

ABSTRACT
Degradation of mitochondria is an important cellular quality control mechanism mediated by two distinct pathways: one involving Parkin-mediated ubiquitination and the other dependent on mitophagy receptors. It is known that mitochondria are degraded by the autophagy pathway; however, we recently reported that the small GTPase Rab5 and early endosomes also participate in Parkin-mediated mitochondrial clearance. Here, we have developed a protocol to isolate Rab5-positive vesicles from cells for proteomics analysis and provide additional data confirming that mitophagy regulators and mitochondrial proteins are present in these vesicles. We also demonstrate that the mitophagy receptor BNIP3 utilizes the Rab5-endosomal pathway to clear mitochondria in cells. These findings indicate that a redundancy exists in the downstream degradation pathways to ensure efficient mitochondrial clearance.
KEYWORDS: autophagy, BNIP3, endosomes, mitochondria, mitophagy, Parkin, Rab5
Introduction
Mitochondria play a critical role in multiple cellular processes, such as oxidative phosphorylation, metabolism, and apoptosis. However, when damaged, mitochondria may become cytotoxic which can lead to activation of cell death. The efficient and selective elimination of damaged or excessive mitochondria is critical for maintaining a healthy and appropriate population of mitochondria and preventing unnecessary cell death. Mitochondrial autophagy, or mitophagy, is considered to be a central mechanism of mitochondrial quality and quantity control.1 In this process, a mitochondrion that has been labeled for degradation is sequestered in a double membrane autophagosome which is then delivered it to the lysosome for degradation.2 Formation of autophagosomes requires autophagy-related proteins (Atg), such as Atg5 and Atg7.3,4
Mitophagy is mediated by two distinct pathways: one involves Parkin-mediated ubiquitination5 and the other depends on mitophagy receptors in the mitochondrial outer membrane.6,7 The first pathway involves recruitment of the E3 ubiquitin ligase Parkin to depolarized mitochondria where it proceeds to ubiquitinate diverse mitochondrial outer membrane proteins. Adaptor proteins, such as p62 and NBR1, contain both an ubiquitin-binding domain and a LC3-interacting region (LIR) motif. They bind to ubiquitinated proteins on the mitochondria and to LC3 on the autophagosome tethering the 2 together. The second pathway involves proteins that are integrated in the outer mitochondrial membrane that harbor a LIR. These so-called mitophagy receptors can directly interact with LC3 on the autophagosomal membrane.8-11 BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) is an atypical pro-apoptotic protein and a member of the BH3-only protein family that can also function as a mitophagy receptor.12 It interacts directly with LC3 via its LIR motif to promote mitophagy, and this function is independent of its pro-death activity.11
Recent studies have uncovered that additional pathways to traditional Atg-dependent autophagy exist in cells that can degrade mitochondria. For instance, an alternative Atg5/7-independent autophagy pathway13 participates in clearing mitochondria during maturation of erythrocytes14 and reprogramming iPS cells.15 We recently reported that the endosomal-lysosomal degradation pathway can also mediate the clearance of mitochondria and that this process is independent of traditional and alternative autophagy.16 Members of the small GTPases family are key regulators of the endosomal pathway, and various Rab proteins regulate different steps in the pathway.17 Rab5 is mainly associated with early endosomes and controls biogenesis and function of this compartment, whereas Rab7 defines the late endosomes and is involved in their fusion with lysosomes. We discovered that depolarized mitochondria that have been marked for degradation by Parkin were sequestered in Rab5-positive early endosomes.16 These early endosomes matured into late endosomes before fusing with lysosomes.
Here, we have developed a protocol to isolate Rab5-positive vesicles from cells for proteomic analysis and provide additional data confirming that mitochondrial proteins are present in these vesicles. In addition, we demonstrate that the mitophagy receptor BNIP3 also utilizes the Rab5-endosomal pathway to clear dysfunctional mitochondria in cells. Overall, these findings indicate that multiple pathways use the endosomal-lysosomal degradation pathway ensuring efficient mitochondrial quality and quantity control.
Results
The small GTPase Rab5 functions in the biogenesis and homotypic fusion of early endosomes.18 Mutating the glutamine to leucine at amino acid residue 79 in the GTPase domain results in a constitutively active Rab5 that is unable to hydrolyze bound GTP. Thus, overexpression of Rab5Q79L in cells leads to fusion of early endosomes resulting in oversized endosomes with an average size of 2–5 µm.19,20 We recently reported that depolarized mitochondria labeled for degradation by the E3 ubiquitin ligase Parkin are sequestered in Rab5-positive endosomes for subsequent delivery to lysosomes, a process that is independent of autophagy.16 Mouse embryonic fibroblasts (MEFs) derived from Atg5−/− embryos are deficient in autophagy as confirmed by the lack of LC3II and accumulation of the autophagy substrate p62 (Fig. 1A). Here, we confirmed that overexpression of Rab5Q79L led to formation of enlarged endosomes (EE) in both wild type (WT) and the autophagy-deficient Atg5−/− MEFs overexpressing Parkin (Fig. 1A and B). We also observed that the majority of the enlarged endosomes did not contain mitochondria at baseline. However, we found that a significant number of mitochondria accumulated inside the enlarged endosomes in both WT and Atg5−/− MEFs after treatment with the mitochondrial uncoupler FCCP (Fig. 1B and C). We have previously found that about 6–10 Rab5-positive endosomes co-localize with mitochondria after FCCP treatment and that there is an average of one mitochondrion per vesicle.16 Here, we observed that an increased number of Rab5Q79L-positive vesicles co-localized with mitochondria but more importantly, we found multiple mitochondria per vesicle after FCCP treatment. The increased number of mitochondria per vesicle is most likely due to the homotypic fusion of the Rab5Q79L-positive vesicles and their inability to mature into late endosomes.
Figure 1.
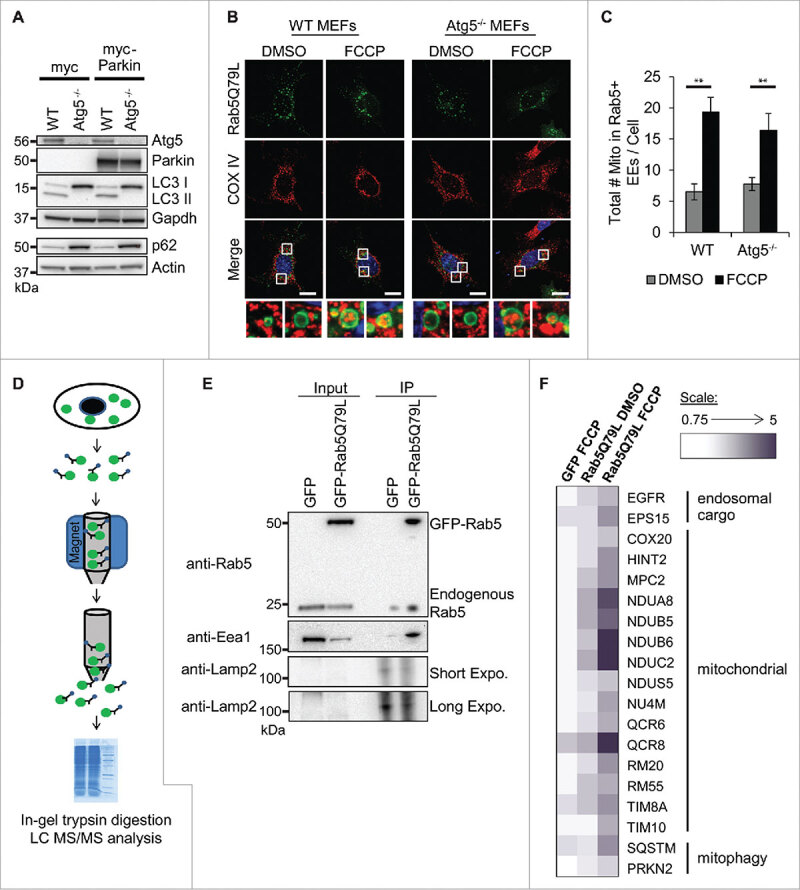
Mitochondria are sequestered inside Rab5-positive endosomes in WT and Atg5−/− MEFs. (A) Western blot for Atg5, Parkin, LC3, and p62 levels in WT and Atg5−/− MEFs overexpressing myc or myc-Parkin. (B) Representative images of WT and Atg5−/− MEFs overexpressing GFP-Rab5Q79L plus myc-Parkin. Cells were treated with DMSO or 25 µM FCCP for 4 h, and then fixed and stained with anti-COX IV to visualize mitochondria. Nuclei were counterstained with Hoechst 33342 (blue). Scale bars = 20 µm. (C) Quantification of total number of mitochondria inside GFP-Rab5Q79L positive vesicles per cell (n = 3, **p < 0.01 vs DMSO). EE = enlarged endosomes. All values are means ± s.e.m. (D) Scheme for isolation of GFP-labeled vesicles. (E) Immunoprecipitation (IP) of GFP or GFP-Rab5Q79L with anti-GFP coupled to magnetic beads confirms enrichment for GFP-Rab5Q79L as well as endogenous Rab5. Pulldown also enriches for the early endosome marker Eea1. The lysosomal protein Lamp2 was undetectable unless blot was subjected to a long (10 min) exposure. (F) Heatmap of proteins from endosome isolation identified by mass spectrometry. Values were generated using a ratio of each sample to the GFP DMSO control. Scale is in fold change over GFP DMSO control.
Next, we aimed to perform proteomics analysis of isolated Rab5-positive endosomes to confirm mitochondrial content. In our previous study, we found that once the mitochondria have been sequestered inside Rab5-positive endosomes, these vesicles mature very quickly into late endosomes.16 Due to the transient number of early endosomes containing mitochondria present in cells at any given time, it was challenging to isolate a sufficient number Rab5-positive vesicles for proteomics analysis. Thus, we took advantage of cells overexpressing Rab5Q79L which accumulate the enlarged early endosomes containing mitochondria. We have previously isolated intact GFP-positive autophagosomes using antibody against GFP conjugated to magnetic beads.11 Here, we adapted this protocol to enrich for Rab5-positive endosomes. We used the anti-GFP conjugated to magnetic beads to capture the enlarged Rab5-positive vesicles from Atg5−/− MEFs (Fig. 1D). After confirming that we successfully pulled down endosomes by blotting for the markers Rab5 and Eea1 (Fig. 1E), but not lysosomal protein Lamp2, the purified endosomes were resolved on one-dimensional SDS-PAGE, subjected to in-gel tryptic digestion and analysis by LC MS/MS. The mass spectrometry confirmed the presence of mitochondrial proteins in GFP-Rab5Q79L labeled endosomes. The number of mitochondrial proteins was also increased after FCCP treatment (Fig. 1F). GFP-Rab5Q79L-labeled endosomes also contained higher levels of proteins related to endosomal cargo and mitophagy than DMSO controls after FCCP treatment (Fig. 1F).
Mitochondrial clearance can also be mediated by mitophagy receptors such as BNIP312. In our previous study, we found that overexpression of BNIP3 led to an increase in the number of Rab5-positive vesicles and amount of mitochondrial clearance in both WT and Atg5−/− MEFs.16 Here, we confirmed the accumulation of mitochondria inside enlarged Rab5Q79L-positive endosomes in both WT and Atg5−/− MEFs overexpressing BNIP3 (Fig. 2A-C). This suggests that mitophagy receptors can also utilize the endosomal pathway to clear mitochondria. Furthermore, BNIP3 is known to induce mitochondrial dysfunction and cell death when overexpressed in cells.21-23 Therefore, to examine whether removal of mitochondria by Rab5-positive endosomes was a protective response activated by the cell, we examined the effects of modulating Rab5 activity on cell viability. We found that overexpression of BNIP3 caused a significant increase in cell death as measured by BOBO-3 uptake in cells with compromised plasma membranes in both WT and Atg5−/− MEFs (Fig. 2D and E). We also found that the presence of GFP-Rab5Q79L did not change BNIP3-mediated cell death in either WT or Atg5−/− MEFs (Fig. 2D), suggesting that merely sequestering mitochondria in vesicles is not sufficient to protect against cell death. In contrast, overexpression of BNIP3 plus the Rab5 dominant negative, Rab5S34N, led to a significant increase in BNIP3-mediated cell death in Atg5−/− MEFs but not in WT MEFs (Fig. 2E). This is most likely due to the fact that the WT still have intact autophagy to clear mitochondria.
Figure 2.
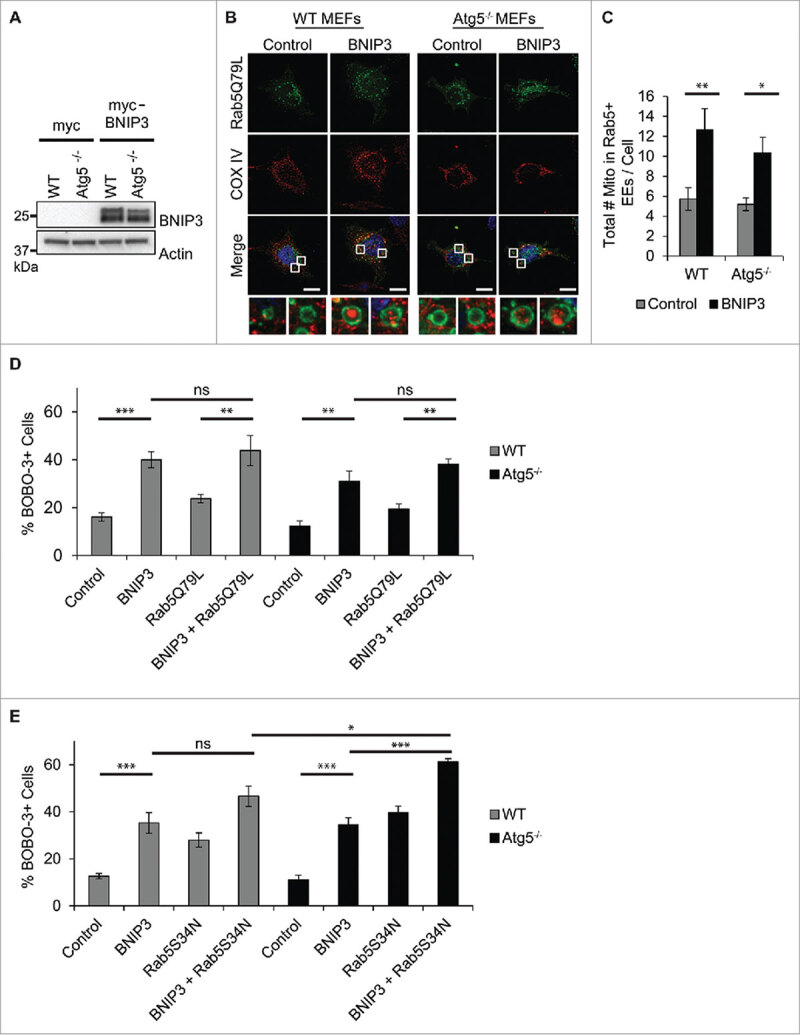
BNIP3 promotes engulfment of mitochondria into Rab5-positive endosomes. (A) Western blot for BNIP3 in WT and Atg5−/− MEFs overexpressing myc or myc-BNIP3. (B) Representative images of WT and Atg5−/− MEFs overexpressing GFP-Rab5Q79L plus empty vector or BNIP3. 24 h post-transfection, the cells were fixed and stained with anti-COX IV to label mitochondria. (C) Quantification of total number of mitochondria inside GFP-Rab5Q79L positive vesicles per cell (n = 4, *p < 0.05, **p < 0.01 vs DMSO). EE = enlarged endosomes. (D) Quantification of BOBO-3 positive (dead) cells. Sequestration of mitochondria in Rab5Q79L-positive vesicles has no effect on BNIP3-mediated cell death in WT and Atg5−/− MEFs. (E) Quantification of BOBO-3 positive (dead) cells. Abrogation of Rab5 activity increases susceptibility to BNIP3-mediated cell death in Atg5−/− MEFs. Cell death was assessed by BOBO-3 Iodide uptake in cells overexpressing the indicated constructs (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant). All values are means ± s.e.m.
Discussion
In this study, we demonstrate that if we use Rab5Q79L to enrich for early endosomes, a sufficient number of Rab5-positive vesicles can be isolated for proteomic analysis from cultured cells. The ability to isolate vesicles from cells provides investigators the potential to analyze the content of the vesicles which might vary depending on the extracellular or intracellular environments. Because the vesicles were isolated based on their GFP label, it is possible to adapt this protocol to other vesicles such as autophagosomes. In fact, we have used a similar protocol to isolate GFP-labeled autophagosomes from cells.11 Thus, adaptation of our protocol will allow investigators to compare contents and compositions of different vesicles, such as early endosomes and autophagosomes, in response to various stressors. Also, an outstanding question is still whether Rab5-positive endosomes involved in sequestering mitochondria are different from the Rab5-positive endosomes involved in receptor degradation. The average size of an early endosome is about 20–50 nm,24 whereas we found that the Rab5-positive vesicles containing mitochondria are about 500 nm in size.16 Whether the early endosomes undergo homotypic fusion to accommodate the larger mitochondria still needs to be confirmed. Interestingly, previous studies have reported that early endosomes are heterogeneous in terms of morphology, localization, and function.25
In this study, we have also confirmed that the mitophagy receptor BNIP3 can promote sequestration of mitochondria into Rab5-positive endosomes and that this process occurs independently of autophagy. We also found that abrogation of Rab5 led to increased susceptibility to BNIP3-mediated cell death in both WT and autophagy-deficient Atg5−/− MEFs, suggesting that this is a protective mechanism activated by the cells. BNIP3 is known to induce autophagy and mitophagy when overexpressed in cells.21,26,27 Thus, additional studies are needed to examine if BNIP3 simultaneously activates autophagy and endosomal-mediated mitochondrial clearance or if their timing of activation is different. In our previous study, we found that treatment of cells with a mitochondrial uncoupler led to increased formation of Rab5-positive early endosomes before formation of autophagosomes.16 The cells contain a large number of Rab5-positive endosomes even under baseline conditions, whereas autophagosomes must be generated de novo upon demand. This suggests that mitochondria can be sequestered more quickly into existing early endosomes and may represent a first line of defense.
Mitophagy receptors such as BNIP3 directly target mitochondria to autophagosomes via their LIR motifs, therefore bypassing the need for Parkin and ubiquitination.8,11,28 Interestingly, our data suggest that BNIP3 can also target mitochondria to the early endosomes, but how this occurs is still unclear. Internalization of cargo into endosomes is mediated by the endosomal sorting complex required for transport (ESCRT) machinery.29 The ESCRT complexes bind to ubiquitinated cargo and pull them into the endosome through invagination and scission.29 Parkin labels mitochondria for degradation by ubiquitinating mitochondrial proteins, and we found that Parkin-mediated removal of mitochondria by the Rab5-positive endosomes involves the ESCRT machinery.16 Here, we found that BNIP3 promotes engulfment of mitochondria into endosomes in the absence of Parkin. However, whether BNIP3 utilizes the ESCRT machinery in capturing mitochondria needs to be investigated further. It is possible that BNIP3 recruits a different E3 ubiquitin ligase to the mitochondria or activates a resident mitochondrial E3 ubiquitin ligase, such as MITOL/MARCH5.30 Future studies need to focus on how BNIP3 targets mitochondria to early endosomes.
In conclusion, mitochondrial clearance is mediated by two distinct processes, and multiple downstream degradation pathways exist to degrade the mitochondria. These include traditional1 and alternative autophagy,13,31 microautophagy,32 and endosomal-lysosomal degradation pathway.16 Thus, future studies should focus on characterizing the mechanisms underlying BNIP3-mediated targeting of mitochondria to early endosomes, and determining whether other mitophagy receptors can utilize this pathway. Clearly, mitochondrial quantity and quality control comprises an intricate network of proteins and pathways. It is very likely that future studies will reveal additional proteins and pathways involved in this important process. Mitochondrial dysfunction plays a role in the development of many neurodegenerative and cardiovascular diseases. Increased knowledge into the pathways that regulate mitochondrial health will allow for identification of new therapeutic targets to treat various diseases.
Materials and methods
Cells and culture conditions
Mouse embryonic fibroblasts (MEFs) were cultured in MEF media (DMEM (Thermo Fisher Scientific, 10569044) supplemented with 10% FBS (Thermo Fisher Scientific, 16000044), 100 U/mL penicillin and 100 μg/mL streptomycin (Gemini, 400–401)) and maintained in a 5% CO2 atmosphere at 37°C. WT and Atg5−/− MEFs were generously provided by Dr. Noboru Mizushima (The University of Tokyo, Japan).4
Immunofluorescence
Cells were transiently transfected with DNA using FuGENE 6 Transfection Reagent (Promega, E2691) according to the manufacturer's instructions with myc-Parkin (gift from Ted Dawson, Addgene plasmid #17612) and GFP-Rab5Q79L (gift from Qing Zhong, Addgene plasmid #28046). For imaging of mitochondria in BNIP3 experiments, cells were transfected with myc vector or myc-BNIP333 plus GFP-Rab5Q79L and cultured in the presence of 50 µM ZVAD (Millipore, 627610) to inhibit apoptosis. Cells undergoing FCCP treatment were treated with DMSO or 25 µM FCCP (Sigma, C2920) for 4 h and fixed with 4% paraformaldehyde (Ted Pella Inc., 18505) in Dulbecco's Phosphate Buffered Saline without calcium chloride and magnesium chloride (PBS; Thermo Fisher Scientific, 14200–075). BNIP3 cells were fixed 24 h after transfection. All cells were permeabilized with 0.2% Triton X-100 in PBS, blocked in 5% normal goat serum, and incubated with the primary antibody OxPhos Complex IV subunit 1 (1:100, Thermo Fisher Scientific, 459600) at 4oC overnight, rinsed with PBS, incubated with secondary antibody Alexa Fluor 594 goat anti-mouse (1:200, Thermo Fisher Scientific, A11032) at 37oC for 1 h, and counter-stained for nuclei with Hoechst 33342 (Thermo Fisher Scientific, H3570). Fluorescence images were captured using a Carl Zeiss Axio Observer Z1 fitted with a motorized Z-stage with a 63X Plan-Apochromat (oil immersion) objective. Z stacks separated by 0.6 µm along the z-axis were acquired with an ApoTome using a high-resolution AxioCam MRm digital camera and Zeiss AxioVision 4.8 software (Carl Zeiss). Scoring of mitochondria was performed manually by counting the total number of mitochondria found within the enlarged GFP-Rab5Q79L-positive endosomes within a given cell. A minimum of 30 cells were counted per condition in 3–4 independent experiments.
Western blot analysis
Cells were homogenized in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton-X 100, and Complete protease inhibitor cocktail (Roche)) and centrifuged at 20,000 x g for 20 mins at 4°C. Proteins were separated by gel electrophoresis on NuPAGE Bis-Tris gels (Life Technologies) and transferred to nitrocellulose membranes. Membranes were probed for Actin (1:2000, Sigma, A4700), Atg5 (1:1000, Abgent, AP1812a), BNIP3 (1:1000, Sigma, B7931), Gapdh (1:2000, Genetex, GTX627408), LC3 (1:1000, Cell Signaling, 4108), p62 (1:1000, ARP, 03-GP62-C), or Parkin (1:1000, Cell Signaling, 4211) primary antibodies and imaged on a Bio-Rad ChemiDoc XRS+ imager. Immunoblots for the GFP-Rab5Q79L pulldown were probed with Eea1 (1:1000, Cell Signaling, 3288), Lamp2 (1:1000, Santa Cruz Biotechnology, sc-18822), or Rab5 (1:1000, Cell Signaling, 3547) primary antibodies.
Endosome pull down and mass spectrometry
GFP-Rab5Q79L-positive endosomes were isolated from Atg5−/− MEFs using a protocol adapted from Hanna et. al. 201211. Briefly, MEFs were infected with mCherry-Parkin (MOI: 75) plus GFP or GFP-Rab5Q79L (MOI: 50) for 12 h. After treatment with DMSO or 25 µM FCCP for 4 h, MEFs were homogenized in ice-cold buffer (0.25 M sucrose, 20 mM Tris-HCl, pH 7.0) supplemented with protease inhibitors (Roche) and then centrifuged for 5 min at 600 x g. The supernatants were incubated with a monoclonal mouse GFP antibody coupled to MACS magnetic beads (Miltenyl Biotech, 130-091-125) overnight before being applied to a MACS MS separation column (Miltenyl Biotech, 130-042-201). Endosomes were eluted in a buffer containing 50 mM Tris-HCl (pH 6.8), 120 mM DTT, 1% SDS, 1 mM EDTA, 0.005% bromophenol blue, and 10% glycerol. The constituent proteins were separated on SDS-PAGE, subjected to in-gel trypsin digestion, and analyzed by LC-MS/MS as described.34 The proteomics data were obtained from a Q Exactive instrument at the Rutgers University Neuroproteomics Core Facility. Heat map was generated as a ratio of the spectra counts for a given condition over the spectra counts for GFP DMSO for each protein. A value of 2 was added to each spectra count to generate ratios.
Cell death assays
WT and Atg5−/− MEFs were transiently transfected with empty myc vector, myc-BNIP3, empty pEGFP-C1 (Clontech, 6084-1), GFP-Rab5Q79L, or GFP-Rab5S34N (generously donated by JoAnn Trejo, UCSD) using FuGENE 6 according to the manufacturer's instructions. Cells were transfected for 24 h. To assess permeability of cell membranes, cells were stained with BOBO-3 Iodide (1:1000, Thermo Fisher Scientific, B3586) plus Hoechst 33342 for 20 minutes. BOBO-3 is taken up by dead cells but impermeant to live cells. BOBO-3 positive cells were divided by total number of Hoechst 33342 positive cells as described.2
Statistical analysis
Statistical significance was calculated using ANOVA followed by Tukey's posthoc test for multiple comparison. Differences were considered to be significant when p < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by an AHA Established Investigator Award (ABG), NIH R01HL087023 (ABG), the UCSD Graduate Training Program in Cellular and Molecular Pharmacology grant T32GM007752 (SES), NIH predoctoral training grant T32 GM008666 (BCH) and AHA Predoctoral Fellowship (BCH).
References
- [1].Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 2012; 111:1208-21; PMID:23065344; https://doi.org/ 10.1161/CIRCRESAHA.112.265819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol: CB 2012; 22:R29-34; PMID:22240478; https://doi.org/ 10.1016/j.cub.2011.11.034 [DOI] [PubMed] [Google Scholar]
- [3].Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr., Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A 2007; 104:14489-94; PMID:17726112; https://doi.org/ 10.1073/pnas.0701311104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; https://doi.org/ 10.1038/nature03029 [DOI] [PubMed] [Google Scholar]
- [5].Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795-803; PMID:19029340; https://doi.org/ 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 2009; 17:98-109; PMID:19619495; https://doi.org/ 10.1016/j.devcel.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ney PA. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta 2015; 1853:2775-83; PMID:25753537; https://doi.org/ 10.1016/j.bbamcr.2015.02.022 [DOI] [PubMed] [Google Scholar]
- [8].Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al.. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010; 11:45-51; PMID:20010802; https://doi.org/ 10.1038/embor.2009.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielmann Y, Stangler T, Hersch N, Hoffmann B, Merkel R, Willbold D. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 2009; 5:690-8; PMID:19363302; https://doi.org/ 10.4161/auto.5.5.8494 [DOI] [PubMed] [Google Scholar]
- [10].Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, Han Z, Chen L, Gao R, Liu L, et al.. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016; 12:689-702; PMID:27050458; https://doi.org/ 10.1080/15548627.2016.1151580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 2012; 287:19094-104; PMID:22505714; https://doi.org/ 10.1074/jbc.M111.322933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 2009; 16:939-46; PMID:19229244; https://doi.org/ 10.1038/cdd.2009.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654-8; PMID:19794493; https://doi.org/ 10.1038/nature08455 [DOI] [PubMed] [Google Scholar]
- [14].Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, Shimizu S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun 2014; 5:4004; PMID:24895007; https://doi.org/ 10.1038/ncomms5004 [DOI] [PubMed] [Google Scholar]
- [15].Ma T, Li J, Xu Y, Yu C, Xu T, Wang H, Liu K, Cao N, Nie BM, Zhu SY, et al.. Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat Cell Biol 2015; 17:1379-87; PMID:26502054; https://doi.org/ 10.1038/ncb3256 [DOI] [PubMed] [Google Scholar]
- [16].Hammerling BC, Najor RH, Cortez MQ, Shires SE, Leon LJ, Gonzalez ER, Boassa D, Phan S, Thor A, Jimenez RE, et al.. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun 2017; 8:14050; PMID:28134239; https://doi.org/ 10.1038/ncomms14050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005; 122:735-49; PMID:16143105; https://doi.org/ 10.1016/j.cell.2005.06.043 [DOI] [PubMed] [Google Scholar]
- [18].Zeigerer A, Gilleron J, Bogorad RL, Marsico G, Nonaka H, Seifert S, Epstein-Barash H, Kuchimanchi S, Peng CG, Ruda VM, et al.. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature 2012; 485:465-70; PMID:22622570; https://doi.org/ 10.1038/nature11133 [DOI] [PubMed] [Google Scholar]
- [19].Stenmark H, Valencia A, Martinez O, Ullrich O, Goud B, Zerial M. Distinct structural elements of rab5 define its functional specificity. EMBO J 1994; 13:575-83; PMID:8313902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J 1994; 13:1287-96; PMID:8137813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, Gustafsson ÅB. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ 2011; 18:721-31; PMID:21278801; https://doi.org/ 10.1038/cdd.2010.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 2002; 91:226-31; PMID:12169648; https://doi.org/ 10.1161/01.RES.0000029232.42227.16 [DOI] [PubMed] [Google Scholar]
- [23].Vande VC, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. MolCell Biol 2000; 20:5454-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J Cell Biol 1983; 97:508-21; PMID:6309862; https://doi.org/ 10.1083/jcb.97.2.508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Huotari J, Helenius A. Endosome maturation. EMBO J 2011; 30:3481-500; PMID:21878991; https://doi.org/ 10.1038/emboj.2011.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Tan J, Tan Y, Han H, Tian R, et al.. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J Biol Chem 2016; 291:21616-29; PMID:27528605; https://doi.org/ 10.1074/jbc.M116.733410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 2007; 14:146-57; PMID:16645637; https://doi.org/ 10.1038/sj.cdd.4401936 [DOI] [PubMed] [Google Scholar]
- [28].Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al.. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14:177-85; PMID:22267086; https://doi.org/ 10.1038/ncb2422 [DOI] [PubMed] [Google Scholar]
- [29].Campsteijn C, Vietri M, Stenmark H. Novel ESCRT functions in cell biology: spiraling out of control? Curr Opin Cell Biol 2016; 41:1-8; PMID:27031044; https://doi.org/ 10.1016/j.ceb.2016.03.008 [DOI] [PubMed] [Google Scholar]
- [30].Nagashima S, Tokuyama T, Yonashiro R, Inatome R, Yanagi S. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J Biochem 2014; 155:273-9; PMID:24616159; https://doi.org/ 10.1093/jb/mvu016 [DOI] [PubMed] [Google Scholar]
- [31].Hirota Y, Yamashita S, Kurihara Y, Jin X, Aihara M, Saigusa T, Kang D, Kanki T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 2015; 11:332-43; PMID:25831013; https://doi.org/ 10.1080/15548627.2015.1023047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hwang S, Disatnik MH, Mochly-Rosen D. Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington's disease. EMBO Mol Med 2015; 7:1307-26; PMID:26268247; https://doi.org/ 10.15252/emmm.201505256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol 2008; 295:H2025-31; PMID:18790835; https://doi.org/ 10.1152/ajpheart.00552.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wu C, Jain MR, Li Q, Oka S, Li W, Kong AN, Nagarajan N, Sadoshima J, Simmons WJ, Li H. Identification of novel nuclear targets of human thioredoxin 1. Mol Cell Proteomics: MCP 2014; 13:3507-18; https://doi.org/ 10.1074/mcp.M114.040931 [DOI] [PMC free article] [PubMed] [Google Scholar]