

ABSTRACT
Neoantigens present unique and specific targets for personalized cancer immunotherapy strategies. Given the low mutational burden yet immunotherapy responsiveness of malignant mesothelioma (MM) when compared to other carcinogen-induced malignancies, identifying candidate neoantigens and T cells that recognize them has been a challenge. We used pleural effusions to gain access to MM tumor cells as well as immune cells in order to characterize the tumor-immune interface in MM. We characterized the landscape of potential neoantigens from SNVs identified in 27 MM patients and performed whole transcriptome sequencing of cell populations from 18 patient-matched pleural effusions. IFNγ ELISpot was performed to detect a CD8+ T cell responses to predicted neoantigens in one patient. We detected a median of 68 (range 7–258) predicted neoantigens across the samples. Wild-type non-binding to mutant binding predicted neoantigens increased risk of death in a model adjusting for age, sex, smoking status, histology and treatment (HR: 33.22, CI: 2.55–433.02, p = .007). Gene expression analysis indicated a dynamic immune environment within the pleural effusions. TCR clonotypes increased with predicted neoantigen burden. A strong activated CD8+ T-cell response was identified for a predicted neoantigen produced by a spontaneous mutation in the ROBO3 gene. Despite the challenges associated with the identification of bonafide neoantigens, there is growing evidence that these molecular changes can provide an actionable target for personalized therapeutics in difficult to treat cancers. Our findings support the existence of candidate neoantigens in MM despite the low mutation burden of the tumor, and may present improved treatment opportunities for patients.
KEYWORDS: Malignant mesothelioma, neoantigen, pleural effusion, tumor-immune interface
Background
Examination of cancer genomes has revealed sets of mutations that could be exploited in immunotherapy, either as neoantigen vaccines as well as the ability to track specific anti-cancer immunity.1 The immunogenic potential of tumor neoantigens derives from the difference between the mutated sequence and the normal non-mutated sequence to which the host is largely tolerant. While neoantigen identification has been successful in many human tumors,2-12 neoantigen vaccination using this knowledge is in its infancy. It is hampered by a number of challenges, including tumor sampling and identification of which neoantigens to target, especially in low mutation tumors.
Malignant mesothelioma (MM) is an aggressive cancer of the cells of the pleura and peritoneum caused predominantly by exposure to asbestos, and has a median survival of 9–12 months post-diagnosis.13,14 Current treatment options for pleural MM are limited, and offer only modest improvement in survival.15 In early studies, clinical immunotherapy for MM appears promising.16 Novel personalized therapeutic strategies based on the highly individual molecular and immunological characteristics of each MM tumor including neoantigen vaccination is an attractive avenue for improving treatment outcomes.
Over 90% of MM patients develop pleural effusion during the course of their disease, which can be a rich source of cells shed from the primary tumor17 and we have previously shown that the tumor cells found in the malignant pleural effusions of MM patients carry many of the hallmark mutations found in solid MM tumors, as well as a wealth of novel individual mutations.18
In our murine MM model, we have previously demonstrated a strong spontaneous endogenous CD8+ T cell response to a predicted MM tumor neoantigen.19 A recent sequencing study of 98 primary human MM tumor samples found that 59% of all identified somatic mutations resulted in predicted MHC class I-binding neoantigens.20
Analysis of the mutations and gene expression patterns of the cells found in MM pleural effusions may give us novel insights into the tumor-immune interface of MM and allow us to identify candidate neoantigens that can be used to develop a personalized vaccination approach for MM patients. This study characterizes the predicted neoantigen burden and the immune environment plus the specific T cell response to a set of predicted neoantigens from the malignant pleural effusions of MM patients and reports the identification of a candidate neoantigen specific CD8+ T cell response in one of the subjects.
Results
Patient characteristics
Potential neoantigens were studied in 27 MM patient pleural effusions.18 The median age of the group at diagnosis was 65 years (range 45–88 years). Median survival was 15 months (range 1–61 months) (Table 1). The median number of protein altering mutations per sample was 50 (range 16–134; Table 1). Comprehensive patient demographics have been detailed previously.18 Somatic mutation burden was not associated with a survival benefit.18
Predicted neoantigens identified in all patient samples
Purity of the isolated tumor cells was estimated to be a median of 98.1% (range 91.1–99.9%) using ESTIMATE analysis.21 A total of 2236 patient HLA specific neoantigens were predicted across all of the MM tumor cell samples from 48% (607/1256) of the previously identified protein coding somatic mutations (Figure 1).18 A median of 68 (range 7–258) predicted neoantigens were detected across the samples (Figure 1). Of these, a median of 32 (range 15–93) predicted neoantigens were observed to bind (IC50 ≤ 500 nM) to MHC class I in the mutated but not the wild-type state (Figure 1). There was a median of 2 (range 0–8) predicted neoantigens that were observed to bind to MHC class I with strong affinity (IC50 ≤ 50 nM) across the samples (Figure 1). There was no correlation between tumor purity and the number of predicted neoantigens. Somatic mutations affecting known oncogenic MM genes, BAP1, NF2 and TP53 resulted in predicted neoantigens. We found that 63.2% (range 21.9–85.7%) of predicted neoantigens were expressed in the isolated tumor cells (counts per million > 0.5).
Figure 1.
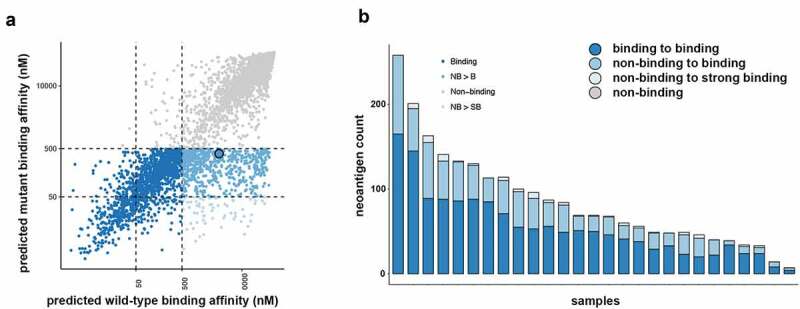
Neoantigen landscape of tumor cells from malignant mesothelioma pleural effusions. (a) Neoantigens were predicted for each patient based on their specific HLA type. Overview of the 2236 predicted neoantigens that were observed to bind to MHC class I highlighted by their binding change from the wild-type to the mutant state. (b) Predicted neoantigens identified in each sample colored by their binding change from the wild-type to the mutant state.
Predicted neoantigen burden and survival
Survival analysis was performed using data for 26 of the patients. As previously determined, the primary key factors associated with survival for this cohort were histology (non-epithelial versus epithelial) and treatment status (untreated versus treated). We previously found that the total number of mutations identified in each sample had no association with survival.18 In univariate and in multivariate analysis adjusting for age, sex, smoking status, histology and treatment the number of predicted neoantigens determined for each patient based on their specific HLA type with a high differential agretopicity index (DAI) (i.e. predicted to bind in the mutated state and not in the wild-type state) increased the risk of death (HR: 33.2, CI: 2.5–433; p < .01) (Table 2). Although statistically significant, the negative prognostic association of the DAI is observed only after adjustment for histological subtype. A non-epithelial histology is relatively rare but it is also a strong negative prognostic indicator. In this study six of the seven non-epithelial tumors were in the upper 50% of the DAI values, therefore the strong association of these relatively small number of non-epithelial tumors associated with a poor prognosis is likely to have resulted in a somewhat skewed cohort-dependent positive HR.
Table 2.
Cox regression analysis. Hazard ratio (HR) shown with 95% CI. Significant p-values are shown in bold. Multivariate analysis is adjusted for age, sex smoking status, histology and treatment and dichotomized at the mean. Significant p-values are shown in bold.
| Parameter | Univariate HR | p-Value | Multivariate HR | p-Value |
|---|---|---|---|---|
| Age | 1.00 (0.96–1.05) | .91 | ||
| Sex (male vs. female) | 0.67 (0.22–2.04) | .48 | ||
| Smoking status (ever vs. never) | 0.88 (0.37–2.09) | .78 | ||
| Histology (non-epithelial vs. epithelial) | 6.14 (1.79–21.09) | .004 | ||
| Treatment (treated vs. non-treated) | 0.17 (0.06–0.47) | <.001 | ||
| Non-binding to binding neoantigens (high vs. low) | 0.38 (0.16–0.92) | .03 | 33.22 (2.55–433.02) | 0.007 |
| All binding neoantigens (high vs. low) | 0.45 (0.18–1.07) | .07 | 0.14 (0.02–1.16) | 0.068 |
When expression of predicted neoantigens was examined no association between the number of expressed predicted neoantigens and survival were seen. Higher expression of HLA-C was significantly associated with worse survival (log-rank p < .01) and while the expression of HLA-A and –B were not significantly associated with survival, the relationship between higher gene expression and worse survival trended toward significance (Additional Files 1 and 2).
Pleural effusions from MM patients are a rich source of informative immune cells
The immune infiltrate was determined for 18 patient-matched pleural effusions using gene expression data of the total cell populations. Tumor purity in the pleural effusions ranged from 34.5% to 70.1% (median 47%). The non-tumor cell compartment was found to contain 0–36.5% immune cells (median 23.8%) and 0–31.5% stromal cells (median 30.1%) (Figure 2a). There was a significant negative correlation between the tumor purity and size of the immune component as measured by the immune score given by the ESTIMATE algorithm (Pearson’s r = −0.67; p = .002) but not between the tumor purity and stromal component (Additional File 3). There was no association between the immune score and survival nor between the size of the immune compartment and predicted neoantigen burden.
Figure 2.
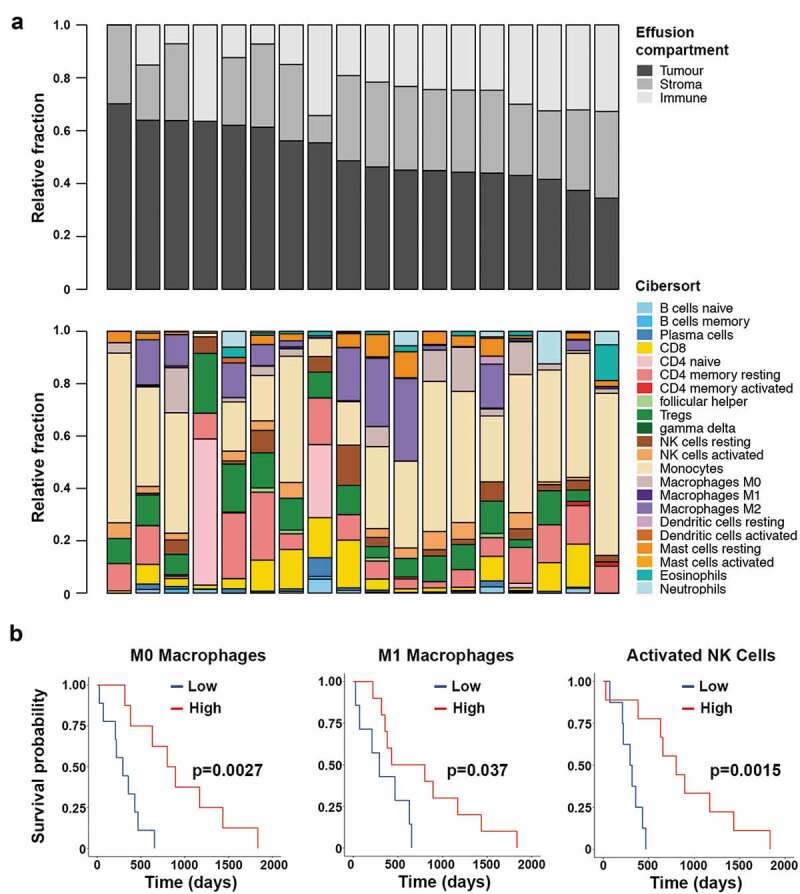
Assessment of the immune compartment of the pleural effusions from 18 patients. Top panel shows the estimation of the size of each of the cell compartments found in the effusions. Bottom panel shows the deconvolution of the immune compartment of the pleural effusions into immune cell type (a). Kaplan-Meier analysis of M0 macrophage, M1 macrophage and activated NK cell proportions within the pleural effusions. The threshold value denoting low and high groups is the median value as determined by CIBERSORT (b).
The composition of the immune infiltrates varied substantially between patients (Figure 2a). Deconvolution analysis of gene expression data into 22 immune cell subsets showed that regulatory T cells were prevalent in all but one sample, and only half of the samples had high proportions of natural killer (NK) cells, CD8+ and CD4+ resting T cells. Few samples had B cells or plasma cells. Monocytes comprised the largest of the immune cell fractions and around half of the samples contained M2 macrophages and a quarter M0 macrophages. The two samples with the highest immune cell compartments had a high proportion of naïve CD4+ T cells, while this signature was absent in all other samples. Survival analysis showed that patients who had a high proportion of M0 or M1 macrophages in their pleural effusions had better survival (log-rank p < .01 and p < .05), and we also observed a survival benefit for patients who had a high proportion of activated NK cells in their pleural effusions (log-rank p < .01; Figure 2b). There was no significant prognostic benefit associated with the presence or absence of any of the other 19 immune cell subsets analyzed.
For the patients where transcriptome data were available for both the whole pleural effusion total cell population as well as the isolated tumor cells (n = 18), we sought to analyze the dynamics of the tumor-immune interface. We found that there was no correlation between the predicted neoantigen burden and the size of the immune compartment regardless of the change in predicted binding affinity of the predicted neoantigens. There was a correlation between the expression of PD-L1 on the isolated tumor cells and the expression of PD-1 on the immune cells in the pleural effusion (p < .05; Additional File 4). The expression of PD-1 and CTLA4 was not associated with the number of predicted neoantigens. There was no association between normalized expression of PD-1 or CTLA4 in the pleural effusions and survival.
When clonality of T cells, as determined by TCR analysis, was studied we found that TCRβ sequences were detectible in all pleural effusion samples. A total of 617 sequences were detected, with a median of 18 unique sequences found per sample (range 3–194). CDR3 sequence length ranged from 6 to 35 amino acids (median 15 amino acids), and the distribution of the lengths was Gaussian (Figure 3a). The most frequently observed motif across the CDR3 sequences was the highly conserved CASS motif which was detected in 54.9% of unique sequences (Figure 3b).22 The Gini coefficient for each sample was low, with a median of 0.25 over the cohort (range 0–0.4), indicating that the diversity of clonotypes within each sample was even.
Figure 3.
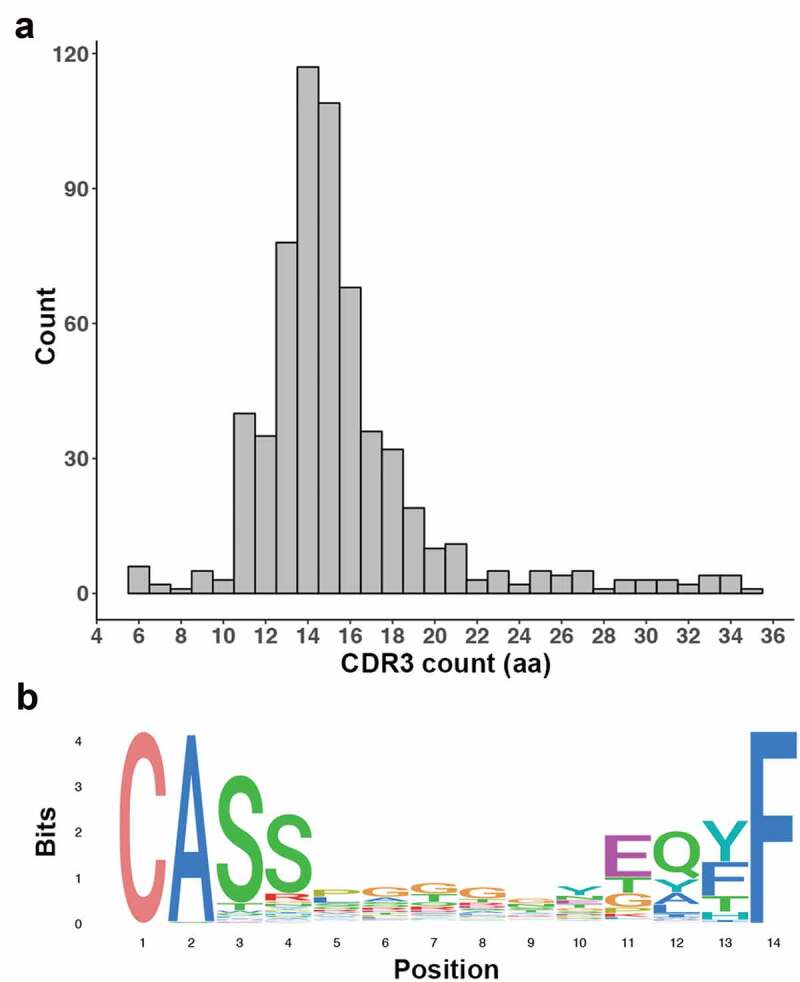
CDR3 detection in immune cells derived from MM pleural effusions. (a) Histogram shows a Gaussian distribution for CDR3 length. (b) Sequence logo of the 14aa CDR3 sequences show a distinct domination of the CASS and EQYF motifs at each end of the sequence.
The number of TCRβ sequences detected increased as the immune compartment size and expression of CD8A increased in the samples (p < .001 and p < .05, respectively). An increase in the number of detectable TCRβ sequences correlated with a higher neoantigen burden (p < .05). There was no correlation between the number of detectible TCRβ sequences and the expression of MHC class I genes in the corresponding isolated tumor samples. None of the detected TCRβ sequences have been previously described in the VDJdb browser database (https://vdjdb.cdr3.net/).
Identification of a CD8+ T cell response to a predicted neoantigen
One subject, patient 09567, was chosen for further study to test for evidence of T cell responses directed at predicted neoantigens due to the availability of sequential samples taken over the course of disease (Table 1). To control recurrent accumulation of pleural effusion an indwelling pleural catheter was inserted approximately 8 months after diagnosis and remained in place for nearly 2 years. The tumor showed evidence of slow progression over the following 12 month period. Chemotherapy was given as clinically indicated and as a surrogate for tumor burden serum mesothelin levels monitored (Figure 4a). The patient died from disease nearly 4 years after the initial diagnosis.
Table 1.
Characteristics of patient cohort.
| Sample ID | Age at Dx (years) | Sex | Histology | Smoking (pack years) | Survival (days) | Missense mutations | Neoantigen burden |
|---|---|---|---|---|---|---|---|
| MMCL_00068 | 66 | M | Epithelial | 39 | 517 | 51 | 84 |
| MMCL_00184 | 77 | M | Sarcomatoid | 0 | 188 | 43 | 81 |
| MMCL_01570 | 80 | M | Epithelial | 95 | 290 | 44 | 128 |
| MMCL_01605 | 68 | M | Epithelial | 37 | 301 | 16 | 32 |
| MMCL_01679 | 58 | M | Sarcomatoid | 0 | 71 | 39 | 97 |
| MMCL_02053 | 77 | M | Biphasic | 32 | 193 | 49 | 133 |
| MMCL_02305* | 48 | M | Biphasic | 0 | 70 | 32 | 42 |
| MMCL_02475 | 53 | M | Epithelial | 48 | 629 | 45 | 38 |
| MMCL_02749 | 52 | M | Epithelial | 0 | 467 | 134 | 155 |
| MMCL_03716 | 63 | M | Epithelial | 0 | 654 | 41 | 57 |
| MMCL_03778 | 69 | M | Epithelial | 15 | 293 | 53 | 195 |
| MMCL_05250 | 55 | M | Epithelial | 16 | 316 | 69 | 89 |
| MMCL_05750 | 76 | M | Epithelial | 5 | 26 | 35 | 54 |
| MMCL_07127 | 67 | M | Epithelial | 62 | 1835 | 36 | 14 |
| MMCL_07427 | 70 | F | Epithelial | 40 | 430 | 36 | 46 |
| MMCL_07675 | 60 | M | Epithelial | 0 | 801 | 60 | 40 |
| MMCL_09298 | 79 | M | Epithelial | 32 | 752 | 66 | 132 |
| MMCL_09300 | 52 | M | Epithelial | 12 | 219 | 32 | 48 |
| MMCL_09446 | 62 | M | Epithelial | 0 | 894 | 43 | 68 |
| MMCL_09566 | 72 | M | Epithelial | 8 | 1171 | 38 | 48 |
| MMCL_09567 | 70 | M | Epithelial | 10 | 1435 | 41 | 67 |
| MMCL_09624 | 68 | F | Epithelial | 7 | 209 | 31 | 31 |
| MMCL_09780 | 70 | F | Epithelial | 12 | 653 | 31 | 68 |
| MMCL_10006 | 88 | M | Epithelial | 0 | 356 | 72 | 258 |
| MMCL_10121 | 59 | M | Sarcomatoid | 0 | 381 | 29 | 39 |
| MMCL_10305 | 45 | F | Epithelial | 10 | 452 | 58 | 113 |
| MMCL_11202 | 57 | M | Epithelial | 11 | 491 | 51 | 7 |
* Patient died due to surgical complications.
Figure 4.
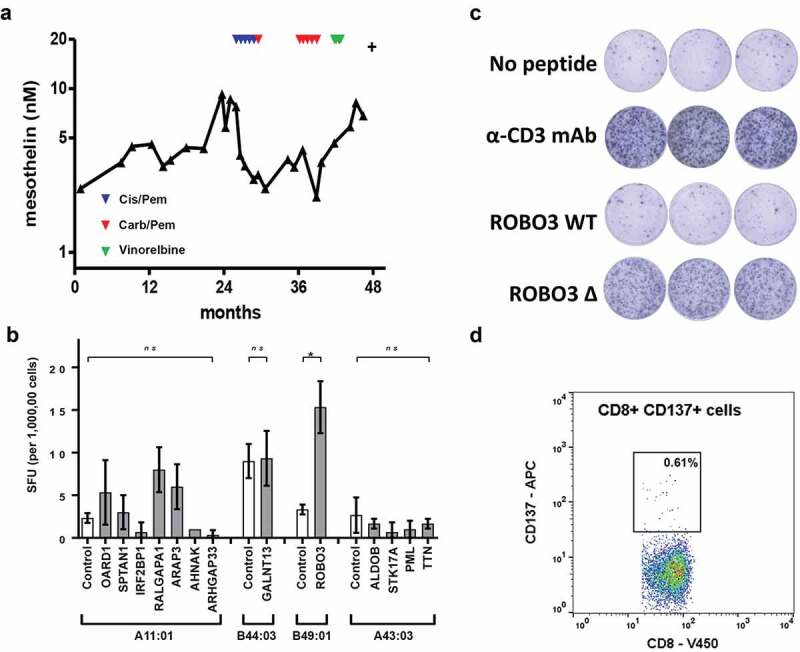
Description of biomarker levels and neoantigen identification in a single human patient (ID 9567). (a) Longitudinal serum mesothelin levels. Indicated by triangles are the time points for first-line chemotherapy (red -cisplatin and pemetrexed); second line (blue – carboplatin and pemetrexed) and third line (green – vinorelbine). (b) PBMC stimulated for 1 h with mutant peptide pool, then expanded in the presence of IL2 and CD3 for 14 days. T cell reactivity was assessed by ELISpot using the indicated mutant peptide exposed HLA expressing single antigen (SAL) lines. Controls show results for indicated HLA expressing SAL without peptides. Controls for HLA expressing SAL with wild-type peptides were performed for each mutant peptide of interest (Additional File 7 and data not shown). Difference between groups determined using non-parametric tests Kruskal-Wallis with Dunn’s multiple correction or Mann-Whitney as appropriate. *p < .05. SFU, spot forming units. (c) PBMC (200,000 cells per well) stimulated for 1 h with mutant ROBO3 peptide, then expanded in the presence of IL2 and CD3 for 14 days. T-cell reactivity was assessed by ELISpot assay. Negative control was T cells and B49:01SAL without peptide. Positive control was T cells plus αCD3 antibody. (d) CD137 cell surface expression measured using flow cytometry after stimulation of T cells with ROBO3 mutant peptide Wild-type controls are included in additional file 6.
Tumor cells were purified from cells collected from the pleural fluid. In total, 41 somatic protein-coding mutations were detected which were used to generate 67 predicted neoantigens (Additional File 5). Thirteen candidate neoantigens were selected for further interrogation based on the predicted binding affinities for the wild-type and the mutated epitopes. Peptides corresponding to the mutated and wild-type sequences were synthesized and tested initially using a blood sample collected after the first line of chemotherapy.
No IFN-γ responses were detected by ELISpot in non-expanded PBMCs to either pooled mutated or wild-type peptides. The cells were retested following 3 weeks of T-cell expansion. The mutated roundabout guidance receptor 3 (ROBO3) epitope (SEHSPVSEPV), which had a predicted binding affinity of 392 nM (highlighted, Figure 1a), generated a strong response as measured by IFN-γ production, whereas the wild-type ROBO3 epitope (SEPSPVSEPV), that had a predicted MHC class I binding affinity of 3192 nM (highlighted, Figure 1a), generated no significant response (Figure 4b). Specific T-cell clones were generated and were confirmed to be specific for ROBO3P640H peptide when assayed using ELISpot (Figure 4c). FACS analysis of these clones demonstrated an activated CD8+ T cell phenotype (Figure 4d), but showed no response for wild-type peptide pulsed T cells (Additional File 6).
Discussion
MM is a highly heterogeneous tumor and studies that rely on single-sample biopsies may not give a full and accurate representation of the interplay between the tumor and immune environments.23 Pleural effusions are a rich source of both tumor and immune cells and offer the ability to monitor the dynamic characteristics of the underlying MM tumor over time, and present the opportunity to mine the immune milieu for information regarding the tumor-immune interface. We found that MM tumor cells contain somatic mutations that may be exploited as potential neoantigens for personalized immunotherapeutic treatment strategies, and that the immune cells within pleural effusions are dynamic, containing both immune suppressive and tumor suppressive milieu. We showed that the TCR repertoire broadens as predicted neoantigen burden increases, and for the first time, we have described a CD8+ T cell response to a neoantigen predicted from a somatic mutation in an MM patient.
Given the relatively low mutation rate in MM when compared to other carcinogen-induced cancers,20,24 neoantigen identification has proven to be a difficult challenge. Not only is the neoantigen prediction process confounded by sequencing inefficiencies, algorithm inaccuracies and the fact that peptide-MHC affinities are themselves predicted, but these predictions do not account for whether the peptide is processed and presented by the tumor cell or recognized by the T cell. We used isolated tumor cells from pleural effusions to increase the sensitivity of mutation detection algorithms,18 leading to the detection of an immune response to a single predicted neoantigen generated from a mutation in the ROBO3 gene. By including APCs in the immunoassay, we have demonstrated the recognition of the peptide-MHC complex by the CD8+ T cell. While ROBO3 has not been previously implicated in MM, these results show that neoantigens are an accessible and promising target for therapy in MM whilst demonstrating the need for high sensitivity methods in order to properly perform robust identification. Recently, T cell responses to gene fusion-generated neoantigens were identified in MM,25 indicating that the predicted neoantigen landscape is a complex and multifaceted target for personalized immunotherapy.
We observed both immunosuppressive and tumor suppressive factors within pleural effusions, based on the presence of CD4+ and CD8+ T cells, T regulatory cells and M2 macrophages, but showed that survival was increased when patients had a tumor-suppressive gene expression signature. This has been seen before in other cancers such as in bowel cancer where increased T regulatory cell numbers are thought to be a control mechanism for an active immune response.26 Other evidence suggesting an ongoing response to neoantigens is that T cells found in MM pleural effusions have been shown to possess high levels of inhibitory receptors demonstrating an exhausted phenotype when compared to peripheral blood T cells, suggesting possible chronic antigen stimulation. They behave similarly to tumor infiltrating lymphocytes based on expression profiles of relevant markers.27 The success of any neoantigen-based immunotherapeutic strategy will be dependent on the tumor microenvironment and tumor-immune interface, as an immune suppressive environment can inhibit T-cell activation. Demonstrable immune responses to neoantigens have previously been identified and the magnitude of these responses has been shown to increase following immunotherapy.28,29 As immunotherapies such as checkpoint blockade inhibitors can increase the breadth of neoantigen response,30,31 a combinatory approach when using neoantigens to treat MM is likely needed. Given the presence of both immune suppression and activation within the effusions, future studies analyzing the function of the neoantigen-specific T cells present in the effusions will elucidate the outcome of these factors.
We were able to predict putative TCR clonotypes within the pleural effusion samples using in silico methods, and the repertoires increased in relation to predicted neoantigen burden. Diversity in the T-cell repertoire is required in order for a CD8+ T cell to recognize and engage with a broad range of neoantigens, suggesting that breadth of TCR clonotypes in tumor fluids reflects neoantigen recognition and suggests that such fluids might be a useful source for dynamic analysis of dynamic changes in T cell responses over time. While we found no association between checkpoint blockade molecules and predicted neoantigen burden in this cohort, it is predicted that a combinatory approach involving a neoantigen vaccine alongside checkpoint blockade inhibition would be an effective strategy.11,32
We observed that low expression of MHC class I genes were associated with increased survival. While this finding could be attributed to the small sample size used in this study, there is evidence of this phenomenon in other cancer types.33,34 This is nevertheless surprising as higher expression of the genes that make up the machinery of immune recognition and activation mechanisms, such as the MHC class I genes, have typically been correlated with better prognosis when compared to lower expression levels of these genes35-37 therefore this observation warrants further study.
Conclusion
We have identified a CD8+ T-cell response to a predicted neoantigen in an MM patient as well as characterized the predicted neoantigen landscape and tumor-immune interface in the pleural fluids of 27 MM patients. Though the identification of bonafide neoantigens is more difficult in a tumor with such a low mutation burden, these promising results indicate that predicted neoantigen-specific T-cells can be identified within the blood of the patient. Identification of the mutations responsible for neoantigens as well as indication of the tumor-immune characteristics of the patient can be easily performed using pleural effusions at earlier stages than when biopsies are typically performed, and could lead to improved treatment opportunities for patients.
Materials and methods
Samples
Pre-treatment pleural effusion specimens were collected from subjects with pathologically diagnosed MM. A fraction of the original pleural effusion cell pellet collected from 50 mL undiluted effusion was homogenized in 1 mL UltraSpecTM RNA Isolation Reagent (Biotecx Laboratories, Inc., BL-10050) and stored at −80°C. The remainder of the cell pellet was placed into culture as previously described38 and resulting enriched primary tumor cells were passaged a median of 7 times (range 4–10) before harvest for tumor nucleic acid extraction. All cultures were confirmed to be Mycoplasma spp. free by polymerase chain reaction. Personal HLA types of individual patients were determined by serotyping.
Blood for germline DNA extraction was collected from all individuals in either Vacutainer K2EDTA plasma or sodium heparin tubes (BD Bioscience, 366643 and 367886). Blood for immunological testing was collected in Vacutainer K2EDTA plasma (BD Bioscience, 366643) and processed by Ficoll-Paque (GE Healthcare Life Sciences, 45-001-751) for isolation of peripheral blood mononuclear cells (PBMC) as previously described.39 Mesothelin is a biomarker for MM13 and its concentrations were determined following the manufacturer’s instructions using the MESOMARK kit (Fujirebio Diagnostics, 801–900). This study was approved by the Human Research Ethics Committee of at Sir Charles Gairdner Hospital and the University of Western Australia, Perth, Western Australia. Written informed consent from all participants was obtained.
Neoantigen prediction
Germline and tumor DNA were extracted, whole exome and transcriptome sequencing were performed and somatic single nucleotide variants (SNVs) were identified as previously described.18 Briefly, sequencing data were generated on the Ion Torrent system to a depth of 114x per sample for exomes and 35–40 million reads per sample for transcriptomes. SNVs were identified in the exomes using both VarScan240 and SomaticSniper41 to produce a high confidence set. SNVs were excluded from neoantigen prediction if only the wild-type allele was present in the corresponding transcriptome sequence data. SNVs were annotated using snpEff42 and converted from variant call format (VCF) to mutation annotation format (MAF) using vcf2maf (https://github.com/mskcc/vcf2maf). HLA alleles were determined by sequencing-based typing (PathWest, Fiona Stanley Hospital, Murdoch, Western Australia) or predicted from transcriptome data using HLAminer v1.3.1.43 Neoantigen prediction was performed as previously described44 for the patient specific HLA-A, -B and -C alleles for all 8-11mer peptides using NetMHCpan v2.8.45 A half-maximal inhibitory concentration (IC50) value of <500 nM was used as the threshold for determining if mutated sequences were classed as potential neoantigens; an IC50 value of <50 nM was used to denote predicted high binding affinity neoantigens. Candidate neoantigens are defined in this study as potential neoantigens chosen for further analysis.
To examine CD8+ T cell reactivity 13 synthetic peptides were selected from the predicted neoantigen list for Patient ID 09567 and purchased from Mimotopes (Melbourne, Victoria, Australia) based on the wild-type epitope IC50 being >500 nM and the mutant epitope IC50 being <500nM.
Whole transcriptome sequencing of total cell population from pleural effusions
In order to examine the MM immune microenvironment and its relationship to predicted MM neoantigen loads, RNA was isolated and whole transcriptome sequencing performed as previously described18 on the whole cell population of the pleural effusion from time of sample collection. HTseq was used to collect raw alignment counts for the human (hg19) RefSeq reference transcriptome.46 CIBERSORT was used to deconvolute immune subsets within samples on a per sample basis.47 The CIBERSORT parameters used quantile normalization and 500 permutations to define immune types within samples. Expression values for CD8A and the geometric mean of expression values for GZMA and PRF148 were used as surrogate markers for level of CD8+ T cell and cytolytic activity, respectively. ESTIMATE software was used to determine tumor purity and to score the size of the immune and stromal compartments in each sample using a gene signature matrix to determine expression of tumor-, immune- and stroma-specific genes.21 Genes were considered to be expressed when they generated more than 0.5 counts per million, equivalent to 10 reads, in a given sample.
Identification of TCR sequences
In order to determine if there was evidence of oligoclonality of the MM T cell response based on expression of clonal T cell receptors (TCRs) and to relate that to predicted neoantigen load TCR β sequences were identified from sequencing data using MiTCR.49 Parameters were optimized to minimize the number of false positives by adjusting the minAlignmentMatches parameter to 14 and 16 for the V and J genes, respectively.50 Sequences featuring stop codons or ambiguous V, J and D gene calls were filtered out of the final dataset. The Gini coefficient, which has been used to determine the diversity of a TCR repertoire,51 was generated to give a diversity score of between 0 and 1.
CD8+ T cell isolation and polyclonal T-cell expansion
CD8+ T cells were isolated from PBMC using the EasySep Human CD8+ T cell Enrichment Kit (STEMCELL Technologies, 19053), following the manufacturer’s instructions. Enriched T cells were expanded with 30 ng/mL of CD3 antibody (Biolegend, 317301); 200 U/mL of human recombinant IL-2 (PeproTech, 200–02) and irradiated allogenic feeder cells as described.52 Peptide titrations were performed and a LogEC50 value of 9.42 (± 0.295) μg/mL was determined (Additional File 7). For peptide specific expansion, cells were incubated for 1 h with either 10 μg/mL of mutant or wild-type peptides before T cell expansion. After 14 days and an approximate 10-fold expansion, cells were pooled, washed and resuspended in media containing 20 U/mL of human-recombinant IL-2 for 2–4 days before testing by ELISpot.
INFγ ELISpot
ELISpot assays were performed according to standard protocols.53 Briefly, wells of nitrocellulose microtiter plates were coated with 1 µg/mL anti-interferon-γ (INFγ) antibody (Mabtech, 3420-3-250) overnight at 4°C. Samples and controls were incubated in RPMI-1640 media with 10% fetal calf serum at 37°C for 20 h and tested in triplicate, where 200,000 cells were assayed per well. PBMCs and expanded T cells to be tested were pulsed with single-antigen cell lines (SALs) expressing specific HLA molecules and 10 μg/mL of mutant or wild-type peptides. SALs were a kind gift from Professors Frans Claas and Ilias Doxiadis (Academic Hospital Leiden, The Netherlands). INFγ responses were detected with 1 mg/mL biotinylated anti-INFγ antibody (Mabtech, 3420-6-250) followed by 1 µg/mL streptavidin horseradish peroxidase (Mabtech, 3310–9) and development with TMB substrate (Mabtech, 3651–10). Immune reactivity was enumerated using the AID ELISpot reader system (Autoimmun Diagnostika GmbH) running count algorithm v.3.2.x and data was generated in Spot Forming Units (SFU) per 1 × 105 cells.
Single cell isolation and monoclonal expansion
Peptide-specific, IFNγ ELISpot reactive, polyclonal T-cell pools were incubated with SALs expressing B49:01 and 10 μg/mL of mutant peptide (SEhSPVSEPV) for 24 h. Cells were stained with CD137-APC and CD8-V450 (BD Biosciences, 560347) and FACS-single-cell sorted using the BD Influx (BD Biosciences) into individual wells in a 96-U shaped well plate, based on the positive expression of CD8 and the CD137 activation marker. CD8+ CD137+ T cells were expanded in supplemented RPMI-1640 with 200 U/mL of human-recombinant IL-2 and allogenic irradiated feeder cells. Peptide responsive T cell clones were identified by ELISpot assay.
Statistical analysis
Statistical analyses were performed using IBM® SPSS® statistics version. Summary results were reported as the median and range and groups were compared using the non-parametric Mann-Whitney U test. Correlations were performed using the Spearman correlation function. For survival analysis, continuous variables were analyzed as both trend variables and also as median dichotomized groups where indicated. Smoking status (ever versus never smokers) and histology (epithelioid or non-epithelioid) were analyzed as categorical.
Patient survival was calculated from the time of diagnosis and analyzed using the Cox proportional hazards regression model, when indicated results were also adjusted by tumor histology as categorical variable and age as a continuous variable. Survival analysis was also performed using the Kaplan-Meier product limit procedure with patients dichotomized into groups based on median predicted neoantigen burden. One patient (ID 02305) was excluded from survival analysis as death was due to complications of surgery unrelated to their mesothelioma. A p-value of less than 0.05 was considered significant.
Funding Statement
This work was supported in part by the Housing Industry of Australia Charitable Foundation, the Insurance Commission of Western Australia and the Australian National Health and Medical Research Council (NHMRC) (APP1089404 and APP1107043). SS receives a scholarship from the Ross Divett Foundation. BR is supported by a NHMRC Practitioner fellowship (APP1108638). The funding agencies played no part in study design, data analysis, interpretation of data or manuscript preparation.
Abbreviations
| MM | Malignant mesothelioma |
| TCR | T cell receptor |
| MHC | Major histocompatibility complex |
| HLA | Human leukocyte antigen |
| SAL | Single antigen-expressing cell line |
| APC | Antigen presenting cell |
Acknowledgments
The authors would like to acknowledge Frans H. J. Claas and Ilias I. N. Doxiadis for supplying the single antigen-expressing lines used in this study.
Ethics approval and consent to participate
This study was approved by the Human Research Ethics Committee of at Sir Charles Gairdner Hospital and the University of Western Australia (Perth, Western Australia). Written informed consent from all participants was obtained.
Availability of data and materials
The dataset supporting the conclusions of this article is available in the NCBI SRA repository, [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA419420].
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SS, JC and BWSR conceived the study. RA performed the sequencing. CR and SM performed the ELISpot assay and T cell culture. SS, ID and JC performed the data analysis. SB and RA provided the neoantigen prediction pipeline and assisted with transcriptome analysis. SS and ID performed the statistical analysis. SS and JC drafted the manuscript. All authors read and approved the final manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Schumacher TN, Schreiber RD.. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–10. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 2.Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, Zhang W, Sougnez C, Cibulskis K, Sidney J, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124(3):453–462. doi: 10.1182/blood-2014-04-567933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wick DA, Webb JR, Nielsen JS, Martin SD, Kroeger DR, Milne K, Castellarin M, Twumasi-Boateng K, Watson PH, Holt RA, et al. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clin Cancer Res. 2014;20(5):1125–1134. doi: 10.1158/1078-0432.CCR-13-2147. [DOI] [PubMed] [Google Scholar]
- 6.Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21(1):81–85. doi: 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- 7.Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125(10):3981–3991. doi: 10.1172/JCI82416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536(7614):91–95. doi: 10.1038/nature18945. [DOI] [PubMed] [Google Scholar]
- 10.Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017;7(3):264–276. doi: 10.1158/2159-8290.CD-16-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–239. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. Lancet. 2005;366(9483):397–408. doi: 10.1016/S0140-6736(05)67025-0. [DOI] [PubMed] [Google Scholar]
- 14.Robinson BW, Lake RA. Advances in malignant mesothelioma. N Engl J Med. 2005;353(15):1591–1603. doi: 10.1056/NEJMra050152. [DOI] [PubMed] [Google Scholar]
- 15.Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S, Manegold C, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21(14):2636–2644. doi: 10.1200/JCO.2003.11.136. [DOI] [PubMed] [Google Scholar]
- 16.Ye L, Ma S, Robinson BW, Creaney J. Immunotherapy strategies for mesothelioma - the role of tumor specific neoantigens in a new era of precision medicine. Expert Rev Respir Med. 2019;13(2):181–192. [DOI] [PubMed] [Google Scholar]
- 17.Lee YCG, Light RW. Management of malignant pleural effusions. Respirology. 2004;9(2):148–156. doi: 10.1111/res.2004.9.issue-2. [DOI] [PubMed] [Google Scholar]
- 18.Sneddon S, Dick I, Lee YCG, Musk AW, Patch A-M, Pearson JV, Waddell N, Allcock RJN, Holt RA, Robinson BWS, et al. Malignant cells from pleural fluids in malignant mesothelioma patients reveal novel mutations. Lung Cancer. 2018;119:64–70. doi: 10.1016/j.lungcan.2018.03.009. [DOI] [PubMed] [Google Scholar]
- 19.Creaney J, Ma S, Sneddon SA, Tourigny MR, Dick IM, Leon JS, Khong A, Fisher SA, Lake RA, Lesterhuis WJ, et al. Strong spontaneous tumor neoantigen responses induced by a natural human carcinogen. Oncoimmunology. 2015;4(7):e1011492. doi: 10.1080/2162402X.2015.1011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, Gnad F, Nguyen TT, Jaiswal BS, Chirieac LR, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48(4):407–416. doi: 10.1038/ng.3520. [DOI] [PubMed] [Google Scholar]
- 21.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res. 2009;19(10):1817–1824. doi: 10.1101/gr.092924.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiyotani K, Park JH, Inoue H, Husain A, Olugbile S, Zewde M, Nakamura Y, Vigneswaran WT. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology. 2017;6(2):e1278330. doi: 10.1080/2162402X.2016.1278330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mansfield AS, Peikert T, Smadbeck JB, Udell JBM, Garcia-Rivera E, Elsbernd L, CL Erskine, Van Keulen VP, Kosari F, SJ Murphy, et al. Neoantigenic potential of complex chromosomal rearrangements in mesothelioma. J Thorac Oncol. 2019;14(2):276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009;27(2):186–192. doi: 10.1200/JCO.2008.18.7229. [DOI] [PubMed] [Google Scholar]
- 27.Chee J, Watson MW, Chopra A, Nguyen B, Cook AM, Creaney J, Lesterhuis WJ, Robinson BW, Lee YCG, Nowak AK, et al. Tumour associated lymphocytes in the pleural effusions of patients with mesothelioma express high levels of inhibitory receptors. BMC Res Notes. 2018;11(1):864. doi: 10.1186/s13104-018-3953-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJA, Behjati S, Hilkmann H, El Atmioui D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–42. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, van Rooij N, Linnemann C, van Buuren MM, Urbanus JHM, et al. TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology. 2012;1(4):409–418. doi: 10.4161/onci.18851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, Welters MJP, van der Burg S, Kapiteijn E, Michielin O, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med. 2014;6(254):254ra128. doi: 10.1126/scitranslmed.3008918. [DOI] [PubMed] [Google Scholar]
- 32.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 33.Menon AG, Morreau H, Tollenaar RA, Alphenaar E, Van Puijenbroek M, Putter H, Janssen-van Rhijn CM, van de Velde CJH, Fleuren GJ, Kuppen PJK, et al. Down-regulation of HLA-A expression correlates with a better prognosis in colorectal cancer patients. Lab Invest. 2002;82(12):1725–1733. doi: 10.1097/01.LAB.0000043124.75633.ED. [DOI] [PubMed] [Google Scholar]
- 34.Ramnath N, Tan D, Li Q, Hylander BL, Bogner P, Ryes L, Ferrone S. Is downregulation of MHC class I antigen expression in human non-small cell lung cancer associated with prolonged survival? Cancer Immunol Immunother. 2006;55(8):891–899. doi: 10.1007/s00262-005-0085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turcotte S, Katz SC, Shia J, Jarnagin WR, Kingham TP, Allen PJ, Fong Y, D’Angelica MI, DeMatteo RP. Tumor MHC class I expression improves the prognostic value of T-cell density in resected colorectal liver metastases. Cancer Immunol Res. 2014;2(6):530–537. doi: 10.1158/2326-6066.CIR-13-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Speetjens FM, de Bruin EC, Morreau H, Zeestraten EC, Putter H, van Krieken JH, van Buren MM, van Velzen M, Dekker-Ensink NG, van de Velde CJH, et al. Clinical impact of HLA class I expression in rectal cancer. Cancer Immunol Immunother. 2008;57(5):601–609. doi: 10.1007/s00262-007-0396-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goeppert B, Frauenschuh L, Zucknick M, Roessler S, Mehrabi A, Hafezi M, Stenzinger A, Warth A, Pathil A, Renner M, et al. Major histocompatibility complex class I expression impacts on patient survival and type and density of immune cells in biliary tract cancer. Br J Cancer. 2015;113(9):1343–1349. doi: 10.1038/bjc.2015.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manning LS, Whitaker D, Murch AR, Garlepp MJ, Davis MR, Musk AW, Robinson BWS. Establishment and characterization of five human malignant mesothelioma cell lines derived from pleural effusions. Int J Cancer. 1991;47(2):285–290. doi: 10.1002/(ISSN)1097-0215. [DOI] [PubMed] [Google Scholar]
- 39.Cook AM, McDonnell AM, Lake RA, Nowak AK. Dexamethasone co-medication in cancer patients undergoing chemotherapy causes substantial immunomodulatory effects with implications for chemo-immunotherapy strategies. Oncoimmunology. 2016;5(3):e1066062. doi: 10.1080/2162402X.2015.1066062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28(3):311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: sNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warren RL, Choe G, Freeman DJ, Castellarin M, Munro S, Moore R, Holt RA. Derivation of HLA types from shotgun sequence datasets. Genome Med. 2012;4(12):95. doi: 10.1186/gm396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24(5):743–750. doi: 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2008;61(1):1. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anders S, Pyl PT, Huber W. HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics. 2014;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolotin DA, Shugay M, Mamedov IZ, Putintseva EV, Turchaninova MA, Zvyagin IV, Britanova OV, Chudakov DM. MiTCR: software for T-cell receptor sequencing data analysis. Nat Methods. 2013;10(9):813–814. doi: 10.1038/nmeth.2555. [DOI] [PubMed] [Google Scholar]
- 50.Brown SD, Raeburn LA, Holt RA. Profiling tissue-resident T cell repertoires by RNA sequencing. Genome Med. 2015;7:125. doi: 10.1186/s13073-015-0248-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas PG, Handel A, Doherty PC, La Gruta NL. Ecological analysis of antigen-specific CTL repertoires defines the relationship between naive and immune T-cell populations. Proc Natl Acad Sci U S A. 2013;110(5):1839–1844. doi: 10.1073/pnas.1222149110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26(4):332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mobs C, Schmidt T. Research techniques made simple: monitoring of T-cell subsets using the ELISPOT assay. J Invest Dermatol. 2016;136(6):e55–e9. doi: 10.1016/j.jid.2016.04.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset supporting the conclusions of this article is available in the NCBI SRA repository, [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA419420].