

ABSTRACT
Acute myeloid leukemia (AML) is a devastating malignancy for which novel treatment approaches are desperately needed. Ras signaling is an attractive therapeutic target for AML because a large proportion of AMLs have mutations in NRAS, KRAS, or genes that activate Ras signaling, and key Ras effectors are activated in virtually all AML patient samples. This has inspired efforts to develop Ras-targeted treatment strategies for AML. Due to the inherent difficulty and disappointing efficacy of targeting Ras proteins directly, many have focused on inhibiting Ras effector pathways. Inhibiting the major oncogenic Ras effectors, the mitogen-activated protein kinase (MAPK) and/or phosphatidylinositiol-3-kinase (PI3K) pathways, has generally demonstrated modest efficacy for AML. While this may be in part related to functional redundancy between these pathways, it is now clear that other Ras effectors have key oncogenic roles. Specifically, the Ras-like (Ral) GTPases have emerged as critical mediators of Ras-driven transformation and AML cell survival. Our group recently uncovered a critical role for RALB signaling in leukemic cell survival and a potential mediator of relapse following Ras-targeted therapy in AML. Furthermore, we found that RALB signaling is hyperactivated in AML patient samples, and inhibiting RALB has potent anti-leukemic activity in preclinical AML models. While key questions remain regarding the importance of RALB signaling across the genetically diverse spectrum of AML, the specific mechanism(s) that promotes leukemic cell survival downstream of RALB, and how to pharmacologically target RALB signaling effectively – RALB has emerged as a critical Ras effector and potential therapeutic target for AML.
KEYWORDS: AML, MAPK, PI3K, Ral, RALB, Ras, targeted therapy
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy characterized by genetic mutations that promote proliferation and prevent differentiation of myeloid progenitors. Despite aggressive cytotoxic chemotherapy, the majority of adults with AML die of relapsed or treatment refractory disease.1 Furthermore, a large proportion of older adults with AML are not fit for intensive treatment approaches, and have only palliative treatment options. While the genetic landscape of AML has been extensively characterized,2,3 effective genetically based therapies have yet to be realized. The toxicity and disappointing outcomes associated with conventional approaches have driven interest in developing safer and more effective targeted treatments.
The RAS proto-oncogenes – HRAS, NRAS, and KRAS – are among the most frequently mutated genes in human cancer. Ras small GTPases act as molecular switches to modulate signal transduction by cycling between active guanine triphosphate (GTP)-bound and inactive guanine diphosphate (GDP)-bound states.4 Ras activation is catalyzed by guanine exchange factors (GEFs) that promote the exchange of GDP for GTP in response to growth factor receptor activation, and negatively regulated by the effects of GTPase activating proteins (GAPs) to greatly enhance the inefficient intrinsic Ras GTPase activity.5 Oncogenic mutations in RAS genes most commonly involve amino acid substitutions at codons 12, 13, or 61 that disrupt the coordination of the catalytic glutamine residue at codon 61 and impair GTP hydrolysis, thereby leading to constitutive activation of Ras effector pathways and cellular transformation.6 Ras-GTP regulates diverse cellular processes including proliferation, motility, and survival by interacting with a complex array of effector enzymes (Fig. 1).7
Figure 1.
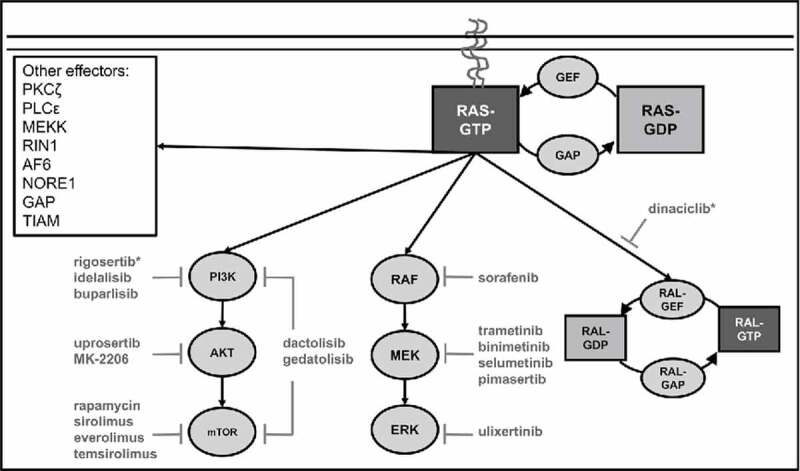
Canonical Ras signaling. Ras acts as a molecular switch that transduces signals from growth factor receptors to a variety of effector enzymes. Ras proteins are activated by guanine-exchange factors (GEFs) that promote the exchange of GDP for GTP leading to membrane localization and activation of effector enzymes. Ras proteins are negatively regulated by GTPase activating proteins (GAPs) that catalyze Ras's intrinsic GTPase activity resulting in the hydrolysis of GTP to GDP. The major oncogenic Ras effector pathways include the phosphatidylinositiol-3-kinase (PI3K), mitogen-activated protein kinase (MAPK), and Ras-like (Ral) small GPTase signaling pathways. The role of other Ras effectors in oncogenesis remains unclear. Selected inhibitors of Ras effector signaling that have been evaluated in clinical trials for AML are included. A complete list of clinical trials can be found at ClinicalTrials.gov. * Dinaciclib also inhibits CDK1, CDK2, CDK5, CDK9 and rigosertib also inhibits polio-like kinase 1 (PLK1).
The mitogen-activated protein kinase (MAPK) and phosphatidylinositiol-3-kinase (PI3K) signaling pathways are the Ras effector pathways with the most well established roles in cancer. Activation of MAPK signaling involves Ras-GTP binding of RAF kinases resulting in plasma membrane localization and activation of their serine/threonine kinase activity.8,9 Subsequently, active RAF phosphorylates and activates the mitogen-activated kinase kinases, MEK1 and MEK2, that phosphorylate and activate the mitogen-activated kinases, ERK1 and ERK2. Primary ERK targets include the ETS family transcription factors, JUN, and ultimately drive AP1-mediated proliferation.10 Similarly, Ras-GTP induces PI3K signaling through interactions with type I PI3K catalytic subunits resulting in localization to the membrane and kinase activation leading to phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) to produce phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 then acts as a second messenger activating AKT-dependent and AKT-independent signaling pathways that modulate diverse cellular processes including proliferation, survival, motility, and metabolism.11,12
Approximately 15–25% of AMLs harbor activating mutations in NRAS or KRAS.3,13,14 Unlike many solid tumors, both NRAS and KRAS are mutated in AML, although NRAS mutations predominate. While RAS mutations are seen across the spectrum of genetically heterogeneous AMLs, they are more common in specific AML sub-sets. For example, NRAS mutations occur in approximately 40% of AML with inv(16) or t(16;16) and 20% of AML with t(8;21), t(9;11), inv(3), or t(3;3).13,15,16 Similarly, KRAS mutations are found in approximately 15% of AML with inv(16) or t(16;16) and 20% of AML with t(6;9).13,15,16 NRAS mutations are also found in about 30% of AML with biallelic mutation of CEPBA and 20% of AML with mutated NPM1.13,15 While RAS mutations do not have a clear impact on clinical outcomes for AML patients, there is a suggestion that AML with oncogenic RAS mutations benefit more from cytarabine containing chemotherapy regimens than AML with wild-type RAS.17,18 In addition to AML with mutant RAS, proteins that regulate the activation of Ras (e.g. PTPN11 and NF1) and signaling receptors that rely on Ras for their oncogenic effects (e.g., FLT3 and KIT) are also frequently mutated in AML.3,13,14,19,20 While oncogenic RAS mutations occur at similar frequencies across the age spectrum of AML, pediatric AMLs exhibit a distinct pattern of mutations in upstream regulators of RAS with an increased frequency of KIT mutations and fewer FLT3-ITD mutations than adult AML, reflective of the distinct pathogenesis of AML in children compared with adults.14 Together with the prevalence of RAS-associated mutations described above, the almost ubiquitous activation of MAPK and PI3K signaling in AML further supports a key role for Ras signaling in the growth and survival of leukemic cells. Together, these observations have fueled intense interest in the development of Ras-targeted AML therapy.21,22
Ras's picomolar affinity for GTP and the challenge of designing a small molecules capable of restoring mutant Ras's defective GTPase activity have thwarted the successful development of direct inhibitors of oncogenic Ras.23 Although the recent development of a specific small molecule inhibitor of KRAS(G12C) suggests that these hurdles may not be insurmountable.24 An alternative approach to overcome the inherent difficulty of targeting Ras directly is targeting the post-translational processing and localization of Ras. Unfortunately, farnasyltransferase inhibitors (FTIs), such as tipifarnib, demonstrated impressive preclinical activity, but subsequent clinical studies yielded disappointing results due to resistance driven by alternative prenylation pathways for Ras.5 Alternatively, targeting the palmitoylation/depalmitoylation cycle of Ras with the acyl protein thioesterase (APT) inhibitor palmostatin B can disrupt the localization of oncogenic Ras and inhibit the growth and clonogenicity of murine haematopoietic cells expressing oncogenic Nras, but the translational potential of this strategy remains to be determined.25 While renewed efforts fueled by the National Cancer Institute's Ras Initiative are challenging the paradigm that Ras is an undruggable cancer target, drugs that directly target Ras have yet to make their way into clinical practice.
The struggle to directly inhibit Ras has motivated intense efforts to target Ras effector pathways in AML. These efforts have largely focused on the MAPK and/or PI3K pathways, and have generally demonstrated modest and predominately cytostatic effects in a variety of AML models.26-30 For example, inhibition of MEK alone or in combination with PI3K in a mouse model of Nras mutant AML inhibited proliferation but failed to induce leukemic cell death, suggesting that MAPK and PI3K pathways drive AML proliferation but may be dispensable for AML survival.30 Similarly, our group found that inhibition of MAPK and/or PI3K signaling led to G0/G1 cell cycle arrest of human AML cell lines with negligible effects on apoptosis, and led to predominately static effects in vivo in a murine NRAS(G12V)-driven AML model.26 The clinical experience targeting MAPK and PI3K have been similar. Inhibition of MEK with selumetinib had modest and transient activity for patients with relapsed/refractory AML, and inhibition of AKT with MK-2206 had essentially no activity against AML in phase II clinical trials.31,32 Strategies that combined inhibitors of MEK and MDM2 or MEK, mTOR, and BCL2 have demonstrated synergistic anti-leukemic activity and induced leukemic cell apoptosis in vitro, suggesting that combined inhibition of Ras signaling together with key survival pathways may be advantageous.28,29 The modest efficacy of targeting the MAPK or PI3K pathways alone is likely related to functional redundancy and/or feedback loops that compensate for the loss of a single effector pathway. Indeed, biopsy specimens from patients with advanced solid tumors that were treated with the mTOR inhibitor everolimus exhibited higher levels of MAPK signaling.33 Another potential explanation for the lack of efficacy of MAPK and/or PI3K inhibitors is that alternate Ras effectors may play important roles in cancer growth and survival. Supporting the later, elegant studies investigating the essential oncogenic signals downstream of Ras revealed that activation of MAPK and PI3K signaling are not sufficient to transform human fibroblasts.34 Similarly, we found that combined inhibition of MAPK and PI3K could not reproduce the apoptotic effects of NRAS oncogene withdrawal in an NRAS(G12V)-addicted AML mouse model, indicating that other Ras effector(s) provide critical support to leukemic cells.26
Other clues to the key mediators and specific vulnerabilities of Ras-driven cancer cells come from several large-scale synthetic lethality screens. These screens are based on the premise that specific mutations can have insignificant or beneficial effects in isolation, but can be lethal when combined. Transcriptome-scale loss of function screens have identified genes and pathways that exhibit synthetic lethality with mutant Ras.35 For example, a recent CRISPR/Cas9-based screen identified synthetic lethal interactions between genes involved in Ras processing and MAPK signaling and oncogenic RAS mutations in human and murine leukemia cells.36 While such screens have uncovered putative Ras-associated cancer genes and pathways, comparisons between these studies are complicated by the differences in technology, conditions, and model systems used. These differences undoubtedly contribute to the lack of overlap observed across studies, but may also indicate that Ras's vulnerabilities are greatly influenced by the cellular and molecular context. Furthermore, functional validation in relevant and robust model systems including primary patient-derived cancer cells will be essential to validate candidate genes and pathways identified in large-scale synthetic lethal screens to determine their true translational potential.
There is mounting evidence that Ras-like (Ral) proteins are critical effectors of Ras in cancer (Fig. 2). Like Ras, the Ral proteins, RALA and RALB, are small GTPases that are activated by Ral guanine exchange factors (RalGEFs) that promote the exhange of GDP for GTP and are inactivated by Ral GTPase activating proteins (RalGAPs) that catalyze their instrinsic GTPase activity. Ral-GTP, and in some cases Ral-GDP, interacts with various effectors to regulate diverse cellular processes. The best characterized effectors of Ral signaling include RALBP1/RLIP and the SEC5 and EXO84 subunits of the hetero-octomeric exocyst complex, which has exocytic and non-exocytic cellular functions.37 Seminal studies uncovered an essential role for Ral proteins in the transformation of murine fibroblasts downstream of Ras.38,39 Subsequent studies confirmed that Ral activation downstream of Ras was sufficient for transformation of human cells.34 RALA and RALB appear to have unique roles in anchorage-independent growth and survival, respectively;40 however, either RALA or RALB, but not both, are required for proliferation of murine Kras oncogene-driven non-small cell lung cancer cells, suggesting some degree of functional redundancy.40,41 The divergent functional roles of RALA and RALB, which can interact with similar effectors in vitro, are primarily mediated by their unique subcellular localization. While the functional roles for specific Ral effectors in cancer are not well understood, a synthetic lethal screen identified an essential role for the RALB-SEC5-TBK1 signaling axis in the maintenance of several KRAS oncogene-driven human epithelial cancers.42 RALB appears to be required for the survival of malignant cells, but not normal cells, making it an attractive therapeutic target.40 Our group uncovered a critical role for RALB in human AML cell survival, and confirmed that RALB-TBK1 signaling is hyperactivated in AML patient samples.26 We also demonstrated that the clinically relevant drug, dinaciclib, has RALB-dependent anti-leukemic effects in murine and human preclinical AML models including patient derived AML mouse xenografts (PDX mice) with negligible effects on normal blood progenitor cells.43 A central role for RALB signaling in AML was corroborated by other work by our group that discovered that Ras oncogene-independent activation of RALB signaling is a targetable mechanism of relapse after suppression of oncogenic Ras expression in a mouse model of NRAS(G12V)-addicted AML.43 The specific mechanism that drives Ras oncogene-independent activation of RALB signaling in this model and the role of RALB in human AML relapse are areas of active investigation. These studies support a central role for RALB in the pathophysiology of AML and as a promising therapeutic target.
Figure 2.
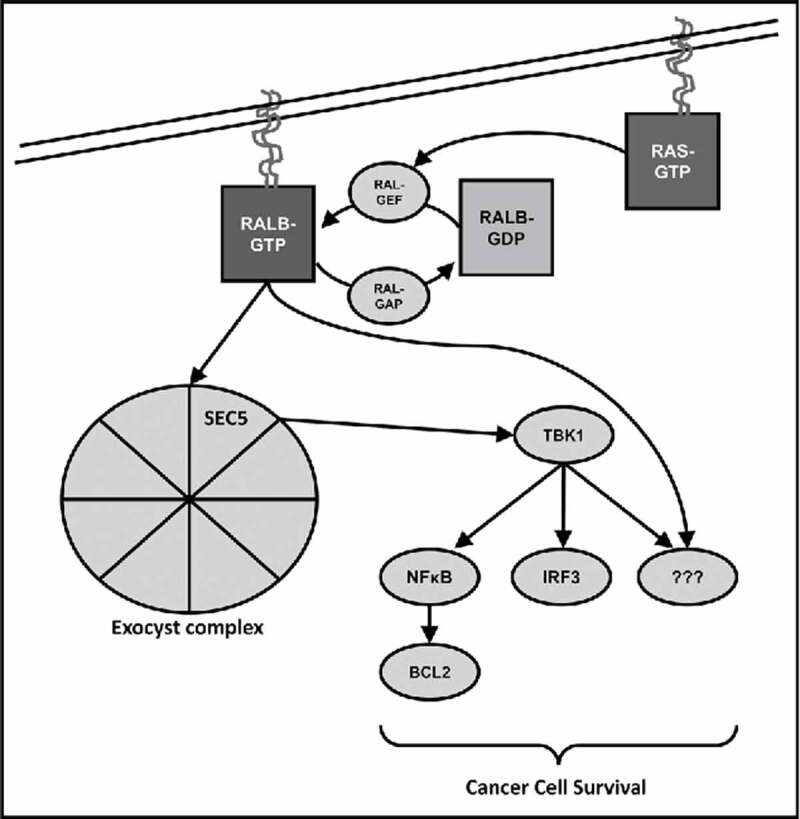
Oncogenic RALB signaling. RALB transduces signals to effector enzymes downstream of Ras. Like Ras, RALB activation is regulated by Ral-specific guanine-exchange factors (Ral GEFs) and GTPase activating proteins (Ral GAPs). The specific oncogenic mechanism(s) of RALB are not well understood, but is thought to involve interaction with the SEC5 subunit of the exocyts complex to recruit and activate the non-canonical IκB kinase TANK-binding kinase 1 (TBK1) that promotes cancer cell survival through NFκB and interferon response factor 3 (IRF3).
While Ral GTPases and their effectors have emerged as important drivers of cancer and RALB appears to play a key role in AML, several unanswered questions remain. Our findings demonstrate that RALB signaling is hyperactivated in several diverse primary AML patient samples, but whether RALB-dependence is a general feature of AMLs or is limited to specific genetic subsets (e.g., AML with oncogenic Ras mutations) has not been systematically evaluated.26 From a translational perspective, this has important implications for identifying specific AML patients that might benefit from RALB-based therapy. Another challenge is to develop clinically relevant strategies to target RALB. Similar to Ras, there are technical hurdles to directly targeting Ral GTPases related to its structure and affinity for GTP. While the clinically relevant cyclin dependent kinase (CDK) inhibitor dinaciclib has RALB-dependent effects against AML, it also has RALB-independent effects through inhibition of CDK1, CDK2, CDK5, and CDK9.43 Ral specific small molecule inhibitors have recently been developed with encouraging preclinical results, but have not yet made the leap into clinical development.44,45 Pharmacologic targeting of Ral effector pathways represents another potential therapeutic strategy, but the detailed mechanisms that support AML survival downstream of RALB have not been characterized. We have shown that knockdown of RALB leads to decreased expression of BCL2 in leukemic cells, but the mechanism and functional consequence of this association remains to be seen.26 Given the virtually ubiquitous development of treatment resistance seen with clinical targeting of single oncogenic pathways, it seems likely that potent and durable anti-leukemic responses will require combined targeting of multiple signaling nodes. A more comprehensive evaluation of RALB survival signaling will be critical to understand its pathophysiology, and will likely uncover novel drug targets. A better understanding of Ras and key effectors like RALB will be essential to guide the rational development of safer and more effective targeted cancer treatments.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, et al.. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010; 115:453-74; PMID:19880497; https://doi.org/ 10.1182/blood-2009-07-235358 [DOI] [PubMed] [Google Scholar]
- [2].Patel JP, Levine RL. How do novel molecular genetic markers influence treatment decisions in acute myeloid leukemia? Hematology Am Soc Hematol Educ Program 2012; 2012:28-34; PMID:23233557; https://doi.org/ 10.1182/asheducation-2012.1.28 [DOI] [PubMed] [Google Scholar]
- [3].Network CGAR . Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368:2059-74; PMID:23634996; https://doi.org/ 10.1056/NEJMoa1301689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science 2001; 294:1299-304; PMID:11701921; https://doi.org/ 10.1126/science.1062023 [DOI] [PubMed] [Google Scholar]
- [5].Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3:11-22; PMID:12509763; https://doi.org/ 10.1038/nrc969 [DOI] [PubMed] [Google Scholar]
- [6].Bos JL. Ras oncogenes in human cancer: A review. Cancer Res 1989; 49:4682-9; PMID:2547513 [PubMed] [Google Scholar]
- [7].Malumbres M, Barbacid M. RAS oncogenes: The first 30 years. Nat Rev Cancer 2003; 3:459-65; PMID:12778136; https://doi.org/ 10.1038/nrc1097 [DOI] [PubMed] [Google Scholar]
- [8].Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 1994; 369:411-4; PMID:8196769; https://doi.org/ 10.1038/369411a0 [DOI] [PubMed] [Google Scholar]
- [9].Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J 1995; 14:3136-45; PMID:7542586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yordy JS, Muise-Helmericks RC. Signal transduction and the Ets family of transcription factors. Oncogene 2000; 19:6503-13; PMID:11175366; https://doi.org/ 10.1038/sj.onc.1204036 [DOI] [PubMed] [Google Scholar]
- [11].Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994; 370:527-32; PMID:8052307; https://doi.org/ 10.1038/370527a0 [DOI] [PubMed] [Google Scholar]
- [12].Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2014; 15:7-24; PMID:25533673; https://doi.org/ 10.1038/nrc3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al.. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374:2209-21; PMID:27276561; https://doi.org/ 10.1056/NEJMoa1516192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shiba N, Yoshida K, Shiraishi Y, Okuno Y, Yamato G, Hara Y, Nagata Y, Chiba K, Tanaka H, Terui K, et al.. Whole-exome sequencing reveals the spectrum of gene mutations and the clonal evolution patterns in paediatric acute myeloid leukaemia. Br J Haematol 2016; 175:476-89; PMID:27470916; https://doi.org/ 10.1111/bjh.14247 [DOI] [PubMed] [Google Scholar]
- [15].Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol 2017; 35:934-46; PMID:28297624; https://doi.org/ 10.1200/JCO.2016.71.2208 [DOI] [PubMed] [Google Scholar]
- [16].Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, Radtke I, Chao JR, Walsh MP, Song G, et al.. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet 2016; 48:1551-6; PMID:27798625; https://doi.org/ 10.1038/ng.3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kadia TM, Kantarjian H, Kornblau S, Borthakur G, Faderl S, Freireich EJ, Luthra R, Garcia-Manero G, Pierce S, Cortes J, et al.. Clinical and proteomic characterization of acute myeloid leukemia with mutated RAS. Cancer 2012; 118:5550-9; PMID:22569880; https://doi.org/ 10.1002/cncr.27596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Neubauer A, Maharry K, Mrozek K, Thiede C, Marcucci G, Paschka P, Mayer RJ, Larson RA, Liu ET, Bloomfield CD. Patients with acute myeloid leukemia and RAS mutations benefit most from postremission high-dose cytarabine: A cancer and leukemia group B study. J Clin Oncol 2008; 26:4603-9; PMID:18559876; https://doi.org/ 10.1200/JCO.2007.14.0418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fröhling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: Pathogenetic and clinical implications. J Clin Oncol 2005; 23:6285-95; PMID:16155011; https://doi.org/ 10.1200/JCO.2005.05.010 [DOI] [PubMed] [Google Scholar]
- [20].Braun BS, Shannon K. Targeting Ras in myeloid leukemias. Clin Cancer Res 2008; 14:2249-52; PMID:18413813; https://doi.org/ 10.1158/1078-0432.CCR-07-1005 [DOI] [PubMed] [Google Scholar]
- [21].Ricciardi MR, McQueen T, Chism D, Milella M, Estey E, Kaldjian E, Sebolt-Leopold J, Konopleva M, Andreeff M. Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia 2005; 19:1543-9; PMID:16001087; https://doi.org/ 10.1038/sj.leu.2403859 [DOI] [PubMed] [Google Scholar]
- [22].Martelli AM, Nyåkern M, Tabellini G, Bortul R, Tazzari PL, Evangelisti C, Cocco L. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia 2006; 20:911-28; PMID:16642045; https://doi.org/ 10.1038/sj.leu.2404245 [DOI] [PubMed] [Google Scholar]
- [23].Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 2014; 13:828-51; PMID:25323927; https://doi.org/ 10.1038/nrd4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013; 503:548-51; PMID:24256730; https://doi.org/ 10.1038/nature12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xu J, Hedberg C, Dekker FJ, Li Q, Haigis KM, Hwang E, Waldmann H, Shannon K. Inhibiting the palmitoylation/depalmitoylation cycle selectively reduces the growth of hematopoietic cells expressing oncogenic Nras. Blood 2012; 119:1032-5; PMID:22144181; https://doi.org/ 10.1182/blood-2011-06-358960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Eckfeldt CE, Pomeroy EJ, Lee RD, Hazen KS, Lee LA, Moriarity BS, Largaespada DA. RALB provides critical survival signals downstream of Ras in acute myeloid leukemia. Oncotarget 2016; 7:65147-56; PMID:27556501; https://doi.org/ 10.18632/oncotarget.11431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Milella M, Estrov Z, Kornblau SM, Carter BZ, Konopleva M, Tari A, Schober WD, Harris D, Leysath CE, Lopez-Berestein G, et al.. Synergistic induction of apoptosis by simultaneous disruption of the Bcl-2 and MEK/MAPK pathways in acute myelogenous leukemia. Blood 2002; 99:3461-4; PMID:11964319; https://doi.org/ 10.1182/blood.V99.9.3461 [DOI] [PubMed] [Google Scholar]
- [28].Zhang W, Konopleva M, Burks JK, Dywer KC, Schober WD, Yang JY, McQueen TJ, Hung MC, Andreeff M. Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase and murine double minute synergistically induces Apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Res 2010; 70:2424-34; PMID:20215498; https://doi.org/ 10.1158/0008-5472.CAN-09-0878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang W, Ruvolo VR, Gao C, Zhou L, Bornmann W, Tsao T, Schober WD, Smith P, Guichard S, Konopleva M, et al.. Evaluation of apoptosis induction by concomitant inhibition of MEK, mTOR, and Bcl-2 in human acute myelogenous leukemia cells. Mol Cancer Ther 2014; 13:1848-59; PMID:24739393; https://doi.org/ 10.1158/1535-7163.MCT-13-0576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Burgess MR, Hwang E, Firestone AJ, Huang T, Xu J, Zuber J, Bohin N, Wen T, Kogan SC, Haigis KM, et al.. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood 2014; 124:3947-55; PMID:25361812; https://doi.org/ 10.1182/blood-2014-05-574582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jain N, Curran E, Iyengar NM, Diaz-Flores E, Kunnavakkam R, Popplewell L, Kirschbaum MH, Karrison T, Erba HP, Green M, et al.. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: A University of Chicago phase II consortium trial. Clin Cancer Res 2014; 20:490-8; PMID:24178622; https://doi.org/ 10.1158/1078-0432.CCR-13-1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Konopleva MY, Walter RB, Faderl SH, Jabbour EJ, Zeng Z, Borthakur G, Huang X, Kadia TM, Ruvolo PP, Feliu JB, et al.. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clin Cancer Res 2014; 20:2226-35; PMID:24583795; https://doi.org/ 10.1158/1078-0432.CCR-13-1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al.. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118:3065-74; PMID:18725988; https://doi.org/ 10.1172/JCI34739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, Der CJ, Counter CM. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev 2002; 16:2045-57; PMID:12183360; https://doi.org/ 10.1101/gad.993902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Downward J. RAS synthetic lethal screens revisited: Still seeking the elusive prize? Clin Cancer Res 2015; 21:1802-9; PMID:25878361; https://doi.org/ 10.1158/1078-0432.CCR-14-2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, Sabatini DM. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 2017; 168:890-903 e15; PMID:28162770; https://doi.org/ 10.1016/j.cell.2017.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gentry LR, Martin TD, Reiner DJ, Der CJ. Ral small GTPase signaling and oncogenesis: More than just 15minutes of fame. Biochim Biophys Acta 2014; 1843:2976-88; PMID:25219551; https://doi.org/ 10.1016/j.bbamcr.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Urano T, Emkey R, Feig LA. Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J 1996; 15:810-6; PMID:8631302 [PMC free article] [PubMed] [Google Scholar]
- [39].White MA, Vale T, Camonis JH, Schaefer E, Wigler MH. A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J Biol Chem 1996; 271:16439-42; PMID:8663585; https://doi.org/ 10.1074/jbc.271.28.16439 [DOI] [PubMed] [Google Scholar]
- [40].Chien Y, White MA. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep 2003; 4:800-6; PMID:12856001; https://doi.org/ 10.1038/sj.embor.embor899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Peschard P, McCarthy A, Leblanc-Dominguez V, Yeo M, Guichard S, Stamp G, Marshall CJ. Genetic deletion of RALA and RALB small GTPases reveals redundant functions in development and tumorigenesis. Curr Biol 2012; 22:2063-8; PMID:23063435; https://doi.org/ 10.1016/j.cub.2012.09.013 [DOI] [PubMed] [Google Scholar]
- [42].Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al.. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009; 462:108-12; PMID:19847166; https://doi.org/ 10.1038/nature08460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pomeroy EJ, Lee LA, Lee RD, Schirm DK, Temiz NA, Ma J, Gruber TA, Diaz-Flores E, Moriarity BS, Downing JR, et al.. Ras oncogene-independent activation of RALB signaling is a targetable mechanism of escape from NRAS(V12) oncogene addiction in acute myeloid leukemia. Oncogene 2016; 36(23):3263-73; PMID:27991934; https://doi.org/ 10.1038/onc.2016.471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yan C, Liu D, Li L, Wempe MF, Guin S, Khanna M, Meier J, Hoffman B, Owens C, Wysoczynski CL, et al.. Discovery and characterization of small molecules that target the GTPase Ral. Nature 2014; 515:443-7; PMID:25219851; https://doi.org/ 10.1038/nature13713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yan C, Jones DN, Theodorescu D. Drugging the Ral GTPase. Small GTPases 2015; 6:157-9; PMID:26280620; https://doi.org/ 10.1080/21541248.2015.1018403 [DOI] [PMC free article] [PubMed] [Google Scholar]