

ABSTRACT
Expression of the disialoganglioside GD2 has been identified as a marker antigen associated with a breast cancer stem-like cell (BCSC) phenotype. Here, we report on the evaluation of GD2 as a BCSC-specific target antigen for immunotherapy. GD2 expression was confirmed at variable degree in a set of breast cancer cell lines, predominantly in triple-negative breast cancer (TNBC). To target GD2, we have generated novel anti-GD2 chimeric antigen receptors (GD2-CAR), based on single-chain variable fragments (scFv) derived from the monoclonal antibody (mAb) ch14.18, also known as dinutuximab beta. Expressed on T cells, GD2-CARs mediated specific GD2-dependent T-cell activation and target cell lysis. In contrast to previously described GD2-CARs, no signs of exhaustion by tonic signaling were found. Importantly, application of GD2-CAR expressing T cells (GD2-CAR-T) in an orthotopic xenograft model of TNBC (MDA-MB-231) halted local tumor progression and completely prevented lung metastasis formation. In line with the BCSC model, GD2 expression was only found in a subpopulation (4-6%) of MDA-MB-231 cells before injection. Significant expansion of GD2-CAR-T in tumor-bearing mice as well as T-cell infiltrates in the primary tumor and the lungs were found, indicating site-specific activation of GD2-CAR-T. Our data strongly support previous findings of GD2 as a BCSC-associated antigen. GD2-targeted immunotherapies have been extensively studied in human. In conclusion, GD2-CAR-T should be considered a promising novel approach for GD2-positive breast cancer, especially to eliminate disseminated tumor cells and prevent metastasis formation.
KEYWORDS: GD2, CAR-T cells, breast cancer stem-like cells, triple-negative breast cancer, metastasis
Introduction
Breast cancer is the most common cancer in women and second leading cause of cancer-related mortality.1 Whereas overall survival rates improved over the past decades, the prognosis of patients diagnosed with metastatic disease, either de novo (5-10%) or progressed in the cause of their disease (20–30%), remains poor with an estimated 5 year survival rate of below 30%.2 Cumulating evidence suggests that metastatic spread occurs early at a pre-symptomatic stage of the disease.3,4 Consequently, the incidence of metastatic or disseminated disease at initial diagnosis is likely to be underestimated. The introduction of adjuvant and neoadjuvant therapy did not substantially reduce the rates of distant or systemic recurrence and death associated with metachronous metastatic relapse, indicating an intrinsic resistance of disseminated tumor cells (DTCs) to conventional therapies.5–7 Resistance of DTCs was attributed to dormancy, characterized by cessation of proliferation as well as active protection by the microenvironment in the perivascular niche.8,9 There is broad consent, that dissemination of cancer cells is initiated by epigenetic changes leading to epithelial-to-mesenchymal transition (EMT) of tumor cells.10 Importantly, EMT was shown to be associated with stem-like traits.11,12 The cancer stem cell (CSC) model postulates that tumor cells are hierarchically organized with minor subpopulations essential for continued tumor growth, metastasis formation, resistance to therapy and recurrence after therapy.13–16 Consequently, identifying and specifically targeting DTCs, possessing a BCSC phenotype, is of paramount importance to improve outcomes of patients with metastatic or disseminated disease.
Battula and colleagues have identified the disialoganglioside GD2 as a novel BCSC-associated antigen.17 GD2 was found to be expressed on a subpopulation of cells in human breast cancer cell lines and patient samples that were capable of forming mammospheres and initiating tumors in vivo. GD2 expression correlated with expression of established BCSC markers CD44highCD24low and was dramatically increased by induction of EMT. Moreover, they were able to demonstrate that knockdown or pharmacological inhibition of GD3 synthase (GD3S), an enzyme involved in the biosynthesis of GD2, abrogated tumor formation after transplantation and reduced metastasis in vivo.17,18 GD2 expression on breast cancer and BCSC was independently verified by different groups.19,20
Notably, GD2 is an excellent and extensively studied target antigen for antibody-based immunotherapy.21 We and others have clinically evaluated GD2 mAbs to treat patients suffering from metastatic neuroblastoma, leading to significantly improved survival and FDA/EMA approval of ch14.18, also known as dinutuximab beta.22,23 In the present study, we used a GD2-directed immunotherapy by GD2-CAR-T to target BCSCs. CARs are synthetic receptors consisting of an extracellular binding domain, usually a scFv derived from a mAb, a transmembrane domain as well as one or multiple co-stimulatory and signaling domains.24 Expressed on T cells, CARs are capable to mediate surface antigen-dependent T-cell activation and subsequent target cell lysis. Our hypothesis was that GD2-CAR-T will circulate the body to identify and eliminate BCSC phenotypic DTCs and prevent metastasis formation.
Materials and methods
Cell lines and culturing conditions
Human breast cancer cell lines MCF7, T-47D, MDA-MB-468, MDA-MB-231, HS 578T and BT-549 as well as human ovarian cancer cell line OVCAR-8 were purchased from ATCC. Human neuroblastoma cell line LS (ACC 675) and LAN-1 (ACC 655) were purchased from DSMZ. Neuroblastoma cell lines were maintained in RPMI 1640 media supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific), 2 mM L-glutamine (Biochrom) and 1 mM sodium pyruvate (Biochrom). Breast cancer and ovarian cancer cell lines were maintained in Dulbecco's VLE DMEM media supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific). All media contained 100 units/mL of penicillin and 100 µg/mL of streptomycin (Biochrom).
Evaluation of GD2 expression on tumor cell lines
GD2 expression was assessed by flow cytometry on a BDTM LSR II flow cytometer. Antibody staining was done according to standard operating procedure at 4°C in PBS buffer. Gating strategy is demonstrated in Supplementary Figure 2a. Doublets and dead cells, identified by Viability Dye eFluor™ 780 (ThermoFischer), were excluded before assessment of GD2 positivity. Antigen positivity was defined by staining of tumor cells using PE labeled GD2 mAb (14G2a, BioLegend) compared to PE labeled mouse IgG2a, κ isotype control (BioLegend). Overton positivity was calculated by integral subtraction (specific fluorescence minus fluorescence of isotype control) using FlowJo 10.4 software. Median fluorescence intensity ratio (MFIR) was calculated by (sample MFI/isotype control MFI) for each cell line separately to facilitate comparison antigen expression intensity between the different cell lines and tumor entities.
CAR design and vector production
GD2-targeted scFvs were designed in silico, based on the protein sequence of the GD2 mAb ch14.18. Variable regions of the heavy and light chain were paired using a G4Sx3 linker. The anti-CD19 scFv was derived from the CD19 mAb 4G7. scFvs were assembled on a second-generation CAR backbone incorporating a CD8 hinge, CD8 transmembrane domain, as well as the cytoplasmic domain of 41BB and CD3zeta and truncated EGFR (EGFRt) after a P2A site.25 Custom synthesis of CAR constructs was done by GeneArt. The CAR constructs were subcloned into a third generation lentiviral vector transfer plasmid for luciferase/mCherry expression kindly provided by Irmela Jeremias, Helmholtz Centre Munich, Germany.26 Lentivirus (LV) was produced in Lenti-XTM 293T (Clontech) after 3 plasmid transfection using Lipofectamine 3000 (Thermo Fisher) of the second-generation packaging plasmid psPAX2 (Addgene), VSV-G envelope plasmid pMD2.G (Addgene) and the CAR containing transfer plasmid.27 LV supernatants were harvested, concentrated using Lenti-X concentrator (TaKaRa) and cryopreserved at −80°C.
T-cell transduction
PBMC were isolated from whole peripheral blood, acquired from healthy volunteer donors at the University Children’s Hospital Tuebingen, by Ficoll-Paque density gradient centrifugation (Biocoll, Biochrom). T cells were sequentially isolated using CD4 and CD8 microbeads (Miltenyi Biotec) and mixed at a 1:1 ratio. T cells were activated with TransActTM (anti-CD3 and anti-CD28 agonistic signal, Miltenyi Biotec) and cultivated in TexMACS media (Miltenyi Biotec) supplemented with 10 ng/mL IL7 and 5 ng/mL IL15 (Miltenyi Biotec). After 24 h, activated T cells were transduced at a multiplicity of infection (MOI) of 3. Transduced T cells were maintained at 0.5–2 × 106 cells/mL in IL7/IL15 containing TexMACS® media. On day +7, CAR transduction efficiency and CD4/CD8 ratio were determined by flow cytometry using the antibodies: CD4-BUV395 (SK3, BD Bioscience), CD8-APC (BW135/80, Miltenyi Biotec) and EGFR-PE (AY13, BioLegend). Tonic signaling was assessed by expression of the exhaustion markers PD-1, TIM-3, and LAG-3 via flow cytometry on day +21 of culturing using the antibodies: EGFR-FITC (13/EGFR (RUO), BD Bioscience); PD-1-PE (PD1.3.1.3, Miltenyi); TIM-3-PE-/Dazzle594 (F38-2E2, BioLegend); LAG-3-APC (7H2C65, BioLegend) and LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (ThermoFisher).
Real-time impedance-based cytotoxicity assay
For label-free real-time cytotoxicity assessment, the xCELLigence device, an impedance-based Real-Time Cytotoxicity Analyzer (RTCA) (ACEA Biosciences Inc.) allowing continuous assessment of cell growth and cytolysis, was used. The adherent growing breast cancer, neuroblastoma and ovarian cancer cell lines were plated at 30.000 cells per well except for the cell lines BT549 and HS578T (20.000 cells per well) in the regular culture media for the respective cell line (see above) in 96-well electronic microtiter plates E-Plate® 96 (ACEA Biosciences Inc.). After cell attachment for 24 h to achieve a robust cell index, effector cells were added at indicated effector to target (E:T) ratio. The total volume per well was 200 µl. Plates were incubated in a HERAcell incubator (Heraeus) under 37°C, 95% humidity and 5% CO2 and impedance was assessed every 15 min for 48 h.
Quantification of cytokine release
For determination of secreted cytokines, 200.000 GD2-CAR-T or CD19-CAR-T were incubated for 12 h with 100.000 target cells per well in a 96-well plate in regular media (see above) in a HERAcell incubator (Heraeus) at 37°C, 95% humidity and 5% CO2. After incubation supernatants were collected. Quantification of the cytokines INFγ, IL-2 and TNFα in the supernatants was performed via cytokine capture beads using the MACSPlex® custom cytokine assay with indicated specificities, the MACSQuant® Analyzer and the MACSQuantify® software according to the manufacturer’s instructions (Miltenyi Biotec).
Animals and in vivo model
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, originally obtained from Jackson Laboratories, were bred at our institution. All procedures described herein were approved by the authorities for Biomedical Research Institutional Animal Care and Use Committee responsible for the University Children’s Hospital Tuebingen, Germany and performed according to national and institutional guidelines for the humane treatment of animals. Six to eight-week-old female NSG mice were injected into the 4th mammary fat pad with 5 × 106 MDA-MB-231 cells, stably expressing luciferase (Vector kindly provided by Irmela Jeremias, Helmholtz Centre Munich, Germany). Tumor growth was monitored by in vivo bioluminescence imaging (BLI) and caliper measurement. Tumor volume was calculated: Tumor volume = (width)2 x length/2 in mm3.28 Seven days after injection, mice were randomized and injected intravenously with PBS or 5 × 106 CAR-T. Tumor growth was monitored weekly by BLI and every other day by caliper. Body weight was monitored daily. Twenty-one days after tumor inoculation mice were sacrificed because of reaching end point criteria. Primary tumors and lungs were isolated and fixed in 4.5% buffered formalin. Blood was drawn for further analysis.
Optical imaging
Optical Imaging was performed using an IVIS Spectrum Optical Imaging system (Perkin Elmer). For weekly tumor burden monitoring, mice were injected 75 mg/kg XenoLightTM D-Luciferin (Perkin Elmer, Waltham, MA, USA) i.p. and luciferase activity was detected using bioluminescence imaging (BLI) following a 5-min uptake. Images were acquired using a 5-s exposure, f-Stop 8 and binning 8 with a field of view of C-14. Signals above a 10% threshold were normalized for the photon radiance [photons/second/cm2/sr] and quantified using Living Image Software 4 (Perkin Elmer).
Mouse pathology and histology
Mouse lungs and tumors were fixed in 4.5% buffered formalin and paraffin embedded. For histology 3–5 µm-thick sections were cut and stained with hematoxylin and eosin (H&E). Immunohistochemistry was performed on an automated immunostainer (Ventana Medical Systems, Inc.) according to the company’s protocols for open procedures with slight modifications. The slides were stained with the antibodies AE1/3 (M3515, DAKO), GD2 (MAB2052, EMD Millipore Corporation, Temecula, USA), human CD3 (CI597C01, DCS Innovative Diagnostik-Systeme GmbH u. Co. KG), CD4 (503–3354, Zytomed), CD8 (M7103, DAKO, Glostrup), B220 (550286, BD Biosciences) and MAC3 (550292, BD Biosciences). Appropriate positive and negative controls were used to confirm the adequacy of the staining.
CAR-T in vivo monitoring
The in vivo expansion and phenotype of CAR-T was analyzed postmortem in peripheral blood of CAR-T treated mice. Red blood cells were lysed with aqua followed by 10x PBS. Remaining cells were stained with the following fluorophore conjugated antibodies: murineCD45-APC-eflour780 (30F11, eBioscience); EGFR-FITC (13/EGFR(RUO), BD Bioscience); CD25-BUV737 (2A3, BD Bioscience); PD-1-PE (PD1.3.1.3, Miltenyi); TIM-3-PE-/Dazzle594 (F38-2E2, BioLegend); LAG-3-APC (7H2C65, BioLegend) and LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (ThermoFisher). To enumerate the number of T cells (mCD45−hCD3+) and CAR-T (mCD45−hCD3+hEGFRt+) per microlitre of blood, all tubes were recorded exhaustively on a BDTM LSR II flow cytometer. Gating strategy is demonstrated in Supplementary Figure 6a. CAR-T activation was determined by CD25 expression. Terminal exhaustion was defined as expression of the three inhibitory receptors PD-1, TIM-3, and LAG-3.
Software
Flow cytometric data were analyzed by FlowJo 10.4. Optical Imaging analysis was conducted using Living Image Software 4 (Perkin Elmer). Graphs and statistics were generated using the GraphPad Prism 7.0 software.
Statistics
Significance of in vitro real-time impedance-based cytotoxicity assay was determined by two-sided multiple T-tests. Significance of differences in tumor burden in vivo, measured by BLI and caliper, was calculated by 2way ANOVA Tukey’s multiple comparison test. Significance of differences in CAR-T expansion in vivo was calculated by paired T-test. p < .05 was considered statistically significant.
Results
Design and functionality of novel GD2-CAR-T
In order to target GD2 on breast cancer and BCSCs, we have developed novel GD2-targeted CARs. For antigen binding, two scFvs in heavy chain/light chain (HL) and light chain/heavy chain (LH) configuration divided by a G4Sx3 linker were designed, derived from the chimeric monoclonal GD2 antibody ch14.18.29 ch14.18, also known as dinutuximab beta, has been extensively studied at our institution in clinical trials in neuroblastoma patients (NCT02258815).22 The scFvs were assembled via a CD8 hinge and CD8 transmembrane domain on a 4-1BB costimulatory and CD3zeta signaling domain second generation CAR backbone (Figure 1a,b). Truncated EGFR (EGFRt) was fused after a P2A site (Figure 1a,b) for detection, enrichment and as a possible safety switch.25 GD2-CARs were expressed on activated CD4 and CD8 positive T cells at a ratio of 1:1 by lentiviral transduction. Our 10-day expansion protocol in TexMACS® in the presence of IL7 and IL15 leads to a balanced expansion of CD4 and CD8 positive T cells (Supplementary Figure 1a). Surface expression of CARs was indirectly determined by flow cytometry against EGFRt (Figure 1c and Supplementary Figure 1a). The HL design resulted in slightly superior expression and lysis at a low E:T ratio of 2:1 against the highly GD2 expressing neuroblastoma cell line LS (Figure 1c,d). Consequently, HL design was used for further evaluation, referred to as GD2-CAR. Importantly, we did not find any signs of exhaustion due to tonic signaling as described by Long et al.30 for CD28 co-stimulated GD2-CAR-T derived from the murine GD2 antibody clone 14G2a, even after 21 days of culture (Supplementary Figure 1b).
Figure 1.
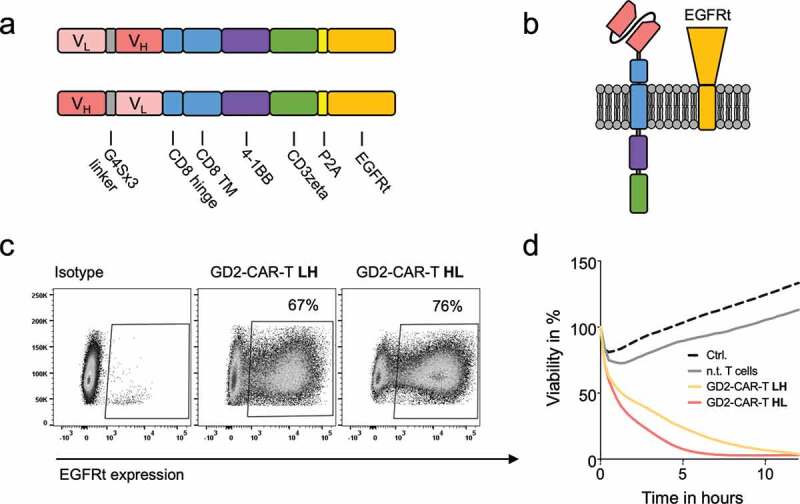
Design and functionality of novel GD2-CAR-T. a) Schematic illustration of GD2-CAR constructs in heavy chain/light chain (HL) and light chain/heavy chain (LH) configuration. b) Schematic illustration of GD2-CAR and EGFRt expression on the plasma membrane. In c), GD2-CAR expression was determined by flow cytometry against EGFRt, coexpressed with the GD2-CAR. Representative plots for non-transduced T cells (left), GD2-CAR-T LH (middle) and GD2-CAR-T HL (right) transduced T cells (middle) are shown. In d), GD2-CAR-T were incubated with GD2+ neuroblastoma cell line LS at an E:T ratio of 2:1 for 12 h. Target-cell lysis was determined by impedance-based real-time cytotoxicity assay xCELLigence® RTCA. Data shown represent mean of two independent experiments in triplicates.
GD2 is expressed at variable degree on breast cancer cell lines
To determine GD2 expression on breast cancer cell lines and validate previously published findings, we have evaluated a small but representative panel of six breast cancer cell lines,31–33 MCF7 (luminal A), T-47D (luminal A), MDA-MB-468 (TNBC basal A, basal-like), MDA-MB-231 (TNBC basal B, mesenchymal stem-like, claudin-low), HS 578T (TNBC basal B, mesenchymal stem-like, claudin-low) and BT-549 (TNBC basal B, mesenchymal-like, claudin-low) from the NCI-60 panel34 by flow cytometry. As reported by Battula et al.,17 GD2 expression was found in a subpopulation of cells in two cell lines, 4.5% (range 3.8–6.2%) positive cells in MDA-MB-231 and 6.5% (4.7–8.1%) positive cells in T-47D (Figure 2a,b). No significant expression was found in MCF7 and MDA-MB-468 (Figure 2a). Unexpectedly, uniform GD2 expression was found in HS 578T and BT-549 (Figure 2a) with a low positive MFIR of 6.7 (MFI 1791/isotype MFI 269) and MFIR 16.4 (MFI 4074/isotype 249), respectively, compared to high GD2 expression in the neuroblastoma cell lines LAN-1 and LS with a MFIR 1364 (MFI 42036/isotype MFI 30.8) and MFIR 2885 (MFI 15321/isotype MFI 5.31) (Figure 2a), respectively. Screening for other gynecological cancer entities revealed high uniform GD2 expression MFIR 70.2 (MFI 8775/isotype MFI 125), in the ovarian cancer cell line OVCAR-8 (Supplementary Figure 2b).
Figure 2.
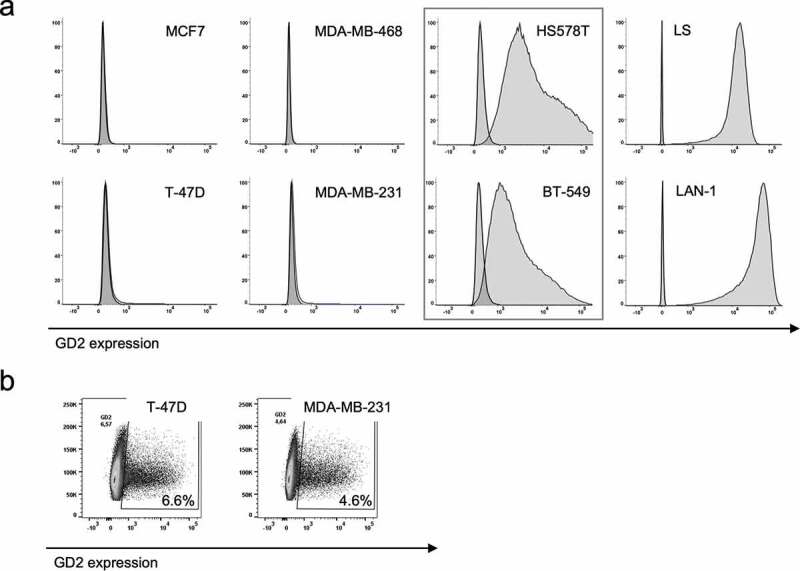
GD2 expression on breast cancer cell lines. GD2 expression on indicated breast cancer cell lines was determined by flow cytometry using an APC-labeled GD2 mAb. In a), representative histograms of three individual analyses are shown. GD2 positivity was calculated by Overton subtraction of histograms, sample data minus corresponding isotype control. In b), representative plots SSC vs. GD2 expression of three individual analyses are shown to illustrate GD2 expression in a subpopulation of cells in the cell lines T-47D and MDA-MB-231.
GD2-CAR-T cells specifically lyse GD2-positive breast cancer cells in vitro
After establishing functional GD2-CAR-T, specific T-cell activation and target cell lysis of breast cancer cell lines was evaluated. Activated and CAR-transduced T cells are known to execute some degree of unspecific effector function depended on the susceptibility of the target cell. To exclude antigen-independent effects, CD19-CAR-T cells served as negative control. In line with GD2 expression, we found highly specific target cell lysis by GD2-CAR-T at a low E:T ratio of 2:1 against the TNBC cell lines HS 578T and BT-549 (Figure 3). Target cell lysis was complete but delayed in comparison to the neuroblastoma cell line LS with higher GD2 expression (Supplementary Figure 3a). No specific lysis was detected in a 48-h cytotoxicity assay targeting MDA-MB-231 and T-47D, both expressing GD2 on a minor subpopulation as well as MCF7 and MDA-MB-468, shown to be GD2 negative (Figure 3). Of note, both MDA-MB-231 and MCF7 appear susceptible to target antigen-independent effects, demonstrated by growth arrest induced by GD2-CAR-T and CD19-CAR-T and unspecific target cell lysis likewise (Supplementary Figure 3b). Paradoxically, proliferation of the breast cancer cell line T-47D was enhanced in the presence of CAR-T. Specific lysis was also found against GD2-positive OVCAR-8 cells, underscoring the universal functionality of our GD2-CAR-T against GD2-expressing cells (Supplementary Figure 3c). Confirming the results for specific lysis, cytokine secretion of INFγ, IL-2 and TNFα by GD2-CAR-T was specifically induced in the presence of GD2 positive HS 578T cells, similar to LS cells, but not by MDA-MB-468 cells (Supplementary Figure 3d).
Figure 3.
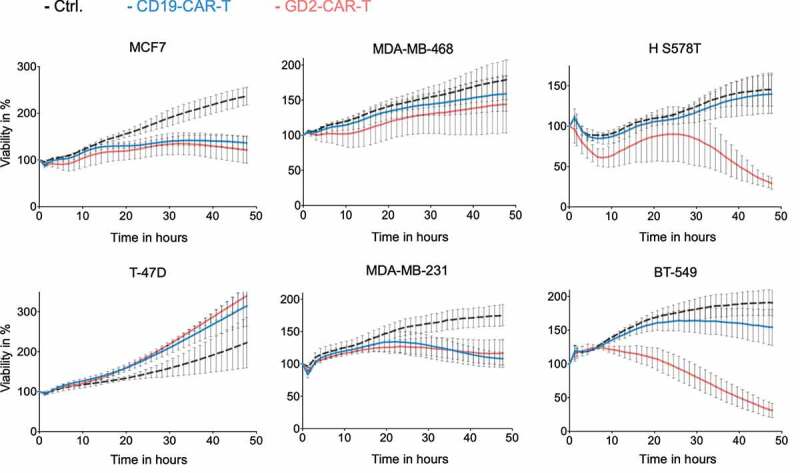
Specific lysis of breast cancer cell lines by GD2-CAR-T. GD2-CAR-T (red) or CD19-CAR-T (blue) were incubated with indicated breast cancer cell lines at an E:T ratio of 2:1 for 48 h. Target cell lysis was determined by impedance-based real-time cytotoxicity assay xCELLigence® RTCA. Kinetics over 48 h are demonstrated. Data shown represent mean ± SD of three independent experiments in triplicates.
GD2-CAR-T cells arrest tumor growth and prevent metastasis formation in vivo
GD2 was identified as a BCSC-associated antigen.17,18 To test this hypothesis, we made use of a well-established orthotopic xenograft mouse model of reliable spontaneous dissemination and metastasis formation.28,35 5 × 106 MDA-MB-231 TNBC cells were injected in the mammary fat pad of 6–8 weeks old female NSG mice. After 7 days, tumor establishment was verified (Supplementary Figure 4a). Tumor-bearing mice were randomized and treated with either 5 × 106 GD2-CAR-T or 5 × 106 CD19-CAR-T. Tumor growth was monitored by noninvasive in vivo BLI and caliper measurement. Mice in the control groups (PBS and CD19-CAR-T) had to be sacrificed at day +21 because of reaching end point criteria, tumor diameter of 10 mm, stipulated by the local authorities. To compare the effects on metastasis formation, day +21 was chosen as the endpoint in all groups. All mice in the experiment, including the ones treated with GD2-CAR-T, were in excellent condition during the observation period without demonstrating any signs of neurological symptoms that could be attributed to neuroinflammation or neurotoxicity. Treatment with GD2-CAR-T led to a highly significant reduction in tumor growth compared to CD19-CAR-T cells (p < .001) and the PBS-injected control (p < .001) (Figure 4a–c and Supplementary Figure 4a,b). This is remarkable, since GD2 is only expressed in a subpopulation of MDA-MB-231 cells in tissue culture and in the xenograft primary tumor as well as lung metastasis (Supplementary Figure 5a). Moreover, histological analysis demonstrated that the lungs of all GD2-CAR-T treated mice were free of metastasis, whereas nests of metastatic cells, positive for pan-cytokeratin staining AE1/3, were detectable in the lung parenchyma of all CD19-CAR-T treated mice (Figure 5a,b and Supplementary Figure 5b). Of note, lung metastasis in CD19-CAR-T treated mice were smaller compared to PBS-injected mice (Supplementary Figure 5a,b), in line with antigen-independent effects seen in vitro.
Figure 4.
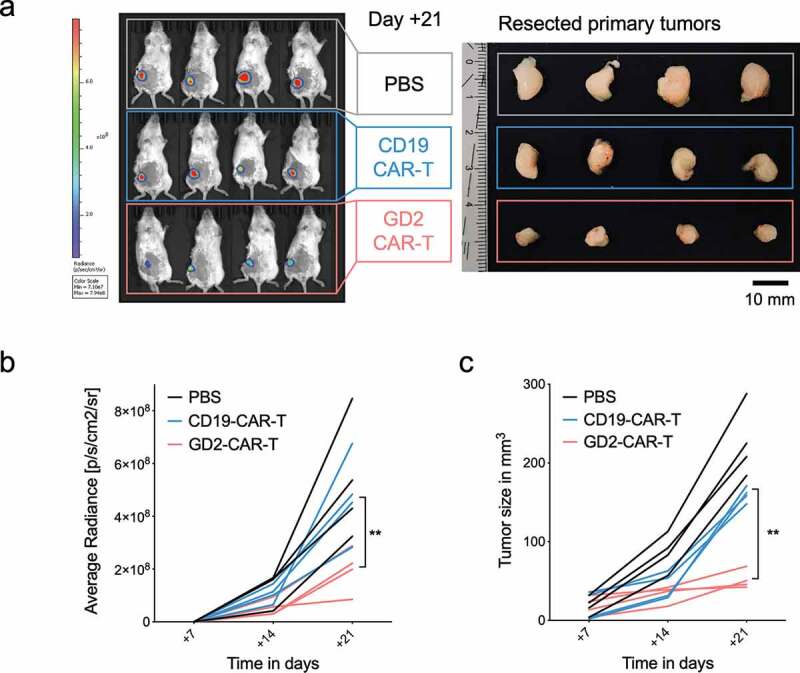
GD2-CAR-T cells arrest tumor growth in vivo. NSG mice were injected into the fourth mammary fat pad with 5 × 106 MDA-MB-231 cells, stably expressing luciferase on day 0. On day +7, tumor-bearing mice were treated with either PBS, 5 × 106 CD19-CAR-T or 5 × 106 GD2-CAR-T. Mice were sacrificed on day +21. In a), noninvasive BLI images of luciferase activity at day +21 are shown for tumor burden assessment (left panel, n = 4). Resected tumors of the corresponding mice are shown in the right panel. In b), BLI signals above a 10% threshold were quantified as average radiance [photons/second/cm2/sr]. Timepoints day +7, injection of CAR-T, day +14 and day +21, end of the experiment, are shown. (Of note, for day +7 values were set 0 as no signals above a 10% threshold could be calculated. Corresponding optical imagines are given in Suppl. Figure 4a). In c), tumor growth kinetics are demonstrated as determined by caliper measurement. Data shown represent mean ± SD of 4 mice each group. (** = p < .001).
Figure 5.
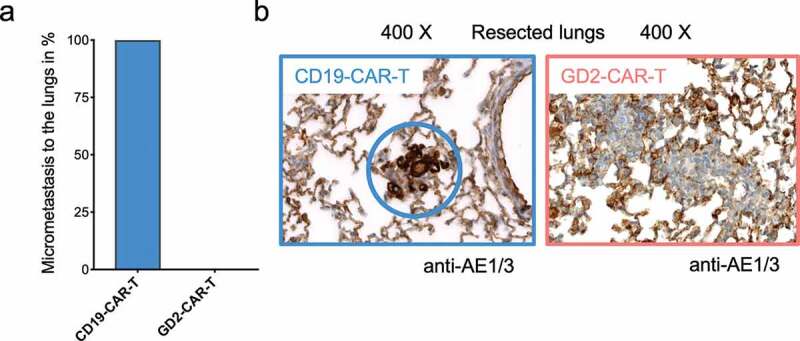
GD2-CAR-T cells prevent metastasis formation in vivo. Lungs of NSG mice injected with 5 × 106 MDA-MB-231 cells into the fourth mammary fat pad on day 0, treated on day +7 with either 5 × 106 CD19-CAR-T or 5 × 106 GD2-CAR-T and sacrificed on day +21 were fixed and stained with the pan-cytokeratin staining AE1/3 to determine metastatic infiltration. In a), the presence of AE1/3 positive cells in the lungs of mice was quantified, 4 mice each group. In b), representative images for both groups are shown, demonstrating the presence and absence of metastatic cells.
GD2-CAR-T specifically expand and infiltrate in tumor and metastatic tissue
After demonstrating in vivo activity, transferred CAR-T cells were further analyzed by flow cytometry. We found a highly significant (p < .001) specific expansion of GD2-CAR-T in tumor-bearing mice, compared to CD19-CAR-T (Figure 6a). GD2-CAR-T expressed CD25 as a marker of activation but no signs of terminal exhaustion, assessed by expression of PD1, LAG-3, and TIM-3 (Figure 6b,c). Histological analysis demonstrated a diffuse infiltration of CD3 positive human T cells in the primary tumor in GD2-CAR-T, but not in CD19-CAR-T treated mice (Figure 6d). Moreover, in the histological analyses of the lungs, we found a prominent infiltrate of human T cells, positive for CD3, CD4 and CD8, mainly around the bronchi and the blood vessels in GD2-CAR-T but not CD19-CAR-T treated mice (Figure 6d and Supplementary Figure 6b,c).
Figure 6.
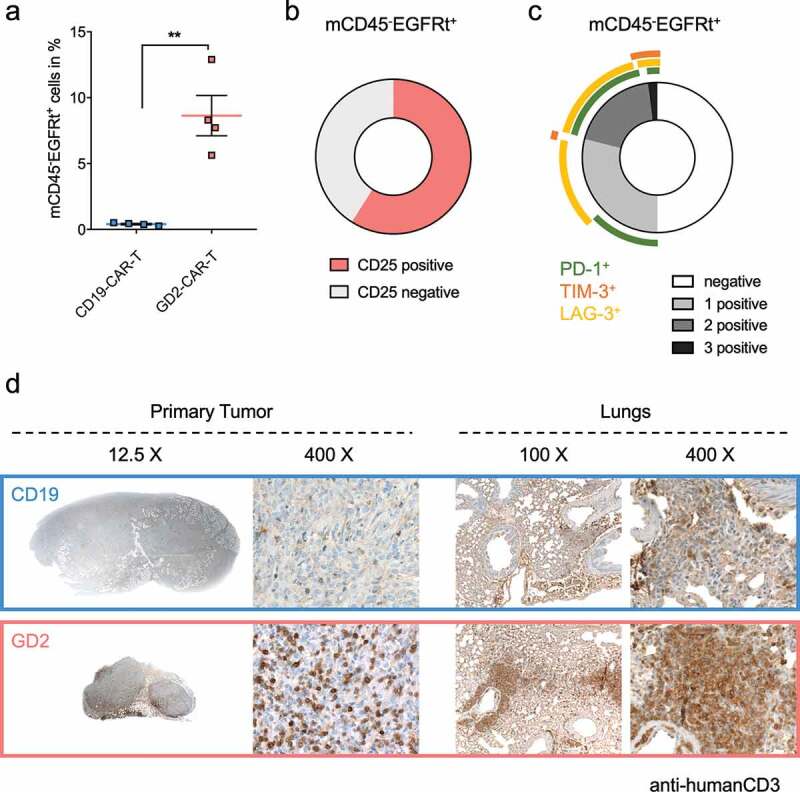
GD2-CAR-T specifically expand and infiltrate in tumor and metastatic tissue. NSG mice were injected with 5 × 106 MDA-MB-231 cells into the fourth mammary fat pad on day 0, treated on day +7 with either 5 × 106 CD19-CAR-T or 5 × 106 GD2-CAR-T and sacrificed on day +21. In a), blood was taken on day +21 and analyzed for the presence of CAR-T by flow cytometry. CAR-T were identified being negative for mCD45 and positive for hEGFRt expression. Data shown represent mean ± SD of 4 mice each group. In b) and c), GD2-CAR-T were further analyzed for the activation marker CD25 and the exhaustion markers PD-1, TIM-3 and LAG-3 by flow cytometry. In c), expression of single and multiple exhaustion markers at the same time is demonstrated. Data shown in b) and c) represent mean of 4 mice in the GD2-CAR-T treated group. In d), primary tumors and lungs of mice were fixed after scarification on day +21 and stained with a hCD3 antibody to identify infiltrateing CAR-T. Representative images for 4 mice each group at different magnification are shown, demonstrating the absence or presence of infiltrating CAR-T. (** = p < .001).
Discussion
In the present study, we report on the development of novel GD2-targeted CAR-T cells to treat breast cancer and BCSCs. Generating functional GD2-CAR-T has been challenging. Long et al. elegantly demonstrated antigen-independent clustering of anti-GD2 scFvs leading to tonic signaling, early CAR-T exhaustion and impaired function.30 To circumvent this problem, we generated novel scFvs derived from the anti-GD2 mAb ch14.18, also known as dinutuximab beta. Additionally, we used a 4-1BB costimulatory domain in our second generation CAR construct, demonstrated to reduce exhaustion in comparison to CD28.30 Applying these modifications, we were able to generate fully functional GD2-CAR-T as demonstrated in vitro for target cell lysis and cytokine production as well as in vivo. GD2-CAR-T mediated sufficient target cell lysis even at low antigen expression levels. Importantly, we did not find any signs of tonic signaling and subsequent terminal exhaustion, neither in an ex vivo culture system, nor after 14 days in tumor-bearing mice.
Several reports have identified GD2 as a relevant antigen target in breast cancer. Batulla et al. and subsequently Liang et al. demonstrated GD2 expression on BCSCs phenotypically, by coexpression with established BCSC markers like CD44hiCD24lo as well as functionally, looking at mammosphere formation and tumorigenicity in vivo.17,19 The same group also stained 15 patient samples on GD2 expression, reporting highest expression (35.8%) in a metastatic TNBC sample. Orsi et al. analyzed 63 patient samples of different breast cancer subtypes for GD2 expression. They found GD2 expression in 59% of samples with increased prevalence toward highly aggressive breast cancer subtypes, such as TNBC and metaplastic variants.20 In our study, we analyzed GD2 expression on a small panel of breast cancer cell lines. In line with Batulla et al., we found GD2 expression in a subpopulation of cells, possibly BCSCs, in the cell lines T-47D (luminal A) and MDA-MB-231 (TNBC basal B). In contrast, the two TNBC cell lines HS 578T and BT-549, both basal B and claudin-low, presented with a uniform GD2 expression. This expression pattern was also found in the ovarian cancer cell line OVCAR-8 (G3 adenocarcinoma). Despite the small number of tested cell lines, we can also see a tendency toward GD2 expression in highly aggressive subtypes across entities. Importantly, our GD2-CAR-T demonstrated excellent cytolytic activity against GD2 positive cell lines, independent of the tumor entity.
GD2 expression was additionally demonstrated to be linked to EMT. This observation was mainly driven by studies in HMLER cells. HMLER cells are genetically transformed primary human mammary epithelial cells (HMECs) overexpressing hTERT, SV40 T/t, and H-RasV12. Batulla et al. identified a subpopulation of around 5% GD2 positive cells, possessing BCSC properties. Additional ectopic expression of the EMT-associated transcription factors Snail or Twist expanded this population to 40% and 100%, respectively.17 GD2 positivity was associated with an increased expression of GD3S, a rate limiting enzyme in GD2 biosynthesis. Supporting the hypothesis that GD3S plays an essential role in the regulation of EMT and mediation of BCSC traits, knockdown or pharmacological inhibition of GD3S in the human MDA-MB-231 and murine 4T1 cell lines abrogated tumorigenicity and reduced metastasis formation in xenograft and syngeneic mouse models.17,18 Moreover, high expression of GD3S in patient samples of TNBC was correlated with poor prognosis.18 Interesting additional observations underscore the relationship of EMT, aggressive phenotype and GD2 expression. An EMT core gene-expression signature was shown to be associated with the prognostic dismal basal-like, claudin-low and metaplastic breast cancer subtypes,36,37 which were found to have highest GD2 expression. In line with this observation, cancer entities with the highest EMT scores, expression of EMT-associated genes plus infiltrative and metastatic, mesenchymal phenotype, namely neuroblastoma, glioma, osteosarcoma, Ewing’s sarcomas, and melanoma express GD2 in a high proportion.38 Further, high GD2 expression was found to correlate with metastatic potential in melanoma, osteosarcoma and bladder cancer and enforced GD2 expression, by overexpression of GD3S, leads to enhanced invasiveness and migratory potential in small cell lung cancer and osteosarcoma.39–43
Based on the outlined findings, we hypothesized that depletion of GD2 positive cells would impair dissemination and metastasis formation. Moreover, GD2-targeted therapy should be able to eliminate BCSC phenotypic DTCs. To test this hypothesis we used the well-evaluated TNBC cell line MDA-MB-231, demonstrated by us and others to possess a GD2 positive subpopulation of 5–10% of cells,17,18 in a xenograft model of spontaneous metastasis formation after orthotopic injection.28,35 In line with our hypothesis, GD2-CAR-T therapy completely prevented metastasis formation to the lungs. Moreover, growth of the primary tumor was substantially arrested, possibly by eliminating the BCSC compartment. This was associated with specific activation and expansion of GD2-CAR-T as well as infiltration into the primary tumor. Moreover, we found infiltrates of GD2-CAR-T in the lungs, indicating stimulation at the site of metastasis formation. Of note, application of CD19-CAR-T, serving as a negative control, did have an antigen-independent effect on tumor growth. CD3/CD28 activated and IL15 cytokine expanded T cells correspond to cytokine-induced killer cells (CIK cells) that are known to mediate lysis of tumor cells by NK-like effector function in a MHC non-restricted manner.44 However, in contrast to GD2-CAR-T, nests of tumor cells were found in the lungs of all mice in the CD19-CAR-T group. We conclude that GD2-CAR-T therapy specifically eliminates GD2-expressing disseminated BCSCs, thereby preventing metastasis formation as schematically illustrated in Figure 7.
Figure 7.
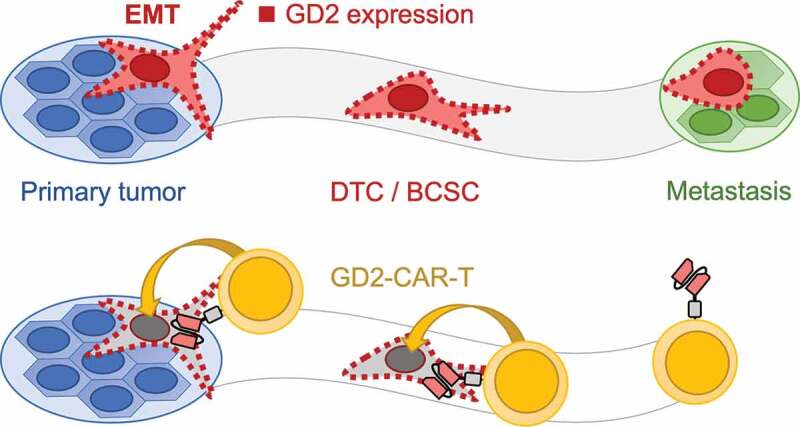
Schematic illustration of the mechanism how GD2-CAR-T prevent metastasis formation. Metastasis formation is a process in which tumor cells dissociate from the primary tumor after undergoing EMT. These DTCs or BCSCs disseminate throughout the body and finally form distant metastasis. GD2 expression, indicated by red squares, is associated with EMT and the acquisition of a BCSC phenotype (upper panel). Targeting GD2 on BCSCs by GD2-CAR-T leads to the elimination of BCSCs and prevents metastasis formation (lower panel).
GD2-targeted immunotherapies are well established and clinically evaluated. There are extensive data demonstrating anti-tumor activity of GD2 mAb in human.21–23,45 The major side effects identified in these studies were severe pain symptoms as a result of GD2 expression on peripheral nerves and possibly engagement of the complement system.46 Recently, Richman et al. reported on rapid mortality in a xenograft model of neuroblastoma treated with high affinity, introducing an E101K mutation into a 14G2a derived scFv, GD2-CAR-T.47 The authors attributed this lethal side effect to specific GD2-CAR-T-mediated neurotoxicity due to low-level expression of GD2 in the central nervous system (CNS). In contrast, we did not observe any neurological symptoms in our study. This is in line with other reports applying 14G2a, the parental mAb ch14.18 was chimerized from, derived GD2-CAR-T.30,48 GD2-CAR-T have been and are currently investigated in a number of clinical trials in neuroblastoma, sarcoma, glioma and melanoma. Early work in the group of Malcolm Brenner, using a 1st generation GD2-CAR expressed in EBV-specific cytotoxic T cells, demonstrated induction of complete remission in 3 out of 11 neuroblastoma patients with active disease. No dose-limiting toxicities and no pain symptoms were reported.49,50 More recently, the same group reported on a clinical trial applying third generation GD2-CAR-T. Again, administration was safe. However, therapeutic effects were modest, which was attributed to the expansion of suppressive myeloid cells.51 Further engineering will improve the efficacy of GD2-CAR-T, for example by coexpression of IL15.52 Moreover, GD2-CAR-T effector function might be enhanced with additional immune checkpoint blockade.53
In conclusion, we report for the first time, that GD2-directed immunotherapy by GD2-CAR-T can prevent metastasis formation in a highly aggressive model of TNBC. This observation is in line with previous reports identifying GD2 as an EMT- and BCSC-associated antigen. Our results have major implications: first, our and other’s work is giving a strong rationality to routinely screen for GD2 expression on breast cancer specimens and possibly on circulating tumor cells to gain more knowledge on the prevalence of GD2 expression in breast cancer. Second, GD2-targted therapy, e.g. using the FDA/EMA approved mAb dinutuximab beta or GD2-CAR-T, should be considered as an adjuvant consolidation therapy for patients at high risk for metastatic relapse to eradicate persisting DTCs not accessible and/or refractory to conventional therapeutics. Feasibility of adjuvant immunotherapy has been demonstrated over two decades in the setting of HER2-positive breast cancer.54 Third, GD2-CAR-T might be a valuable therapeutic option for patients with high-risk breast cancer subtypes like TNBC and metaplastic variants, lacking curative treatment strategies to date. Clinical trials will have to demonstrate clinical efficacy translating GD2-CAR-T cell therapy into improved survival.
Funding Statement
This work was supported by the Deutsche Krebshilfe (German Cancer Aid) - DKH 70111975 (RH), the Stiftung des Fördervereins für krebskranke Kinder Tübingen, Germany (CMS, PL, RH, PS) and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2180 – 390900677 (BP, RH, PS).
Acknowledgments
The authors gratefully acknowledge Binje Vick and Irmela Jeremias for generously providing the lentiviral transfer plasmids (luciferase/mCherry and luciferase/GFP) that were used for subcloning and in vivo (BLI) experiments. The authors thank Stephanie Zug, Dennis Haupt, Renu Karthika Murugesan and Petra Lehnert for excellent technical assistance.
Disclosure of potential conflicts of interest
The authors have no disclosures and no conflicts of interests related to the present study.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Siegel RL, Miller KD, Jemal A.. Cancer statistics. CA Cancer J Clin. 2018;68:7–12. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Mariotto AB, Etzioni R, Hurlbert M, Penberthy L, Mayer M. Estimation of the number of women living with metastatic breast cancer in the United States. Cancer Epidemiol Biomarkers Prev. 2017;26:809–815. doi: 10.1158/1055-9965.EPI-16-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hosseini H, Obradovic MM, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, Nanduri LK, Werno C, Ehrl C, Maneck M, et al. Early dissemination seeds metastasis in breast cancer. Nature. 2016;540. doi: 10.1038/nature20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, Avivar-Valderas A, Nagi C, Girnius N, Davis RJ, et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature. 2016;540. doi: 10.1038/nature20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapiro CL, Recht A. Side effects of adjuvant treatment of breast cancer. N Engl J Med. 2001;344:1997–2008. doi: 10.1056/NEJM200106283442607. [DOI] [PubMed] [Google Scholar]
- 6.Asselain B, Barlow W, Bartlett J, Bergh J, Bergsten-Nordström E, Bliss J, Boccardo F, Boddington C, Bogaerts J, Bonadonna G. Early Breast Cancer Trialists’ Collaborative G . Long-term outcomes for neoadjuvant versus adjuvant chemotherapy in early breast cancer: meta-analysis of individual patient data from ten randomised trials. Lancet Oncol. 2018;19:27–39. doi: 10.1016/S1470-2045(17)30777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun S, Kentenich C, Janni W, Hepp F, de Waal J, Willgeroth F, Sommer H, Pantel K. Lack of effect of adjuvant chemotherapy on the elimination of single dormant tumor cells in bone marrow of high-risk breast cancer patients. J Clin Oncol. 2000;18:80–86. doi: 10.1200/JCO.2000.18.1.80. [DOI] [PubMed] [Google Scholar]
- 8.Pantel K, Schlimok G, Braun S, Kutter D, Lindemann F, Schaller G, Funke I, Izbicki JR, Riethmuller G. Differential expression of proliferation-associated molecules in individual micrometastatic carcinoma cells. J Natl Cancer Inst. 1993;85:1419–1424. doi: 10.1093/jnci/85.17.1419. [DOI] [PubMed] [Google Scholar]
- 9.Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, Shor RE, Goddard ET, Dai J, Schweitzer EM, et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol. 2019;21:238–250. doi: 10.1038/s41556-018-0267-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 11.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69:4116–4124. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 14.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 16.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, Guerra R, Sahin AA, Marini FC, Hortobagyi G, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest. 2012;122:2066–2078. doi: 10.1172/JCI59735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarkar TR, Battula VL, Werden SJ, Vijay GV, Ramirez-Pena EQ, Taube JH, Chang JT, Miura N, Porter W, Sphyris N, et al. GD3 synthase regulates epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene. 2015;34:2958–2967. doi: 10.1038/onc.2014.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang YJ, Ding Y, Levery SB, Lobaton M, Handa K, Hakomori SI. Differential expression profiles of glycosphingolipids in human breast cancer stem cells vs. cancer non-stem cells. Proc Natl Acad Sci U S A. 2013;110:4968–4973. doi: 10.1073/pnas.1302825110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orsi G, Barbolini M, Ficarra G, Tazzioli G, Manni P, Petrachi T, Mastrolia I, Orvieto E, Spano C, Prapa M, et al. GD2 expression in breast cancer. Oncotarget. 2017;8:31592–31600. doi: 10.18632/oncotarget.16363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed M, Cheung NK. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett. 2014;588:288–297. doi: 10.1016/j.febslet.2013.11.030. [DOI] [PubMed] [Google Scholar]
- 22.Handgretinger R, Anderson K, Lang P, Dopfer R, Klingebiel T, Schrappe M, Reuland P, Gillies SD, Reisfeld RA, Niethammer D, et al. A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. Eur J Cancer. 1995;31A:261–267. doi: 10.1016/0959-8049(94)00413-Y. [DOI] [PubMed] [Google Scholar]
- 23.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vick B, Rothenberg M, Sandhofer N, Carlet M, Finkenzeller C, Krupka C, Grunert M, Trumpp A, Corbacioglu S, Ebinger M, et al. An advanced preclinical mouse model for acute myeloid leukemia using patients’ cells of various genetic subgroups and in vivo bioluminescence imaging. PLoS One. 2015;10:e0120925. doi: 10.1371/journal.pone.0120925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puchalapalli M, Zeng X, Mu L, Anderson A, Hix Glickman L, Zhang M, Sayyad MR, Mosticone Wangensteen S, Clevenger CV, Koblinski JE, et al. NSG mice provide a better spontaneous model of breast cancer metastasis than athymic (Nude) mice. PLoS One. 2016;11:e0163521. doi: 10.1371/journal.pone.0163521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillies SD, Lo KM, Wesolowski J. High-level expression of chimeric antibodies using adapted cDNA variable region cassettes. J Immunol Methods. 1989;125:191–202. doi: 10.1016/0022-1759(89)90093-8. [DOI] [PubMed] [Google Scholar]
- 30.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai X, Cheng H, Bai Z, Li J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J Cancer. 2017;8:3131–3141. doi: 10.7150/jca.18457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 35.Iorns E, Drews-Elger K, Ward TM, Dean S, Clarke J, Berry D, Ashry DE, Lippman M. A new mouse model for the study of human breast cancer metastasis. PLoS One. 2012;7:e47995. doi: 10.1371/journal.pone.0047995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]
- 37.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107:15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, Thiery JP. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med. 2014;6:1279–1293. doi: 10.15252/emmm.201404208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hersey P, Jamal O, Henderson C, Zardawi I, D’Alessandro G. Expression of the gangliosides GM3, GD3 and GD2 in tissue sections of normal skin, naevi, primary and metastatic melanoma. Int J Cancer. 1988;41:336–343. doi: 10.1002/(ISSN)1097-0215. [DOI] [PubMed] [Google Scholar]
- 40.Roth M, Linkowski M, Tarim J, Piperdi S, Sowers R, Geller D, Gill J, Gorlick R. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer. 2014;120:548–554. doi: 10.1002/cncr.28461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vantaku V, Donepudi SR, Ambati CR, Jin F, Putluri V, Nguyen K, Rajapakshe K, Coarfa C, Battula VL, Lotan Y, et al. Expression of ganglioside GD2, reprogram the lipid metabolism and EMT phenotype in bladder cancer. Oncotarget. 2017;8:95620–95631. doi: 10.18632/oncotarget.21038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida S, Fukumoto S, Kawaguchi H, Sato S, Ueda R, Furukawa K. Ganglioside G(D2) in small cell lung cancer cell lines: enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 2001;61:4244–4252. [PubMed] [Google Scholar]
- 43.Shibuya H, Hamamura K, Hotta H, Matsumoto Y, Nishida Y, Hattori H, Furukawa K, Ueda M, Furukawa K. Enhancement of malignant properties of human osteosarcoma cells with disialyl gangliosides GD2/GD3. Cancer Sci. 2012;103:1656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J, Introna M. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. 2011;118:3301–3310. doi: 10.1182/blood-2011-02-336321. [DOI] [PubMed] [Google Scholar]
- 45.Ladenstein R, Potschger U, Valteau-Couanet D, Luksch R, Castel V, Yaniv I, Laureys G, Brock P, Michon JM, Owens C, et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1617–1629. doi: 10.1016/S1470-2045(18)30578-3. [DOI] [PubMed] [Google Scholar]
- 46.Sorkin LS, Otto M, Baldwin WM 3rd, Vail E, Gillies SD, Handgretinger R, Barfield RC, Yu HM, Yu AL. Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain. 2010;149:135–142. doi: 10.1016/j.pain.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, Barrett DM, Grupp SA, Milone MC. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. 2018;6:36–46. doi: 10.1158/2326-6066.CIR-17-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Majzner RG, Weber EW, Lynn RC, Xu P, Mackall CL. Neurotoxicity associated with a high-affinity GD2 CAR-letter. Cancer Immunol Res. 2018;6:494–495. doi: 10.1158/2326-6066.CIR-18-0089. [DOI] [PubMed] [Google Scholar]
- 49.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. 2017;25:2214–2224. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin Cancer Res. 2019. [DOI] [PubMed] [Google Scholar]
- 53.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 54.von Minckwitz G, Huang CS, Mano MS, Loibl S, Mamounas EP, Untch M, Wolmark N, Rastogi P, Schneeweiss A, Redondo A, et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N Engl J Med. 2019;380:617–628. doi: 10.1056/NEJMoa1814017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.