMKIIa did not contain a repressor (Mora et al., 2013). The fluorophore label was exchanged to contain the following tags in place of GFP: CaMKIIα-FingR-YFP2, PSD-95-FingR-mCh, and gephyrin-FingR-mTurquois (Cook et al., 2019). These modifications were performed using Gibson Assembly (Gibson et al., 2009).
CA/CPR
Cardiac arrest and cardiopulmonary resuscitation (CA/CPR) was performed as previously described (Deng et al., 2017; Orfila et al., 2014; Shimizu et al., 2016). Briefly, asystolic cardiac arrest was induced by KCl injection via jugular catheter. 6 min later, CPR was initiated by injecting 0.5–1 mL epinephrine (16 mg/mL in saline solution), chest compressions at 300/min, and ventilation with 100% O2. If return of spontaneous circulation was not achieved within 2 min of CPR, the animal was excluded from the study. Tissue from was collected 72 h after CA/CPR for assessment cell death in the hippocampal CA1 region.
Cell death assays
H&E staining of hippocampal slices was performed as previously described (Deng et al., 2017). 72 h after CA/CPR, brains were removed, post-fixed with paraformaldehyde, and embedded in paraffin. Coronal sections 6 mm thick were serially cut and stained with H&E. Three levels of the hippocampal CA1 region were analyzed (100 μm apart), beginning from 1.5 mm bregma. Nonviable neurons were determined by the presence of hypereosinophilic cytoplasm and pyknotic nuclei. The percentage of nonviable neurons was calculated for each brain region (average of three levels per region). The investigator was blinded to treatment before analyzing neuronal damage.
Western analysis
Protein concentration was determined using the Pierce BCA protein assay (Thermo-Fisher). Before undergoing SDS-PAGE, samples were boiled in Laemmli sample buffer for 5 min at 95°C. Proteins were separated in a resolving phase polymerized from 7%–9% acrylamide, then transferred to a polyvinylidene difluoride membrane at 24 V for 1–2 h at 4C. Membranes were blocked in 5% milk or BSA and incubated with anti-CaMKIIa (1:4000, CBa2, available at Invitrogen but made in house), pT286-CaMKII (1:2500, Phospho-Solutions), anti-GluN2B (1:1000, Cell Signaling), DAPK1 (1:800, Sigma-Aldrich), followed by either Amersham ECL goat anti-mouse HRP-linked secondary 1:10000 (GE Healthcare) or goat anti-rabbit horseradish peroxidase conjugate 1:10000 (Bio-Rad). Blots were developed using chemiluminescence (Super Signal West Femto, Thermo-Fisher), imaged using the Chemi-Imager 4400 system (Alpha-Innotech), and analyzed by densitometry (ImageJ). Variations in sample loading were corrected using b-actin (1:2000, Cell Signaling) as a loading control. Relative band intensity was normalized as a percent of control conditions on the same blot, which was set at a value of one to allow for comparison between multiple experiments.
CaMKII and DAPK1 binding to GluN2B in vitro
CaMKII and DAPK1 binding to immobilized GST-GluN2Bc (residues 1122 to 1482) was performed as described previously and detected by Western analysis of the eluted protein (Goodell et al., 2017). For CaMKII binding, GST-GluN2Bc was immobilized on antiGST-coated microtiter plates (Bayer et al., 2001; O’Leary et al., 2011); for DAPK1 binding, GST-GluN2Bc was instead immobilized on glutathione-coated magnetic beads (Goodell et al., 2017).
CaMKII and DAPK1 colocalization with GluN2B in HEK cells
HEK culture, transfection, imaging, and analysis was performed as described previously (Goodell et al., 2017). mCh-DAPK1 or mChCaMKIIα and pDisplay-eGFP-GluN2Bc (1:10) were transfected using Ca2+ phosphate and expressed for 16–24 hours prior to imaging. During imaging, cells were maintained in a climate-controlled chamber at 32°C in HEPES buffered imaging solution containing (in mM): 130 NaCl, 5 KCl, 20 Glucose, 2 CaCl2, 1 MgCl2, 10 HEPES pH 7.4. Images were acquired on a ZeissAxiovert 200M inverted microscope (Carl Zeiss GmbH, Oberkochen, Germany) fitted with a 63x objective (1.4NA, plan-Apo). Cells were stimulated with ionomycin (10 μM) at 1 min and imaged every 30 s over 6 min using 0.3 μm steps over 1.8 μm of the cell center. Images were deconvolved, collapsed across z-plane, and analyzed using Slidebook 6.0 software. A Pearson’s correlation of fluorescent overlap for each time point was calculated using hand drawn masks encompassing the entire cell and excluding the nucleus. Raw Pearson’s correlations are shown for starting values. Due to blebbing and loss of cell area, a slight but significant increase in Pearson’s correlation was seen between mCh vector only control and GluN2Bc constructs. Thus, the Pearson’s correlation obtained from mCh cell-fill was subtracted from data with ionomycin stimulation and the resulting value was termed “adjusted colocalization.” All image analysis was done blinded to experimental condition.
Non-radioactive kinase assay
A kinase assay coupling the conversion of ATP to ADP during a phosphorylation event to the loss of absorbance (Abs) at 340 nM upon the oxidation of NADH to NAD by pyruvate kinase lactate dehydrogenase upon ADP accumulation was used to verify inhibitor effects on CaMKII. A reaction containing 50 mM PIPES, pH 7.12, 0.4 mM NADH, 2 mM CaCl2, 20 mM MgCl2, 2 mM Phos-phen pyruvate, 19 units of pyruvate kinase, 27.4 units of lactate dehydrogenase, 75 μM peptide substrate, 1 μM inhibitor (tatCN21 or TC-DAPK-6) and 20 nM CaMKIIα was initiated by the addition of 1 mM ATP. Abs was measured every 10 s for 10 min on a SpectraMax 340PC plate reader (Molecular Devices). All values were normalized to starting Abs for a given reaction.
Transfection and live imaging of primary hippocampal cultures
At 12–14 DIV, neurons were transfected with the intrabodies using a 1:1:1 ratio, at a concentration of 1 μg total DNA/well, using Lipofectamine 2000 (3 μL/well, Invitrogen) according to the manufacturer’s recommendations. Two to three days following transfection (DIV 14–17), neurons were imaged using spinning disk confocal microscopy as described above and analyzed using Slidebook 6.0. During image acquisition, neurons were maintained in a climate-controlled chamber at 34°C in conditioned feeding media buffered with 5 mM HEPES, pH 7.4. Glutamate excitotoxicity was induced by treating with 100 μM glutamate for 5 min. Hippocampal neurons were selected based on pyramidal shaped soma and presence of spiny apical dendrites, and tertiary dendritic branches were selected for analysis to maintain consistency. Images were acquired before stimulation and 1 min or 5 mins after glutamate excitotoxicity. 2D maximum intensity projection images were then generated and analyzed using Slidebook 6.0. The average YFP intensity (CaMKIIα) at excitatory and inhibitory synapses was quantified using threshold masks of PSD-95 and gephyrin signal to calculate a puncta-to-shaft ratio (mean YFP intensity at PSD-95 or gephyrin puncta, divided by the mean YFP intensity of the adjacent dendritic shaft).
Acute hippocampal slices
Acute hippocampal slices were collected from wild-type adult mice as previously described (Goodell et al., 2017). Mice were anesthetized by isoflurane and quickly decapitated. Hippocampi were dissected and sliced (400 μm) in ice-cold cutting solution (in mM: 220 sucrose, 12 MgSO4, 10 glucose, 0.2 CaCl2, 0.5 KCl, 0.65 NaH2 PO4, 13 NaHCO3, and 1.8 ascorbate) before transfer to were transferred to artificial cerebral spinal fluid (ACSF; in mM: 124 NaCl, 2 KCl, 1.3 NaH2PO4, 26 NaHCO3, 10 glucose, 2 CaCl2, 1 MgSO4, and 1.8 ascorbate) at 32°C. After at least 1 h recovery, slices were treated with 100 μM glutamate for 5 min and subsequently either collected or replaced with fresh ACSF for 2 h. All solutions were saturated with 95% O2/5% CO2.
Neuronal fractionation
Following treatment, DIV16 hippocampal neurons from rat or acute hippocampal slices from mice were homogenized as follows: To make the extra-synaptic fraction, cells were homogenized in ice-cold 1% TX100 buffer also containing (in mM): 10 Tris, 10 EDTA, 10 EGTA, 150 NaCl, plus protease and phosphatase inhibitors and cleared by ultracentrifugation (100,000 x g) for 20 min at 4°C. To make the crude synaptic fraction, the remaining insoluble pellet (including synaptosomes and lipid rafts) was solubilized in 1% SDS buffer also containing (in mM): 10 Tris, 10 EDTA, 10 EGTA, 150 NaCl, plus protease and phosphatase inhibitors, then heated at 65C for 15 min, quenched with 1.5% TX100, and cleared by centrifugation (20,000 x g) for 20 min.
Immunoprecipitation
After treatment, acute hippocampal slices were homogenized in TX100 to isolate the extra-synaptic fraction (described above). Cleared material was incubated on a head-over-head tilter with protein A-Sepharose beads and 2 μg of anti-GluN2B (Cell Signaling) or nonspecific rabbit immunoglobulin G (IgG, Jackson) control for 4 h at 4°C. Beads were washed three times with 0.1% Triton X-100 wash buffer also containing (in mM): 150 NaCl, 10 EDTA, 10 EGTA, 10 Tris base, pH 7.4.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are shown as mean ± SEM. Statistical significance is indicated in the figure legends. Statistics were performed using Prism (GraphPad) software. Imaging experiments were obtained and analyzed using SlideBook 6.0 software. Immunoblots were analyzed using ImageJ (NIH). All data were tested for their ability to meet parametric conditions, as evaluated by a Shapiro-Wilk test for normal distribution and a Brown-Forsythe test (3 or more groups) or an F-test (2 groups) to determine equal variance. All comparisons between two groups met parametric criteria, and independent samples were analyzed using unpaired, two-tailed Student’s t tests. Comparisons between pre- and post-treatment images from the same neurons were analyzed using paired, two-tailed Student’s t tests. Comparisons between three or more groups meeting parametric criteria were done by one-way ANOVA with specific post hoc analysis indicated in figure legends. Non-parametric data comparisons between 3 or more groups were done by Kruskal-Wallice followed by Bonferroni post hoc analysis. Comparisons to control group used Dunn’s post hoc analysis.
DATA AND CODE AVAILABILITY
The datasets generated during this study are available through Mendeley (https://dx.doi.org/10.17632/5y58k87rw6.1).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CaMKIIα | Made in House | CBα2 |
| CaMKIIα pT286 | Phospho-Solutions | p1005–286; RRID:AB_2492051 |
| DAPK1 | Sigma-Aldrich | D1319; RRID:AB_1078622 |
| GluN2B | Cell Signaling | D8E10; RRID:AB_2798506 |
| GluN2B pS1303 | Millipore | 07–398; RRID:AB_310582 |
| GluN1 | BD Biosciences | 556308; RRID:AB_2314954 |
| PSD-95 | NeuroMab | 73–028; RRID:AB_10698024 |
| Beta Actin | Cell Signaling | 4970; RRID:AB_2223172 |
| Goat anti-Rabbit | GE Healthcare | NA934V; RRID:AB_2722659 |
| Goat anti-Mouse | GE Healthcare | NA931V; RRID:AB_772210 |
| Rabbit IgG | Jackson | 011–000–003; RRID:AB_2337118 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Papain | Worthington | LS 03126 |
| Lipofectamine 2000 | Invitrogen | 11668027 |
| B-27 supplement | GIBCO | 17504044 |
| cOmplete protease inhibitor cocktail (EDTA-free) | Roche | 1187380001 |
| Microcystin-LR | Calbiochem | 475815 |
| tat2BC | Chi Scientific; Tu et al., 2010 | N/A (custom peptide) |
| DAPK-TC6 | Tocris | 19401 |
| tatCN21 | Chi Scientific; Vest et al., 2007 | N/A (custom peptide) |
| Glutathione-coated magnetic beads | Pierce | 78601 |
| Protein A-Sepharose beads | RepliGen | 10–1003–01 |
| Critical Commercial Assays | ||
| Pierce BCA protein assay | Thermo-Fisher | 23225 |
| Deposited Data | ||
| Raw and analyzed data | This paper | https://dx.doi.org/10.17632/5y58k87rw6.1 |
| Experimental Models: Cell Lines | ||
| Primary hippocampal cultures | Laboratory of K. Ulrich Bayer | N/A |
| HEK293 cells | ATCC | CRL-1573 |
| Experimental Models: Organisms/Strains | ||
| Rat: Sprague-Dawley | Charles River Labs | N/A |
| Mouse: Wild type: C57BL/6 | Charles River Labs | N/A |
| Mouse: GluN2BΔCaMKII: C57BL/6 | Halt et al., 2012 | N/A |
| Recombinant DNA | ||
| CaMKIIα-FingR-GFP | Dr. Donald Arnold (USC) Mora et al., 2013 | N/A |
| PSD-95-FingR-GFP | Gross et al., 2013 | Addgene #46295 |
| Gephyrin-FingR-GFP | Gross et al., 2013 | Addgene #46296 |
| YFP-CaMKIIα | Cook et al., 2019 | Addgene #15214 |
| mCherry-DAPK1 | Goodell et al., 2017 | Clonetech #632524 |
| Software and Algorithms | ||
| Slidebook 6.0 | Intelligent Imaging Innovations (3i) | RRID:SCR_014300 |
| Prism 7.0 | Graphpad | RRID: SCR_002798 |
| AlphaEase FC 4.0 | Alpha Innotech | N/A |
| ImageJ | NIH | RRID:SCR_003070 |
Highlights.
Disrupting CaMKII/GluN2B binding protects from neuronal injury after cardiac arrest
Neuroprotective CaMKII and DAPK1 inhibitors are selective for their respective kinase
CaMKII, but not DAPK1, accumulates at synaptic GluN2B during excitotoxic insults
Extra-synaptic GluN2B decreases during excitotoxic and ischemic insults
ACKNOWLEDGMENTS
The Roatan Tech Center kindly allowed us to derive the skull and crossbones image from their logo. This work was supported by NIH grants F31AG062160 (to S.G.C.), F31NS092265 (to D.J.G.), T32GM007635 (supporting J.E.T. and S.R.), T32AG000279 (supporting N.L.R.), R01NS081248 (to K.U.B.), and R01NS080851 (to P.S.H. and K.U.B.).
Footnotes
DECLARATION OF INTERESTS
The University of Colorado holds the patent rights for tatCN21, its derivatives, and their uses (PCT/US08/077934 “Compositions and methods for improved CaMKII inhibitors and uses thereof”). K.U.B. is founder of Neurexus Therapeutics, LLC.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.076.
REFERENCES
- Allen D, Nakayama S, Kuroiwa M, Nakano T, Palmateer J, Kosaka Y, Ballesteros C, Watanabe M, Bond CT, Luján R, et al. (2011). SK2 channels are neuroprotective for ischemia-induced neuronal cell death. J. Cereb. Blood Flow Metab. 31, 2302–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashpole NM, and Hudmon A (2011). Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol. Cell. Neurosci. 46, 720–730. [DOI] [PubMed] [Google Scholar]
- Barcomb K, Buard I, Coultrap SJ, Kulbe JR, O’Leary H, Benke TA, and Bayer KU (2014). Autonomous CaMKII requires further stimulation by Ca2+/calmodulin for enhancing synaptic strength. FASEB J. 28, 3810–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcomb K, Hell JW, Benke TA, and Bayer KU (2016). The CaMKII/ GluN2B protein interaction maintains synaptic strength. J. Biol. Chem 291, 16082–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, and Malinow R (2005). NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301. [DOI] [PubMed] [Google Scholar]
- Bayer KU, and Schulman H (2019). CaM kinase: still inspiring at 40. Neuron 103, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, and Schulman H (2001). Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805. [DOI] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O’Leary H, Schulman H, and De Koninck P (2006). Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci 26, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SG, Goodell DJ, Restrepo S, Arnold DB, and Bayer KU (2019). Simultaneous live imaging of multiple endogenous proteins reveals a mechanism for Alzheimer’s-related plasticity impairment. Cell Rep. 27, 658–665.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Vest RS, Ashpole NM, Hudmon A, and Bayer KU (2011). CaMKII in cerebral ischemia. Acta Pharmacol. Sin. 32, 861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G, Orfila JE, Dietz RM, Moreno-Garcia M, Rodgers KM, Coultrap SJ, Quillinan N, Traystman RJ, Bayer KU, and Herson PS (2017). Autonomous CaMKII activity as a drug target for histological and functional neuroprotection after resuscitation from cardiac arrest. Cell Rep. 18, 1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Raghavachari S, and Lisman J (2011). Quantitative estimates of the cytoplasmic, PSD, and NMDAR-bound pools of CaMKII in dendritic spines. Brain Res. 1419, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, and Silva AJ (1998). Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873. [DOI] [PubMed] [Google Scholar]
- Goodell DJ, Zaegel V, Coultrap SJ, Hell JW, and Bayer KU (2017). DAPK1 mediates LTD by making CaMKII/GluN2B binding LTP specific. Cell Rep. 19, 2231–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GG, Junge JA, Mora RJ, Kwon HB, Olson CA, Takahashi TT, Liman ER, Ellis-Davies GC, McGee AW, Sabatini BL, et al. (2013). Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 78, 971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, and Traystman RJ (2009). Role of animal studies in the design of clinical trials. Front Neurol. Neurosci 25, 10–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, and Hell JW (2012). CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 31, 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, and Traynelis SF (2014). Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/ GluN2B NMDA receptors. Neuron 81, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, and Bading H (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci 11, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, and Bading H (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci 5, 405–414. [DOI] [PubMed] [Google Scholar]
- Horn M, and Schlote W (1992). Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol. 85, 79–87. [DOI] [PubMed] [Google Scholar]
- Kennedy MB (2000). Signal-processing machines at the postsynaptic density. Science 290, 750–754. [DOI] [PubMed] [Google Scholar]
- Kostandy BB (2012). The role of glutamate in neuronal ischemic injury: the role of spark in fire. Neurol. Sci. 33, 223–237. [DOI] [PubMed] [Google Scholar]
- McQueen J, Ryan TJ, McKay S, Marwick K, Baxter P, Carpanini SM, Wishart TM, Gillingwater TH, Manson JC, Wyllie DJA, et al. (2017). Pro-death NMDA receptor signaling is promoted by the GluN2B C-terminus independently of Dapk1. eLife 6, e17161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SG, and Kennedy MB (1986). Regulation of brain type II Ca2+/ calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell 44, 861–870. [DOI] [PubMed] [Google Scholar]
- Mora RJ, Roberts RW, and Arnold DB (2013). Recombinant probes reveal dynamic localization of CaMKIIa within somata of cortical neurons. J. Neurosci. 33, 14579–14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T, Graham DI, Adams JH, and Ford I (1989). Changes in the hippocampus and the cerebellum resulting from hypoxic insults: frequency and distribution. Acta Neuropathol. 78, 438–443. [DOI] [PubMed] [Google Scholar]
- O’Leary H, Liu WH, Rorabaugh JM, Coultrap SJ, and Bayer KU (2011). Nucleotides and phosphorylation bi-directionally modulate Ca2+/ calmodulin-dependent protein kinase II (CaMKII) binding to the N-methyl-Daspartate (NMDA) receptor subunit GluN2B. J. Biol. Chem 286, 31272–31281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orfila JE, Shimizu K, Garske AK, Deng G, Maylie J, Traystman RJ, Quillinan N, Adelman JP, and Herson PS (2014). Increasing small conductance Ca2+-activated potassium channel activity reverses ischemia-induced impairment of long-term potentiation. Eur. J. Neurosci 40, 3179–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Shang Y, Jin H, Wang S, Wei N, Yan H, Wu Y, Yao C, Wang X, Zhu LQ, and Lu Y (2014). DAPK1-p53 interaction converges necrotic and apoptotic pathways of ischemic neuronal death. J. Neurosci. 34, 6546–6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulsinelli WA (1985). Selective neuronal vulnerability: morphological and molecular characteristics. Prog. Brain Res 63, 29–37. [DOI] [PubMed] [Google Scholar]
- Quillinan N, Grewal H, Deng G, Shimizu K, Yonchek JC, Strnad F, Traystman RJ, and Herson PS (2015). Region-specific role for GluN2Bcontaining NMDA receptors in injury to Purkinje cells and CA1 neurons following global cerebral ischemia. Neuroscience 284, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillinan N, Herson PS, and Traystman RJ (2016). Neuropathophysiology of brain injury. Anesthesiol. Clin 34, 453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauner C, and Kö hr, G. (2011). Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. J. Biol. Chem 286, 7558–7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, and Attwell D (2000). Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403, 316–321. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, and Freund TF (1991). Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 40, 599–636. [DOI] [PubMed] [Google Scholar]
- Shamloo M, Soriano L, Wieloch T, Nikolich K, Urfer R, and Oksenberg D (2005). Death-associated protein kinase is activated by dephosphorylation in response to cerebral ischemia. J. Biol. Chem 280, 42290–42299. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Quillinan N, Orfila JE, and Herson PS (2016). Sirtuin-2 mediates male specific neuronal injury following experimental cardiac arrest through activation of TRPM2 ion channels. Exp. Neurol 275, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, McNeill RB, and Colbran RJ (2000). Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem 275, 23798–23806. [DOI] [PubMed] [Google Scholar]
- Takata K, Takeda Y, Sato T, Nakatsuka H, Yokoyama M, and Morita K (2005). Effects of hypothermia for a short period on histologic outcome and extracellular glutamate concentration during and after cardiac arrest in rats. Crit. Care Med 33, 1340–1345. [DOI] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ, and Westbrook GL (2013). Triheteromeric NMDA receptors at hippocampal synapses. J. Neurosci 33, 9150–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, et al. (2010). DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 140, 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velentza AV, Wainwright MS, Zasadzki M, Mirzoeva S, Schumacher AM, Haiech J, Focia PJ, Egli M, and Watterson DM (2003). An aminopyridazine-based inhibitor of a pro-apoptotic protein kinase attenuates hypoxia-ischemia induced acute brain injury. Bioorg. Med. Chem. Lett 13, 3465–3470. [DOI] [PubMed] [Google Scholar]
- Vest RS, Davies KD, O’Leary H, Port JD, and Bayer KU (2007). Dual mechanism of a natural CaMKII inhibitor. Mol. Biol. Cell 18, 5024–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, O’Leary H, Coultrap SJ, Kindy MS, and Bayer KU (2010). Effective post-insult neuroprotection by a novel Ca(2+)/ calmodulin-dependent protein kinase II (CaMKII) inhibitor. J. Biol. Chem 285, 20675–20682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira MM, Schmidt J, Ferreira JS, She K, Oku S, Mele M, Santos AE, Duarte CB, Craig AM, and Carvalho AL (2016). Multiple domains in the C-terminus of NMDA receptor GluN2B subunit contribute to neuronal death following in vitro ischemia. Neurobiol. Dis 89, 223–234. [DOI] [PubMed] [Google Scholar]
- Waxham MN, Grotta JC, Silva AJ, Strong R, and Aronowski J (1996). Ischemia-induced neuronal damage: a role for calcium/calmodulin-dependent protein kinase II. J. Cereb. Blood Flow Metab. 16, 1–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available through Mendeley (https://dx.doi.org/10.17632/5y58k87rw6.1).