

Abstract
Pyridines are valuable motifs in a number of bioactive and functional molecules. The chemoselective functionalization of these structures from stable and widely available starting materials is a meaningful goal. We have demonstrated selective formation of pyridyl radicals at any position (2-, 3-, 4-pyridyl), through the action of a reducing photoredox catalyst. These radicals readily engage alkenes to deliver high-value products. Alteration of the reaction medium has enabled the use of a diverse range of alkene subtypes in a highly divergent and chemoselective manner.
Keywords: alkenes, catalysis, chemoselectivity, heterocycles, photochemistry, radicals, regioselectivity
Graphical Abstract
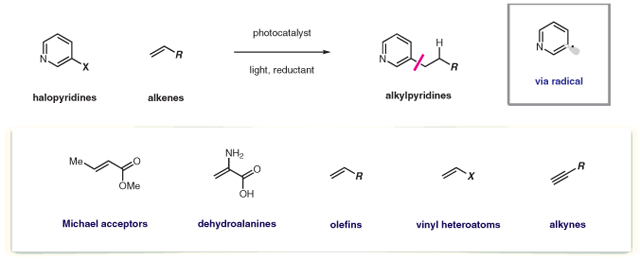
1. Introduction
Pyridines hold a privileged role within medicinal and agrochemical discovery, often providing improved physicochemical properties and metabolic stability over their arene counterparts.1 Pyridines are the most prevalent aromatic heterocycle in pharmaceuticals and, as reported in 2013, 10% of approved pharmaceutical agents contained at least one pyridine unit.2 Shown in Figure 1 are selected examples of alkylpyridine-containing medicines, where in addition to the Lewis basic heteroaromatic function, acidic X–H bonds abound. As such, direct methods for the construction of the alkylpyridine motif are of great value to the medicinal chemistry community. Furthermore, the modular ability to forge pyridyl C–C bonds with control over regiochemistry (with respect to both the pyridyl and alkyl units) under mild conditions with the capacity to tolerate a variety of polar functional groups are particularly powerful.
Figure 1.
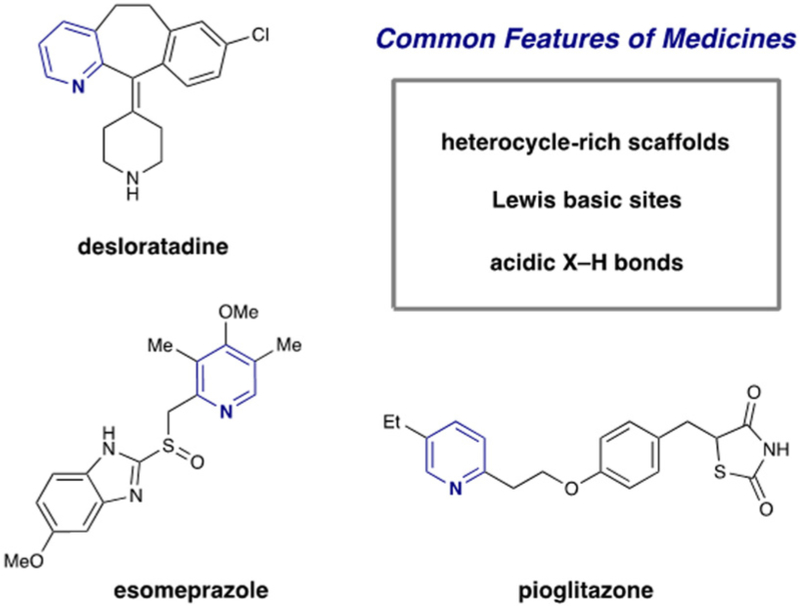
Selected examples of pyridine-containing drugs
Although there are many effective processes for de novo pyridine synthesis (e.g., condensation reactions of acyclic precursors), these strategies are suboptimal in medicinal chemistry because they do not typically allow for modular alteration of the pyridine substituents.1 Transition-metal-catalyzed cross-coupling is a mainstay strategy in complex pyridine synthesis, where a wide range of robust, chemoselective methods for the arylation of pyridines have emerged.3 However, this strategy is limited in the ability to forge the alkylpyridine motif (through formation of sp2–sp3 bonds). This is primarily due to competitive β-hydride elimination pathways of the organometallic intermediates.4 Radical-based methods are particularly well suited to heterocyclic chemistry due to their tolerance of polar, Lewis basic functionality, which can prove difficult to overcome under a two-electron paradigm.5 Accordingly, a large number of radical processes have demonstrated the ability to accomplish late-stage functionalization processes on pharmaceutical scaffolds. These processes have been largely based on the classical Minisci reaction, where radical addition to azaarenes under oxidative conditions allows for the direct installation of a diverse array of alkyl units.
2. Minisci-Type Pyridine Alkylation
As first detailed in 1968, the Minisci radical arylation process operates through interception of alkyl radical species by protonated heteroarenes.6 Upon C–C bond formation, oxidative rearomatization delivers the desired alkylated heteroaromatic products in a manner that allows for variation of both the alkyl unit and the heteroaromatic core (generally illustrated in Scheme 1). Building on this mechanistic principle, the scope of the radical precursor has been greatly expanded to include carboxylic acids and derivatives,7 alkyl halides,8 boronates,9 Barton esters,10 and xanthates,11 among others. Less conventional precursors have also been used where extrusion of sulfur dioxide through reduction of sulfonyl halides or oxidation of sulfinate salts have also been employed.12 These processes have been particularly successful in the installation of high-value fluoroalkyl units (trifluoromethyl, etc.) onto heteroaromatics.13,14 In addition, a range of unactivated substrates have been shown to smoothly participate in this process, where homolytic C–H activation can be leveraged in the radical-forming step (see below).
Scheme 1.

Current paradigm for the radical alkylation of pyridines and an alternative complementary method utilizing pyridyl radicals
As mentioned above, a main advantage of this strategy stems from the highly chemoselective nature of the radical mechanism. Because carbon radicals react selectively with heteroaromatics, very complex substrates containing other structural elements that are common in drugs (e.g., alcohols or amides) undergo selective alkylation of the heteroaromatic unit. A small collection of bioactive scaffolds that exemplify this attribute is shown in Scheme 1, where radical addition occurs without the need for protecting or activating groups. For example, Baran showed that unfunctionalized/unactivated drug scaffolds could be directly ‘tagged’ using a range of fluoroalkylsulfinate salts.14 Building on early work from the MacMillan lab,13 DiRocco and Kyrska demonstrated that late-stage methylation of medicinal compounds could be accomplished under mild conditions using photoredox catalysis.15
While tremendously enabling, this strategy for alkylpyridine synthesis is limited primarily by poorly controlled regiochemical outcomes. Two structures from the Merck study (given in Scheme 1) illustrate this point. While alkylation occurs selectively at heteroaromatic sites, each of these structures contains multiple reactive sites (indicated by the centers highlighted in blue) where regioisomeric mixtures of monoalkylated (as well as difunctionalized) products were observed. While altering reaction solvent and conditions can impact the obtained product ratios,12–15 selectivity (inherently biased for radical addition to the 2- or 4-position of the azaarene) is, so far, primarily determined by substrate structure. Moreover, this strategy does not allow for functionalization at other positions (i.e., 3- or 5-positions, indicated in red in Scheme 1). We were motivated to develop a general and complementary approach to pyridine alkylation that would address this regiochemical limitation, while retaining the excellent chemoselectivity and functional group compatibility of the Minisci radical arylation.
3. An Alternate Approach – Reductive Radical Formation
As a complement to Minisci-type processes, we envisioned a strategy involving direct intermolecular coupling of pyridyl radicals with olefins (Scheme 1). This design was particularly attractive because it would enable a collection of C–C bond constructions that utilize stable, readily available olefins as alkyl sources. Aryl radicals are highly reactive species that effectively engage a wide range of unsaturated systems (i.e., Meerwein arylation processes).16 Because these intermediates can be accessed in a regiospecific fashion and radical addition to olefins occurs with anti-Markovnikov selectivity, this mechanistic design would deliver alkylated pyridines with complete regiocontrol.
While regiospecific aryl radical formation can be accomplished using a range of common aryl precursors, pyridyl halides were the most promising radical sources in this context because they are more stable and widely available (as starting materials in cross-coupling and nucleophilic aromatic substitution) than the corresponding pyridyldiazonium salts or pyridylboronic acids. Under classical tin-mediated atom-transfer conditions, Snieckus showed that pyridyl radicals effectively participate in intramolecular cyclization with olefins (Scheme 2).17 However, the rapid rates of hydrogen atom transfer (HAT) by aryl radicals from tin-hydrides has precluded effective intermolecular reaction of these intermediates.18 These intermediates can also be accessed via electrochemical reduction of pyridyl halides, where single-electron transfer (SET) is followed by rapid dissociation of the resulting radical anion. These radicals are known to undergo further reduction under electrochemical conditions, thus producing the corresponding anions.19 We hypothesized that the use of photoredox catalysis (where potent reductants can be generated in minute quantities) would allow us to realize a general reaction pathway that could be leveraged in a series of new processes.
Scheme 2.

State of the art in reductive dehalogenation
Pioneering works by Stephenson,20 Read de Alaniz and Hawker,21 Weaver,22 and König23 have shown that photoredox catalysis can be utilized in reductive radical formation of aryl and heteroaryl halides. While much of this work has utilized the propensity of these intermediates to undergo HAT, resulting in net protodehalogenation (Scheme 2), this reactivity can also achieve carbon–carbon bond formation. Stephenson and Read de Alaniz detailed effective conditions for dehalogenation of a range of organic halides, where the combination of amine reductants with organometallic, or organic photoredox catalysts, serves as attractive nontoxic alternatives to tin hydrides. In 2014, König demonstrated that aryl radicals produced in this manner can perform intermolecular arylation processes, although large excesses (≥20 equiv) of pyrrole radical traps were required to effectively compete with HAT.23 In addition, Weaver has demonstrated a number of elegant intermolecular coupling processes that utilize 2-haloazoles and perfluorinated aromatics or heteroaromatics.22 Indeed, the resulting electrophilic perfluoropyridyl radicals were shown to undergo efficient intermolecular coupling with alkenes. Building on these findings, we sought to develop a general approach for complex pyridine functionalization. We were motivated to identify mild conditions that would generally enable activation of pyridines for programmed alkylation at any position for coupling with a wide range of alkene subtypes. Successful implementation of this strategy would provide an alternative to the Minisci coupling particularly in the context of building block synthesis.
4. Conjugate Addition of Pyridyl Radicals
We began our investigations by assessing the union of 2-iodopyridine with the ethylidene malonate that is shown in Scheme 3. We observed trace amounts of the desired coupling product A under previously reported conditions for aryl halide reduction, but radical protodehalogenation (to afford pyridine, B) was the major pathway. While a range of alkylamines are routinely utilized as stoichiometric reductants, we found that selectivity for intermolecular radical conjugate addition was significantly improved when Hantzsch ester (HEH) was employed as reductant in aqueous mixtures of DMSO. As shown in Scheme 3, increasing the amount of water cosolvent was accompanied by an increase in selectivity for the desired product A. Solubility of the reductant (HEH) is markedly decreased with increased water content, and we propose that limited solubility of reductant is key to increasing intermolecular reaction efficiency. Under optimized conditions, we found that the scope of this radical conjugate addition process is broad with respect to both the pyridyl unit and Michael acceptor.24 Importantly, coupling was effectively performed at the 2-, 3-, and 4-positions on a range of pyridine substrates that contain electron-donating or -withdrawing groups.
Scheme 3.
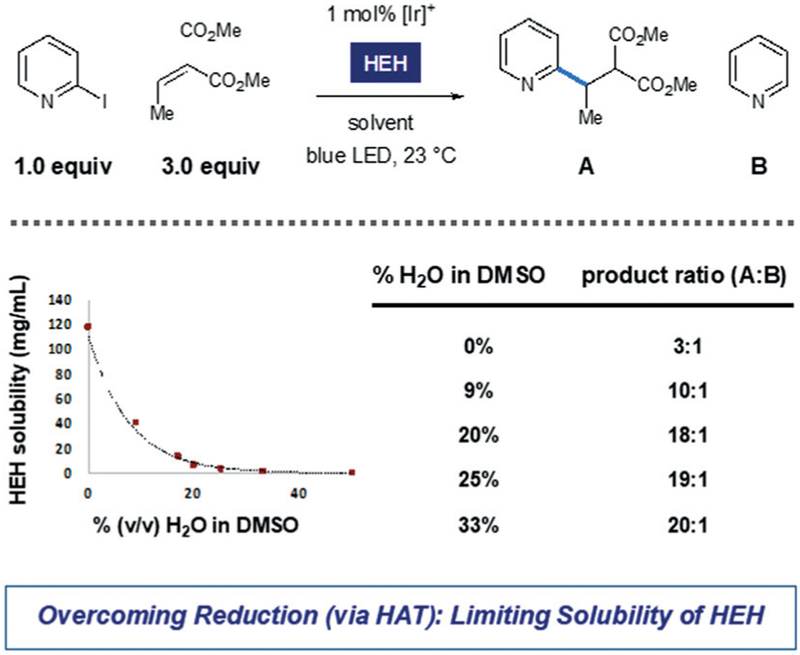
Aqueous reaction medium enables effective conjugate addition
This programmable regiocontrol provides access to alkylated pyridine isomers that are inaccessible under Minisci conditions. Effective radical acceptors within this reaction manifold include α,β-unsaturated ketones, esters, amides, and carboxylic acids (Figure 2, 1a–f). Substrates with some degree of substitution are generally more successful in this system, as unsubstituted acrylate derivatives undergo radical polymerization. Incorporation of minimal steric bulk in either the α- or β-position was sufficient to eliminate unwanted oligomeric byproducts arising from competitive conjugate addition from the α-carbonyl radical to another equivalent of acceptor.
Figure 2.
Proposed reversal of radical polarity through protonation
The mild, catalytic conditions proved to be highly effective for amino acid synthesis using dehydroalanine derivatives as radical acceptors.25 Control of the amino acid α-stereocenter could be achieved through the use of a chiral dehydroalanine derivative first reported by Karady and co-workers at Merck Sharpe and Dohme (Scheme 4).26 We found that this chiral conjugate acceptor was an efficient coupling partner for pyridyl radicals, delivering heterocyclic amino acids 1g–j with complete diastereoselectivity. Besides pyridine derivatives, this versatile radical trap could be leveraged towards the synthesis of other unnatural amino acids through the use of alternative radical precursors. Alkyl groups could be incorporated effectively using either activated alkyl halides (allyl bromide 1k, CF3I 1m) or through the use of redox active NHPI esters 1l, all precursors were incorporated with excellent stereocontrol. As shown in Scheme 4, acidic hydrolysis of the products delivered the free amino acids in high yields and enantiopurities. This strategy provides a versatile platform for the synthesis of a broad range of amino acids under mild conditions. Additionally, the method could be performed on a 25 mmol scale using 0.1 mol% Ir catalyst and 1.1 equivalents of acceptor with minimal erosion of isolated yield.
Scheme 4.

Conjugate addition of pyridyl radicals and its application to the synthesis of unnatural amino acid derivatives
5. Radical Hydroarylation of Neutral and Rich Olefins
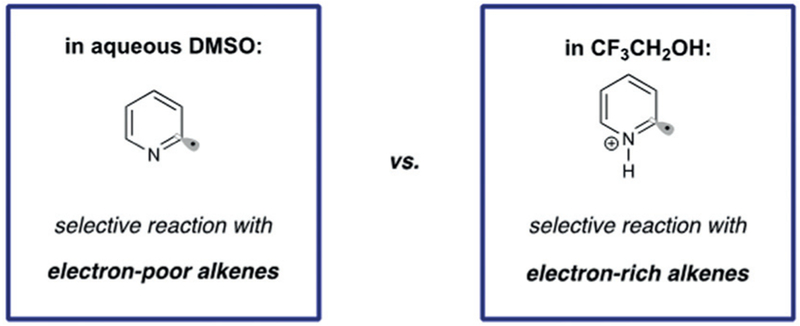
We next turned our attention to reactions of less activated electron-neutral and electron-rich olefins, which are challenging substrates for intermolecular radical addition. Balancing the kinetics of alkene addition with HAT in this system presented a significant challenge. Indeed, under our previously optimized conditions, 1-octene provided only 4% yield of the desired product with the remainder of the mass balance consisting of hydrodehalogenated starting material. This indicated a slow rate of radical addition to the unactivated alkene in comparison to the rate of HAT. We suggest that this rate difference is caused, in part, by a polarity mismatch between the nucleophilic pyridyl radical and the more electron-rich olefin. To resolve this, we proposed that inversion of the of radical polarity through protonation could enable selective reactivity with nucleophilic alkenes over the electrophilic H-atom donor (Figure 2).
We found that use of the mildly acidic fluorinated solvent 2,2,2-trifluoroethanol (TFE) was uniquely effective in mediating the addition to unactivated alkenes. Consistent with our hypothesis, weakly acidic additives (NH4Cl) further increased the yield, providing an effective method for the anti-Markovnikov addition of pyridyl radicals to simple alkenes (Scheme 5, 3a–e).27 The method was tolerant of alkenes bearing protic functionality such as unprotected alcohols and carbamates without significant loss in yield. Additionally, reducible functions such as alkyl chlorides and ketones were retained with perfect chemoselectivity. The alkylated pyridines synthesized through this method are typically accessed through cross-coupling of reactive organometallic nucleophiles (RMgX, RZnX),28 unselective Minisci couplings,6 or through multistep sequences such as Heck coupling followed by hydrogenation. The mild nature of the reaction conditions is exemplified by the use of alkenes bearing polar, protic functionality, which is typically poorly tolerated in metal-catalyzed methods.
Scheme 5.

Hydroarylation of neutral and rich olefins using pyridyl radicals in 2,2,2-trifluoroethanol
We then looked to extend this method towards the synthesis of more biologically relevant motifs such as phenethylamines or phenethyl alcohols. We envisioned access to these substructures through the application of our radical-addition manifold to heteroatom-substituted alkenes. Selective anti-Markovnikov radical addition to these olefins would lead to a stabilized carbon-centered radical α to a heteroatom. HAT from this intermediate provides the desired product, however, we found that in many cases oligomeric byproducts were observed that arose from slow HAT. Key to successful implementation of this reactivity was the addition of a polarity reversal catalyst. A more nucleophilic α-hetero radical is polarity mismatched for the delivery of an H-atom from Hantzsch ester, but well matched for an electrophilic HAT catalyst such as a thiol. Addition of 5 mol% CySH was sufficient to reduce oligomeric byproducts that arise from slow HAT and provide synthetically useful yields across a broad range of alkene subtypes (Scheme 5, 3f–i).29 This subtle addition allowed for the effective use of alkenes substituted with halides (F, Cl, Br), providing a novel route towards compounds typically accessed through elimination prone nucleophilic displacement. Vinyl metalloids and main group elements were readily incorporated, β-boryl, silyl and phosphoryl pyridines could be forged in this manner in a single synthetic step.
Additionally, terminal alkynes could be used to synthesize alkenyl pyridines in excellent yield as mixtures of cis/trans isomers. Enol ethers and enol acetates were also shown to be viable substrates, providing phenethyl alcohol derivatives in good yield. We present a new disconnection to these common pharmacophores as demonstrated by a highly convergent synthesis of the diabetes medication pioglitazone from a simple halopyridine and a vinyl phenolic ether. In order to validate our method for use in an industrial setting, pyridyl enol acetate 3k was shown to be amenable to mmol scale synthesis in both batch and flow regimes without any effect on the isolated yield (Scheme 5).
Deuterium-labeling studies confirmed that HAT from the thiol catalyst was the primary source of radical termination. The presence of cationic intermediates arising from oxidation were ruled out through the use of a cation-trap substrate, which did not cyclize under the reaction conditions.30
6. Solvent-Based Chemoselectivity
Competition experiments performed during the course of this work revealed a remarkable effect where highly selective radical addition to either electrophilic or nucleophilic olefins can be dictated by the employed reaction solvent. Treating equistoichiometric amounts of both 1-octene and ethylcrotonate with bromopyridine with the optimized conditions in either TFE or DMSO/H2O led to divergent reactivity.
As indicated in Scheme 6, complete selectivity is observed for the conjugate addition product 4a in aqueous DMSO. Conversely, pyridyl radical addition to 1-octene proceeded with very high selectivity to afford hydroarylation product 5b. These experiments were replicated in an additional competition experiment where two different olefin types were present on a single molecule (Scheme 6, 6a,b). Again, these results illustrate that the reaction solvent can enforce different polarities on the pyridyl radical intermediate; the pyridyl radical is electrophilic in TFE and nucleophilic in aqueous DMSO. This effect is proposed to stem from the unusual coordinating properties of TFE, which is known to complex with even the poorest Lewis bases.31 In this context, protonation of the pyridyl radical by TFE presumably renders it electrophilic in its reactivity towards alkenes. This effect could be perturbed though the addition of superstoichiometric quantities of H2O, which is able to break up the coordination sphere and provide a mixture of both products.
Scheme 6.

Mechanistic divergence in radical polarity based on solvent selection
7. Summary and Outlook
Highly substituted pyridines have been an attractive target for method development due to their widespread use and relative difficulty in synthesis. Radical-based methods are ideal for these motifs because of the functional group compatibility offered by a single-electron regime. Alteration of the classical Minisci disconnection has allowed for the programmed, regioselective alkylation of a diverse array of halogenated pyridine units. This was achieved through single-electron reduction of halopyridines using a highly reducing photoredox catalyst, which selectively generates a single sp2 pyridyl radical that can engage either electron-poor olefins (in DMSO/H2O) or electron-neutral/rich olefins (in TFE).
We anticipate that this new disconnection will lead to a diverse array of reactions based upon the same pyridyl radical scaffold. Currently, we are exploring new reactivity modes within this research theme and further studying the mechanistic aspects of this chemistry.
Acknowledgments
Funding Information
Financial support for this work was provided by startup funds from Emory University and the National Institutes of Health (GM129495).
Biography

Nathan T. Jui (right) grew up in Colorado and received a BSc in chemistry at CSU under the guidance of Prof. Tomislav Rovis. He then moved to Princeton University to perform graduate studies with Prof. David MacMillan developing radical methodologies for enantioselective aldehyde alkylation. In 2011, Nate was appointed at as a NIH postdoctoral fellow at MIT working in the group of Prof. Steve Buchwald. In 2014 he started his independent career at Emory university. Dr. Jui's research interests lie in method development and the application of chemical tools to the study of human disease. Ciaran P. Seath (left) grew up in the highlands of Scotland and received his MChem degree at the University of Strathclyde, Glasgow in 2014. Staying at the same institution, he received his PhD in 2017 under the guidance of Dr. Allan Watson working on chemoselective transition-metal catalysis. He then joined the Jui Lab at Emory University where his research is focused on developing novel photochemical methods.
References
- (1) (a).Baumann M; Baxendale IR Beilstein J. Org. Chem 2013, 9, 2265. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Joule JA; Mills K Heterocyclic Chemistry, 4th ed; Blackwell: Malden, MA, 2000. [Google Scholar]; (c) Eicher T; Hauptmann S The Chemistry of Heterocycles: Structures, Reactions, Synthesis and Applications, 2nd ed; Wiley-VCH: Weinheim, 2003. [Google Scholar]; (d) Grimmett MR Adv. Heterocycl. Chem 1993, 58, 271. [Google Scholar]
- (2).Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- (3).Almond-Thynne J; Blakemore DC; Pryde DC; Spivey AC Chem. Sci 2017, 8, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhao S; Gensch T; Murray B; Niemeyer ZL; Sigman MS; Biscoe MR Science 2018, 362, 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5) (a).Smith JM; Harwood SJ; Baran PS Acc. Chem. Res 2018, 51, 1807. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yan M; Lo JC; Edwards JT; Baran PS J. Am. Chem. Soc 2016, 138, 12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Minisci F; Galli B; Cecere M; Malatesta V; Caronna T Tetrahedron Lett. 1968, 54, 5609. [Google Scholar]
- (7).Minisci F; Bernardi R; Bertini F; Galli R; Perchinummo M Tetrahedron 1971, 27, 3575. [Google Scholar]
- (8).Minisci F; Vismara E; Fontana F; Morini G; Serravalle M; Giordano C J. Org. Chem 1986, 51,4411. [Google Scholar]
- (9).Molander GA; Colombel V; Braz VA Org. Lett 2011, 13, 1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Barton DHR; Garcia B; Togo H; Zard SZ Tetrahedron Lett. 1986, 27, 1327. [Google Scholar]
- (11).Coppa F; Fontaria F; Minisci F; Pianese G; Tortoreto P; Zhao L Tetrahedron Lett. 1992, 33, 687. [Google Scholar]
- (12).Seiple IB; Su S; Rodriguez RA; Gianatassio R; Fujiwara Y; Sobel AL; Baran PS J. Am. Chem. Soc 2010, 132, 13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nagib DA; MacMillan DWC Nature 2011,480, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Fujiwara Y; Dixon JA; O’Hara F; Funder ED; Dixon DD; Rodriguez RA; Baxter RD; Herlé B; Sach N; Collins MR; Ishihara Y; Baran PS Nature 2012, 492, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).DiRocco DA; Dykstra K; Krska S; Vachal P; Conway DV; Tudge M Angew. Chem. Int. Ed 2014, 53, 4802. [DOI] [PubMed] [Google Scholar]
- (16).Hari DP; König B Angew. Chem. Int. Ed 2013, 52, 4734. [DOI] [PubMed] [Google Scholar]
- (17).Shankaran K; Sloan CP; Snieckus V Tetrahedron Lett. 1985, 26, 6001. [Google Scholar]
- (18).Garden SJ; Avila DV; Beckwith ALJ; Bowry VW; Ingold KU; Lusztyk J J. Org. Chem 1996, 61, 805. [DOI] [PubMed] [Google Scholar]
- (19).Birch AJ J. Chem. Soc 1944, 430. [Google Scholar]
- (20).Nguyen JD; D'Amato EM; Narayanam JMR; Stephenson CR J. Nat. Chem 2012, 4, 854. [DOI] [PubMed] [Google Scholar]
- (21).Discekici EH; Treat NJ; Poelma SO; Mattson KM; Hudson ZM; Luo Y; Hawker CJ; de Alaniz JR Chem. Commun 2015, 51, 11705. [DOI] [PubMed] [Google Scholar]
- (22) (a).Senaweera S; Weaver JD J. Am. Chem. Soc 2016, 138, 2520. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Arora A; Weaver JD Org. Lett 2016, 18, 3996. [DOI] [PubMed] [Google Scholar]; (c) Singh A; Fennell CJ; Weaver JD, Chem. Sci 2016, 7, 6796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Singh A; Kubik JJ; Weaver JD Chem. Sci 2015, 6, 7206. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Arora A; Teegardin KA; Weaver JD Org. Lett 2015, 17, 3722. [DOI] [PubMed] [Google Scholar]
- (23) (a).Ghosh I; König B Angew. Chem. Int. Ed 2016, 55, 7676. [DOI] [PubMed] [Google Scholar]; (b) Marzo L; Ghosh I; König B ACS Catal. 2016, 6, 6780. [Google Scholar]; (c) Ghosh I; Ghosh T; Bardagi JI; König B Science 2014, 346, 725. [DOI] [PubMed] [Google Scholar]
- (24).Aycock RA; Wang H; Jui NT Chem. Sci 2017, 8, 3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Aycock RA; Vogt DB; Jui NT Chem. Sci 2017, 8, 7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Karady S; Amato S; Weinstock M Tetrahedron Lett. 1984, 25, 4337. [Google Scholar]
- (27).Boyington AJ; Riu M-LY; Jui NT J. Am. Chem. Soc 2017, 139, 6582. [DOI] [PubMed] [Google Scholar]
- (28).Jana R; Pathak TP; Sigman MS Chem. Rev 2011, 111, 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Seath CP; Vogt DB; Xu Z; Boyington AJ; Jui NT J. Am. Chem. Soc 2018, 140, 15525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Campbell JM; Xu H-C; Moeller KD J. Am. Chem. Soc 2012, 134, 18338. [DOI] [PubMed] [Google Scholar]
- (31).Bégué J-P; Bonnet-Delpon D; Crousse B Synlett 2004, 18. [Google Scholar]