

ABSTRACT
Immune checkpoint blockade (ICPB) is a powerfully effective cancer therapy in some patients. Tumor neo-antigens are likely main targets for attack but it is not clear which and how many tumor mutations in individual cancers are actually antigenic, with or without ICPB therapy and their role as neo-antigen vaccines or as predictors of ICPB responses. To examine this, we interrogated the immune response to tumor neo-antigens in a murine model in which the tumor is induced by a natural human carcinogen (i.e. asbestos) and mimics its human counterpart (i.e. mesothelioma). We identified and screened 33 candidate neo-antigens, and found T cell responses against one candidate in tumor-bearing animals, mutant UQCRC2. Interestingly, we found a high degree of inter-animal variation in the magnitude of neo-antigen responses in otherwise identical mice. ICPB therapy with Cytotoxic T-lymphocyte-associated protein (CTLA-4) and α-glucocorticoid-induced TNFR family related gene (GITR) in doses that induced tumor regression, increased the magnitude of responses and unmasked functional T cell responses against another neo-antigen, UNC45a. Importantly, the magnitude of the pre-treatment draining lymph node (dLN) response to UNC45a closely corresponded to ICPB therapy outcomes. Surprisingly however, boosting pre-treatment UNC45a-specific T cell numbers did not improve response rates to ICPB. These observations suggest a novel biomarker approach to the clinical prediction of ICPB response and have important implications for the development of neo-antigen vaccines.
KEYWORDS: Neo-antigen, checkpoint blockade, T cells, mesothelioma, biomarker
Introduction
Immunotherapies such as anti-cytotoxic T lymphocyte associated antigen (CTLA)-4 and anti-programmed death (PD)-1 antibodies can cause long term, durable tumor regression in some cancers. Tumor mutation burden correlates with immune checkpoint blockade (ICPB) outcomes in multiple studies1–3 and as such, favorable responses to ICPB have been reported in patients with tumors caused by carcinogen exposure1 with consequently high-predicted numbers of neo-antigens.1–3 However, the actual role of neo-antigens in these responses remains uncertain.
Neo-antigens, altered peptides derived from somatically mutated tumor DNA, can be predicted from whole exome and RNA sequencing in combination with in silico MHC binding affinity algorithms. However, ex vivo testing often finds that only a small proportion of predicted neo-antigen candidates are immunogenic.4–7 Whilst this can be partly attributed to limitations in current prediction methods and neo-antigen presentation, the absence of neo-antigen responses could be due to the failure of the immune system to generate a detectable response in tumor bearing individuals. A recent study demonstrated that tumor neo-antigen specific T cells can be expanded from HLA matched healthy donor lymphocytes, but not from cancer patient lymphocytes.7 This suggests that some tumor bearing individuals have a limited capacity to generate a detectable neo-antigen response which may be caused by a negative regulatory tumor microenvironment.
ICPB removes some of the negative regulatory pressures exerted on T cells. It has been observed that ICPB increases the magnitude of T cell responses against tumor neo-antigens, enabling detection of neo-antigen specific immune responses not detectable prior to treatment.1,8,9 To explore this notion and assess whether ICPB response rates could be improved we used a mouse mesothelioma model, AB1-HA.
This model was chosen for these studies because it is one of the few tumor models that is induced by the relevant human carcinogen (i.e. asbestos) and exhibits histological, clinical, immunological and mutational characteristics similar to the equivalent human cancer, mesothelioma.10 In addition, AB1-HA is susceptible to immunotherapy11–14 despite not having a high mutation burden,15,16 as preliminary studies are now suggesting for the human counterpart.12,17
AB1-HA has two known, tractable tumor antigens, a previously described neo-antigen, UQCRC218 and hemagglutinin19 previously transfected into the cell line as a model neo-antigen.19 We hypothesized that ICPB would increase the magnitude of these neo-antigen specific T cell responses as well as unmasking responses to additional neo-antigens from in silico predicted candidates. Furthermore, we examined whether pre-existing immune recognition of neo-antigens reflected response rates to ICPB. This has been hard to determine directly because of the inherent variability in response to ICPB which of course can only be assessed after therapy. In order to overcome this limitation, we used a recently developed dual-tumor model that enables the status of the tumor to be assessed prior to therapy and this crucial question to be addressed.
Materials and methods
Mice
Eight to 10-week old female BALB/c and C57J/BL6 mice were purchased from the Animal Resource Center, Murdoch, Australia and maintained under standard specific pathogen-free housing conditions at the Harry Perkins Institute of Medical Research. All animal experiments were conducted with the approval of Harry Perkins Institute of Medical Research Animal Ethics Committee.
Murine mesothelioma and lung cancer models
The murine mesothelioma cell line AB1 was previously generated as described.20 H1N1/PR8 influenza hemagglutinin HA was transfected as a model neo-antigen to generate AB1-HA.19 Cells were maintained as previously described.18 The AE17 murine mesothelioma cell line was established in 2003 by exposing C57BL/6J mice to crocidolite asbestos.21 The Line1 murine alveolar carcinoma (Line1) was established in 1974 .22 Original stocks were obtained from Professor Najat Eglimez, University of Louisville (KY, USA). Line1 was cultured with DMEM (Gibco) containing 20mM HEPES and supplemented with 10% FCS. Cells were used at below 20 passages for experiments and were confirmed to be negative for Mycoplasma spp by PCR.
5 x 105 tumor cells were injected subcutaneously in the flank of mice (right flank for single tumor model, bilateral flanks for dual-tumor model). Mice were euthanized at indicated timepoints. Ipsilateral axillary and inguinal tumor draining lymph nodes (dLN) were studied because they are the primary location of neo-antigen cross-priming in this and other murine models,18 optimizing our chances of detecting significant and biologically relevant differences.
Combination immune checkpoint blockade
Mice bearing AB1/AB1-HA were administered intraperitoneally (ip) with 100 µg anti-CTLA-4 (clone 9H10; Bioceros; Netherlands) and 50 µg anti-GITR (clone DTA-1; BioXcell, NH, USA) antibodies in a total volume of 200 µL PBS at indicated time points. Animals were dosed at concentrations previously shown to induce AB1-HA regression12 when tumor are between 16 mm2 and 20 mm2 in size. An equivalent volume of PBS was injected ip in control animals. Mice were randomized to treatment or control arms and tumor measurements were made blinded to treatment group.
Tumor and DLN resection
We utilized a version of the dual-tumor model developed by Lesterhuis et al.,13 in which bilateral tumors progress or regress synchronously and symmetrically during therapy with ICPB. For experiments with bilateral tumors, the right flank tumor and its corresponding inguinal lymph nodes were surgically resected at day 11 as previously described.23 The following day, mice received 100 µg anti-CTLA-4 and 50 µg anti-GITR ip and growth of the left flank tumor was monitored.
Mutation calling and neo-antigen prediction
AB1/AB1-HA whole exome sequencing (WES) and RNA sequencing (RNA-seq) data were reanalyzed for this study.18 WES and RNA-seq of Line1 and AE17 were performed under contract by Australian Genome Research Facility. DNA and RNA were extracted from in vitro cultured cells. DNA was processed with SureSelectXT Mouse All Exon kit (Agilent, Australia) and RNA with Illumina’s TruSeq Stranded mRNA library preparation protocol. Paired-end sequencing (2x100bp) was performed on the Illumina HiSeq 2500 where >50 million total reads per sample were generated.
WES data were aligned to the updated murine reference genome mm10 (UCSC mm10/NCBI GRCm38) using Burrows-Wheeler Aligner.24 RNA-seq data were aligned to mm10 using Hisat2 v2.04.25 Expression analysis of aligned RNA-seq data was performed with Stringtie v1.3.26
Somatic single nucleotide variants (SNVs) were identified from WES using both Varscan 2.3.627 and Somatic sniper 1.0.5.28 High-confidence SNVs as defined by Varscan (at least 10% mutant reads in tumor, < 5% in normal and a one-tailed Fisher’s exact test P-value < 0.07) and Somatic sniper (variant supporting reads with an average mapping quality of ≥ 40 and a somatic score of ≥ 40) were pooled. Variants were annotated with Annovar29 and SNVs were filtered to identify non-synonymous, exonic variants.
The MHC-I binding affinity of mutated sequences were predicted as previously described18 to respective MHC-1 haplotype. MHC-I binding affinity of AB1/AB1-HA and Line1 SNVs were predicted for H2-Dd, H2-Kd and H2-Ld, and AE17 SNVs for H2-Db and H2-Kb. Briefly, predicted MHC I binders were selected based on their relative ranking in NetMHCpan 2.8 where the top 0.5% of ranked peptides were considered strong binders, top 2% ranked peptides weak binders, and those ranked > 2% non-binders.30 The expression of predicted strong and weak binders was confirmed with RNA-seq data and candidates with > 1 FPKM and the mutant allele expressed were selected.
A number of potential MHC binders can be predicted from a peptide sequence. The epitope with the top predicted binding affinity for each variant sequence was selected for peptide synthesis. Short peptides of 8 to 11 amino acid length were synthesized. Long peptides (27 amino acids long with the point mutation at position 13) for 9 of the top 10 expressed candidates were synthesized. Peptides (Mimotopes, VIC, Australia) were dissolved in 1% DMSO (Sigma Aldrich, NSW, Australia) and Ultrapure™ DNAse/RNase-free distilled water (Invitrogen, VIC, Australia) to a concentration of 1 mg/mL and stored at −20°C.
IFN-γ ELISPOT
IFNγ production in response to neo-antigen was examined by ELISPOT. Single-cell suspensions of the dLN were prepared by mechanical disruption. Tumors were cut into 1 mm2 pieces with a scalpel, and digested with a mouse tumor dissociation kit (Miltenyi Biotec, NSW, Australia) on the GentleMACSTM Octo Dissociator (Miltenyi Biotec). Tumor cell suspensions underwent CD8+ T cell selection with CD8 Microbeads (Miltenyi Biotec) as per manufacturer’s instructions. 1 × 105 freshly isolated dLN cells or 1 × 104 post-CD8-selection tumor infiltrating lymphocytes (TILs) were incubated per well in a microtiter plate and stimulated with 1 µg/mL of individual peptides. Peptide pools consisting of 8–25 peptides, with each individual peptide at a final concentration of 1 µg/mL, were also tested. As a positive control, 0.5 μL/well of Dynabeads® Mouse T-activator CD3/CD28 beads (Thermo Fisher Scientific, NSW, Australia) were used. The volume of beads used was empirically calculated to enable enumeration of the positive control. All ELISPOT assays were performed in duplicate and results presented as IFNγ Spot Forming Units (SFU) per 105 cells, unless stated otherwise. IFNγ SFU made in response to mutant peptides were compared to those made in responses to corresponding wild type peptides. In situations where the wild type peptide was unavailable or not applicable (such as for HA responses), the media only negative control was used as a comparison.
In vivo cytotoxic t lymphocyte (CTL) assay
Splenocytes were prepared from non-tumor bearing BALB/c mice and resuspended at 1 × 107 cells per mL in RPMI containing 2% fetal calf serum. Target splenocytes were pulsed with either 1 μg/mL HA518-526 (IYSTVASSL), UNC45a730-738 mutant (IYEVVRSLV), UQCRC2405-413 wildtype (SYMPPSTVL) or UQCRC2405-413 mutant (SYMAPSTVL) peptides for 1 h at 37°C. After washing, cells pulsed with two different peptides were labeled with 2.5 μM of either CFSE (Invitrogen, VIC, Australia) (CFSEhi) or Violet-Tag (Biolegend, CA, USA) (Violethi) for 10 min at 37°C . Control unpulsed cells were labeled with 0.25 μM CFSE (CFSElo) or Violet-Tag (Violetlo). Cells were resuspended to 1x108/mL. 1 × 107 peptide pulsed cells and 1 × 107 unpulsed cells were injected intravenously into tumor-bearing mice treated with ICPB. dLNs were collected 18–20 h later and single-cell suspensions were prepared. Cells were stained with anti-CD8a (clone 53.67, Biolegend, CA, USA) for 20 min at room temperature. Cells were washed and analysis was performed on a BD LSRFortessa (San Jose, CA, USA). Data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA). Percent-specific lysis was calculated as: (1−(RBALB)/RAB1HA-BALB) × 100, where R = %CFSElo/%CFSEhi.
Neo-antigen immunization
UNC45a mutant synthetic long peptide (MTFPGERIYEVVRSLVSLLHLSCSGLQ) was emulsified in MontanideTM ISA 51 VG (Seppic, Paris, France). A total of 40 μg peptide per mouse was made up to 100 μL volume with PBS and emulsified in a 1:1 ratio with 100 μL Montanide in accordance to the manufacturer’s instructions. Animals were injected with 100 μL of the vaccine subcutaneously at the base of tail on both the left and the right flank (equivalent to 20 μg peptide per flank).
Statistical analysis
Data were analyzed with Graph Pad Prism Software (Graphpad software, CA, USA) with results presented as mean ± SD. Paired student’s T-test was used to compare IFNγ responses of individual mice to mutant peptide or controls. A p value of <0.05 was considered significant and indicates a positive response to the test peptide. To assess the variability of responses between mice, results were presented as median ± IQR and the Levene’s test was used to determine the homogeneity variances of the neo-antigen responses. One-way ANOVA with Dunnett’s multiple comparison was performed to compare long-peptide responses with media negative control. Linear regression was performed to determine correlations. Fisher’s exact test was used to compare UNC45a response in treated and untreated mice. The effect of immunotherapy on the magnitude of neo-antigen T-cell response was analyzed in a mixed model ANOVA. The background effect was adjusted in the analysis as a covariate, the treatment was entered as a fixed effect and the experiment was entered as a random effect. The dependent variable was computed as the difference between mutant and wildtype peptide response. The log rank test (Mantel Cox) was performed to compared survival between different treatment groups.
Results
Few in silico predicted neo-antigen elicit an immune response
In AB1, a total of 2,697 high-confidence SNVs were indentified while there were 1,047 SNVs in AE17 and 3,620 in Line1. Of these, 434, 14 and 361 non-synonmous exonic SNVs were predicted to be MHC class I binders where 78, 13 and 104 were expressed in the three cell lines, respectively.
In our previous study, we described immune responses against 1 out of 20 tested candidate neo-antigens in the murine mesothelioma, AB1. In the present study, we increased the number of tested candidate neo-antigens. A total of 16 new candidates were tested, 8 that were identified previously but not tested and 8 new candidate neo-antigens that were identified. Of these 16 new peptides, 6 were predicted to be strong MHC-I binders, and the other 10 were predicted to be weak MHC-I binders (Supplementary table I).
We screened for ex vivo IFNγ production in response to a total of 33 neo-antigen short peptides (17 previously tested and 16 new candidates) from AB1 murine mesothelioma, 31 top candidates based on predicted MHC class I binding affinity from Line1 lung carcinoma and 13 from AE17 murine mesothelioma. There was no significant response against any of the 33 mutant peptides in the dLN of AB1-HA bearing animals (Figure 1a and b) yet strong, spontaneous responses to mutated UQCRC2 and HA peptides were again detected (Figure 1b). No responses were detected to the 31 neo-antigen candidates in Line1 lung carcinoma (Figure 1c) and 13 neo-antigen candidates in AE17 murine mesothelioma (Figure 1d). Tested peptide sequences are listed in supplementary figure II. Thus, a measurable immune response was only detected in AB1-HA tumor bearing animals to one of the panel of in silico predicted neo-antigens.
Figure 1.
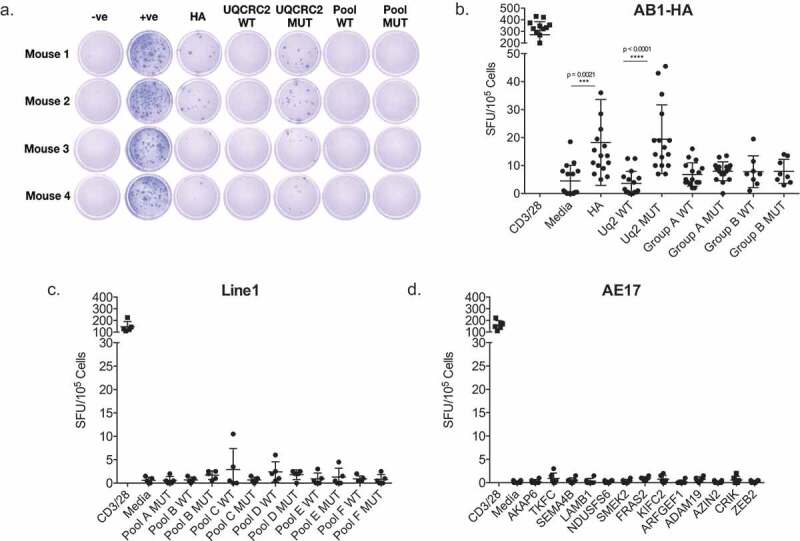
Screening of immune responses against a panel of predicted neo-antigen candidates reveals oligoreactivity. a. Representative ELISPOT wells showing IFNγ production in response to HA; UQCRC2 wild type (WT) and mutant (MUT) peptides; WT and MUT neo-antigen peptide pools. b. Dot plot summarizing responses against individual peptides, and peptide pools in AB1 murine mesothelioma bearing mice. Group A: 17 peptides, Group B: 16 peptides. (n = 8–16, 3 separate experiments). c.Responses against peptide pools of 5–6 neo-antigen candidates in Line1 Alveolar lung carcinoma bearing mice (n = 4). d. Responses against peptides of neo-antigen candidates in AE17 murine mesothelioma bearing mice (n = 5). Each dot represents mean dLN responses from an individual animal. Error bars show mean ± SD. Paired Student’s T-test.
Variability in immune responses to neo-antigens
Neo-antigen responses in outbred humans are variable. To examine the stochasticity of neo-antigen resonses we studied inbred mice that are genetically identical, handled identically and bear identical tumors, examining the range of responses to these neo-antigens. Substantial variability in immune responses against HA and mutant UQCRC2 peptides were observed between AB1-HA bearing animals (median response to HA: 17 SFU/105 cells, IQR 7–52; Mutant UQCRC2: 22 SFU/105 cells, IQR 11–43) (Figure 2a). Variability in IFNγ SFU was also observed in unstimulated or wild-type UQCRC2 stimulated wells, and mice with high levels of background reactivity were generally more likely to respond to neo-antigen peptide. The number of IFNγ SFU in response to HA and UQCRC2 mutant peptide correlated with each other (r2 = 0.7308, p < 0.0001) (Figure 2b). There was no association between increasing tumor size and the magnitude of the neo-antigen responses (HA r2 = 0.03294, p = 0.1192; Mutant UQCRC2 r2 = 0.01283, p = 0.3365). Thus neo-antigen responses to identical tumors varied hugely between identical animals with some mice generating strong neo-antigen specific T cell responses but others generating none at all.
Figure 2.
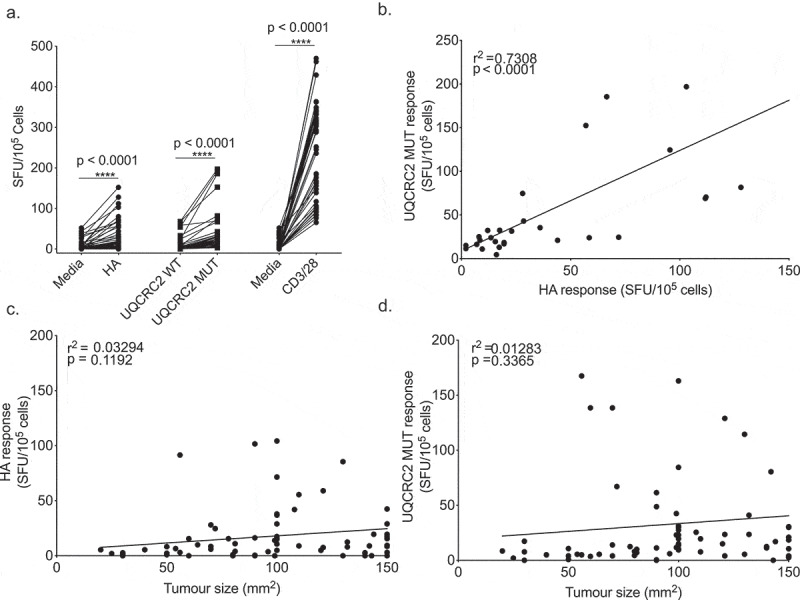
Neo-antigen specific responses vary stochastically between otherwise identical animals. a. IFNγ production in response to HA, UQCRC2 peptides and CD3/28 stimulation (n = 52). Each dot represents mean IFNγ SFU from the dLN of a tumor bearing mouse. Levene’s test for homogeneity of variance. b. Correlation between HA-specific IFNγ responses and UQCRC2 specific responses in mice with AB1-HA tumor of more than 100 mm2 in size (n = 33). Association of c. HA responses and tumor size (n = 75) and d. mutant UQCRC2 responses and tumor size (n = 75). Pearson’s correlation coefficient determined by linear regression.
One new neo-antigen response was detected following ICPB
As there were minimal detectable immune responses against our panel of candidate neo-antigens, we hypothesised that ICPB would increase the magnitude and substantially broaden the range of neo-antigen responses, as suggested by some studies, albeit in outbred humans.31 AB1-HA bearing animals were treated with a combination of anti-CT
LA-4 and anti-GITR, which results in reliable immune-mediated tumor regression. We screened for responses in the dLN against neo-antigen candidate peptides of various lengths in treated animals (Figure 3a).
Figure 3.
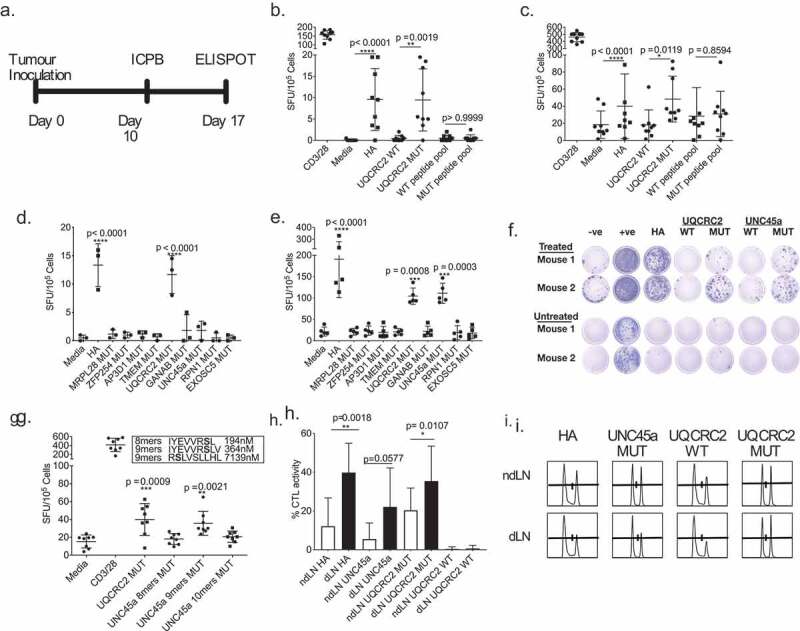
Combination ICPB therapy has limited capacity to broaden the range of neo-antigen specific T cell responses. a. Experimental schema; tumor-bearing mice received 100 µg anti-CTLA-4 and 50 µg anti-GITR on day 10 and dLN were harvested seven days later . b and c. IFNγ responses in the dLN against short HA, UQCRC2 and peptide pools (33 short peptides) in b. untreated and c. treated mice (n = 9). Paired Student’s T-test. d and e. dLN responses against long neo-antigen peptides in D. untreated (n = 3) and e. treated mice (n = 5). One-way ANOVA with Dunnett’s multiple comparison f. Representative ELISPOT wells of neo-antigen long-peptide responses in dLN from treated and untreated mice. g. dLN IFNγ responses against predicted UNC45a minimal peptides in treated mice (n = 8). h. Percentage of HA, UQCRC2 and UNC45a specific killing in dLN and ndLN of treated mice (n = 5). Paired Student’s T-test was performed to compared responses between ndLN and dLN. Error bars represent mean ± SD. i. Representative histogram of CTL assay in the dLN and ndLN of one mouse.
We first tested for dLN responses against short peptides of the 33 neo-antigen panel and found significant responses against HA and mutant UQCRC2 peptides in both treated and untreated mice. No responses were observed to any of the other short peptides (Figures 3b and c).
CD8+ T cells typically respond to 8-11mer short peptide epitopes in an ELISPOT assay, whereas both CD4+ and CD8+ T cells can respond to 27mer long peptides in an ELISPOT assay with antigen-presenting cells.32 To further explore responses to our neo-antigen candidates, we tested long peptides encoding the top nine expressed neo-antigen candidates (listed in Supplementary table III). Responses to mutant UQCRC2 long peptide were again detected and seen in both untreated and treated mice (Figure 3d, e). We detected immune responses to an additional neo-antigen, mutant UNC-45 homolog A (UNC45a) in the dLN of treated animals, but not untreated animals (Figures 3e and g). All other neo-antigen long peptides failed to elicit any IFNγ production regardless of treatment. Thus ICPB unmasked one additional neo-antigen rather than many as we had hypothesized.
Minimal UNC45a epitope is not the strongest predicted binder
We did not detect responses to the 8mer mutated UNC45a peptide (IYEVVRSL), which was predicted to be the strongest MHC H2-Kd binder with a binding affinity of 194 nM, in our peptide pool (Figure 3b). We tested two more predicted minimal epitopes within the mutant UNC45a sequence, and observed reactivity to the 9mer IYEVVRSLV (H2-Kd, 364nM), but not RSLVSLLHL (H2-Dd, 7139nM)(Figure 3g). This suggests that IYEVVRSLV is a minimal mutant UNC45A epitope. Thus selection based on predicted binding affinity alone could be insufficient in selecting an immunogenic neo-antigen epitope.
In vivo cytotoxic activity of neo-antigen specific T cells in DLN
We examined the cytotoxic function of neo-antigen specific T cells within the dLN in ICPB treated mice and found significantly higher HA-specific CTL activity in the dLN (40 ± 15%) compared to the non-dLN (12 ± 15%) (p = 0.0018; n = 5), strongly suggesting that, as predicted, the dLN was the dominant site of neo-antigen specific CTL activity (Figures 3h and 3i). A significantly higher UQCRC2 specific CTL activity was also observed in the dLN (36 ± 18% vs 20 ± 12%; p = 0.0107). The UNC45a specific CTL activity was not significantly different between the two sites (22 ± 20% vs 6 ± 8%; p = 0.057). This supports previous observations that the examination of the dLN cells can provide a useful profile of the neo-antigen reactivities.18,33
Icpb-induced neo-antigen specific T cells are not excluded from the tumor
We have previously shown that not all cross-primed neo-antigen specific CD8+ T cells in the dLN enter the tumor microenvironment,34 raising the concern that the typical studies of neo-antigen specific T cells obtained from tumor tissue rather than dLN may miss some of the relevant responses. When we investigated this using the neo-antigens described above, we found that ICPB immunotherapy increased the magnitude of all three neo-antigen T cell responses in the dLN and in the tumors. Responses to HA, mutant UQCRC2 and UNC45a in the dLN of treated mice were significantly higher compared to untreated mice (Figure 4a-c) (HA, 96 ± 101 vs 10 ± 35 SFU/105, p = 0.03; UQCRC2, 73 ± 71 vs 19 ± 22 SFU/105 p = 0.021; UNC45a, 83 ± 88 vs 15 ± 28 SFU/105, p = 0.03) and this increase in magnitude was also observed in the tumor (Figure 4d-f) (HA, 107 ± 68 vs 34 ± 32 SFU/104 CD8+ TILs, p = 0.002; UQCRC2, 172 ± 97 vs 53 ± 44 SFU/104 CD8+ TILs, p = 0.014; UNC45a, 159 ± 86 vs 18 ± 18 SFU/104 CD8+ TILs, p < 0.001). ICPB increased the proportion of individual mice that made a significant response to UNC45a in both the dLN (15% to 73%) and tumor (20% to 90%) (Table 1).
Figure 4.
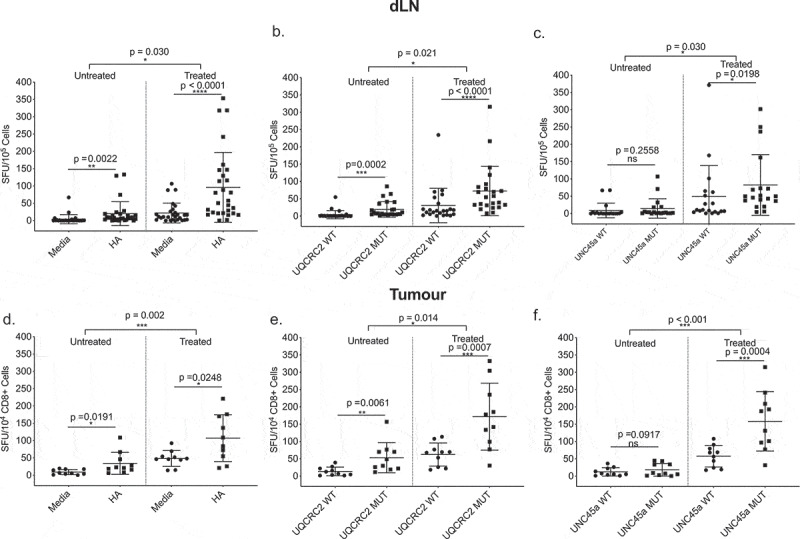
Combination ICPB-induced neo-antigen specific T cells are not excluded from tumors. IFNγ responses to a. HA, b. UQCRC2, and c. UNC45a short peptides in dLNs of treated and untreated mice (n = 23–28). Tumor CD8+ tumor infiltrating lymphocyte responses to d. HA, e. UQCRC2 and f. UNC45a peptides in treated and untreated mice (n = 10). Mixed model ANOVA compared T-cell responses between treated and untreated mice.
Table 1.
Percentage of mice with positive responses to indicated neo-antigens in the draining lymph nodes and the tumor of treated and untreated mice.
| dLN |
Tumor |
|||||
|---|---|---|---|---|---|---|
| Untreated (PBS) |
Treated (α-CTLA-4 + α-GITR) |
p-value | Untreated (PBS) |
Treated (α-CTLA-4 + α-GITR) |
p-value | |
| HA | 57% (16/28) | 78% (22/28) | ns p = 0.32 |
70% (7/10) |
70% (7/10) | ns p = 1.0 |
| UQCRC2 | 68% (17/25) | 82% (19/23) | ns p = 0.32 |
100% (10/10) | 90% (9/10) | ns p = 1.0 |
| UNC45A | 15% (3/19) |
73% (14/19) | ** p = 0.008 |
20% (2/10) |
90% (9/10) | ** p = 0.005 |
A positive response was defined as a significant difference (p < 0.05) between indicated neo-antigen and media control (for HA) and wild-type peptide (for UQCRC2 and UNC45A) using paired student’s T-test.
Two-tailed Fisher’s exact test was performed to compare between treated and untreated mice.
Strong DLN responses to UNC45a prior to ICPB therapy predict successful treatment outcome
As the endogenous neo-antigen response varied between identical animals, we hypothesized that those with stronger mutant UQCRC2 and UNC45a ELISPOT responses would successfully respond to ICPB therapy. To determine if pre-treatment neo-antigen responses correlated with therapy outcome, we utilized an established bilateral tumor model.13 The symmetry in this model allowed us to resect a tumor and dLN before treatment to determine the level of neo-antigen response, whilst leaving the other tumor as a readout of subsequent therapy response (Figure 5a and b). Around half of the mice treated with ICPB responded to treatment as evidenced by the complete regression of tumor in 12 out of 22 mice. Pre-treatment dLN UQCRC2 response was significantly higher in responders (n = 12, 17 ± 12 SFU/105) compared to non-responders (n = 10, 7 ± 5 SFU/105, p = 0.0182) (Figure 5c). Similarly, the UNC45a response was significantly higher in the responders (24 ± 14 SFU/105 vs 5 ± 2 SFU/105, p = 0.0004) (Figure 5d). This suggests that the variability of neo-antigen immune responses between mice is biologically significant and responses to some neo-antigens could predict subsequent response to ICPB therapy.
Figure 5.
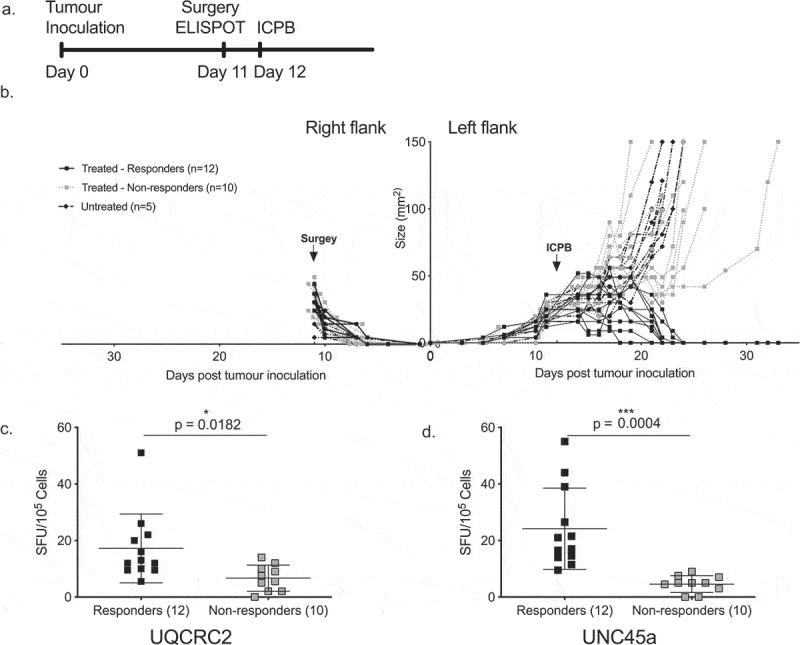
Strong dLN neo-antigen responses to UNC45a predict combination ICPB outcomes.
a. Schema of treatment and surgery. b. Bilateral tumor growth curve over time. Arrows represent surgery and treatment time (n = 22). Responses to therapy are depicted by black solid lines. Pre-treatment reactivity to c. mutant UQCRC2, and d. mutant UNC45a in the dLN of responders (n = 12) and non-responders (n = 10). Student’s T-test; error bars mean ± SD.
Boosting unc45a immune responses did not improve ICBP outcomes
As low UNC45a specific T cell responses pre-treatment was closely associated with failure to respond to ICPB therapy, we hypothesized that ICPB outcomes would be improved by increasing the magnitude of pre-treatment UNC45a responses through peptide immunization (Figure 6a). Administration of UNC45a synthetic long peptide in montanide significantly increased the immune response to UNC45a in vaccinated mice (n = 19, 34 ± 21 SFU/105) compared to both unvaccinated mice (n = 15, 8 ± 8 SFU/105, p < 0.0001) and to mice that received montanide alone (n = 18, 13 ± 15 SFU/105, p = 0.0009) (Figure 6b). There was no difference in the response to UQCRC2 neo-antigen which was not included in the vaccine (Figure 6b).
Figure 6.
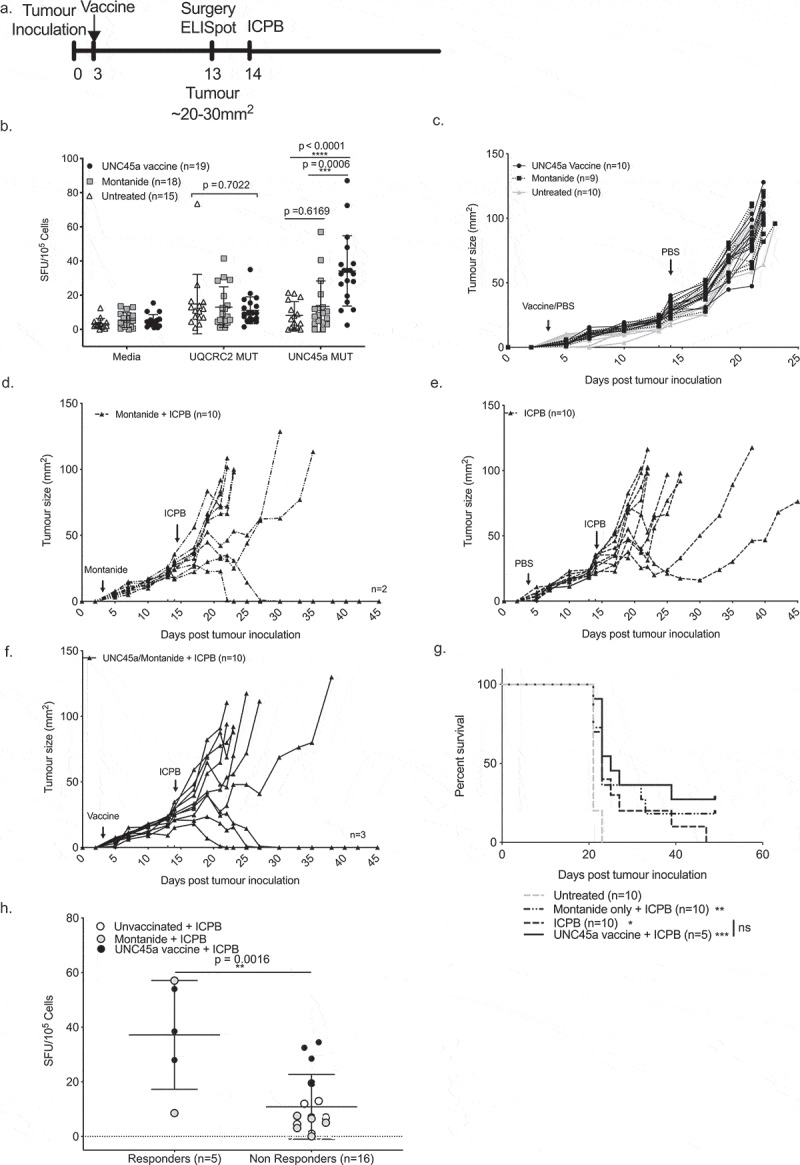
Boosting UNC45a specific T cells does not improve responses to combination ICPB.
a. Schema of tumor inoculation, vaccination, surgery and treatment. b. Post-vaccination IFNγ production in response to neo-antigen peptides. Tumor growth curves of c. control, d. Montanide vehicle control + ICPB, e. ICPB only and f. UNC45a vaccine + ICPB groups. g. Survival plot and log-rank (Mantel Cox). h. IFNγ responses to UNC45a peptide in responders and non-responders to ICPB. Unpaired Student’s T-test.
Importantly, UNC45a vaccination alone did not have any effect on tumor growth (Figure 6c). Of mice that received both montanide and ICPB, 20% (2/10) had a complete response (Figure 6d). Mice that received ICPB alone had 20% (2/10) partial responders where tumor growth was hampered after treatment but tumors eventually grew out (Figure 6e). Of those that received both the UNC45a vaccine and ICPB, 30% (3/10) were complete responders (Figure 6f). As before, ICPB treatment significantly improved survival (p = 0.0101) compared to untreated mice (Figure 6g). Whilst there were some long-term survivors in mice that received combination vaccine and ICPB therapy, the effect size was small and there was no significant benefit in survival compared to those that received ICPB alone (p = 0.2188). Again, mice that responded to ICPB had a higher UNC45a response pre-treatment compared to non-responders (37 ± 20 SFU/105 vs 11 ± 12 SFU/105 , p = 0.0016) (Figure 6g). This result suggests that a strong UNC45a response could predict a therapeutic response to ICPB treatment but it is not a sufficient individual component of an effective ICPB-induced response.
Discussion
Tumor mutations can be targets of anti-tumor immunity, and mutational load has been associated with response to ICPB in the clinic. Whilst few spontaneous immune responses against genomically predicted neo-antigens have been detected,4–6,35,36 these responses appear to be important in immune-mediated rejection of tumor.37,38 Thus these neo-antigen responses could represent predictors of response to therapy.
In this and other studies, few sequencing derived neo-antigen candidates elicit immune responses in the presence of tumor. Most published murine neo-antigen reactivities have been identified postvaccination.39,40 Our previously identified UQCRC2 neo-antigen in AB1-HA18 and three neo-antigens identified in a murine colon adenocarcinoma model38 were detected in tumor-bearing mice without treatment. The paucity of neo-antigens identified in this setting could be partly attributed to the limitations of current epitope prediction algorithms and selection, but also to tumor-associated immune suppression.
Relaxing the immune suppressive environment in our model by ICPB increased the magnitude of immune responses against the endogenously recognized neo-antigen UQCRC2 by 3.5-fold and enabled immune responses to a previously undescribed neo-antigen, mutant UNC45a, to be detected. Somewhat surprisingly, there were minimal responses against the rest of the candidate neo-antigens. This suggests that ICPB increased the dynamic range of immune responses which improved our ability to detect the neo-antigen response, a finding that has been observed in other studies.8,9,35 This result also supports the hypothesis that immune responses to neo-antigens in tumor-bearing hosts are attenuated and yet partially reversible by CTLA-4 treatment.41
In this study, we observed significant inter-mouse variation in neo-antigen reactivities before ICPB therapy. Such varability has previously been reported in in-bred mice bearing MC-38 murine colon adenocarcinoma.38 There are many potential factors underlying such T cell response heterogeneity, including stochastic variability in the TCR repertoire in otherwise genetically identical mice. We considered it unlikely that the microbiome was a cause for variability as it has been shown that animals housed in the same cage and within the same animal facility have little variation in microbiomes.42 The issue of variable immune responses is much harder to study in outbred humans. Recent melanoma studies have shown that individual patients have variable responsiveness to neo-antigen in terms of both the number of responses and their magnitude before and after personalized neo-antigen vaccination.5,6 It is impossible to tell if this variability is related to the host immune responsiveness or other factors to do with the candidate selection, vaccine or T-cell repertoire. Our data suggest that an inherent variability in neo-antigen responses may underlie at least some of this interpatient variability. Understanding the determinants that underlie this response variability may enable the selection of a therapeutic strategy that is useful for generating an anti-tumor response in individual patients.
One of the major issues confronting cancer immunologists at the moment is to determine which patients will respond to ICPB. Tumor mutational load and the predicted neo-antigen load have been shown to broadly correlate with outcomes to ICPB in multiple studies.1–3 By utilizing the dual-tumor model, we were able to show that the variation in neo-antigen response can be predictive of subsequent ICPB outcomes. We observed that strong dLN neo-antigen response is often accompanied by a higher background response to media and a strong response to other antigens. Overall it remains unclear if these dLN neo-antigen responses participate, or are even relevant, for a successful anti-tumor response i.e. the immune responses against neo-antigens in the dLN could potentially be secondary to events occurring in the tumor. Lesterhuis et al. showed that by day 6, identical tumors in genetically identical mice exhibit stochastically heterogeneous networks of gene expression that correlate with ICPB outcome.13 These responses may be a surrogate of a ‘hot’ tumor or a more active immune system. One argument that the latter notion is not the whole story is the observation that the neo-antigen response that best correlated with outcome was the one seen after ICPB – if the finding was entirely due to ‘hot tumors’, all neo-antigen reactivities would have been similarly boosted. Regardless, our observations have potential clinical significance. If the presence of a strong neo-antigen response in the dLN predicts response to ICPB therapy, this could be useful as a predictive biomarker in patients with accessible tumor dLN. Once relevant reactivities are identified in lymph nodes, they may then be able to be tracked in blood.
Also, the finding that the dLN is the major site cross-priming neo-antigen specific T-cells43 processes means that local interventions in the region of the dLN may alter systemic anti-tumor immunity44 sufficient to make them a targetable site to boost anti-tumor response. Given this, it was logical to ask if ICPB unresponsiveness could be reversed if the low UNC45a response in some animals was boosted by pre-treatment vaccination. Interestingly, although an UNC45a vaccine boosted UNC45a specific responses, it failed to improve ICPB outcome. It is unclear if this is simply due to an inadequate vaccination response compared to other more aggressive vaccination strategies32,37,38 or that UNC45a is not a target for tumor rejection or that such responses are just a surrogate of the host’s immune responsiveness. It is also possible that a single antigen might be insufficient to induce a complete response alone, as it has been shown that vaccination with two neo-antigens induce optimal tumor rejection compared to either neo-antigen alone.37
Selection of neo-antigen candidates to screen for immune responses is crucial. In our study, our strongly immunogenic neo-antigen UNC45a minimal epitope was not the highest predicted MHC-I binder. This was also evident in other studies where minimal epitopes with intermediate predicted MHC-I binding affinities were immunogenic.36,40 Furthermore, UQCRC2 and UNC45a peptide sequences (Supplementary figure I) did not present any distinct immunological features, such as predicted binding affinity or predicted immunogenicity score. This highlights the challenges in neo-antigen discovery, epitope prediction, and the inherent disadvantage of relying on in silico predictions alone. A potential strategy to partially overcome this is the use long peptides spanning all the possible minimal epitopes.
The predictive value of functional, neo-antigen responses to ICPB outcomes in our study provides the impetus to further examine these neo-antigens as vaccination targets. Importantly, neo-antigen T cell responses could be a potential biomarker in the clinic for ICPB responses whilst neo-antigen vaccination could be used to either treat or to ‘prime’ patients for a potential ICPB response. Further studies in the combination of neo-antigen vaccination strategies are required to provide better insights on how this can be achieved.
Funding Statement
SM was supported by the Australian Research Training Program and The Peter Douglas Swift Scholarship. JCh was supported by a US Department of Defense Cancer Horizon Award. BWSR and JCr received funding from National Health and Medical Research Council, Department of Health WA, Insurance Commission of WA and Cancer Council WA.
Acknowledgments
The authors thank the Harry Perkins Institute Bioresources team for their excellent animal care. The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments.
Disclosure of potential conflicts of interest
The authors declare no potential conflicts of interest.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–12. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99 doi: 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221 doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 7.Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Boschen M-L, Lund-Johansen F, Olweus J, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352(6291):1337–1341. doi: 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- 8.Fehlings M, Simoni Y, Penny HL, Becht E, Loh CY, Gubin MM, Ward JP, Wong SC, Schreiber RD, Newell EW, et al. Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8+ T cells. Nat Commun. 2017;8(1):562. doi: 10.1038/s41467-017-00627-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, Welters MJP, van der Burg S, Kapiteijn E, Michielin O, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med. 2014;6(254):254ra128. doi: 10.1126/scitranslmed.3008918. [DOI] [PubMed] [Google Scholar]
- 10.Sneddon S, Patch AM, Dick IM, Kazakoff S, Pearson JV, Waddell N, et al. Whole exome sequencing of an asbestos-induced wild-type murine model of malignant mesothelioma. BMC Cancer. 2017;17(1):396. doi: 10.1186/s12885-017-3382-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alley EW, Lopez J, Santoro A, Morosky A, Saraf S, Piperdi B, et al. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2017;18(5):623–630. doi: 10.1016/S1470-2045(17)30169-9. [DOI] [PubMed] [Google Scholar]
- 12.Fear VS, Tilsed C, Chee J, Forbes CA, Casey T, Solin JN, Lansley SM, Lesterhuis WJ, Dick IM, Nowak AK, et al. Combination immune checkpoint blockade as an effective therapy for mesothelioma. Oncoimmunology. 2018;7(10):e149411110. ARTN. doi: 10.80/2162402X.2018.1494111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lesterhuis WJ, Rinaldi C, Jones A, Rozali EN, Dick IM, Khong A, Boon L, Robinson BW, Nowak AK, Bosco A, et al. Network analysis of immunotherapy-induced regressing tumours identifies novel synergistic drug combinations. Sci Rep. 2015;5:12298. doi: 10.1038/srep12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nowak AK, Lesterhuis WJ, Hughes BGM, Brown C, PS K, O’Byrne KJ, John T, Pavlakis N, Kao SCH, Yip S. DREAM: a phase II study of durvalumab with first line chemotherapy in mesothelioma-First results. J Clin Oncol. 2018;36(15):8503. doi: 10.1200/JCO.2018.36.15_suppl.8503. [DOI] [Google Scholar]
- 15.Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, Gnad F, Nguyen TT, Jaiswal BS, Chirieac LR, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48(4):407–416. doi: 10.1038/ng.3520. [DOI] [PubMed] [Google Scholar]
- 16.Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, Tarpey P, Danilova L, Drill E, Gibb EA, et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018. doi: 10.1158/2159-8290.CD-18-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisher SA, Aston WJ, Chee J, Khong A, Cleaver AL, Solin JN, Ma S, Lesterhuis WJ, Dick I, Holt RA, et al. Transient treg depletion enhances therapeutic anti-cancer vaccination. Immun Inflamm Dis. 2017;5(1):16–28. doi: 10.1002/iid3.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Creaney J, Ma S, Sneddon SA, Tourigny MR, Dick IM, Leon JS, Khong A, Fisher SA, Lake RA, Lesterhuis WJ, et al. Strong spontaneous tumor neoantigen responses induced by a natural human carcinogen. Oncoimmunology. 2015;4(7):e1011492. doi: 10.1080/2162402X.2015.1011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marzo AL, Lake RA, Robinson BW, Scott B. T-cell receptor transgenic analysis of tumor-specific CD8 and CD4 responses in the eradication of solid tumors. Cancer Res. 1999;59:1071–1079. [PubMed] [Google Scholar]
- 20.Davis MR, Manning LS, Whitaker D, Garlepp MJ, Robinson BW . Establishment of a murine model of malignant mesothelioma. Int J Cancer. 1992;52(6):881––886.. [DOI] [PubMed] [Google Scholar]
- 21.Jackaman C, Bundell CS, Kinnear BF, Smith AM, Filion P, van Hagen D, Robinson BWS, Nelson DJ, et al. IL-2 intratumoral immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J Immunol. 2003;171(10):5051–5063. doi: 10.4049/jimmunol.171.10.5051. [DOI] [PubMed] [Google Scholar]
- 22.Yuhas JM, Pazmino NH. Inhibition of subcutaneously growing line 1 carcinomas due to metastatic spread. Cancer Res. 1974;34(8):2005–2010 [PubMed] [Google Scholar]
- 23.Brown MD, van der Most R, Vivian JB, Lake RA, Larma I, Robinson BWS. Currie AJ. Loss of antigen cross-presentation after complete tumor resection is associated with the generation of protective tumor-specific cd8(+) t-cell immunity. Oncoimmunology. 2012;1(7):1084–1094 doi doi: 10.4161/onci.20924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28(3):311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61(1):1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schumacher TN, Schreiber RD.. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69-74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 32.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al . Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692––696. doi doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D, Fu Y, Alexandrov A, Mardis ER, Artyomov MN, et al. Endogenous neoantigen-specific cd8 t cells identified in two glioblastoma models using a cancer immunogenomics approach. Cancer Immunology Research 2016;4(12):1007––1015. doi doi: 10.1158/2326-6066.CIR-16-0156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BWS, Scott B. Tumor-specific CD4(+) T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol. 2000;165(11):6047–6055. doi: 10.4049/jimmunol.165.11.6047. [DOI] [PubMed] [Google Scholar]
- 35.van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJA, Behjati S, Hilkmann H, El Atmioui D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–42. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125(10):3981–3991. doi: 10.1172/JCI82416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 39.Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–1091. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 40.Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, Blanchard T, McMahon D, Sidney J, Sette A, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med. 2014;211(11):2231–2248. doi: 10.1084/jem.20141308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 42.Hufeldt MR, Nielsen DS, Vogensen FK, Midtvedt T, Hansen AK. Variation in the gut microbiota of laboratory mice is related to both genetic and environmental factors. Comparative Med. 2010;60:336–342. [PMC free article] [PubMed] [Google Scholar]
- 43.Marzo AL, Lake RA, Lo D, Sherman L, McWilliam A, Nelson D, Robinson BW, Scott B. Tumor antigens are constitutively presented in the draining lymph nodes. J Immunol. 1999;162(10):5838–5845. [PubMed] [Google Scholar]
- 44.Jackaman C, Lansley S, Allan JE, Robinson BWS, Nelson DJ. IL-2/CD40-driven NK cells install and maintain potency in the anti-mesothelioma effector/memory phase. Int Immunol. 2012;24(6):357–368. doi: 10.1093/intimm/dxs005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.