

ABSTRACT
Oncogenic KRAS-mutant lung cancers remain treatment refractory. A better understanding of the immune response of KRAS-mutant lung cancers is required to facilitate the development of potential therapeutic strategies. Regulatory T cells (Tregs) are a subset of immune cells that promote tumor progression through suppressing anti-tumor immune response. Here, we used KrasG12D lung cancer mice to examine the characteristics of tumor-infiltrating Tregs. In tumor-bearing animals, Tregs are increased during tumor progression. Of note, a majority of Tregs that localized in lung tumors of Kras-mutant mice expressed ST2, a receptor for IL-33, which are different from Tregs in secondary lymphoid organs. To investigate the function of local Tregs influencing immune response in primary lung tumor development, we used anti-ST2 antibody to deplete Tregs in lung tumors of Kras-mutant mice. Treatment of Kras-mutant mice with anti-ST2 antibody resulted in depletion of activated Tregs in lung tumor while leaving Tregs in secondary lymphoid organs intact. Also, localized Tregs depletion led to a significant reduction in lung tumor burden. Immune response after Tregs depletion in tumors showed restoration of NK cell activity and enhanced Th1 activity, with increased CD8 cytotoxic T cell response. In addition, we found that the M2 macrophage signature in lung tumors was suppressed upon Tregs depletion, accompanied by upregulation of surface expression of MHC-II molecules and reduced expression of Arg1, Mmp12, Cxcl2, and Chi3l3. These data suggest that therapeutic strategies targeting activated Tregs in lung cancer have the potential to restrain tumor progression by enhancing anti-tumor immunity.
KEYWORDS: Lung cancer, Kras, ST2, Tregs
Introduction
Lung cancer is the leading cause of cancer-related deaths, with a 5-year average survival of 15%.1 Lung adenocarcinomas are stratified on the basis of driver mutations such as those in KRAS, epidermal growth factor receptor (EGFR), and anaplastic lymphoma receptor tyrosine kinase (ALK) genes.2 Activated KRAS mutations have been identified in approximately 25% of human lung adenocarcinomas that are primarily associated with smoking.2 Patients with KRAS-mutated lung adenocarcinoma have the poorest overall survival among the subtypes of lung cancer, partly because EGFR and ALK adenocarcinomas are treated with targeted therapies in addition to standard chemotherapy. Still, there is an urgent need to develop more efficacious targeted treatments.
Recent advancements in immunotherapy provide new waves of targeted cancer therapy. The development and testing of new agents using an appropriate preclinical cancer model is critical. Genetically engineered mouse (GEM) models of spontaneous cancer offer an advantage since pathophysiology of tumor growth recapitulates that of human cancer and allows activation of the host immune system against endogenously arising tumor. We have thus used a mouse model of lung cancer, in which an oncogenic form of Kras (KrasG12D) is restrictedly expressed in lung epithelial cells via Clara cell secretory protein (CCSP)cre. In this model, we have shown that Th17 cells and Tregs were enriched in tumor tissues and that Th17 cells mediate a protumorigenic role.3 Since Tregs are associated with hampering anti-tumor immunity during tumor development, a therapeutic approach targeting Tregs is of interest.
Accumulation of Tregs in the early and late stages of human lung adenocarcinoma has been described by others.4,5 Tregs exist as multiple subsets that efficiently suppress respective subsets of effector Th cells and maintain tissue integrity. The function of Tregs that have specific cellular and molecular characteristics seems to be affected by local cues.6 Recent studies using spontaneous cancer animal models demonstrated that short-term depletion of Tregs is effective in reducing tumor burden.7–9 However, complete depletion of Tregs leads to lethal autoimmunity, whereas Tregs-targeted therapies, such as anti-CD25 Ab, can also deplete activated effector T cells due to low specificity.10,11 Therefore, an effective strategy to promote anti-tumor immunity by localized Tregs depletion or preferential targeting of immune suppressive Treg subsets is of interest.
In this study, we examined Tregs signatures in an attempt to ablate tumor-infiltrating Tregs locally. We found that ST2 expressing Tregs are highly enriched in tumor tissues during tumor progression. ST2 is a receptor for IL-33, an IL-1 family member, and has been described in other studies as a signature of activated and tissue-associated Tregs in adipose tissue, intestine, and muscle.12–14 In a pathogenic setting of influenza infection, ST2 expressing Tregs have been shown to be a key mediator maintaining tissue integrity.15 Human lung adenocarcinoma samples expressed increased levels of IL-33,16 suggesting that ST2 expressing Tregs could expand in lung cancer. In this study, we demonstrated that the depletion of locally expanded Tregs using anti-ST2 antibody reduced tumor burden associated with enhanced anti-tumor innate and adaptive immunity. We also showed that lung tumor-associated Tregs promote the M2 phenotype of alveolar macrophages. Therefore, this study suggests that local Tregs response during lung adenocarcinoma can be controlled by ST2 targeting.
Materials and methods
Mice
Mice were maintained in a specific pathogen-free facility, and all animal experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee at MD Anderson Cancer Centre. KrasG12D x CCSPCre mice were described previously.3 C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA).
Treatment with anti-ST2 antibody
For lung infiltrating Tregs depletion, 12-week-old KrasG12D mice were injected i.p. with 150 µg of anti-ST2 antibody (R&D Systems, clone 245707) or isotype control rat Ig (BioXcell) 2 times per week for 3 weeks and euthanized for analysis on week 15.
Assessment of lung tumor burden
Bronchoalveolar lavage fluid (BALF) was obtained by collecting 1 ml PBS through a tracheostomy cannula after euthanization. Total lung weights were measured, and the lungs were prepared for histologic analysis first by perfusion with PBS, and then by inflation with 10% phosphate-buffered formalin (pH 7. 4). BALF was centrifuged at 1,250 × g for 10 min, and the cells were subjected to FACS analysis.
Histological analyses and tumor area quantification data
Sections (4 μm) of fixed tissues were stained with H&E according to standard procedures. Tumor area was quantitated as the total area of four lobes of lung presenting lesions in KrasG12D mice using Image J software.
Isolation of lung resident mononuclear cells
Lungs were harvested after perfusion with PBS. Lungs were first inflated with 0.1 mg/ml collagenase IV for 30 min at 37°C. Single-cell suspensions were prepared by mechanical dissociation of lung tissue through a 70-micron nylon mesh. Lung cells were suspended in PBS and layered on LSM Lymphocyte Separation (Medium MP Biomedical). Cells were centrifuged at room temperature at 900 x g for 20 min. Mononuclear cells were harvested from the gradient interphase. These cells were stimulated with PMA and Ionomycin in the presence of Brefeldin A for intracellular cytokine staining. The resultant isolated cells were subjected to FACS analysis. The following antibodies were used to detect macrophages, Tregs, Th cells and CD8 T cells, and innate lymphoid cells including ILC2s. FACS antibodies were purchased from BioLegend, eBioscience, and BD Biosciences: CD45.2 (104), CD44 (IM7), TCRγδ (GL3), CD25 (PC61), CD206 (C068C2), CD3 (145-2C11), CD4 (RM4–5), TCRβ (H57–597), NK1.1 (PK136), Foxp3 (FJK-16 s), CD11c (N418), CD11b (M1/70), Siglec-F (E50–2440), CD8a (53–6.7), ST2 (RMST2-2), PD1 (J43), MHC-II (M5/114.15.2), ICOS (7E.17G9), CD39 (24DMS1), OX40 (OX-86), CCR6 (CD196), Helios (22F6) CD103 (M290), GITR (DTA-1), GATA3 (16E10A23), RORγt (Q31-378), GRB (GB11), IFNγ (XMG1.2), IL-17 (eBio17B7). Cells were analyzed with the use of the BD FACSAria instrument (Becton Dickinson) and FlowJo software (Tree Star).
RT-PCR
Tissue samples for RNA analysis were collected and mRNA was isolated by homogenization in TRIzol (Invitrogen) using a tissue homogenizer. BALF cells were directly placed in TRIzol. RNA was reverse transcribed by using an Affinity Script Reverse Transcriptase Kit (Invitrogen), according to the manufacturer’s instructions. Transcripts were measured by Q-RTPCR on a Real-Time PCR system (Biorad). All of the primers used in Q-RTPCR are shown in Table S1.
Tumor models
C57BL/6 mice were inoculated intravenously with 1.0 × 105 B16F10, a murine melanoma cell line. Mice were euthanized 2 weeks later, and mononuclear cells from metastasized tumor lungs were analyzed for Tregs. For the ectopic transplantation model, 0.5 × 106 B16F10 cells were subcutaneously injected into B6 mice. Mice were euthanized when the tumor diameters reached 10 mm, and tumor-infiltrating T cells were analyzed for Tregs. Lamina propria T cells were isolated from colons of normal C57BL/6J or C57BL/6J-ApcMin heterozygous mice, which develop multiple intestinal tumors.
Allergic lung inflammation
To induce allergic lung inflammation, we adopted an animal model of proteinase-induced allergic asthma induced by repeated intranasal challenge with fungal proteinase allergens, as previously described.17 In brief, mice were anesthetized with isoflurane and challenged intranasally with 7 μg of proteinase from Aspergillus oryzae (Sigma-Aldrich) in 50 μL of PBS every other day for 6 times (days 0, 2, 4, 6, 8, and 10). A day after the last challenge, lungs were collected and mononuclear cells from lungs were stained for Tregs.
Statistical analysis
Statistical comparisons were performed by using a two-tailed unpaired Student t-test using GraphPad Prism 7. P values less than 0.05 were considered statistically significant; *p < .05, **p < .01, ***p < .001.
Results
Tregs accumulate in KrasG12DCCSPcre lung tumors and have an activated phenotype expressing ST2
We have previously shown that Th17 cells and Tregs increase concurrently in the lungs of KrasG12D x CCSPcre mice.3 As lung tumors progressed with age, we found that Tregs frequency among CD4 T cells also increased over time (Figure 1a). To better understand the characteristics of Tregs that develop upon oncogene activation and local cues driven by the lung microenvironment, we examined the Tregs phenotype by using activation markers. Tregs isolated from lung tumor of KrasG12D mice highly expressed well-described activation markers such as OX40, GITR, ICOS, and Helios; inhibitory markers such as CD39 and PD-1; and migratory markers CCR6 and CD103, compared with those in WT control mice (Figure 1b). We found that Tregs in lung tumors of KrasG12D mice also expressed high levels of CD25 and ST2, a receptor for alarmin IL-33 (Figure 2a).
Figure 1.
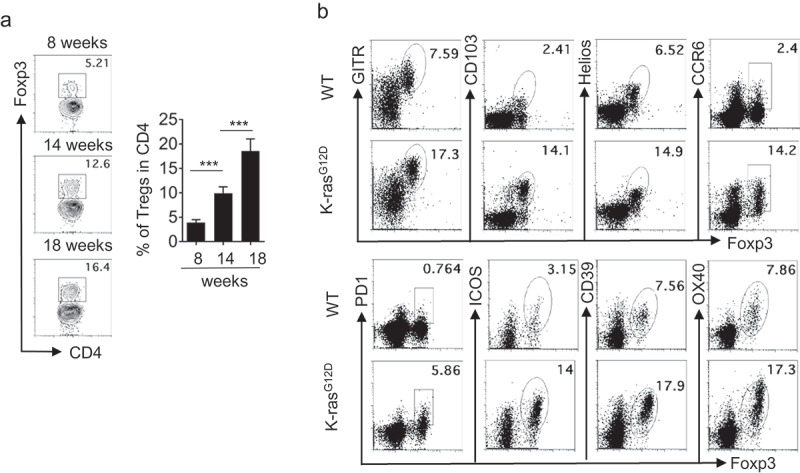
Characterization of tumor-infiltrated Tregs in Kras-mutated lung cancer. Freshly isolated lung mononuclear cells from WT or KrasG12D mice were analyzed by flow cytometry. (a) Representative flow cytometric analysis of Tregs during tumor progression (n = 3–6 mice per group). Data are mean and s.d. (b) Representative flow cytometric analysis of Tregs.
Figure 2.
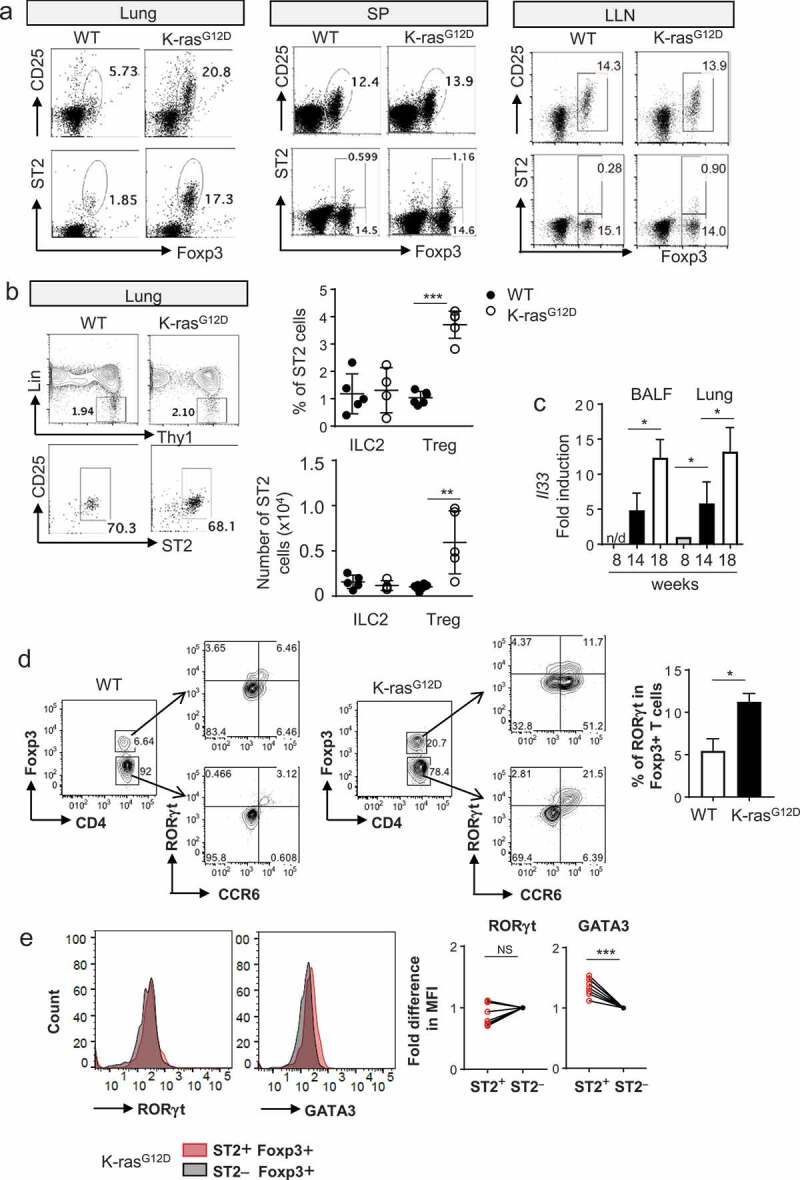
ST2 expressing CD4+Foxp3+ T cells were increased in Kras-mutated lung cancer. (a) Representative flow cytometric analysis of Tregs from tumor lung, spleen (SP), and lung-draining lymph node (LLN). (b) Representative flow cytometric analysis of type 2 innate lymphoid cells (ILC2s) from lung tumor. Percentage and absolute number of ILC2s in tumor. Each dot represents an individual mouse (n = 4–8 mice per group). (c) mRNA expression of IL-33 in tumor lung and BALF during tumor progression at weeks 8, 14, and 18 (n = 3–6 mice per group). n/d, not detected. (d) Representative flow cytometric analysis and percentage of RORγt expressing Tregs from normal lung and tumor tissues. (e) Correlation of ST2 expression with RORγt and GATA3 (n = 8). Data are mean and s.d.
The lungs are also known to harbor another ST2 expressing cell population, type 2 innate lymphoid cells (ILC2s). Lung ILC2s are defined as a population that lacks expression of lineage markers (Lin−) and has high expression of CD90 (Thy-1), IL-7R, CD25, CD44, ICOS, GATA-3, and ST2. We found that the frequency of ILC2s (Lin−CD90+ST2+CD25+) among total CD45+ cells remained constant in lung parenchyma after the tumor developed, and the absolute number of ILC2s was also outnumbered by that of ST2+ Tregs in lungs of KrasG12D mice (Figure 2b). We did not see any measurable expression of ST2 in other Th cells. ST2 expressing T cells in lung tissues were increased as the tumors grew but barely detectable in the lung-draining lymph node or the spleen of tumor-bearing mice (Figure 2a).
Increased levels of ST2 expressing Tregs in Kras-mutant lung raised the question of whether IL-33 is induced in lung tumor as well. IL-33 is known to be produced mainly by lung epithelium and, in some cases, by lung macrophages during allergic asthma.18,19 A few studies have shown an increase in IL-33 in human lung adenocarcinoma16,20 and in the Kras-activated oncogene model.21 We found that IL-33 expression was increased in total lung cells as the tumors grew. We also found that IL-33 was increased in BALF cells (Figure 2c).
RORγt-positive Tregs were also increased in KrasG12D lung tumor (Figure 2d). ST2 expression of Tregs is positively correlated with GATA3 expression in Tregs, as shown in another study,13 but not with RORγt expression, as previously demonstrated (Figure 2e).22 To examine whether ST2 expression in Tregs is confined to an oncogene-driven lung cancer model, we examined ST2 expressing Tregs in various inflammatory settings. During asthma induction, ST2 Tregs were marginally induced but outnumbered by the ST2 expressing CD4+Foxp3¯ effector T cells. In a B16 melanoma lung metastasis model, tumor-infiltrated cells contained some ST2 Tregs, but not as many as those in KrasG12D mice. However, tumor-infiltrated Tregs upon ectopic transplantation of B16 melanoma cells expressed little, if any, ST2, suggesting that the local environment in the lung contributed to ST2 induction in Tregs (Figure 3a,b).
Figure 3.
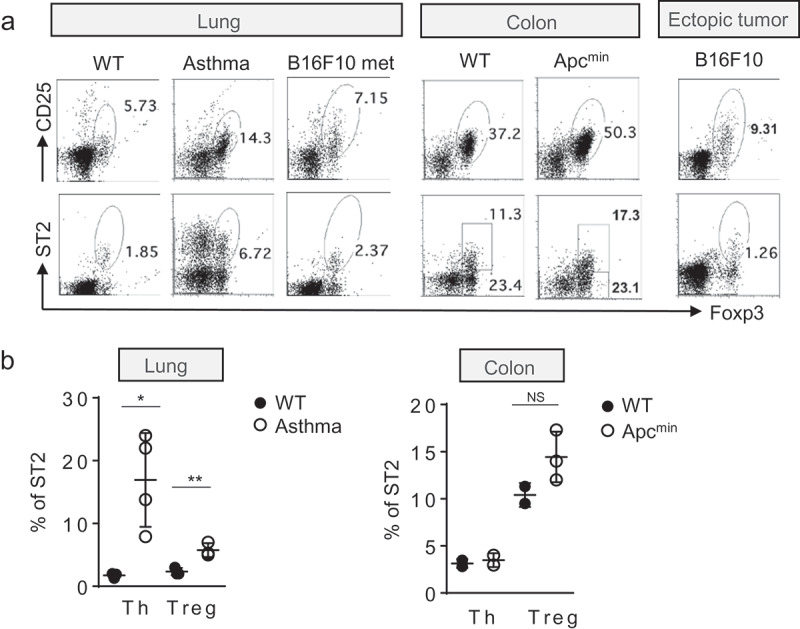
ST2 and CD25 staining of CD4+Foxp3+ T cells from various mouse tumor models and lung inflammation. (a) Representative flow cytometric analysis of Tregs from tumor tissues or lung during allergic inflammation. (b) Percentage of ST2 positive cells in Th (T helper cells) and Tregs. Each dot represents an individual mouse (n = 3–4 mice per group). NS, no significant difference. Data are mean and s.d.
Formation of transplantable tumors likely bypasses the early steps of tumorigenesis, so the immune response may be different from that of the oncogene-induced tumor model. Therefore, we examined another spontaneous model of cancer, Apcmin mouse, an animal model of colorectal cancer. ST2 positive Tregs were, however, resident in normal colon, as reported before,13 and remained at a similar level in the tumor-bearing mouse (Figure 3a,b). Therefore, our data demonstrated an oncogene-driven, lung cancer-specific expansion of ST2 positive Tregs.
Anti- ST2 antibody treatment leads to effective depletion of tumor-infiltrated tregs
Selective expression of ST2 in Tregs in the lungs of Kras-mutant animals presented a unique opportunity to suppress or deplete the cells in the KrasG12D model and to investigate the function of tumor-infiltrated Tregs in spontaneous lung cancer. Removal of Tregs with the use of CD25 antibody23 or with Foxp3-DTR strain7,9 has been considered previously in models of spontaneous cancer. However, systemic Tregs depletion can cause an influx of effector Th cells, which could result in autoimmune disease.
Since ST2 expression in Tregs is confined to lung tumors and almost absent in secondary lymphoid organs, we asked whether tissue-resident Tregs can be inhibited specifically and whether local Tregs in this model are active participants of tumor immunosurveillance. To address this question, we treated tumor-bearing animals for 3 weeks with anti-ST2 antibody. We observed effective depletion of ST2 expressing Tregs in the lungs (Figure 4a).
Figure 4.
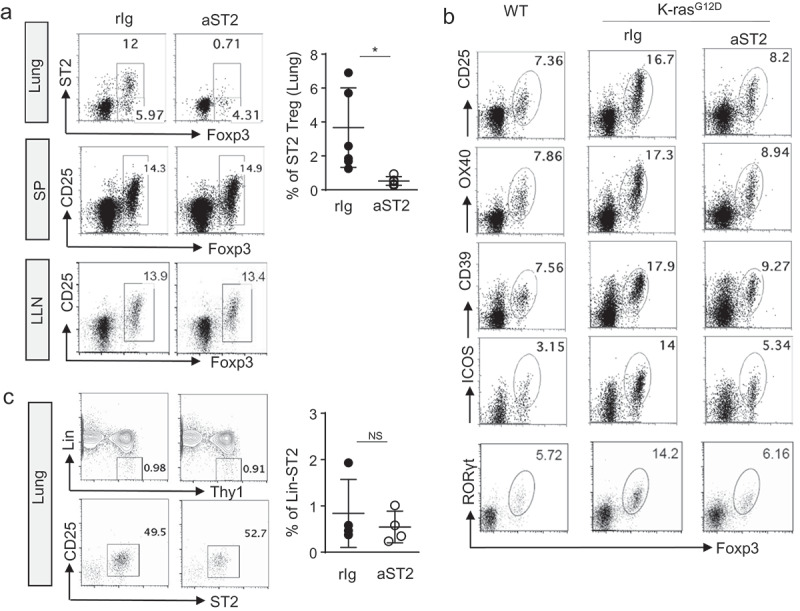
Anti-ST2 antibody is effective in depletion of activated Tregs in lung tumor. (a) Representative flow cytometric analysis of Tregs from lung tumor, SP, and LLN after anti-ST2 antibody treatment. (b) Representative flow cytometric analysis of surface expression of activation markers and RORγt expression of Tregs from tumor lung. (c) Representative flow cytometric analysis of ILC2s from lung tumor. Each dot represents an individual mouse (n = 4–6 mice per group). Error bars represent s.d. from average. rIg; rat IgG
One concern for transient Tregs depletion is a compensatory increase in the remaining populations of Tregs. However, we did not see the increased occurrence of any other subsets of Tregs such as RORγt+ Tregs upon the use of the ST2 depletion strategy in lung tumor. Since RORγt+ Tregs were also reduced after the treatment, general inhibition of activated Tregs infiltrated in lung tumor may have occurred (Figure 4b). Whereas effective depletion of ST2 expressing Tregs was observed in lung tumor, we did not see any differences in the number or activation status of Tregs in terms of CD25 expression from the spleen and lung-draining lymph node after the treatment (Figure 4a).
These data indicate that anti-ST2 antibody is effective in selectively depleting tumor-infiltrating activated Tregs in lung tumor without a compensatory increase in other Tregs subsets in the lung while preserving Tregs in secondary lymphoid organs. We also found that ST2 expressing ILC2s remained intact after anti-ST2 antibody treatment (Figure 4c). Since ILC2s are not proliferating actively in this model, the inhibitory effects of anti-ST2 antibody on ILC2s in KrasG12D mice could be minimal.
Anti-ST2 antibody treatment results in suppression of tumor development
To examine the effects of local Tregs ablation in tumor burden, we evaluated lung weights and lung tumor area after the antibody treatment. Anti-ST2 treatment was sufficient to achieve a significant reduction in tumor burden as shown in Figure 5a,b. The increase in total tumor volume due to the proliferation of cancer cells was reduced by the antibody treatment (Figure 5c). Histological evaluation of whole-lung lobes and high-magnification histological evaluation of hematoxylin and eosin (H&E) lung section also revealed reduced tumor area after the antibody treatment (Figure 5d).
Figure 5.
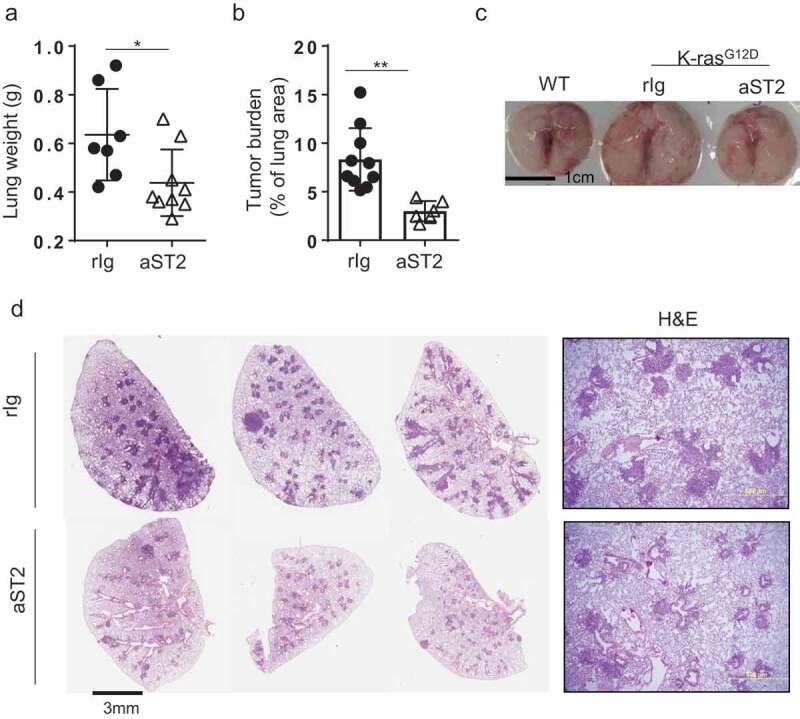
Depletion of ST2 positive Tregs resulted in reduction in lung tumor. (a) Lung weight (rIg; n = 7, anti-ST2; n = 9) and (b) tumor burden, calculated by percentage of lung area (rIg; n = 10, anti-ST2; n = 6). (c) Whole-lung lobes are shown. (d) H&E images of whole lung lobes (left panel) and high-magnification images (right panel). Three representative images per group are shown for whole lung lobes. Error bars represent s.d. from average.
Tregs ablation promotes innate and adaptive anti-tumor immunity
To understand the mechanisms underpinning the observed role of Tregs in tumor development in Kras-mutant lung cancer, we evaluated the phenotype of innate and adaptive immune cells after treatment. Previous studies have shown detailed analyses of immune components of the Kras-mutant model of lung cancer.23,24 To determine whether lymphocytes with cytotoxic potential or anti-tumor immunity are increased upon Tregs depletion by anti-ST2 antibody treatment, we analyzed the lymphocyte population in lung tumors. CD3¯NK1.1 expressing conventional natural killer (NK) cells represented the largest population of granzyme B (GRB) and IFNγ production in the normal lung (Figure 6a). Suppression of NK cells in human lung adenocarcinoma was noted by others,4 and our study is consistent with previous observations in Kras-mutant mice.24
Figure 6.
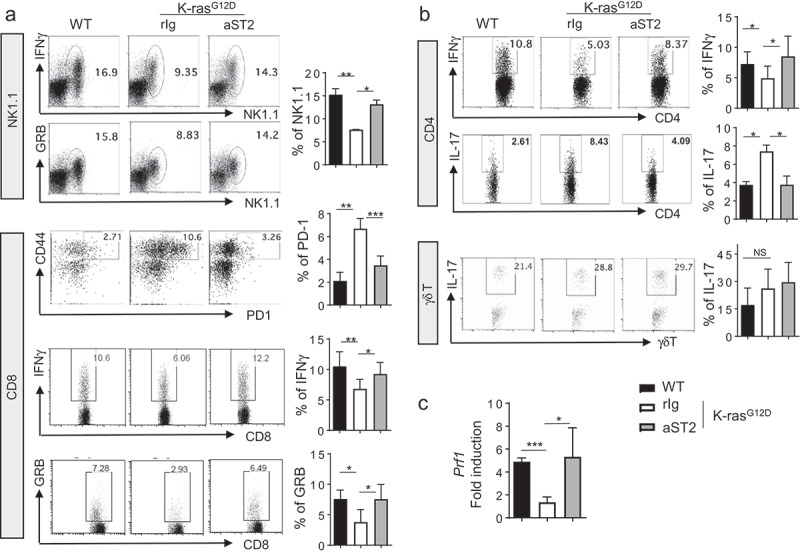
Anti-tumor immune responses are increased after anti-ST2 antibody treatment. (a) Cytokine production of NK1.1 cells and PD-1 expression and cytokine production of CD8 T cells were analyzed by flow cytometry. (b) Flow cytometric analysis of cytokine production by CD4 T cells and γδ T cells. (c) mRNA expression of perforin from mononuclear cells from the whole lung. Error bars represent s.d. from average.
Upon Tregs depletion, IFNγ- or GRB-producing NK cells were significantly increased (Figure 6a). In addition to NK cells, CD8 T cells mediate a cytotoxic T cell response in the tumor environment. In KrasG12D mice, tumor-residing CD8 T cells expressed increased levels of PD-1 and reduced levels of IFNγ compared with those in normal lung. We found that the number of endogenous CD8 T cells was not dramatically altered by Tregs depletion (data not shown). Instead, we found that the percentage of PD-1+CD44hi exhausted CD8 T cells was considerably reduced after treatment (Figure 6a). This result suggests that exhausted CD8 T cells in lung tumor could have been rescued by Tregs ablation.
Consistent with the reduced PD-1 expression, CD8 T cells expressed more IFNγ and GRB after Tregs depletion. Therefore, reduced tumor burden after treatment might be associated with increased activation of CD8 T cells. CD4 T cells also expressed more IFNγ, whereas IL-17 producing CD4 T cells were reduced upon Tregs depletion. However, IL-17 producing γδ T cell numbers remained similar after treatment (Figure 6b). Also, fold induction of Prf1 (encoding perforin) from tumor lungs was increased by the antibody treatment (Figure 6c). Therefore, our study demonstrated that depletion of activated ST2 Tregs specifically in lung tumor of KrasG12D mice resulted in the restoration of NK and CD8 T cell activity and Th1 activity but the reduction of Th17 cell activity.
Tregs ablation resulted in suppression of M2 type of macrophages
Alveolar macrophages constitute a major cell type of the tumor microenvironment in the KrasG12D model of lung cancer.25 The number of alveolar macrophages is associated with the degree of tumor progression, and M2 features of alveolar macrophages are increased during tumor progression.25,26 Therefore, we examined whether changes in the absolute number or the phenotype of alveolar macrophages occur upon depletion of tumor-associated Tregs.
Tregs ablation switched the activation status of BALF macrophages from an immunosuppressive M2-like major histocompatibility complex class II (MHC-II)low to a more inflammatory M1-like (MHC-II+) state (Figure 7a). Anti-ST2 antibody treatment resulted in a reduction of alveolar macrophages (Siglec-F+CD11c+CD11blow) in tumor lung (Figure 7b). We further tested RNA expression of M1 and M2 markers in the tumors after treatment. The expression of prototypic M2 markers (Arg1, Chi3l3) was reduced, whereas expression of the M1 marker (Nos2) remained similar after the treatment. Other macrophage-derived molecules, Mmp12 and Cxcl2, were reduced in the treated tumors (Figure 7c). These results indicate that Tregs expanded upon oncogene activation in the lungs promote M2 polarization of alveolar macrophages associated with a protumorigenic environment.
Figure 7.
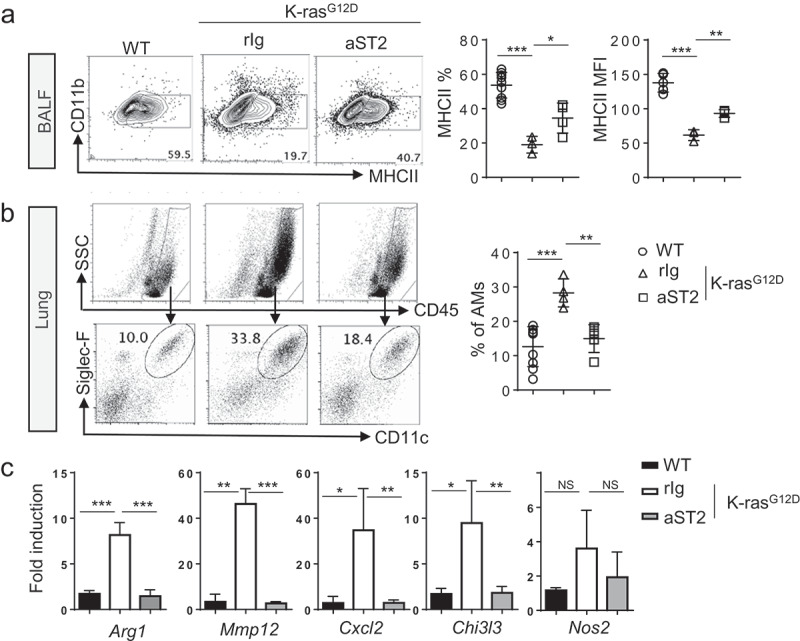
Macrophages are reduced upon anti-ST2 antibody treatment. (a) Flow cytometric analysis of BALF alveolar macrophages (AMs). Representative FACS plot and MHC-II+ cell frequency and mean fluorescence intensity (MFI) values of MHC-II are shown. BALF macrophages were gated first by FSChiSSChiCD45+ and gated on CD11b−GR1−F4/80+CD11c+ cells. (b) Representative flow cytometric analysis and frequency of lung AMs, gated on Siglec-F+CD11c+ among CD45+ lung cells in control or KrasG12D mice. Each dot represents an individual mouse (n = 4–9 per group). (c) mRNA expression of indicated genes in total lung MNCs after the anti-ST2 treatment (n = 3–4 per group). Error bars represent s.d. from average.
Discussion
Oncogenic KRAS mutations are thought to be causal drivers in ∼30% of non-small cell lung carcinoma (NSCLC) and to correlate with poor prognosis without targeted therapy.27,28 This prompted us to investigate distinctive immune components of Kras-mutated lung tumor that can be targeted for therapeutic purposes. We identified a unique signature of Treg-associated molecule, ST2, in Kras-mutated lung tumor. Subsequently, we provided evidence that selective depletion of lung tumor-infiltrated Tregs can be achieved by using anti-ST2 antibody, and the treatment resulted in reduced lung tumor burden. In addition, we demonstrated that Tregs in Kras-mutant lung tumors inhibit an immunosurveillance mechanism by promoting M2 polarization of alveolar macrophages and by retrieving compromised activity of NK cells, CD8 cytotoxic T cells, and CD4 Th1 cells.
The therapeutic potential of suppression or depletion of Tregs during tumor development has been evaluated by others.29 Prevailing technical difficulties have been ineffective depletion of Tregs and a compensatory increase in subsets of Tregs. In addition, prolonged depletion of Tregs could result in increased systemic effector T cell response, leading to undesired inflammation. Paradoxically, systemic activation of immune cells might be needed for a long-lasting immune response.30 Therefore, depletion of local Tregs or transient suppression of Tregs should be further investigated for their therapeutic potential in combination with systemic immunomodulation drugs.
Our data indicated that immune cells in the local lung microenvironment are modulated upon Tregs depletion. Lung tumor-infiltrated Tregs may produce cytokines that promote tumor-associated macrophages. Also, restoration of NK cells in lung tumors upon depletion of Tregs was observed. NK cells and Tregs may compete for IL-2 consumption,31 and TGFβ has been shown to suppress NK cells in mucosal tissues.32 Whether direct interaction between tumor-infiltrated Tregs and specific immune cell subsets is required for Tregs regulation of anti-tumor immunity remains to be elucidated.
As with many solid tumor malignancies, NSCLC is a heterogeneous disease comprising multiple unique histologic subtypes that harbor distinct molecular signatures. Since mutational signatures of NSCLC subtypes differ, immune cell composition and activation status could vary as well,2,33 demonstrating the challenges encountered in searching for an effective targeted therapy.
Several studies have examined the transcriptional landscape of Tregs obtained from human cancer tissues. Although breast cancer and colon cancer Tregs express ST2,5,34 Tregs in lung cancer did not have increased ST2 expression.5 A recent single-cell sequencing study of TILs from a human lung adenocarcinoma patient data set, however, indicated that ST2 expression is associated with Foxp3+ T cells exclusively, but the expression level seemed low.35 Since these studies examined ST2 expression only at the transcript level, protein expression of ST2 in human lung cancer tissues should be examined.
Another confounding factor for ST2 expression is that smoking may reduce ST2 expression.36 Since the current KrasG12D animal model represents an early stage of lung adenocarcinoma prior to metastasis, analysis of human samples in an early stage of cancer, particularly treatment-naïve patient sets or without a smoking history, could be relevant to our current study. While this manuscript was in revision, it was reported that ST2 expression in Tregs promotes an immunosuppressive environment in colorectal cancer by using animal strains of total ST2 deficiency and Tregs-specific ST2 deficiency.37 Although we sought to implement loss of function or elimination of activated Tregs in lung tumor sites with the use of anti-ST2 antibody, but not of ST2 expression in Tregs, both studies corroborate the significance of ST2 Tregs hampering anti-tumor immunity. However, we cannot exclude the possibility that anti-ST2 antibody targets cellular components other than Tregs in Kras-mutant animals and results in tumor suppression in our study.
It remains to be validated whether local Tregs depletion is effective in other preclinical models of lung cancer such as EGFR mutants or advanced model of lung adenocarcinoma accompanying metastasis. For example, it is known that the EGFR mutation-driven lung cancer model has fewer Tregs and CD4 helper T cells.24 The therapeutic window of Tregs depletion may be limited to the early stage of tumorigenesis when tumor burden is low and immune cells are actively recruited to the tumor tissues. This possibility could be explored in the future in combination with other immunotherapies or targeted therapies. A major remaining challenge is to identify biomarkers that can differentiate responders from nonresponders to improve durable response rates. Although peripheral blood profiling may not be appropriate to detect a tumor-infiltrated Tregs signature, noninvasive tools such as BALF collection could determine a suitable population of lung cancer patients.
Despite these drawbacks, an important contribution of the current study is that it helps our understanding of the immune system in the early stage of lung cancer, for which therapeutic intervention is more effective than treatment for late-stage metastasized tumors. Since targeted therapy against KRAS-mutated lung tumors is not currently available, immunotherapy in the subset of patients with this mutation could be helpful. In conclusion, we identified ST2 expressing tumor Tregs as a potential novel therapeutic target and a framework on which to design future studies to incorporate these findings in a clinical setting.
Funding Statement
This research was supported by the Department of Defense (W81XWH-16-1-0100), National Institutes of Health (R21 AI27997), and Center for Inflammation and Cancer Support Grant, The University of Texas MD Anderson Cancer Center to S.H.C.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–10. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS, Moghaddam SJ, Dong C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci U S A. 2014;111(15):5664–5669. doi: 10.1073/pnas.1319051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lavin Y, Kobayashi S, Leader A, Amir EAD, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169(4):750–765. e17. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RP, Provasi E, Sarnicola M, Panzeri I, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016;45(5):1135–1147. doi: 10.1016/j.immuni.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaudhry A, Rudensky AY. Control of inflammation by integration of environmental cues by regulatory T cells. J Clin Invest. 2013;123(3):939–944. doi: 10.1172/JCI57175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joshi NS, Akama-Garren E, Lu Y, Lee D-Y, Chang G, Li A, DuPage M, Tammela T, Kerper N, Farago A, et al. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity. 2015;43(3):579–590. doi: 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, Miranda Rota E, Dahan R, Georgiou A, Sledzinska A, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. 2017;46(4):577–586. doi: 10.1016/j.immuni.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210(11):2435–2466. doi: 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8(2):191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 11.Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, Shimizu J, Nomura T, Chiba T, Sakaguchi S, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005;202(7):885–891. doi: 10.1084/jem.20050940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, Fagarasan S, Mielke LA, Afshar-Sterle S, Masters SL, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. 2015;16(3):276–285. doi: 10.1038/ni.3085. [DOI] [PubMed] [Google Scholar]
- 13.Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, Pott J, Griseri T, Bollrath J, Hegazy AN, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513(7519):564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burzyn D, Kuswanto W, Kolodin D, Shadrach J, Cerletti M, Jang Y, Sefik E, Tan T, Wagers A, Benoist C, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155(6):1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arpaia N, Green J, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting P, Rudensky A. A distinct function of regulatory T cells in tissue protection. Cell. 2015;162(5):1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim MS, Kim E, Heo J-S, Bae D-J, Lee JUW, Lee T-H, Lee HJ, Chang HS, Park JS, Jang AS, et al. Circulating IL-33 level is associated with the progression of lung cancer. Lung Cancer. 2015;90(2):346–351. doi: 10.1016/j.lungcan.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 17.Na H, Lim H, Choi G, Kim B-K, Kim S-H, Chang Y-S, Nurieva R, Dong C, Chang SH, Chung Y, et al. Concomitant suppression of TH2 and TH17 cell responses in allergic asthma by targeting retinoic acid receptor-related orphan receptor gammat. J Allergy Clin Immunol. 2018;141(6):2061–2073. e5. doi: 10.1016/j.jaci.2017.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity. 2015;42(6):1005–1019. doi: 10.1016/j.immuni.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie ANJ, Smith DE, DeKruyff RH, Umetsu DT. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–638. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Chen Z, Bu X, Han Y, Shan S, Ren T, Song W. IL-33 signaling fuels outgrowth and metastasis of human lung cancer. Biochem Biophys Res Commun. 2016;479(3):461–468. doi: 10.1016/j.bbrc.2016.09.081. [DOI] [PubMed] [Google Scholar]
- 21.Lee S, Kang J, Cho M, Seo E, Choi H, Kim E, Kim J, Kim H, Kang GY, Kim KP, et al. Profiling of transcripts and proteins modulated by K-ras oncogene in the lung tissues of K-ras transgenic mice by omics approaches. Int J Oncol. 2009;34(1):161–172. [PubMed] [Google Scholar]
- 22.Kim BS, Lu H, Ichiyama K, Chen X, Zhang Y-B, Mistry NA, Tanaka K, Lee Y-H, Nurieva R, Zhang L, et al. Generation of RORgammat(+) Antigen-Specific T Regulatory 17 Cells from Foxp3(+) Precursors in autoimmunity. Cell Rep. 2017;21(1):195–207. doi: 10.1016/j.celrep.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganesan AP, Johansson M, Ruffell B, Beltran A, Lau J, Jablons DM, Coussens LM. Tumor-infiltrating regulatory T cells inhibit endogenous cytotoxic T cell responses to lung adenocarcinoma. J Immunol. 2013;191(4):2009–2017. doi: 10.4049/jimmunol.1301317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung cancer subtypes generate unique immune responses. J Immunol. 2016;197(11):4493–4503. doi: 10.4049/jimmunol.1600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, Tonon G, McNamara K, Marconcini LA, Hezel A, et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene. 2006;25(14):2105–2112. doi: 10.1038/sj.onc.1209237. [DOI] [PubMed] [Google Scholar]
- 26.Li Q, Anderson CD, Egilmez NK. Inhaled IL-10 suppresses lung tumorigenesis via abrogation of inflammatory macrophage-Th17 Cell Axis. J Immunol. 2018;201(9):2842–2850. doi: 10.4049/jimmunol.1800141. [DOI] [PubMed] [Google Scholar]
- 27.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, Chang MT, Ni A, Kundra R, Jonsson P, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7(6):596–609. doi: 10.1158/2159-8290.CD-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–118. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. Systemic immunity is required for effective cancer immunotherapy. Cell. 2017;168(3):487–502. e15. doi: 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gasteiger G, Hemmers S, Bos PD, Sun JC, Rudensky AY. IL-2-dependent adaptive control of NK cell homeostasis. J Exp Med. 2013;210(6):1179–1187. doi: 10.1084/jem.20122571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viel S, Marçais A, Guimaraes FSF, Loftus R, Rabilloud J, Grau M, Degouve S, Djebali S, Sanlaville A, Charrier E, et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. 2016;9(415):ra19. doi: 10.1126/scisignal.aad1884. [DOI] [PubMed] [Google Scholar]
- 33.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Kadara H, Parra ER, Canales JR, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5(8):860–877. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva E, Chudakov D, Rudensky A. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45(5):1122–1134. doi: 10.1016/j.immuni.2016.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24(7):978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 36.Kearley J, Silver J, Sanden C, Liu Z, Berlin A, White N, Mori M, Pham T-H, Ward C, Criner G, et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity. 2015;42(3):566–579. doi: 10.1016/j.immuni.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 37.Pastille E, Wasmer M-H, Adamczyk A, Vu VP, Mager LF, Phuong NNT, Palmieri V, Simillion C, Hansen W, Kasper S, et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunology. 2019;12(4):990–1003. doi: 10.1038/s41385-019-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.