

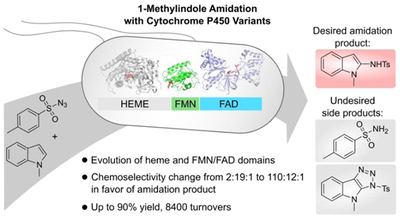
Abstract
Here we report a cytochrome P450 variant that catalyzes C2-amidation of 1-methylindoles with tosyl azide via nitrene transfer. Before evolutionary optimization the enzyme exhibited two undesired side reactivities resulting in reduction of the putative iron-nitrenoid intermediate or cycloaddition between the two substrates to form triazole products. We speculated that triazole formation was a promiscuous cycloaddition activity of the P450 heme domain, while sulfonamide formation likely arose from surplus electron transfer from the reductase domain. Directed evolution involving mutagenesis of both the heme and reductase domains delivered an enzyme providing the desired indole amidation products with up to 8400 turnovers, 90% yield, and a shift in chemoselectivity from 2:19:1 to 110:12:1 in favor of nitrene transfer over reduction or triazole formation. This work expands the substrate scope of hemoprotein nitrene transferases to heterocycles and highlights the adaptability of the P450 scaffold to solve challenging chemoselectivity problems in non-natural enzymatic catalysis.
Keywords: Biocatalysis, Cytochrome P450, Nitrene Transfer, Indole Amidation, Chemoselectivity
Graphical Abstract
One of the hallmarks of enzyme catalysis is the ability to precisely control reaction selectivity. Through finely tuned active site geometries, high levels of substrate binding specificity and preferential stabilization of transition states can be achieved, often resulting in excellent chemo-, regio-, and stereoselectivity. Furthermore, enzyme selectivity can be engineered using methods of directed evolution to access different products from the same set of substrates.1–5
We and others previously engineered native hemoproteins to become selective catalysts for synthetically valuable non-natural carbene and nitrene transfer reactions.6 Hemoprotein “nitrene transferases” catalyze intra- and intermolecular C-H amination,7–13 sulfimidation,14,15 aziridination,16 aminohydroxylation,17 and most recently highly selective intramolecular C-H amination to yield β-, γ- or δ-lactams.18
In our ongoing effort to develop enzymes for new nitrene transfer reactions and inspired by recent reports of hemoprotein-catalyzed carbene transfer for C-H functionalization of indoles,19–21 we investigated indole amidation with sulfonyl azides. Although this reaction can proceed without catalyst,22,23 in recent years Pd, Ir, Rh, and Co-based catalysts have been developed for regioselective indole amidations.24–32 Hemoproteins would offer the advantages of functioning with an earth-abundant iron cofactor in aqueous media at ambient temperatures.
We found that several cytochrome P450 variants catalyze 1-methylindole C(sp2)-H amidation with tosyl azide, delivering the C2-amidated product in low yields and turnovers. However, the enzymatic reactions yielded two additional products: tosyl sulfonamide, likely originating from over-reduction and protonation of the iron-nitrenoid intermediate,6,9,14 and a triazole likely arising from cycloaddition of the two substrates.33 Thus, we were confronted with a three-way chemoselectivity problem (Figure 1A). Building on the adaptability of the P450 scaffold for selectivity challenges,18,34 we report here a comprehensive directed evolution strategy targeting both the P450 heme and reductase domains (Figure 1B) to engineer a variant having high indole amidation activity and chemoselectivity over the undesired reduction and cyclization pathways. This work highlights the potential of enzyme engineering to achieve precise control over non-natural substrates and reaction pathways and expands the scope of enzymatic nitrene transfer to synthetically valuable heterocycles.
Figure 1: Engineering chemoselectivity in P411-catalyzed indole amidation.

(A) Proposed reaction scenarios: tosyl azide 1 can react with the P411 iron heme cofactor to form an iron-nitrenoid intermediate (i). The nitrene can react with 1-methylindole 2a to yield the desired amidation product 3a (ii) or it can be reduced to give sulfonamide product 4 (iii). Alternatively, 1 and 2a can react to give cycloaddition product 5a (iv); while this reaction is likely accelerated by the P411 active site (vide infra), the involvement of the heme cofactor remains to be determined. (B) P450-BM3 domain structure; we targeted both the heme and reductase domains for directed evolution of chemoselective formation of 3a.
We started this work by testing a range of P450 variants, expressed in Escherichia coli and utilized as whole-cell catalysts, for the amidation of 1-methylindole 2a with tosyl azide 1 (TsN3) as the nitrene precursor (Table S1). The C2 amidation product 3a was formed in very low yield by several P450 variants, with the serine-ligated P450 variant P411-CIS35 exhibiting the highest activity (2.1% yield, 100 total turnovers (TTN)). However, the predominant reaction product was tosyl sulfonamide 4. This was expected, as nonproductive over-reduction and protonation of the heme-bound nitrene yielding the sulfonamide has been a recurring problem for hemoprotein-catalyzed nitrene transfer (Figure 1A).6,9,14 More surprisingly, we observed an additional product in whole-cell reaction mixtures (Figure S1), which we identified as triazole 5a, presumably arising from cycloaddition of TsN3 and 1-methylindole.33 Confronted with this three-way chemoselectivity problem, we sought to use directed evolution to increase enzyme activity and selectivity in the P411-catalyzed formation of 3a.
Using P411-CIS as the parent (P), site-saturation mutagenesis and screening at active site positions I263 and T438, previously shown to be important for P411-catalyzed nitrene transfer,7,14,16 yielded variant P411-CIS I263Y T438S (P-YS) (Figure 2, Tables S2, S3). This variant delivered almost 3-fold higher yields of 3a, but concomitantly delivered over 7-fold higher yields of cycloaddition product 5a. This strong influence of P411 active site mutations on the yield of triazole 5a, with all other reaction parameters identical, indicated that 5a formation took place within (or close to) the P411 active site (vide infra). We assumed that mutations I263Y and T438S served to better accommodate both substrates 1 and 2a within the active site, thereby enhancing the yields of both 3a and 5a without discriminating between nitrene formation/amidation versus cycloaddition reaction pathways. We reasoned that further directed evolution should allow enzymatic discrimination of the two pathways. Indeed, subsequent screening of site-saturation mutagenesis libraries (encompassing active-site residues A82, V87, L181, E267, A268, T327 and A328; Table S2) revealed mutations A328V and A82W, which dramatically shifted the 3a:5a product ratio from 0.8:1 to over 25:1 for the desired amidation product. Lastly, mutation A268G further increased activity by ca. 1.6-fold and resulted in variant P-YSVWG, delivering 3a in 68% yield, 5730 TTN, and a 97:1 ratio of 3a to 5a (Figure 2, Figure S2). Thus, directed evolution of the P450 heme domain delivered a variant showing high catalytic indole amidation activity and excellent amidation versus cycloaddition discrimination.
Figure 2: Directed evolution for 1-methylindole amidation.

Evolutionary lineage from P411-CIS to P411-IA. Reactions were performed on 400 μL scale with whole-cell catalysts; HPLC yields of 3a, 4, and 5a are given. Data were derived from two independent experiments performed in duplicate.
Formation of azide reduction product 4 decreased as P411 catalysts gained efficiency in the amidation reaction, presumably as better binding of the indole acceptor substrate increased the rate of the desired transformation relative to nonproductive nitrene reduction.14 However, 4 still represented 14% of the product (5:1 3a:4) in reactions with variant P-YSVWG (Figure 2, Table S3), and we asked whether unproductive nitrene reduction could be suppressed further. Nitrene reduction is presumably driven by electron transfer from the P450 reductase domain,9,14 which accepts electrons from NADPH and relays them to the heme domain, a prerequisite for P450-BM3’s native oxo-transfer chemistry.36 However, non-natural P411 nitrene transfer catalysis only requires a single electron to reduce the heme cofactor from the FeIII to the catalytically competent FeII state, at which point catalysis can proceed without additional electron supply. While surplus reductant is likely required to reduce any cofactor that is inadvertently oxidized to the FeIII state, excessive electron supply aggravates undesired reduction of the iron-nitrenoid intermediate.6,9,14 Thus, we set out to engineer the reductase domain toward lower rates of electron transfer and presumably lower rates of nitrene reduction, anticipating that this would translate into higher amidation yields.
We first attempted a coarse approach of truncating the entire reductase domain or the FAD subdomain in variant P-YSVWG (Figure S3). Removal of the reductase domain proved deleterious for amidation activity, while FAD domain deletion (a mutation recently shown to be beneficial for P411 carbene transfer catalysis)21,37 was slightly less active than P-YSVWG, showing that full domain deletions are insufficient to enhance P411 nitrene transfer activity. Next, we used site-saturation mutagenesis of reductase domain residues involved in electron relay from NADPH to the heme domain36,38,39 in an attempt to fine-tune electron transfer rates (Table S2). This identified beneficial mutation W1046P, located close to the C-terminus of the reductase domain (Figure S4). W1046 stacks over the isoalloxazine ring of the FAD cofactor, and replacement with proline likely alters the NADPH-FAD interaction.39 The W1046P mutation increased amidation yield by 10% while reducing sulfonamide yield by 6%, representing an increase in 3a:4 product selectivity from 5:1 to 9:1 (Figure S3, Table S3). Attempts to further improve the 3a:4 ratio by targeting additional residues in the FAD-FMN-heme electron transfer chain were unsuccessful, and we refer to variant P-YSVWG W1046P as P411-IA (Indole Amidation). P411-IA catalyzes 3a formation in 78% yield, 8400 TTN, and a 3a:4:5a product ratio of 110:12:1 at 25 mM substrate loading (Figure 2), while over 90% yield of 3a and even higher selectivity were observed at 10 mM substrate loading (Figure S3).
To test whether the W1046P mutation in P411-IA affects rates of electron transfer and nitrene reduction, we measured both rates in vitro using purified protein. P411-IA delivered 2060 TTN in vitro, demonstrating that the enzyme functions in purified form (Figure S5). Next, we used a cytochrome c reduction assay, which utilizes reduction of cytochrome c by the P450 reductase domain to provide a spectrophotometric read-out, to estimate rates of electron transfer from NADPH across the reductase domain.40 Strikingly, in comparison to P411-CIS (P) and P-YSVWG, P411-IA showed a ca. 10-fold reduced electron transfer rate (Figure 3A). However, rates of nitrene reduction, measured by assessing formation of 4 in the absence of 1-methylindole (Figure S6), were only reduced ca. 2-fold, indicating that large decreases in electron transfer rate do not translate linearly into lower rates of nitrene reduction. Nonetheless, the data support the hypothesis that engineering the reductase domain to reduce the electron transfer rate can be beneficial for P411-catalyzed nitrene transfer. Underscoring the generalizability of this concept, we found that introducing the W1046P mutation into variant P411-CHA, evolved for intermolecular C-H animation,12 improved its activity in the amination of ethylbenzene (Figure S7).
Figure 3: Rates of nitrene reduction and indole amidation.

(A) Rates of P411 nitrene reduction to TsNH2 and electron transfer (measured with the cytochrome c reduction assay) obtained with purified enzyme in vitro. (B) Amidation kinetics of P411-IA and P411-CIS whole-cell reactions.
In small-scale biocatalytic whole-cell reactions, P411-IA catalyzed 1-methylindole amidation with an initial turnover frequency of 176 min”1 (Figure 3B) and total turnovers (> 8000) that rank among the highest achieved for hemoprotein-catalyzed intermolecular nitrene transfer. P411-IA showed ca. 7-fold lower activity in the presence of oxygen (Figure S8). Notably, however, P411-IA whole-cell reactions did not require the oxygen depletion system used in previous hemoprotein nitrene transfer studies.7,12,14–17
In addition to the screening substrate 2a, P411-IA efficiently amidated N-methylindoles with substitutions at C5 or C6, delivering the C2-amidated products in 64% to 91% yield and 2300 to 3300 TTN (Figure 4, 3c–3f). In contrast, P411-IA showed only low amidation activity on unprotected indole; curiously, P411-IA predominantly delivered the indole cycloaddition product 5b (Figure S9). However, one additional round of site-saturation mutagenesis and screening yielded P411-IA W82C, showing 6-fold higher indole amidation activity and delivering the C2-amidated product 3b in 45% yield with 1220 TTN (Figure 4, Figure S9).
Figure 4: Scope of P411-catalyzed indole amidation.

HPLC yield and TTN for P411-catalyzed amidation or cycloaddition of indoles are given. Biocatalytic reactions for 3a were performed with 25 mM substrate loading, while all other reactions were performed with 10 mM substrate loading; see Table S4 for product selectivities.
Lastly, we were curious to further investigate the apparent indole-azide cycloaddition reactions leading to products 5a and 5b. We hypothesized that these reactions take place in the P411 active site, which may serve as a hydrophobic pocket for orienting the substrates in the proper orientation for cycloaddition. We therefore screened site-saturation mutagenesis libraries of variants P-YS and P411-IA for enhanced triazole formation with 1-methylindole and indole, respectively. This resulted in variants P-YS W82S and P411-IA W82F that shifted the amidation:cycloaddition ratio toward triazole products 5a and 5b (Figure 4, Figure S10, Table S2). This further illustrates how P411 active-site engineering allows routing of substrates along different reaction trajectories. Furthermore, the data point to a critical role for residue 82 in controlling indole amidation versus cycloaddition selectivity; residue 82 may act to orient substrate in the active site and steer reaction specificity. While additional work is required to elucidate the enzymatic cycloaddition reaction mechanism, the variants obtained here may serve as starting points to evolve indole-azide cycloaddition activity.
In summary, we have demonstrated directed evolution of a cytochrome P411 variant to catalyze indole C2-amidation with greater than 8000 TTNs, 90% yield, and excellent selectivity for one out of three possible reaction products. Targeting both the heme and reductase domains for mutagenesis was key to achieving high enzyme activity and selectivity. Five mutations in the P411 heme domain were sufficient to enhance indole amidation activity ca. 20-fold and achieve a 97:1 ratio of indole amidation versus cycloaddition. Moreover, our finding that a single mutation in the reductase domain decreases unproductive nitrene reduction highlights the efficacy of mechanism-inspired reductase domain engineering for P411 nitrene transfer catalysis. Suppression of nitrene reduction was previously reported with iridium-porphyrin substituted hemoproteins.13 The variants reported here are fully genetically encoded and function with the native iron-porphyrin cofactor, broadening the scope of the nascent “nitrene transferase” enzyme family.
Supplementary Material
Acknowledgments
O.F.B. acknowledges support from the Deutsche Forschungsgemeinschaft (DFG grant BR 5238/1-1) and the Swiss National Science Foundation (SNF grant P300PA-171225). D.C.M. was supported by a Ruth Kirschstein NIH Postdoctoral Fellowship (F32GM128247). The authors thank Dr. Christopher K. Prier for help with initial experiments and comments on the manuscript. We also thank Dr. David K. Romney, Dr. Xiongyi Huang, and Kai Chen for helpful comments and discussions.
Footnotes
Supporting Information. Experimental procedures, supplementary data, calibration curves, and characterization of amidation and cycloaddition products.
References
- (1).Tawfik DS Accuracy-Rate Tradeoffs: How Do Enzymes Meet Demands of Selectivity and Catalytic Efficiency? Curr. Opin. Chem. Biol 2014, 27, 73–80. 10.1016/j.cbpa.2014.05.008. [DOI] [PubMed] [Google Scholar]
- (2).Hedstrom L Enzyme Specificity and Selectivity Encyclopedia of Life Sciences; John Wiley & Sons, Ltd: Chichester, U.K., 2010. 10.1002/9780470015902.a0000716.pub2. [DOI] [Google Scholar]
- (3).Breslow R Biomimetic Selectivity. Chem. Rec 2001, 1, 3–11. https://doi.org/l0.1002/1528-0691(2001)1:1<3::AID-TCR3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- (4).Renata H; Wang ZJ; Arnold FH Expanding the Enzyme Universe: Accessing Non-Natural Reactions by Mechanism-Guided Directed Evolution. Angew. Chem. Int. Ed 2015, 54, 3351–3367. 10.1002/anie.201409470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang J; Li G; Reetz MT Enzymatic Site-Selectivity Enabled by Structure-Guided Directed Evolution. Chem. Commun 2017, 53, 3916–3928. 10.1039/C7CC00368D. [DOI] [PubMed] [Google Scholar]
- (6).Brandenberg OF; Fasan R; Arnold FH Exploiting and Engineering Hemoproteins for Abiological Carbene and Nitrene Transfer Reactions. Curr. Opin. Biotechnol 2017, 47, 102–111. 10.1016/j.copbio.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).McIntosh JA; Coelho PS; Farwell CC; Wang ZJ; Lewis JC; Brown TR; Arnold FH Enantioselective Intramolecular C-H Amination Catalyzed by Engineered Cytochrome P450 Enzymes In Vitro and In Vivo. Angew. Chem. Int. Ed 2013, 52, 9309–9312. 10.1002/anie.201304401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Singh R; Kolev JN; Sutera PA; Fasan R Enzymatic C(Sp3)-H Amination: P450-Catalyzed Conversion of Carbonazidates into Oxazolidinones. ACS Catal 2015, 5, 1685–1691. 10.1021/cs5018612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Singh R; Bordeaux M; Fasan R P450-Catalyzed Intramolecular Sp3 C-H Amination with Arylsulfonyl Azide Substrates. ACS Catal 2014, 4, 546–552. 10.1021/cs400893n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bordeaux M; Singh R; Fasan R Intramolecular C(Sp3)H Amination of Arylsulfonyl Azides with Engineered and Artificial Myoglobin-Based Catalysts. Bioorg. Med. Chem 2014, 22, 5697–5704. 10.1016/j.bmc.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hyster TK; Farwell CC; Buller AR; McIntosh JA; Arnold FH Enzyme-Controlled Nitrogen-Atom Transfer Enables Regiodivergent C-H Amination. J. Am. Chem. Soc 2014, 136, 15505–15508. 10.1021/ja509308v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Prier CK; Zhang RK; Buller AR; Brinkmann-Chen S; Arnold FH Enantioselective, Intermolecular Benzylic C-H Amination Catalysed by an Engineered Iron-Haem Enzyme. Nat. Chem 2017, 9, 629–634. 10.1038/nchem.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dydio P; Key HM; Hayashi H; Clark DS; Hartwig JF Chemoselective, Enzymatic C-H Bond Amination Catalyzed by a Cytochrome P450 Containing an Ir(Me)-PIX Cofactor. J. Am. Chem. Soc 2017, 139, 1750–1753. 10.1021/jacs.6bll410. [DOI] [PubMed] [Google Scholar]
- (14).Farwell CC; McIntosh JA; Hyster TK; Wang ZJ; Arnold FH Enantioselective Imidation of Sulfides via Enzyme-Catalyzed Intermolecular Nitrogen-Atom Transfer. J. Am. Chem. Soc 2014, 136, 8766–8771. 10.1021/ja503593n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Prier CK; Hyster TK; Farwell CC; Huang A; Arnold FH Asymmetric Enzymatic Synthesis of Allylic Amines: A Sigmatropic Rearrangement Strategy. Angew. Chem. Int. Ed 2016, 55, 4711–4715. 10.1002/anie.201601056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Farwell CC; Zhang RK; McIntosh JA; Hyster TK; Arnold FH Enantioselective Enzyme-Catalyzed Aziridination Enabled by Active-Site Evolution of a Cytochrome P450. ACS Cent. Sci 2015, 1, 89–93. 10.1021/acscentsci.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cho I; Prier CK; Jia Z-J; Zhang RK; Gorbe T; Arnold FH Enantioselective Aminohydroxylation of Styrenyl Olefins Catalyzed by an Engineered Hemoprotein. Angew. Chem. Int. Ed 2019, 58, 3138–3142. 10.1002/anie.201812968. [DOI] [PubMed] [Google Scholar]
- (18).Cho I; Jia Z-J; Arnold FH Site-Selective Enzymatic C-H Amidation for Synthesis of Diverse Lactams. Science 2019, 364, 575–578. 10.1126/science.aaw9068. [DOI] [PubMed] [Google Scholar]
- (19).Vargas DA; Tinoco A; Tyagi V; Fasan R Myoglobin-Catalyzed C-H Functionalization of Unprotected Indoles. Angew. Chem. Int. Ed 2018, 57, 9911–9915. 10.1002/anie.201804779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hock KJ; Knorrscheidt A; Hommelsheim R; Ho J; Weissenborn MJ; Koenigs RM Tryptamine Synthesis by Iron Porphyrin Catalyzed C-H Functionalization of Indoles with Diazoacetonitrile. Angew. Chem. Int. Ed 2019, 58, 3630–3634. 10.1002/anie.201813631. [DOI] [PubMed] [Google Scholar]
- (21).Brandenberg OF; Chen K; Arnold FH Directed Evolution of a Cytochrome P450 Carbene Transferase for Selective Functionalization of Cyclic Compounds. J. Am. Chem. Soc 2019, 141, 8989–8995. 10.1021/jacs.9b02931. [DOI] [PubMed] [Google Scholar]
- (22).Bailey AS; Buckley AJ; Warr WA; Wedgwood JJ The Reactions of Arenesulphonyl Azides with Indole and with 1-Methylindole. J. Chem. Soc., Perkin Trans. 1 1972, 0, 2411–2415. 10.1039/P19720002411. [DOI] [Google Scholar]
- (23).Harmon RE; Wellman G; Gupta SK Reaction of Arylsulfonyl Azides with N-Methylindole. J. Org. Chem 1973, 38, 11–16. 10.1021/jo00941a003. [DOI] [Google Scholar]
- (24).Hu Z; Luo S; Zhu Q Palladium-Catalyzed Intermolecular C-H Amidation of Indoles with Sulfonyl Azides. Sci. China: Chem 2015, 58, 1349–1353. 10.1007/sll426-015-5369-y. [DOI] [Google Scholar]
- (25).Shi J; Zhou B; Yang Y; Li Y Rhodium-Catalyzed Regioselective Amidation of Indoles with Sulfonyl Azides via C-H Bond Activation. Org. Biomol. Chem 2012,10, 8953–8955. 10.1039/C2OB26767E. [DOI] [PubMed] [Google Scholar]
- (26).Sun B; Yoshino T; Matsunaga S; Kanai M Air-Stable Carbonyl(Pentamethylcyclopentadienyl)Cobalt Diiodide Complex as a Precursor for Cationic (Pentamethylcyclopentadienyl)Cobalt(III) Catalysis: Application for Directed C-2 Selective C-H Amidation of Indoles. Adv. Synth. Catal 2014, 356, 1491–1495. 10.1002/adsc.201301110. [DOI] [Google Scholar]
- (27).Lanke V; Prabhu KR Iridium(III) Catalyzed Regioselective Amidation of Indoles at the C4-Position Using Weak Coordinating Groups. Chem. Commun 2017, 53, 5117–5120. 10.1039/C7CC00763A. [DOI] [PubMed] [Google Scholar]
- (28).Song Z; Antonchick AP Iridium(III)-Catalyzed Regioselective C7-Sulfonamidation of Indoles. Org. Biomol. Chem 2016, 14, 4804–4808. 10.1039/C60B00926C. [DOI] [PubMed] [Google Scholar]
- (29).Kim Y; Park J; Chang S A Direct Access to 7-Aminoindoles via Iridium-Catalyzed Mild C-H Amidation ofN-Pivaloylindoles with Organic Azides. Org. Lett 2016,18, 1892–1895. 10.1021/acs.orglett.6b00662. [DOI] [PubMed] [Google Scholar]
- (30).Chen S; Feng B; Zheng X; Yin J; Yang S; You J Iridium-Catalyzed Direct Regioselective C4-Amidation of Indoles under Mild Conditions. Org. Lett 2017,19, 2502–2505. 10.1021/acs.orglett.7b00730. [DOI] [PubMed] [Google Scholar]
- (31).Park Y; Kim Y; Chang S Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017, 117, 9247–9301. 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]
- (32).Sandtorv AH Transition Metal-Catalyzed C-H Activation of Indoles. A civ. Synth. Catal 2015, 357, 2403–2435. 10.1002/adsc.201500374. [DOI] [Google Scholar]
- (33).Sheng G; Huang K; Chi Z; Ding H; Xing Y; Lu P; Wang Y Preparation of 3-Diazoindolin-2-Imines via Cascade Reaction between Indoles and Sulfonylazides and Their Extensions to 2,3-Diaminoindoles and Imidazo[4,5- b ]Indoles. Org. Lett 2014, 16, 5096–5099. 10.1021/ol502423k. [DOI] [PubMed] [Google Scholar]
- (34).Behrendorff JBYH; Huang W; Gillam EMJ Directed Evolution of Cytochrome P450 Enzymes for Biocatalysis: Exploiting the Catalytic Versatility of Enzymes with Relaxed Substrate Specificity. Biochem. J 2015, 467, 1–15. 10.1042/BJ20141493. [DOI] [PubMed] [Google Scholar]
- (35).Coelho PS; Wang ZJ; Ener ΜE; Baril SA; Kannan A; Arnold FH; Brustad EM A Serine-Substituted P450 Catalyzes Highly Efficient Carbene Transfer to Olefins in Vivo. Nat. Chem. Biol 2013, 9, 485–487. 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Whitehouse CJ; Bell SG; Wong L-L P450 BM3 (CYP102A1): Connecting the Dots. Chem. Soc. Rev 2012, 41, 1218–1260. 10.1039/ClCS15192D. [DOI] [PubMed] [Google Scholar]
- (37).Zhang RK; Chen K; Huang X; Wohlschlager L; Renata H; Arnold FH Enzymatic Assembly of Carbon-Carbon Bonds via Iron-Catalysed Sp3 C-H Functionalization. Nature 2019, 565, 67–72. 10.1038/s41586-018-0808-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Verma R; Schwaneberg U; Roccatano D Insight into the Redox Partner Interaction Mechanism in Cytochrome P450BM-3 Using Molecular Dynamics Simulations. Biopolymers 2014, 101, 197–209. 10.1002/bip.22301. [DOI] [PubMed] [Google Scholar]
- (39).Joyce MG; Ekanem IS; Roitel O; Dunford AJ; Neeli R; Girvan ΗM; Baker GJ; Curtis RA; Munro AW; Leys D The Crystal Structure of the FAD/NADPH-Binding Domain of Flavocytochrome P450 BM3. FEBS J 2012, 279, 1694–1706. 10.1111/j.1742-4658.2012.08544.x. [DOI] [PubMed] [Google Scholar]
- (40).Guengerich FP; Martin MV; Sohl CD; Cheng Q Measurement of Cytochrome P450 and NADPH-Cytochrome P450 Reductase. Nat. Protoc 2009, 4, 1245–1251. 10.1038/nprot.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.