

ABSTRACT
We recently showed that Tiam1 expression is induced in pro-inflammatory T helper 17 (Th17) cells differentiated with interleukin (IL)-6 and TGF-β1, and together with Rac1 promote Th17 cell development and autoimmunity in a mouse model of multiple sclerosis. Here we found that STAT3 and Smad3, downstream transcription factors of IL-6 and TGF-β1, respectively, play opposing roles in regulating Tiam1 transcription in CD4+ T-cells. While IL-6-STAT3 signaling promotes Tiam1 expression, TGF-β1-Smad3 induces the opposite outcome. At the Tiam1 promoter, both STAT3 and Smad3 bind to the Tiam1 promoter in Th17 cells. However, STAT3 induces Tiam1 promoter activity whereas Smad3 competes with STAT3 and inhibits its activity. Our findings uncover the complexity of STAT3/Smad3 signaling in regulating Tiam1 expression and Th17 cells.
KEYWORDS: Smad3, STAT3, Th17, Tiam1
T helper (Th)17 cells have been shown to play a major role in multiple sclerosis (MS) and in many autoimmune and other inflammatory diseases.1 They are differentiated from naïve CD4+ T cells in the presence of interleukin (IL)-6 and transforming growth factor (TGF)-β2 and are identified by the production of IL-17A, IL-17F, and IL-22 in addition to the expression of the master transcription factor RAR-related orphan receptor gamma (RORγ)t.3 Signal transducer and activator of transcription 3 (STAT3) plays a critical role in Th17 cell differentiation in humans and mice as it is rapidly activated upon exposure to IL-6 during Th17 cell polarization via IL-6 receptor (IL-6R). STAT3 has been shown to bind to the promoters and enhancers of several genes involved in Th17 development.4 Specifically, STAT3 binds to the promoter of the Rorc gene and STAT3-deficient T cells have impaired expression of RORγt.5
Rac1 is well characterized as a membrane bound signal transducing molecule that is involved in the regulation of cell motility and adhesion as well as cell cycle progression, mitosis, cell death and gene expression.6 Acting principally upstream of Rac1, increased expression of T-lymphoma invasion and metastasis protein (Tiam1) has been reported in highly invasive breast tumors7 and colon carcinomas8 and is mainly involved in the regulation of Rac1 mediated signaling pathways including cytoskeletal activities, cell polarity, endocytosis and membrane trafficking, cell migration, adhesion and invasion, cell growth and survival, metastasis and carcinogenesis.9,10 Recently, we demonstrated that Tiam1/Rac1 complex is involved in the development of human and murine Th17 cells and that targeting this complex either genetically or using pharmacological inhibitors ameliorated autoimmune encephalomyelitis, a mouse model of MS.11 Here, we analyzed the molecular mechanism that is involved in Tiam1 induction in Th17 cells. We identified opposing functions of STAT3 and Smad3 signaling in Tiam1 promoter activation during Th17 cell polarization revealing a direct crosstalk between STAT3 and Smad3 in autoimmunity.
Results
Opposite roles of STAT3 and Smad3 in the regulation of Tiam1 expression
To study the regulation of Tiam1 expression in Th17 cells, we first measured Tiam1 expression under pro- and anti-inflammatory conditions. Naïve CD4+ T cells were purified from spleens of wild-type (WT) mice and cells were stimulated with anti-CD3/CD28 in the presence of IL-6, TGF-β1 or a combination of IL-6 and TGF-β1 (Th17 cell differentiation condition). RNA lysates were prepared at 0, 16 and 24 hours. We found that while IL-6 induces a rapid increase in Tiam1 mRNA expression in CD4+ T cells measured by quantitative PCR (qPCR) (Fig. 1A), TGF-β1 stimulation exerted a weak effect on Tiam1 mRNA expression (Fig. 1A). Interestingly, addition of TGF-β1 to CD4+ T cells in the presence of IL-6 reduced the stimulatory effects of IL-6 on Tiam1 expression suggesting that TGF-β1 provides a dominant signaling that overrides that of IL-6 in the regulation of Tiam1 expression. We confirmed these findings at the protein level where addition of TGF-β1 to IL-6-treated T cells antagonizes the positive regulatory effects of IL-6 on Tiam1 expression. Rac1 protein expression was not altered by IL-6 or TGF-β1 (Fig. 1B). Next, we sought to analyze the direct effects of STAT3 and Smad3 on Tiam1 expression in CD4+ T cells lacking STAT3 or exposed to a Smad3 inhibitor. In agreement with the above data, we found that IL-6 induces Tiam1 expression in WT CD4+ T cells, however Tiam1 expression was impaired in STAT3 deficient CD4+ T cells that were purified from spleens of STAT3 knockout (KO) mice suggesting that IL-6-induced Tiam1 expression is STAT3-dependent (Fig. 1C). In the following experiment, we asked whether Smad3 signaling plays a role in the downregulation of Tiam1 expression by TGF-β1 in T cells. To this end, we used Smad3 selective inhibitor SIS3. Cells were pre-treated with SIS3 (10 μM) for 30 min followed by T cell stimulation with anti-CD3/CD28 for 24 hours in the presence or absence of recombinant TGF-β1. Control cells were pre-treated with the SIS3 solvent, DMSO. After 24 hours, cells were lysed and Tiam1 expression was measured by qPCR. As expected, TGF-β1 reduced Tiam1 expression in activated WT CD4+ T cells, however, this effect was reversed when cells were pre-incubated with SIS3 compound indicating that TGF-β1-induced inhibition of Tiam1 expression is Smad3-dependent (Fig. 1D).
Figure 1.
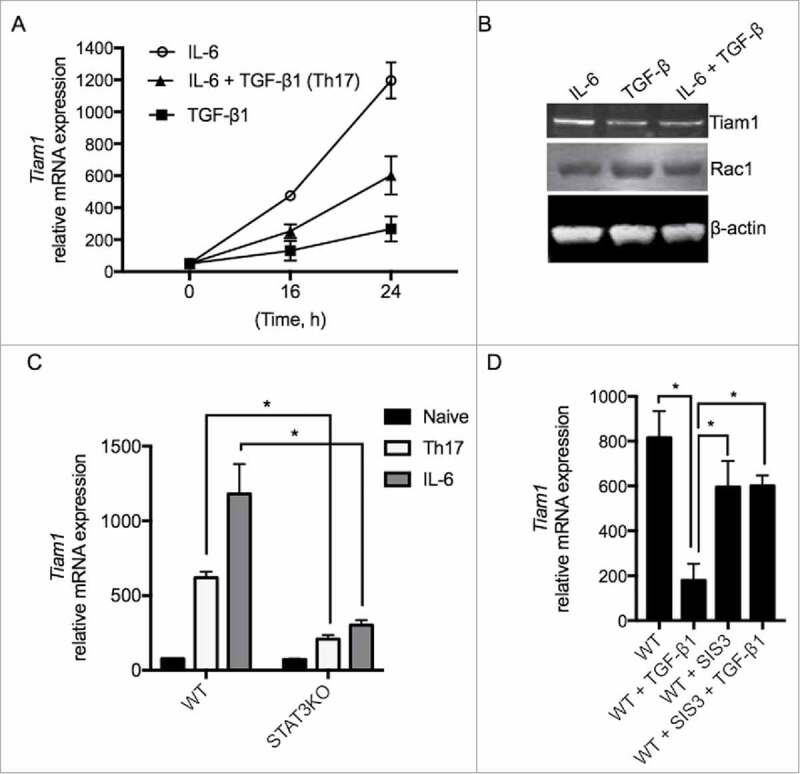
Regulation of Tiam1 Expression in Th17 Cells. (A) A representative gene expression assay of Tiam1 in CD4+ T cells treated with IL-6, TGF-β1 or with IL-6 and TGF-β1 combined. Naïve CD4+ T cells were cultured in the presence of IL-6, TGF-β1 or under Th17 cell polarizing conditions with combined IL-6 plus TGF-β1 for 0, 16 and 24 hours (h) and Tiam1 mRNA expression was assessed by qPCR. (B) Effects of TGF-β1 on IL-6-induced Tiam1 expression. CD4+ T cells were treated with IL-6, TGF-β1 or with IL-6 + TGF-β1 for 24h. Tiam1 and Rac1 protein expression was measured by Western blot. β-actin level is shown as loading control. (C) Tiam1 mRNA expression is reduced in STAT3KO T cells. Naïve CD4+ T cells were prepared from spleens of STAT3KO and control WT mice and cells were activated under IL-6 or Th17 cell condition for 24 hours. Tiam1 expression was analyzed by qPCR. (D) Tiam1 mRNA expression is upregulated in the presence of Smad3 inhibitor. Naïve CD4+ T cells were prepared from spleens of WT mice, cells were kept untreated or were pre-treated with Smad3 inhibitor SIS3 (10 μM) for 30 minutes followed by stimulation under TGF-β1 or Th17 cell condition for 24 hours. Tiam1 expression was analyzed by qPCR.
Competitive binding between STAT3 and Smad3 to the Tiam1 promoter in Th17 cells
Previous reports demonstrated that both STAT3 and Smad3 are key regulators of Th17 cell differentiation.12 Although we found that both STAT3 and Smad3 regulate Tiam1 expression, our above findings did not demonstrate whether these 2 transcription factors regulate Tiam1 expression indirectly or by binding and regulating Tiam1 promoter activity. To test this hypothesis, we searched the Tiam1 promoter for consensus sequence motifs for STAT3 and Smad3 using the sequence analysis algorithm TRANSFAC (TRANScription FACtor database). In silico analysis of the Tiam1 promoter revealed 6 putative Smad3-binding sites (GTCT)13 and 2 STAT3-binding sites (consensus STAT3-binding sequence, TTNCNNNAA where N stands for any nucleotide)14,15 within 1 kb upstream of the transcription start site (TSS). Interestingly, we identified a site at −0.82 kb with potential overlaps between STAT3 and Smad3 binding (Fig. 2A). To determine the Tiam1 promoter occupancies by STAT3 and Smad3, binding motifs were used to design Chromatin immunoprecipitation (ChIP) experiments. Primer sets flanking the STAT3 and Smad3 binding at 4 sites in the Tiam1 promoter (+1 to -111 bp, -450 to -605 bp, -560 to -708 bp and -770 to -930 bp) were designed to amplify the immunoprecipitated ChIP DNA by qPCR. Naïve CD4+ T cells were differentiated under Th17 cell-polarizing conditions for 3 hours and then analyzed by ChIP-qPCR. ChIP analysis of these sites revealed that Th17 cell differentiation condition significantly enhanced the binding of both STAT3 and Smad3 to the Tiam1 promoter at these 4 sites (Fig. 2B). Next, we generated a pGL3-Tiam1 luciferase reporter vector to investigate the ability of STAT3 and Smad3 to regulate the Tiam1 promoter in a luciferase reporter assay. 293T cell line was co-transfected with Tiam1. Unlike Smad3, we found that STAT3 enhanced Tiam1 promoter activity, however, co-transfection of Smad3 with STAT3 reduced STAT3-mediated Tiam1 promoter transactivation (Fig. 2C) which is in agreement with our findings in Fig. 1A showing reduction of Tiam1 expression when T cells were treated with both IL-6 and TGF-β1 compared with IL-6 treated cells. Nonetheless, we measured endogenous Tiam1 protein levels in 293T cells transfected with either STAT3 or in the presence of increasing amounts of Smad3 alone. We found that while STAT3 transfection increased Tiam1 expression, co-transfection with Smad3 plasmid reduces STAT3-mediated increased Tiam1 expression (Fig. 2D). Altogether, the data show that Smad3 competes with STAT3 in the regulation of Tiam1 expression.
Figure 2.
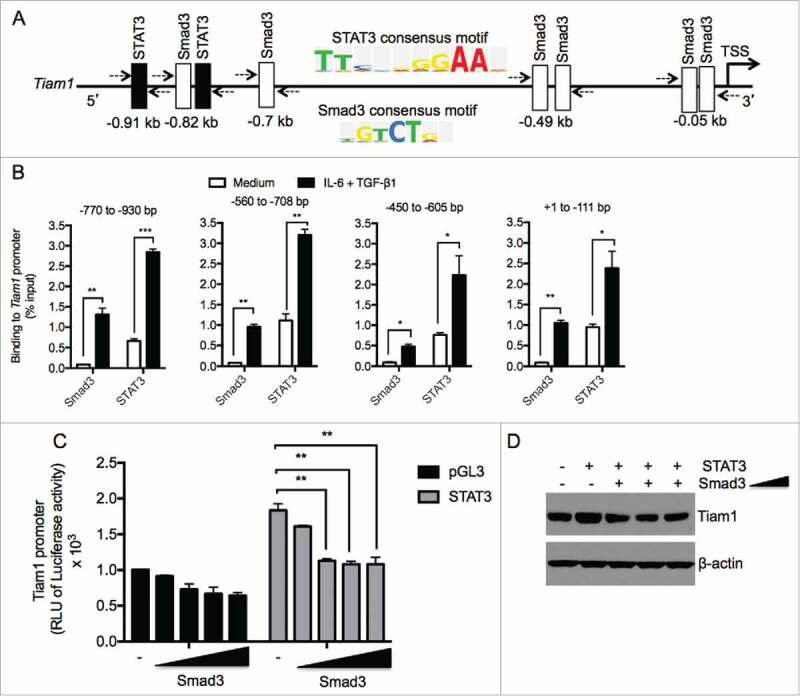
STAT3 and Smad3 bind to the Tiam1 promoter in Th17 cells. (A) Several predicted binding sites for Smad3 (Open box) and STAT3 (filled boxes) were identified upstream of transcription start site (TSS) of the Tiam1 promoter using the sequence analysis algorithm TRANSFAC. Consensus binding sites for STAT3 and Smad3 are shown. (B) ChIP analysis of STAT3 and Smad3 binding to the Tiam1 promoter in Th17 cells. Naïve CD4+ T cells from WT mice were polarized under Th17 cell conditions for 3 hours and ChIP-qPCR was performed to determine STAT3 and Smad3 binding to different regions of the Tiam1 promoter relative to IgG control as indicated above the graphs. Abs used for IP are anti-STAT3, anti-Smad3 and control IgG. Total input DNA before IP was used for normalization of data. Data are presented as average ± s.e.m. of per cent input with subtraction of control IgG. (C) HEK 293T cells were co-transfected with increasing concentrations of Smad3 (0.025 – 0.5 μg/ml) in the presence or absence of STAT3 (0.5 μg/ml), and with a constant amount of Tiam1-pGL3/Renilla reporter constructs. Cells were lyzed 48 hours later and luminescence was measured. RLU, relative luciferase units. Luciferase activities were calculated as fold change relative to empty vector. Data represent mean ± SEM of a representative experiment (n = 3) each performed in triplicate. *P < 0.05; **P < 0.01 by Student t test. (D) HEK 293T cells were co-transfected with increasing concentrations of Smad3 (0.05 – 0.5 μg/ml) in the presence or absence of STAT3 (0.5 μg/ml). Protein lysates were prepared 48 hours later and Tiam1 expression was measured by Western blot. β-actin level is shown as loading control.
Discussion
In summary, our study demonstrates that STAT3 and Smad3 signaling triggered by IL-6 and TGF-β1, respectively, play opposite roles in the direct regulation of Tiam1 promoter activity and Tiam1 expression in Th17 cells. Recently we have shown that Tiam1-Rac1 complex plays a crucial role in the induction of pro-inflammatory Th17 cells and autoimmunity in a mouse model of MS where pharmacological and genetic targeting of Tiam1/Rac1 complex inhibited Th17 cell development and ameliorated disease symptoms and pathology.11 Differentiation of naïve CD4+ T cells into Th17 cells is achieved with the cytokines TGF-β1 and IL-62. This cytokine combination generates Th17 cells, that co-produce IL-9, IL-10 together with IL-17A and do not induce autoimmunity and have therefore been called non-pathogenic Th17 cells.16 To acquire the ability to induce autoimmunity in vivo, IL-17A-producing T cells need to either be exposed to IL-2317 or be generated with alternative cytokine combinations such as IL-1β, IL-6, IL-23 in the absence of TGF-β1.18 Of interest, in addition to IL-6 as an inducer of STAT3 signaling in Th17 cells, both IL-1β and IL-23 modulate STAT3 activation in Th17 cells.19,20 Here, we found that in contrast to the pro-pathogenic STAT3 signaling in Th17 cells that induces Tiam1 expression, Smad3 pathway downregulates Tiam1 expression and is associated with non-pathogenic Th17 cells. Thus, our findings further support the implication of Tiam1/Rac1 complex in the development of pathogenic Th17 cells leading to the exacerbation of autoimmune response in vivo.
It has been previously shown that the Rac1 guanosine triphosphatase can bind to and regulate STAT3 activity. Dominant negative Rac1 inhibited STAT3 activation by growth factors, whereas activated Rac1 stimulated STAT3 phosphorylation on both tyrosine and serine residues.21 This suggests that in Th17 cells, active Rac1 may provide a positive feedback loop to further promote Tiam1 expression in a STAT3-dependent manner. Indeed, we have found that NSC23766, a small molecule that effectively inhibits Tiam1 from binding and activating Rac122 reduced Tiam1 protein expression in Th17 cells (Data not shown) which supports a role of Rac1 downstream signaling in the induction of Tiam1 expression.
TGF-β1 is a pleiotropic cytokine that regulates diverse cellular responses including apoptosis, cell growth inhibition and immune surveillance.23 Several studies, albeit conflicting, reported direct crosstalk between STAT3 and Smad3 signaling.24,25 For instance, TGF-β1 reduces STAT3 phosphorylation and suppresses its downstream target genes.26 On the other hand, a report showed STAT3 induces TGF-β1 expression to promote fibrosis,27 whereas another report showed that STAT3 interacts physically with Smad3 thereby disrupting Smad3-Smad4 complex leading to the attenuation of TGF-β1-mediated Smad3 signaling in epithelial cells.25 Using Tiam1 reporter assay, we found that while Smad3 appears to have a modest effects in Tiam1 promoter activity, co-transfection with STAT3 showed a more prominent repression of Tiam1 promoter suggesting Smad3 may exert both STAT3-dependent and independent effects. It is noteworthy that only one out of the 6 Smad3 predicted binding sites to the Tiam1 promoter overlaps with STAT3 binding site. Recently, the interaction between STAT3 and SMAD signaling pathways was analyzed in Th17 cells. The authors unraveled a complex interaction between these 2 pathways where Smad3 and its highly homologous family member Smad2 play opposite roles by acting as a co-repressor and co-activator of STAT3, respectively, in Th17 cells.12 The findings in our present study are consistent with the oncogenic properties of STAT3 and growth suppressor role of the TGF-β signaling, where STAT3 induces the expression of Tiam1 while TGF-β1 suppresses it.
In conclusion, our study provides additional evidence of the opposing roles of STAT3 and Smad3 in the regulation of T cell function that implicates Tiam1/Rac1 pathway regulation in Th17 cells.
Materials and methods
Mouse T cell differentiation
Naïve CD4+CD62L+ T cells were purified from naïve C57BL/6 wild-type (WT) or STAT3 CD4-Cre-lox conditional knockout mice11 using magnetic-activated cell sorting (MACS) (Miltenyi). Cells (250 – 500 × 103) were stimulated using plate-bound anti-CD3 (4 μg/mL) as well as soluble anti-CD28 (2 μg/mL) in 48-well plates for 4 d in a serum-free medium (X-VIVO-20; Lonza) supplemented with 50 μM 2-mercaptoethanol, 1 mM sodium pyruvate, L-glutamine, nonessential amino acids, and 100 U/mL of penicillin and streptomycin in the presence of recombinant cytokines. Naïve CD4+ T cells were polarized in the presence of recombinant mouse IL-6 (30 ng/mL) and recombinant human TGF-β1 (3 ng/mL) plus anti-IFNγ (10 μg/mL) for Th17. For Smad3 inhibition, cells were pre-treated with Smad3 specific inhibitor SIS3 (10 μM) and treatment was kept for the duration of the assay. All recombinant proteins were purchased from R&D Systems. Neutralizing antibodies were purchased from BD Biosciences.
Expression analysis by real-time quantitative PCR
RNA from 50–100 × 103 T cells was purified using Stratagene RNA kit and directly transferred into the RT reagent using the Applied Biosystems Taqman reverse transcriptase reagents. Samples were then subjected to real-time quantitative PCR (qPCR) analysis on the Applied Biosystems PRISM 7000 Sequencer Detection System (Applied Biosystems, Foster City, CA) using standard conditions. Genes analyzed were detected with commercially available assays (Applied Biosystems). The relative mRNA abundance was normalized against GAPDH.
Western blot
Cells were lysed in RIPA buffer (Thermo Scientific) with a protease inhibitor mixture (Roche Diagnostics); 20 μg total protein was loaded into each well of a SDS-PAGE gel for separation by electrophoresis and then transferred on nitrocellulose membrane. The resulting blots were blocked for 1 h with TBS-Tween 20 containing 5% powder skim milk and then probed for 3 hrs at room temperature with primary antibodies directed against: Tiam1 and Rac1 (Santa Cruz Biotechnology). β-actin mouse mAb was used as the loading control. Anti-mouse and anti-rabbit IRDye® secondary antibodies were purchased from LI-COR.
ChIP-qPCR
Naïve CD4+ CD44lowCD62Lhi T cells were sorted by flow cell sorter and were polarized to Th17 phenotype for 48 hours. ChIP was performed according to the protocol previously described.28 Cell lysates were used for immunoprecipitation with anti-STAT3 and anti-Smad3 (Santa Cruz Biotechnology) and were compared with control IgG. The following primers were used for the detection of STAT3 and Smad3 binding: Fwd: CATCAGAAGCACAATCCAAAA, Rev: TCCCTGTCCTTGCTGTTTCT (+1 to -111 bp); Fwd: CACTGATGGAAGGCACAGAA, Rev: ATCTGGGCAAATAGCAAACG (-450 to -605 bp); Fwd: ATGGACTGAGGGCTGGAGAG, Rev: TACAGGTTGCTAGCCCTTGG (-560 to -708 bp); and Fwd: CAAAACACTTGCGGTACTTGC, Rev: GAGTGTCCAAAGGGGACAGA (-770 to -930 bp). ChIP-qPCR was performed according to our previous report.29
Dual luciferase reporter assay
PGL3 luciferase reporter plasmids encoding the Tiam1 promoter were generated by our group. Briefly, 2kb Tiam1 upstream fragment of transcription start site was cloned from BAC then inserted into pGL3 Basic Vector's multi-cloning site. The following primers were used: PGL3-Tiam1UF-F1: GATCCTAGGGCCGCTGCGGCC TCCAGGACA GCCAGGGCTA CAC; PGL3-Tiam1UF-R1: GATCCTAGGGCCTGTTTGGCC TAGGAAGGATTCCCGGCATGAATC. Final vector was confirmed by BamHI and Bgl2. BglII Fragment sizes 1037, 112, 5707. BamHI Fragment sizes 3363, 3493. For the luciferase reporter assay, Human Embryonic Kidney 293T cells were seeded in culture plates the day before transfection according the manufacturer's instructions. The cells confluency at the day of the transfection was 40–80%. 293T cells were transfected with pGL3 and the Renilla pRL-TK luciferase reporter constructs (0.5 μg/ml) together with the indicated plasmids using Effectene Transfection Reagent kit (Qiagen). Cells were cultured overnight then treated with Luciferase Assay System kit reagents (Promega) and results were acquired on Wallac 1420 Viktor2 plate reader (Perkin-Elmer).
Statistical analysis
Statistical evaluations of gene expression, DNA binding and luciferase activity were performed using the unpaired Student's t test. Values of P < 0.05 were considered to be statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain: J Neurol 2009; 132:3329-41; PMID:19933767; https://doi.org/ 10.1093/brain/awp289 [DOI] [PubMed] [Google Scholar]
- [2].Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006; 441:235-8; PMID:16648838; https://doi.org/ 10.1038/nature04753 [DOI] [PubMed] [Google Scholar]
- [3].Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al.. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008; 28:29-39; PMID:18164222; https://doi.org/ 10.1016/j.immuni.2007.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, Takahashi H, Sun HW, Kanno Y, Powrie F, et al.. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010; 32:605-15; PMID:20493732; https://doi.org/ 10.1016/j.immuni.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al.. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 2007; 282:9358-63; PMID:17277312; https://doi.org/ 10.1074/jbc.C600321200 [DOI] [PubMed] [Google Scholar]
- [6].Boissier P, Huynh-Do U. The guanine nucleotide exchange factor Tiam1: a Janus-faced molecule in cellular signaling. Cell Signal 2014; 26:483-91; PMID:24308970; https://doi.org/ 10.1016/j.cellsig.2013.11.034 [DOI] [PubMed] [Google Scholar]
- [7].Adam L, Vadlamudi RK, McCrea P, Kumar R. Tiam1 overexpression potentiates heregulin-induced lymphoid enhancer factor-1/beta -catenin nuclear signaling in breast cancer cells by modulating the intercellular stability. J Biol Chem 2001; 276:28443-50; PMID:11328805; https://doi.org/ 10.1074/jbc.M009769200 [DOI] [PubMed] [Google Scholar]
- [8].Minard ME, Kim LS, Price JE, Gallick GE. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res Treat 2004; 84:21-32; PMID:14999151; https://doi.org/ 10.1023/B:BREA.0000018421.31632.e6 [DOI] [PubMed] [Google Scholar]
- [9].Arkin M. Protein-protein interactions and cancer: small molecules going in for the kill. Curr Opin Chem Biol 2005; 9:317-24; PMID:15939335; https://doi.org/ 10.1016/j.cbpa.2005.03.001 [DOI] [PubMed] [Google Scholar]
- [10].Stebel A, Brachetti C, Kunkel M, Schmidt M, Fritz G. Progression of breast tumors is accompanied by a decrease in expression of the Rho guanine exchange factor Tiam1. Oncol Rep 2009; 21:217-22; PMID:19082465 [PubMed] [Google Scholar]
- [11].Kurdi AT, Bassil R, Olah M, Wu C, Xiao S, Taga M, Frangieh M, Buttrick T, Orent W, Bradshaw EM, et al.. Tiam1/Rac1 complex controls Il17a transcription and autoimmunity. Nat Commun 2016; 7:13048; PMID:27725632; https://doi.org/ 10.1038/ncomms13048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yoon JH, Sudo K, Kuroda M, Kato M, Lee IK, Han JS, Nakae S, Imamura T, Kim J, Ju JH, et al.. Phosphorylation status determines the opposing functions of Smad2/Smad3 as STAT3 cofactors in TH17 differentiation. Nat Commun 2015; 6:7600; PMID:26194464; https://doi.org/ 10.1038/ncomms8600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell 1998; 1:611-7; PMID:9660945; https://doi.org/ 10.1016/S1097-2765(00)80061-1 [DOI] [PubMed] [Google Scholar]
- [14].Wen Z, Zhong Z, Darnell JE, Jr.. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995; 82:241-50; PMID:7543024; https://doi.org/ 10.1016/0092-8674(95)90311-9 [DOI] [PubMed] [Google Scholar]
- [15].Xu X, Sun YL, Hoey T. Cooperative DNA binding and sequence-selective recognition conferred by the STAT amino-terminal domain. Science 1996; 273:794-7; PMID:8670419; https://doi.org/ 10.1126/science.273.5276.794 [DOI] [PubMed] [Google Scholar]
- [16].Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, et al.. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 2012; 13:991-9; PMID:22961052; https://doi.org/ 10.1038/ni.2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol 2007; 8:1390-7; PMID:17994024; https://doi.org/ 10.1038/ni1539 [DOI] [PubMed] [Google Scholar]
- [18].Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, et al.. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 2010; 467:967-71; PMID:20962846; https://doi.org/ 10.1038/nature09447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, Pear WS, Hatton RD, Weaver CT. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat Immunol 2015; 16:286-95; PMID:25642823; https://doi.org/ 10.1038/ni.3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'Shea JJ. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A 2006; 103:8137-42; PMID:16698929; https://doi.org/ 10.1073/pnas.0600666103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH, Guan KL. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science 2000; 290:144-7; PMID:11021801; https://doi.org/ 10.1126/science.290.5489.144 [DOI] [PubMed] [Google Scholar]
- [22].Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U SA 2004; 101:7618-23; PMID:15128949; https://doi.org/ 10.1073/pnas.0307512101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol 2010; 10:554-67; PMID:20616810; https://doi.org/ 10.1038/nri2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nakashima K, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Kawabata M, Miyazono K, Taga T. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 1999; 284:479-82; PMID:10205054; https://doi.org/ 10.1126/science.284.5413.479 [DOI] [PubMed] [Google Scholar]
- [25].Wang G, Yu Y, Sun C, Liu T, Liang T, Zhan L, Lin X, Feng XH. STAT3 selectively interacts with Smad3 to antagonize TGF-beta signalling. Oncogene 2016; 35:4422; PMID:27345395; https://doi.org/ 10.1038/onc.2016.145 [DOI] [PubMed] [Google Scholar]
- [26].Gunaje JJ, Bhat GJ. Distinct mechanisms of inhibition of interleukin-6-induced Stat3 signaling by TGF-beta and alpha-thrombin in CCL39 cells. Mol Cell Biol Res Commun 2000; 4:151-7; PMID:11281729; https://doi.org/ 10.1006/mcbr.2001.0272 [DOI] [PubMed] [Google Scholar]
- [27].Ogata H, Chinen T, Yoshida T, Kinjyo I, Takaesu G, Shiraishi H, Iida M, Kobayashi T, Yoshimura A. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene 2006; 25:2520-30; PMID:16474852; https://doi.org/ 10.1038/sj.onc.1209281 [DOI] [PubMed] [Google Scholar]
- [28].Bassil R, Orent W, Olah M, Kurdi AT, Frangieh M, Buttrick T, Khoury SJ, Elyaman W. BCL6 controls Th9 cell development by repressing Il9 transcription. J Immunol 2014; 193:198-207; PMID:24879792; https://doi.org/ 10.4049/jimmunol.1303184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang X, Zhang Y, Yang XO, Nurieva RI, Chang SH, Ojeda SS, Kang HS, Schluns KS, Gui J, Jetten AM, et al.. Transcription of Il17 and Il17f is controlled by conserved noncoding sequence 2. Immunity 2012; 36:23-31; PMID:22244845; https://doi.org/ 10.1016/j.immuni.2011.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]