

Abstract
The human pathogens N. gonorrhoeae and N. meningitidis display robust intra- and interstrain glycan diversity associated with their O-linked protein glycosylation (pgl) systems. In an effort to better understand the evolution and function of protein glycosylation operating there, we aimed to determine if other human-restricted, Neisseria species similarly glycosylate proteins and if so, to assess the levels of glycoform diversity. Comparative genomics revealed the conservation of a subset of genes minimally required for O-linked protein glycosylation glycan and established those pgl genes as core genome constituents of the genus. In conjunction with mass spectrometric–based glycan phenotyping, we found that extant glycoform repertoires in N. gonorrhoeae, N. meningitidis and the closely related species N. polysaccharea and N. lactamica reflect the functional replacement of a progenitor glycan biosynthetic pathway. This replacement involved loss of pgl gene components of the primordial pathway coincident with the acquisition of two exogenous glycosyltransferase genes. Critical to this discovery was the identification of a ubiquitous but previously unrecognized glycosyltransferase gene (pglP) that has uniquely undergone parallel but independent pseudogenization in N. gonorrhoeae and N. meningitidis. We suggest that the pseudogenization events are driven by processes of compositional epistasis leading to gene decay. Additionally, we documented instances where inter-species recombination influences pgl gene status and creates discordant genetic interactions due ostensibly to the multi-locus nature of pgl gene networks. In summary, these findings provide a novel perspective on the evolution of protein glycosylation systems and identify phylogenetically informative, genetic differences associated with Neisseria species.
Author summary
Bacteria express a remarkable diversity of sugars and oligosaccharides in conjunction with protein glycosylation systems. Currently however, little is known about the evolutionary processes and selective forces shaping glycan biosynthetic pathways. The closely related bacterial pathogens Neisseria gonorrhoeae and Neisseria meningitidis remain serious sources of human disease and these species express antigenically variable oligosaccharides as components of their broad-spectrum, O‐linked protein glycosylation (pgl) systems. With the exception of isolates of Neisseria elongata subspecies glycolytica, the status of such post-translational modifications in related commensal species colonizing humans remains largely undefined. Here, we exploit new data from further studies of protein glycosylation in Neisseria elongata subspecies glycolytica to address these concerns. Employing comparative genomics and glycan phenotyping, we show that related pgl systems are indeed expressed by all human-restricted Neisseria species but identify unique gene gain and loss events as well as loss-of-function polymorphisms that accommodate a dramatic shift in glycoform structure occurring across the genus. These findings constitute novel perspectives on both the evolution of protein glycosylation systems in general and the macroevolutionary processes occurring in related bacterial species residing within a single host.
Introduction
Bacterial cell surfaces are decorated by diverse oligosaccharides and glycans in the context of capsules, lipopolysaccharides (LPS), glycoproteins and cell wall–associated glycoconjugates. Despite their ubiquity and implicit importance, the evolutionary processes shaping glycan diversity are not fully understood [1]. Such efforts are challenging as oligo- and polysaccharides are generated by the coordinated action of enzymes utilizing diverse monosaccharides and as specific functions of biosynthetic components and the glycans themselves are often undefined. For capsular polysaccharides and LPS, biosynthetic pathways are typically encoded within contiguous gene clusters. This linkage arrangement maintains biosynthetic compatibility allowing wholesale switching via single locus recombination events [2, 3]. Questions of the evolutionary processes and adaptive potential of glycans also apply to bacterial protein glycosylation systems in both their N- and O-linked forms [4]. Although both dedicated and broad-spectrum protein glycosylation are well recognized amongst eubacteria, relatively few studies have comprehensively examined glycan diversity and genotype–phenotype relationships at the genus level [5–8].
The genus Neisseria includes Gram-negative, oxidase-positive bacterial species that are associated with mucosal surfaces of humans and two closely related species are significant human pathogens. Neisseria gonorrhoeae is the agent of the sexually transmitted disease gonorrhea and Neisseria meningitidis is primarily a commensal of the oropharynx that under poorly understood circumstances can lead to invasive disease including meningitis. Despite their differing ecology and mechanisms of transmission, these species display remarkable conservation at the levels of nucleotide sequence, gene content and synteny [9]. Attempts to reconcile the distinctive relationships operating in these species with gene content are further complicated by the likewise, closely related species N. lactamica and N. polysaccharea that are harmless commensals found predominantly in the upper respiratory tracts of infants and children [10]. The genus also includes other less closely related nonpathogenic species that colonize the human oral cavity. Cross-species comparisons of genome sequences are beginning to reveal differences in gene content and organization and provide insights into evolutionary processes operating within the genus. Early studies using limited number of genomes or microarray-based genome hybridization studies concluded that a large number of “virulence” genes were distributed throughout the genus [11–13]. While studies of single gene families may be phylogenetically informative [9], analyses of multiple genes whose products function in concerted biosynthetic and biochemical pathways may be particularly resourceful. Recent examples of this include genus–wide analyses of genes involved in pilus biogenesis [11], determining cell shape (rod to coccus transitions) [14], protein glycosylation [11, 15], cytochrome c-based, electron transfer supporting dissimilatory nitrite reduction [16] and capsular polysaccharide expression [17].
Broad–spectrum, O-linked protein glycosylation (pgl) systems have been defined in N. gonorrhoeae, N. meningitidis and the deeply branching commensal species N. elongata subspecies glycolytica. Based on biochemical and reverse genetic approaches in tandem with mass spectrometry and serotyping for glycan characterization, consensus models for neisserial pgl-dependent protein glycosylation has been identified [15, 18–21]. A NAD+-dependent dehydratase (PglC) and aminotransferase (PglD) generate UDP-2-acetamido-4-amino-2,4,6-trideoxy-α-d-glucose from UDP-GlcNAc [20]. A bifunctional enzyme (PglB) then catalyzes amino acetylation of UDP-2-acetamido-4-amino-2,4,6-trideoxy-α-d-glucose to form UDP-di-N-acetylbacillosamine (diNAcBac) and the subsequent transfer of the phosphosugar to the lipid carrier undecaprenyl phosphate (Und-P) [20]. PglB2, encoded by pglB2 alleles found in some N. meningitidis strains, contains a distinct C-terminal domain proposed to mediate the transfer of a glycerol moiety (in place of the acetyl group) to produce 4-glyceramido-2-acetamido-2,4,6-trideoxy-α-d-hexose (GATDH) [22]. Subsequent elaboration of these undecaprenyl diphosphate (Und-PP) monosaccharides ensues via two pathways using distinct glycosyltransferases. One involves PglH or its allelic variant-encoded PglH2, which attach a Glc or GlcNAc respectively, to the Und-PP-monosaccharides to generate disaccharides [5, 15]. The second pathway utilizes the PglA and PglE glycosyltransferases to add successive Gal units to produce a trisaccharide [21, 23]. As both pathways are active in some N. gonorrhoeae and N. meningitidis isolates, those strains can express simultaneously PglA- and PglH- generated glycoforms[15]. Moreover, PglH/PglH2-generated Und-PP-disaccharides are incapable of being further extended by PglE [5, 15]. Antagonism and potential redundancy involving PglA and PglH in N. gonorrhoeae and N. meningitidis have been hypothesized to account for hypomorphic pglA and pglH alleles as well as a pglH deletion mutation found in some strains of the two species [15, 24]. Studies in N. elongata subspecies glycolytica (that lack the pglA and pglE genes) reported the expression of a di-N-acetylbacillosamine-glucose-di-N-acetyl glucuronic acid-N-acetylhexosamine (diNAcBac-Glc-diNAcHexA-HexNAc) tetrasaccharide [25]. There, the addition of the diNAcHexA moiety at the third position (onto a PglB,C,D and H-dependent Und-PP-diNAcBac-Glc disaccharide) was shown by mutagenesis to require the pglG gene whose product is predicted to be a glycosyltransferase. It also required four genes (pglJ, K, M and N) whose products operate in the step-wise synthesis of the UDP-diNAcGlcA donor [26]. Interestingly, orthologues of pglG are found in most strains of in N. gonorrhoeae and N. meningitidis (where they map just upstream of pglH/H2) but there is no evidence to date there that pglG impacts on glycoform phenotype in those backgrounds [5, 15, 18, 23, 27]. The potential distribution of pglJ, K, M and N gene orthologues in N. gonorrhoeae and N. meningitidis has not been reported.
Another feature distinguishing the pgl systems of N. gonorrhoeae and N. meningitidis from that of N. elongata subspecies glycolytica is their abilities to undergo high frequency, intrastrain glycoform antigenic variation. This phenomenon results from the presence of hypermutable, simple nucleotide repeat elements mapping within the ORFs of the pglA, pglE and pglH glycosyltransferase genes [5, 15, 18, 22, 23, 28, 29]. Stochastic changes in nucleotide repeat copy number there result in on-off glycosyltransferase expression with corresponding alterations in glycoform expression. Such repeat elements are not recognizable in pgl genes from N. elongata subspecies glycolytica. These data combined with the fact that neisserial protein-associated glycoforms possess unique immunogenic and antigenic properties [19] strongly suggests that pgl glycoforms in N. gonorrhoeae and N. meningitidis are subject to diversifying selection [5, 15, 18, 19, 22, 30–32].
In N. gonorrhoeae and N. meningitidis, the most abundant glycoproteins are the PilE pilin proteins which are the major subunit of their type IV pilus colonization (Tfp) factors [29, 33, 34]. Tfp are primary mediators of adherence to human epithelial cells [35, 36] and are required for persistence and disease in experimental gonococcal infection of human male volunteers [37]. Analogous roles for meningococcal Tfp are predicted. The glycosylation status of PilE has been linked with alterations in Tfp-associated phenotypes including autoagglutination, dynamics of organelle extrusion-retraction, adherence to human cells and the proficiency of pilin polymerization [32, 38–40]. Moreover, the glycans of PilE are oriented in a fashion such that they are exposed on the surface of intact Tfp. Furthermore, the PilE subunit protein is subject to extensive antigenic variation (changes in primary structure) in gonococci and a subset of meningococcal strains due to gene conversion-like events between partial, truncated donor alleles and an active expression locus [41]. Thus, PilE glycoproteins are subject to two levels of intrastrain structural variability: one at the level of the protein itself and the other at the level of the attached glycan. PilE intrastrain diversity further complicates attempts to define glycan function as it remains unclear if the effects of glycosylation on Tfp phenotypes are broadly applicable or variant PilE-specific. In N. elongata subspecies glycolytica, PilE is neither subject to antigenic variation nor glycosylated [25].
The complexity and variability of protein glycosylation in this genus and the commonalities of glycosylation in the two pathogenic species prompted us to determine if other human—restricted, Neisseria species similarly glycosylate proteins and if so, to assess the genotype–phenotype relationships acting there. Using comparative genomics and mass spectrometric (MS)-based glycan phenotyping, we identify here gene loss events and loss-of-function polymorphisms at multiple loci that accommodate a shift in glycoform structure occurring across the genus. Using this pgl-centric approach, we also present compelling examples as to how recombination can both reverse epistasis—associated gene inactivation as well as create seemingly discordant gene networks.
Results
Identification of a glycosyltransferase required for N. elongata subsp. glycolytica tetrasaccharide synthesis
To perform a comprehensive comparative analysis of pgl genes in Neisseria, we first determined the complete glycan synthesis pathway of N. elongata subsp. glycolytica. Studies of N. elongata subsp. glycolytica revealed a tetrasaccharide glycoform comprised of di-N-acetylbacillosamine–glucose–di-N-acetylglucuronic acid and N-acetylhexosamine [diNAcBac-Glc-HexNAc(3NAc)A-HexNAc] [25]. There, mutagenesis and glycan profiling defined the role of pglH encoding the glycosyltransferase incorporating glucose at the second position to create a disaccharide (as it does in N. gonorrhoeae and N. meningitidis [5, 15]). This structure was further extended by the PglG glycosyltransferase to generate a diNAcBac-Glc- HexNAc(3NAc)A acid trisaccharide. Synthesis of the UDP–di-N-acetylglucuronic acid precursor entailed the sequential activities of four N. elongata subsp. glycolytica enzymes (encoded by pglJ, K, M and N) starting with UDP-GlcNAc [26]. That work did not identify the glycosyltransferase responsible for addition of the terminating HexNAc moiety. We screened pgl gene clusters in genomes of species closely related to N. elongata subsp. glycolytica to identify potential glycosyltransferases. There, an ORF predicted to encode a glycosyltransferase with an N-terminal glycosyltransferase family 4 domain (PF13439) and a C-terminal glycosyltransferase group 1 domain (PF00534) was identified in an N. oralis strain. Using that nucleotide sequence, queries of N. elongata subsp. glycolytica genomes yielded an ORF sharing significant identity (designated NELON_11110) that mapped just 3´of a gene previously identified as a potential pgl-dedicated flippase (pglF-NELON_11115). Mutagenic disruption of the putative glycosyltransferase ORF led to increased mobility of the NirK glycoprotein in SDS-PAGE and MS-based analyses of purified NirK revealed the presence of a diNAcBac-Glc-HexNAc(3NAc)A trisaccharide (Fig 1). These altered phenotypes were not readily attributable to a polar effect of the mutation on distal gene expression as they were not seen following disruption of the downstream gene (NELON_11105) (Fig 1) nor in a NELON_11110 mutant carrying a short in-frame deletion (Fig 1, S1 Fig). We conclude that NELON_11110 encodes the glycosyltransferase responsible for synthesis of the mature N. elongata subsp. glycolytica tetrasaccharide and termed it pglP. A summary of the bifurcating pathways for glycan biosynthesis as defined here and in earlier studies is shown in Fig 2.
Fig 1. Identification of NELON_1110 as a pgl glycosyltransferase.
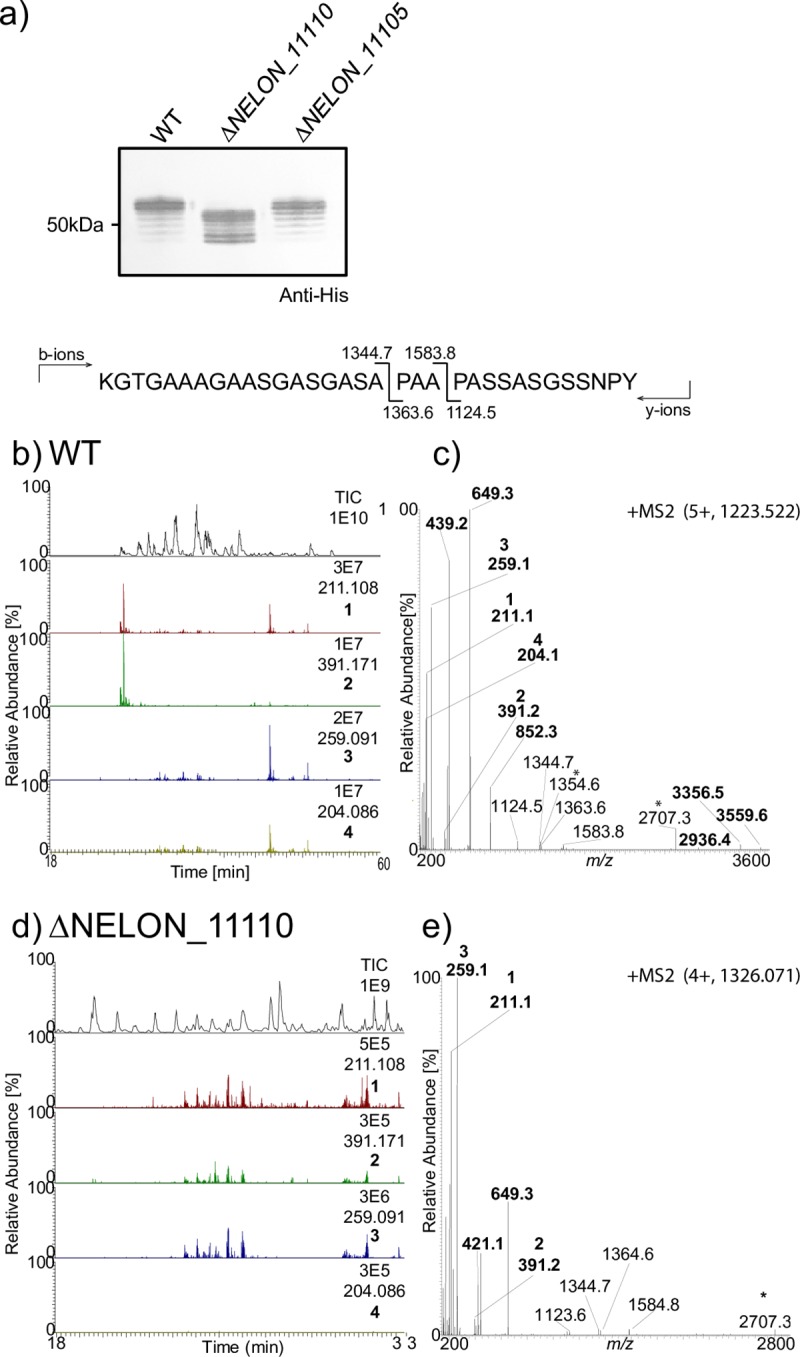
a) Immunoblot of whole-cell lysates from strains expressing NirK-His6x using the wild type (WT, KS992), ΔNELON_11110 (KS1032) and ΔNELON_11105 (NW270) strains (S1 Fig, S1 Table) using a tetra-His epitope recognizing antibody. Multiple isoforms of NirK-His are the result of macroheterogeneity (variable glycan site occupancy) as NirK has five potential sites of glycan addition. b-e) Liquid chromatography tandem MS (LC-MS2) chromatograms of the peptide shown from affinity purified NirK from WT and a ΔNELON_11110 mutant. Total ion chromatogram (TIC) intensity values represents amounts of peptides entering the mass spectrometer. The selected ion chromatograms (SIC) are of the four glycan reporter ions characteristic for a tetrasaccharide; diNAcBac at m/z 211.108 (1), diNAcBac-Hex at m/z 391.170 (2), diNAcHexA at m/z 259.093 (3) and HexNAc at m/z 204.086 (4). The MS2 spectrum demonstrates the presence of glycan reporter ions (marked in bold and numbered as defined above). b) The LC-MS2 chromatogram of the NirK derived peptide from a WT background. c) MS2 spectrum of the peptide from a WT (in panel b) background bearing a diNacBac-Hex-diNAcHexA-HexNAc tetrasaccharide. d) The LC-MS2 chromatogram the peptide from the ΔNELON_11110 background. e) MS2 spectrum of the peptide ΔNELON_11110 background (panel d) carrying the diNacBac-Hex-diNAcHexA trisaccharide.
Fig 2. Divergent pgl pathways for glycan biosynthesis defined in N. gonorrhoeae, N. meningitidis and N. elongata subsp. glycolytica.

The two non-interactive pathways are defined as utilizing the products of the pglA/E genes (top) or the pglG/H/P genes (bottom). The pglA/E pathway requires the product of galE to provide UDP–galactose while the pglG/H/P pathway is absolutely dependent on pglJ (but not pglK, pglM or pglN) to provide UDP–glucuronic acid. (See [26] and text for further details).
Distribution of pgl genes across the genus Neisseria
We analyzed the genomes of temporally and geographically distributed strains of human associated Neisseria species (S1 Dataset) for pgl gene content (Fig 3, n = number of species group genomes included). All genomes contained orthologues of the pglB/pglB2, pglC and pglD, the products of which act in the synthesis of uridine diphosphate (UDP)-sugar (PglD, PglC, and PglB-acetyltransferase domain) and the transfer of the phospho-sugar to undecaprenyl phosphate (Und-P) (PglB-phospho-glycosyltransferase domain [20, 21]). PglB catalyzes the synthesis of Und-PP-N’-diacetylbacillosamine (diNAcBac) while PglB2 catalyzes the synthesis of Und-PP-glyceramido-acetamido trideoxyhexose (GATDH) [20, 22]. Orthologues of the pglG and pglH genes were found in genomes from all human associated species across the genus and that were without exception, arrayed tandemly and mapped just upstream of the pglB/B2, C, and D genes (Fig 3 and S2 Fig). However, in a subset of N. gonorrhoeae, N. meningitidis and N. polysaccharea isolates there is a stereotypic deletion encompassing the 3´ end of pglG and the 5´ end segment of pglH (Fig 3 and S2 Fig). The deletion endpoint sequences in these mutants were highly conserved indicating the likely dissemination of a founder mutation across species by horizontal gene transfer (HGT). Orthologues of pglF (whose product is implicated in translocation of the Und-PP-oligosaccharides from a cytoplasmic to a periplasmic orientation [21] were identifiable as high-quality hits situated within 0.1–5 kB upstream of their pglB/B2, C and D genes. Thus, the synteny of the pglF, pglG, pglH, pglB/B2, pglC and pglD cluster was remarkably conserved across the genus with only limited exceptions involving the pglG/H deletion and a few interspersed ORFs seen in some deeply branching commensal genomes (S2 Fig). Accordingly, we termed this region the pgl core locus. All genomes also contained genes orthologous to pglL (also known as pglO) encoding protein–targeting oligosaccharyltransferases [21, 42] that in each instance mapped outside the core locus (S2 Fig).
Fig 3. Presence and status of pgl genes shaping glycoform diversity in neisserial species groups.

Color boxes indicate gene presence while white boxes indicate gene absence. Boxes with an X denote alleles with ORF-disrupting SNVs and/or CREE insertions while those for pglG and pglH with a diagonal line denote a conserved, inactivating deletion spanning the 3´end of pglG and the 5´end of pglH. The pglE gene with a white circle indicates alleles with the insertion of an IS element. The data are superimposed on a tree of species group relationships established using neighbour‐joining phylogeny (modified from that generated in [9]). n = number of strain genomes included. For each species group, patterns shown are ranked by relative abundance from highest (top) to lowest. Strains used are found in S1 Dataset. Note that N. meningitidis CC269 isolates bearing intact ORF pglP genes are not included here. Also, N. meningitidis pglH alleles carrying IS element insertions have been reported by others but such strains were not in the datasets used for this figure [32]. Neisseria PubMLST loci designations are found in S3 Table.
In line with previous estimates, the pglA and pglE glycosyltransferase genes (unlinked to the core loci and one another) were found in all N. gonorrhoeae, N. meningitidis and N. lactamica strains and there was evident microsynteny conservation at both loci across these species (Fig 3, S2, S3 and S4 Figs). In contrast, these genes were absent from other commensal species save for N. polysaccharea where they were differentially distributed (Fig 3, S2, S3 and S4 Figs). The pglA and pglE genes were present in 61% and 55% of N. polysaccharea isolates respectively. In those N. polysaccharea strains bearing pglE, there was conserved microsynteny with equivalent loci in N. gonorrhoeae, N. meningitidis and N. lactamica. The pglA gene is absent in 39% of N. polysaccharea isolates while for pglE, strains carried an intact allele, an allele with an insertion of an IS element or lacked the gene altogether (Fig 3, S2, S3 and S4 Figs). In those N. polysaccharea strains bearing pglE, there was clear microsynteny conservation between the loci and those in N. lactamica and some N. cinerea isolates while in those lacking pglE, synteny was seen with the equivalent loci in some strains of N. cinerea (S4 Fig). A similar relationship was detected for N. polysaccharea strains lacking pglA where there was shared microsynteny with loci in strains of N. cinerea (S3 Fig).
Next, the status of pglG in N. gonorrhoeae and N. meningitidis was investigated. Given the requirement for a UDP-glucuronic acid donor for PglG to function (as defined in N. elongata subsp. glycolytica [25]), we assessed the genus-wide status of pglJ encoding a dehydrogenase essential to synthesis of the UDP-sugar from UDP-GlcNAcA. In contrast to the widespread presence of pglG, pglJ was absent from the genomes of N. gonorrhoeae, N. meningitidis and N. lactamica species, variably distributed in genomes of N. polysaccharea strains and present in all other commensal genomes examined (Fig 3). Similarly, pglK required for the second step in synthesis of UDP-di-N-acetylglucuronic acid from UDP-glucuronic acid was absent from N. gonorrhoeae, N. meningitidis, N. polysaccharea and N. lactamica genomes but present in those of strains of the N. mucosa, N. oralis and N. elongata species groups (the last three of which all possess the pglJ—encoding dehydrogenase). The pglM and pglN genes (acting downstream of pglJ and pglK Fig 2) were limited to N. oralis and N. elongata species groups (Fig 3). Therefore, the pathogenic species together with N. polysaccharea and N. lactamica appear to be incapable of synthesizing the UDP-sugar donor utilized by PglG.
Analyses of protein glycan diversity in commensal neisserial species groups
We next sought to delineate the prevalence of protein glycosylation across neisserial species and to examine pgl genotype–phenotype relationships. As such connections were already defined for N. gonorrhoeae, N. meningitidis, and N. elongata subsp. glycolytica, focus was placed on remaining commensal species. Using a shot-gun MS approach, oligosaccharides composed of 3–4 sugar residues were identified from all isolates tested (Figs 4 and 5). While MS cannot define glycan stereochemistry, the detection of specific oxonium ions and related fragmentation products are diagnostic for particular sugars. Using this approach, correlations between pgl gene content and oligosaccharide structure were readily observed (Fig 5). For example, the incorporation of hexuronic acid or its modified derivatives at the third position was associated with the presence of pglG, pglH and pglJ while the presence of hexose at the third residue was associated with pglA and pglE. In addition, the presence of either HexNAc or HexN at the fourth position correlated with the presence of pglP together with pglG, pglH and pglJ. These correlations were emphasized by results for two strains of N. polysaccharea differing in pgl gene content and glycan structures (Figs 3, 4 and 5). These data reveal that protein glycosylation is manifest throughout the genus and establish a strong correspondence between pgl gene content and glycoform repertoires.
Fig 4. Targeted MS analysis of glycan structures shows presence of HexNAc/HexN incorporating glycoforms in select commensal strains.

The reporter ions for the protein attached glycans are: diNAcBac at m/z 229.1/211.1, diNAcBac-Hex at m/z 391.2, diNAcBac-Hex-Hex at m/z 553.2, diNAcBac-Hex-HexNAcA at m/z 608.2, diNAcBac-Hex-diNAcHexA at m/z 649.3, diNAcBac-Hex-HexNAcA-HexNAc at m/z 811.3, diNAcBac-Hex-diNAcHexA-HexNAc at m/z 852.3. GATDH at m/z 275.1/257.1, GATDH-Hex at m/z 437.1, GATDH-Hex-HexNAcA at m/z 654.2, GATDH-Hex-diNAcHexA at m/z 695.3, GATDH-Hex-HexNAcA-HexN at m/z 815.3, GATDH-Hex-diNAcHexA-HexNAc at m/z 852.3, HexN at m/z 162.1, HexNAc at m/z 204.1, HexNAcA at m/z 218.1 and diNAcHexA at m/z 259.093. A) MS2 spectrum of a glycopeptide carrying a diNAcBac-Hex-HexNAcA-HexNAc from N. polysaccharea CCUG4790. B) MS2 spectrum of a glycopeptide carrying a diNAcBac-Hex-Hex from N. polysaccharea ATCC43768. C) MS2 spectrum of a glycopeptide carrying a diNAcBac-Hex-Hex from N. lactamica ATCC23970. D) MS2 spectrum of a glycopeptide carrying a diNAcBac-Hex-HexNAcA-HexNAc from N. cinerea ATCC14685. E) MS2 spectrum of a glycopeptide carrying a GATDH-Hex-diNAcHexA-HexNAc from N. oralis CCUG26878. F) MS2 spectrum of a glycopeptide carrying a GATDH-Hex-HexNAcA-HexN from N. mucosa ATCC19696. G) MS2 spectrum of a glycopeptide carrying a diNAcBac-Hex-diNAcHexA-HexNAc from N. elongata ATCC29315. H) MS2 spectrum of a glycopeptide carrying a diNAcBac-Hex-HexNAcA-HexNAc from N. flavescens CCUG17913. All strains used are species type strains.
Fig 5. Genotype / phenotype relationships associated with glycan structures in select commensal strains.

All strains used are species type strains. See Figs 3 and 4 for details.
Species-specific pseudogenization and gene loss of pglP
As pglP acts downstream of pglG and pglJ (as defined in N. elongata subsp. glycolytica [26]), it was of interest to assess its distribution across the genus. Alleles of pglP were identified within all species groups except N. lactamica and a subset of N. meningitidis and N. polysaccharea isolates (Fig 3, S2 and S5 Figs). In these latter instances, there was microsyntenic conservation at the associated loci across all 3 species (S5 Fig). Alleles from commensal species groups N. elongata, N. oralis, N. mucosa, N subflava, and N. cinerea and N. polysaccharea encoded intact ORFs whose corresponding polypeptides were highly related to one another (S6 and S7 Figs). To examine their functional conservation, allelic replacement was used to introduce representative pglP genes from N. oralis and N. cinerea into N. elongata subsp. glycolytica where complementation was observed by immunoblotting of the NirK glycoprotein (S1 Fig).
In stark contrast, all pglP alleles in N. gonorrhoeae and N. meningitidis contained ORF-disrupting mutations (Fig 6). N. gonorrhoeae strains shared a highly conserved allele containing three single nucleotide variants (SNV) generating chain-terminating mutations precluding PglP expression: frameshifts within codons 84 and 387 and a single base substitution creating a nonsense mutation at codon 207 (Fig 6, top panel). These results were confirmed using an additional 833 genomes from gonococcal isolates of diverse geographic and temporal origins (S2 Dataset). Among the set of 107 N. meningitidis genomes representative of disease-causing isolates from the latter half of the 20th century, all but one pglP allele carried a Correia repeat enclosed element (CREE) inserted into the corresponding stop codon (Fig 6, bottom panel). CREEs are short, inverted-repeat containing, transposon-like elements distributed at high copy number within the genomes of all N. gonorrhoeae and N. meningitidis strains [43]. Together with the creation of an AT dinucleotide repeat associated with its insertion, the CREE results in the pglP ORF being extended by 18 amino acid residues. Furthermore, all but two alleles contained pseudogenizing SNVs resulting in ORF disruptions. These included those leading to frameshift mutations at codons 25, 60, 195 and 211 as well as alleles containing an additional CREE insertion between codons 67 and 68 (Fig 6, bottom). Further examination revealed that the vast majority of the N. meningitidis allele population encompassed various admixtures of the ORF-disrupting mutations, results that only can be accounted for by HGT and intragenic recombination (S3 and S4 Datasets). Assuming the presence of an intact allele reflects the ancestral state, the pglP pseudogenes and pglP absence are derived. In addition, isolates lacking pglP likely arose from a seminal deletion event that was subsequently disseminated by HGT across species (S5 Fig).
Fig 6. Pseudogenization of the pglP glycosyltransferase gene.

Positions of ORF-disrupting SNVs in N. gonorrhoeae (top) and of ORF-disrupting SNVs and CREE insertions in N. meningitidis (bottom). For N. gonorrhoeae, the results include those from S1 Dataset as well an additional 833 genomes from gonococcal isolates of diverse geographic and temporal origins (S2 Dataset). For N. meningitidis, the percentage strains carrying particular disruptions are from strains in the 107 isolate strain collection representing global diversity in the latter half of the 20th century (Neisseria PubMLST database) followed in parentheses by those in 3567 isolates in the Meningitis Research Foundation Meningococcus Genome Library. Reductions in the percentage of ORF-disrupting mutations in the latter collection are associated with an over-representation of CC269 strains.
Acquisition of an intact pglP allele in N. meningitidis CC269 isolates from commensal species
The two pglP alleles lacking ORF-disrupting SNVs and the internal ORF-disrupting CREE found in the 107 N. meningitidis strain collection genomes were from clonal complex 269 (CC269) isolates. These two were 100% identical to one another at the nucleotide level and exhibited a distinct pattern of reduced similarity to the other N. meningitidis alleles. We analyzed a larger assemblage of 3567 isolates in which CC269 complex strains genomes were well represented and found over 81% of CC269 genomes carried identical intact pglP alleles (S4 Dataset). BLAST analyses using these alleles revealed higher identities to those from N. polysaccharea and N. cinerea strains than to other N. meningitidis strains. These affinities were confirmed by phylogenetic analysis revealing that CC269 pglP alleles clustered with the N. polysaccharea and N. cinerea alleles (Fig 7, top panel, S7 Fig). These findings were in clear contrast to the species—defined associations generated by core gene-based examination of the same strains (S8 Fig). The shared identities and a likely common source of these CC269 alleles were further confirmed by SNP density plot analysis revealing their nearly identical signatures (Fig 7, bottom panel). Together, the findings indicate that the intact CC269 pglP alleles arose by HGT involving a N. polysaccharea / N. cinerea donor source. Moreover, based on the SNP density analyses of the two flanking genes, the junctions of the DNA integration of this presumed one—time event extend outside the locus defined by the three genes (S9 Fig).
Fig 7. HGT-mediated acquisition of an intact pglP allele in CC269 strains.

ORF nucleotide sequences from pglP alleles (excluding the ORF—extending CREE insertion) were aligned for phylogenetic sequence analyses using Clustal W (top panel). Multiple-alignment view of the variant nucleotide sites detected in pglP with that of FAM18 set as a reference sequence (bottom panel). The FAM18 allele contains the C-terminal, ORF—extending CREE insertion (segment in pink). Each SNP that differed from FAM18 is shown as a single line with thicker lines indicative of multiple neighboring SNPs. The sequence identities of another N. meningitidis pglP pseudogene, two CC269 alleles, two N. polysaccharea alleles and two N. cinerea alleles generated using ParSNP are shown. Note the absence of the CREE sequences in the CC269, N. polysaccharea and N. cinerea alleles and that greater than 80% of all pglP alleles in CC269 strains are 100% identical to those identified here.
Discussion
We used comparative genomics to reconstruct the evolutionary histories of protein glycosylation glycan biosynthesis in N. gonorrhoeae and N. meningitidis. The results show that current glycan repertoires in these species and congeners N. polysaccharea and N. lactamica result from acquisition of new glycosyltransferase genes coincident with loss of gene components of a progenitor pathway. We previously identified a conflict related to potential redundancy and competition for shared pathway intermediates by the PglA and PglH glycosyltransferases [15]. Together with evidence for hypomorphic pglA and pglH alleles, this led to the hypothesis that a conserved deletion inactivating both pglG and pglH in strains of N. gonorrhoeae and N. meningitidis, represented a resolution of this functional redundancy with the consequence of reduced glycan diversity [5, 15, 24]. In retrospect, it is clear that those polymorphisms are a mere subset of a larger number of genetic events associated with epistasis–involved gene decay, gene loss and ultimately replacement of a pre-existing glycan biosynthesis pathway.
Particularly striking in this context is the seeming continuum of inferred gene loss, pseudogenization and gene acquisition spanning the genus Neisseria in a pattern paralleling species group phylogenetic relationships. Here, critical delineating genetic events appear to be the loss of pglJ (encoding the dehydrogenase that generates the substrate UDP-GlcNAc from UDP-GlcNAcA) and the acquisition of pglA and pglE (encoding UDP-Gal utilizing glycosyltransferases) (Figs 2 and 3). Based on earlier findings, the absence of pglJ would be epistatic to both pglG encoding the glycosyltransferase utilizing UDP-GlcNAcA and pglP that targets the Und-PP-oligosaccharide terminating in GlcNAcA (generated by PglG) (Fig 2). Relaxed selection mediated by negative epistasis most likely accounts for the accumulation of ORF-disrupting SNVs and insertion elements in the pglP genes of N. gonorrhoeae and N. meningitidis. It is remarkable given the propensity for HGT between N. gonorrhoeae and N. meningitidis that none of the pglP mutations are shared between strains of these two species. This finding of parallel but independent evolutionary processes in N. gonorrhoeae and N. meningitidis is to our knowledge unprecedented and furthermore, indicates that pglP pseudogenization arose after the divergence of these species from a common ancestor. Although similar relaxed selection should be active on pglG in backgrounds lacking pglJ, none of the N. gonorrhoeae and N. meningitidis alleles have ORF-disrupting mutations. It is possible that such pglG alleles might accumulate missense mutations that preclude function but these are more difficult to infer from genomic sequence data alone. A further complicating factor is that the majority of N. gonorrhoeae and N. meningitidis pglG alleles are subject to high-frequency, on-off expression mediated by hypermutable, homopolymeric polyG repeat tracts. The ability to maintain pglG alleles in an off (out-of-frame) but reversible configuration might buffer against classical gene decay processes.
We suggest two non-exclusive scenarios to account for replacement of the ancestral pglG/H/P pathway by the pglA/E pathway. One would be that the presence of di- and trisaccharides terminating in galactose residues defined by the latter pathway might have altered function with regard to recognition by components of the innate or adaptive immune system or other glycan-associated phenotypes. This shift in function model might also relate to differences in the glycoprotein repertoires manifest in different species. For example, the most abundant glycoproteins in N. gonorrhoeae and N. meningitidis are the pilin subunits of their surface-displayed type IV pilus colonization factors while pilin is not subject to glycosylation in the deeply branching species N. elongata subsp. glycolytica group [25]. Another model would be that the alternate pathways come with variable metabolic costs where differences in the pools of UDP-sugars and/or Und-PP-linked saccharides exert pleiotropic effects on overlapping or converging pathways involving cell wall or LPS biosynthesis. We also examined the distribution of the galE gene encoding UDP-glucose 4-epimerase that carries out the reversible epimerization of UDP-glucose to UDP-galactose. N. gonorrhoeae and N. meningitidis (and likely other Neisseria species) cannot utilize exogenous sources of galactose, and GalE is required to generate the cognate substrates for PglA and PglE galactosyltransferases [44]. Intact galE alleles were found in all genomes of species carrying pglA and pglE but were also present in those of N. cinerea and N. elongata species groups isolates (S1 Dataset). They were differentially distributed in the N. subflava species groups isolates and absent from N. mucosa species group sequences (S1 Dataset). Thus, although galE is necessary for Gal-containing glycoforms, there is no strict correlation between its presence and glycoform status.
Along with work showing that some gonococcal strains can reacquire intact porA alleles from N. meningitidis [45], the results here confirm that intraspecies recombination can have significant consequences for pseudogene structure, distribution and stability. Moreover, the distribution of N. meningitidis pglP alleles bearing anywhere from one to five ORF-disrupting mutations is undoubtedly due to intragenic recombination. It is then impossible to determine the temporal order with which the mutations occurred or to infer the relative age of these pseudogenes by virtue of the number of accumulated mutations [46].
The widespread distribution of inactive or missing pglP alleles in N. gonorrhoeae, N. meningitidis and N. lactamica isolates suggest that as clades, the species groups may have undergone relatively recent reductions in population size (with the altered allele-bearing strain being first to pass the bottleneck). There may have been a selective sweep of the defective or missing allele through the population via natural genetic transformation. While it is difficult to differentiate between these possibilities, it is worthwhile noting that pglP status and allele distribution might be impacted by factors unrelated to PglP function per se [46]. This consideration may especially apply to the situation in N. meningitidis where the most prevalent gene disrupting mutation is the CREE insertion within the stop codon. CREEs contain active promoter elements [43] and hence, their presence there could alter transcription of the downstream gene that in this case is phoH. In fact, RNA-SEQ analyses reported that the phoH transcription start site (TSS) in such a strain occurs within the CREE [47]. It remains unclear what function PhoH serves and if the C-terminal ORF extension resulting from the CREE insertion perturbs PglP activity. Nonetheless, it is plausible that non-neutral forces may be driving the distribution and retention of the pglP pseudogene in N. meningitidis. Although the TSS for wildtype phoH has yet to be determined for any Neisseria species, pglP deletion might likewise impact phoH expression.
The results here also emphasize the potential for HGT to generate unbalanced or discordant polymorphisms due to the multi-locus nature of pgl gene networks. These findings contrast strongly with the genetic events underlying capsule serotype/serogroup switching in Streptococcus pneumoniae and N. meningitidis that involve recombination events spanning a single, large locus in a “plug and play”-type switching process [2, 48]. Discordant gene interactions are particularly evident in the case of N. polysaccharea isolates where some strains carry seemingly incompatible gene sets such as intact pglP alleles in backgrounds lacking pglG/H and/or pglJ. In fact, despite the limited number of genomes examined, N. polysaccharea strains exhibit extreme levels of diversity in pgl gene status that appear to result from interspecies HGT. These findings are consonant with others showing that N. polysaccharea isolates form a polyphyletic group [9, 25]. It is also striking that the pgl gene content of isolates within this single species group encompasses the majority of patterns seen at the macroevolutionary level. Together with the other findings here, we suggest that N. polysaccharea may act as a nexus for gene flow bridging pathogen and commensal species.
Another example of discordant pgl polymorphisms generated by recombination is found in the CC269 strains possessing an intact pglP allele. These strains all carry the pglG/H deletion polymorphism and lack pglJ, conditions that would preclude PglP function (S2 and S3 Datasets). Thus, if there is a selective advantage imparted by the genome import event, it likely relates to a linked gene with which pglP hitchhikes. Given this situation, the current prevalence of CC269 strains as causes of N. meningitidis invasive disease [49] and the seemingly strong selective pressure for pglP gene decay evident in other N. meningitidis strains, it will be of interest to assess the fate of pglP in CC269 lineage isolates over time.
Materials and methods
Bacterial strains and culture conditions
N. elongata subsp. glycolytica ATCC 29315 was used for mutagenesis and genetic complementation studies involving pglP [62]. Other bacterial strains used in this study are described in S1 Table and were grown on conventional GC medium as described previously [50]. Antibiotics were used for selection of Neisseria elongata subsp. glycolytica transformants at the following concentrations: streptomycin, 750 μg/mL; kanamycin, 50 μg/mL; and chloramphenicol, 10 μg/mL.
Directed mutagenesis of Nelon_11110 and Nelon_11105 in N. elongata subsp. glycolytica
The region encompassing Nelon_11110 and flanking sequences from strain KS944 (N. elongata subsp. glycolytica ATCC 29315) was PCR amplified using primers av2934 and av2935 and the resulting product TA-cloned into the pCR2.1-TOPO vector. DNA of the resulting plasmid pAK220 was digested with HincII and StuI (to delete a 587bp intragenic region of the Nelon_11110 ORF) and ligated with the kanR gene cassette from pKan (generated by HincII digestion) to generate AK227. A similar strategy was used to disrupt Nelon_11105 where the kanR cassette was inserted 325bp into the ORF while concurrently deleting the ORF C-terminus to generate strain NW270. Flanking regions to Nelon_11105 were PCR amplified using primers nw180/nw181 and nw184/nw185 and Gibson assembled to the kanR cassette amplified with primers nw188/nw189. These constructs (S1 Fig) were introduced by transformation into N. elongata subsp. glycolytica strain KS992 (that carries a nirK-His allele (in which the NirK ORF is translationally fused to a 6Xhistidine C-terminal extension). The strains, plasmids and oligonucleotide primers used here are found in S1 and S2 Tables.
Allelic exchange of the pglP locus in N. elongata subsp. glycolytica
The introduction of defined, marker-less pglP alleles into N. elongata subsp. glycolytica was performed through modification of a previously established two-step mutagenesis strategy (S1 Fig). The method uses a two-gene cassette containing both a selectable marker and a counter selectable marker (rpsL+). The gene cassette originally employed in N. gonorrhoeae utilizes an ermC´ as a selectable marker [51]. As selection for the erythromycin resistance marker in N. elongata subsp. glycolytica was problematic, a modified gene cassette was constructed in which the ermC´gene was replaced by a kanR gene cassette. This was done by first digesting pFLOB4300 with SacI and NsiI (to release ermC’) and enzymatic treatment to generate blunt ends. This fragment was then ligated to the HincII digestion-generated fragment containing kanR from pKan to generate plasmid pKP79. To generate the streptomycin resistance background in N. elongata subsp. glycolytica, strain KS944 was transformed with rps marker DNA from N. gonorrhoeae strain N400 (that naturally carries the streptomycin resistance point mutation changing amino acid 43 of 30S ribosomal protein S12 from a lysine to an arginine). The ensuing strain (NK2259) was then transformed so as to carry the nirK-His allele from strain KS992 to generate strain NW37. Transformation of NW37 with pKP79 DNA and selection for kanamycin resistance (generating strain NW154) resulted in the replacement of the pglP ORF by the kanR/rpsL+ gene cassette and concurrent streptomycin sensitivity. Transformation of NW154 with DNA bearing homologous sequences flanking pglP and selection for streptomycin resistance results in precise allelic replacement of the pglP ORF. Donor DNAs used for allelic replacement were generated by PCR and Gibson assembly. To generate strain NW180 carrying an in–frame deletion encompassing residues 74–148 of the pglP ORF, primer pairs nw92/nw153 and nw154/nw98 were used to generate overlapping PCR products and Gibson assembled. To generate strain NW182 carrying the pglP allele ORF from N. oralis strain F0314, primer pairs nw92/111, and nw114/98 were used for PCR of the pglP flanking sequences from KS944 while primer pair nw112/113 was used to PCR the N. oralis pglP ORF (HMPREF9016_01275). These fragments were Gibson assembled and amplified by PCR. To generate strain NW212 carrying the pglP allele ORF from N. cinerea strain ATCC 14685, primer pairs nw92/152 and nw122/98 were used for PCR of the pglP flanking sequences from KS944 while primer pair nw151/121 was used to PCR amplify the N. cinerea pglP ORF (NEICINOT_04976). These fragments were Gibson assembled and amplified by PCR. As a positive control to “rescue” the wildtype allele, strain NW254 was generated by transformation using genomic DNA from strain KS944. All constructs were introduced into NW154 by transformation with selection for streptomycin resistance, scored for kanamycin sensitivity and verified by PCR and DNA sequencing. The strains, plasmids and oligonucleotide primers used here are found in S1 and S2 Tables.
Genome analyses and bioinformatics
The presence and status of pgl genes within genomes from isolates across the genus were determined using BLASTn and BLASTp queries of genome sequences using the Neisseria PubMLST (http://pubmlst.org/neisseria/) and Meningitis Research Foundation Meningococcus Genome Library (http://www.meningitis.org/research/genome) databases. Forward searches utilized defined pgl alleles from N. gonorrhoeae and N. elongata subsp. subspecies glycolytica (S3 Table). Specific genomes / strains utilized are found in S1–S4 Datasets. To identify potential distant orthologues, the BLAST E score cutoff was set to 10−5 and sequence alignments were manually examined. Microsynteny at discrete loci was assessed by monitoring Blast hits with nucleotide sequence start and end coordinates within defined sequence bins. Microsynteny was further validated using the compare region viewer function in PATRIC [52], the gene cluster function in KEGG gene database [53] and where necessary, local genome alignment using progressiveMauve [54]. For assessing the relatedness of pglP alleles and associated intraspecies HGT, a reference nucleotide sequence from FAM18 (AM421808.1) comprising the region from the periplasmic protein (NMC0788) to phoH (NMC0784) was used in a BLASTn query with default parameters against a BLAST database built using the contigs from selected PubMLST Neisseria genomes. Top hits were extracted from the contigs. Nucleotide sequences were aligned using MAFFT (version 7.017) [55] and a phylogenetic tree of PglP constructed in Geneious 9.1.7 (https://www.geneious.com) using the RAxML plugin [56]. The phylogeny was visualized and annotated in FigTree (http://tree.bio.ed.ac.uk/software/figtree/). The SNP density plot was generated in and exported from Geneious. Further details of pglP phylogenetic analyses including SNV distribution determination are found in S3 and S4 Datasets.
Analyses of Single Nucleotide Variant (SNV) and CREE distribution in gonococcal and meningococcal pglP Alleles
The distribution of specific ORF-disrupting polymorphisms was determined by BLASTN and BLASTP analyses using a control set of pglP gene and ORF sequences with default settings in BIGSdb / Neisseria PubMLST. In conjunction with these methods, focused BlastN searches using SNV-specific oligonucleotide sequences were employed (S1 Text). 100% scores indicated presence of a SNV or CREE presence. Isolates with poorer hits (90–99%) were manually checked for absence or presence of a mutation. Data for specific strains can be found in S3 and S4 Datasets.
SDS/PAGE, immunoblotting and affinity purification of NirK
Procedures for protein electrophoresis, immunoblotting and purification of NirK-His-tagged proteins have been previously described [26].
Targeted mass spectrometric glycan analyses
Conditions for the MS-based analyses of glycosylated NirK using in-gel protein, reverse-phase liquid chromatography- tandem MS (LC-MS2) analysis of proteolytic peptides, electron transfer dissociation (ETD) experiments and data analyses have been previously defined [26].
LC-MS analysis of protein glycans using membrane extracts from commensal strains
Periplasmic and cytosolic protein fractions were generated as previously described [29] with the following modifications: protein precipitates were washed 10 times with 50mM pH 7.8 TEAB buffer (buffer B) utilizing a 3K cut-of Amicon prior to enzymatic digest. Protein concentration was determined twice on a Qubit and adjusted to 400μg for all protein samples. Adjusted protein samples were resuspended, reduced (DTT 10mM) for 30 min and alkylated (IAA 20mM) for 30 min (dark) in buffer A (buffer B + 6M urea, 1.5M ThioUrea with proteinase Inhibitor (PI, Roche complete EDTA free) and Phosphatase inhibitor (PhosI, Roche phosphostop EasyPack) in 50mM sodium orthovanadate) on a 10KDa cut-of spin filter (Amicon). Reduced and alkylated proteins were washed with Buffer B by spin filtering. Subsequent digest was done in 200μl buffer B, adding 2U of LysC (RT for 2H) before overnight digestion with 3% w/w trypsin at 37°C. Digested peptide samples were moved to a new low bind Eppendorf tube, added 2% FA and spun at 14K g for 10 min to precipitate lipids. The supernatants were transferred to a new tube for TiO2 (titanium dioxide) and SIMAC (sequential elution from Immobilized metal affinity chromatography) purification. TiO2 and SIMAC affinity purification were essentially done as previously described in reverse order. i.e the final TiO2 (high pH eluate was used for the SIMAC affinity purification, leaving a total of 4 samples. Each sample were desalted with R2/R3 as described [57]. Each sample was lyophilized prior to analysis on the Thermo Orbitrap Fusion. The dried peptides were dissolved in 0.1% formic acid and injected into an in-house packed 17 cm × 100 μ m Reprosil-Pur C18-AQ column (3 μ m; Dr. Maisch GmbH, Germany) using an Easy-LC nano-HPLC (Thermo Scientific,Germany).
Further details of MS-based characterization of glycoprotein-derived glycans are available upon request.
Supporting information
(A, B). Detection of the NirK-His glycoprotein in N. elongata subsp. glycolytica pgl mutant / variant backgrounds by immunoblotting with polyHis-epitope recognizing mAb (C). WT: KS944; pglC: KS994, pglPS74-R148: NW180; pglP::kan/rpsL+: NW154; pglP rescue: NW254; pglPN. oralis: NW182 and pglPN. cinerea: NW212. Multiple isoforms of NirK-His are the result of macrohereogeneity (variable glycan site occupancy) as NirK has five sites of glycan occupancy.
(TIF)
Genomes shown as lacking pglG and pglH retain the canonical pglG3´/H5´ spanning deletion. The asterisks for pglP denote a pseudogene. Other genes are annotated as follows: diagonal line fill = O-antigen ligase like (UNIPROT D7N379 in N. oralis); horizontal line fill = HAD hydrolase (UNIPROT D7N385 in N. oralis); vertical line fill = formyl transferase (UNIPROT D7N386 in N. oralis) and blank fill = three unannotated ORFs (NELON_10550, NELON_10555 and NELON_10560 in N. elongata subspecies glycolytica).
(TIF)
% values are percentage of strains in the species group with that configuration. Other genes shown are annotated as encoding 3-oxoacyl-[acyl-carrier-protein] synthase 2 (KASII in Ngo—UNIPROT Q5F603), a transposase (IS in Nme and Npo—IS110) and a potassium transporter (kefC in Npo—UNIPROT E2PBV4).
(TIF)
% values are percentage of strains in the species group with that configuration. Other genes shown are annotated as encoding a putrescine-binding periplasmic protein (potF in Ngo—UNIPROT Q5FA28) and an uncharacterized protein (blue in Nlact—UNIPROT E4ZEM6)
(TIF)
% values are percentage of strains in the species group with that configuration. Other genes shown are annotated as encoding an uncharacterized protein (grey in Ngo–UNIPROT Q5F9H4), a metalloprotease (pmbA in Nsub -UNIPROT C0EPK5), an NADH-dependent flavin oxidoreductase (nox in Nmuc–UNIPROT F9EUJ4) and an uncharacterized protein (brown in Nmuc—UNIPROT F9EUJ7).
(TIF)
Selected PglP alleles/ORFs were aligned with MAFFT using Geneious and subsequently used to generate an identity and a similarity—based matrix. The two tables were imported into Excel and combined into a single panel (top). Alleles of pglP were selected from across the genus as representatives of each species group, aligned using MAFFT and visualized using Jalview (bottom). Strains in which the allele is located within the core pgl locus begin with an asterix (*) and protein regions with >90% conservation are highlighted in red. The locations of the two glycosyltransferase domains (as predicted by NCBI) are underlined in the alignment (blue line = pfam13439 and green line = pfam00534).
(TIF)
A maximum likelihood phylogenetic tree of aligned pglP nucleotide sequences was generated using MEGA (Molecular Evolutionary Genetics Analysis) V7 using the Tamura-Nei model [58]. A total of 500 bootstrap iterations were undertaken allowing a confidence interval for each node to be determined. The resulting consensus tree was then annotated for each Neisseria species. The analysis involved 204 nucleotide sequences.
(TIF)
De novo assemblies from PubMLST were annotated using Prokka (version 1.14) [59]. A genus-level phylogeny was constructed with FastTree (version 2.1.11) [60] using a core genome alignment generated by Roary (version 3.12.0) [61] with a minimum BLASTP identity of 90%.
(TIF)
See Fig 7 and text for further details.
(TIF)
(PDF)
(PDF)
(PDF)
(PDF)
Gonococcal isolates used originate from those used in a prior study of protein antigen distribution across the genus [63]. Meningococcal isolates originate from a public database which represents global meningococcal diversity in the 20th century. The remaining species include all respective isolates with complete genome sequences available at the Neisseria PubMLST database at the time of this work. Colored cells indicate presence of a gene, crossed cells (X) denote out-of-frame alleles (exclusive of those resulting from phase variable mutational hotspots, the presence of transposon elements is marked with TN, diagonal lines (/) indicate that gene is partially present and isolates with two alleles are marked with 2. Isolate ID numbers are derived from the Neisseria PubMLST database.
(XLSX)
Table shows strain name, date and country of isolation and multi-locus sequence type (MLST). Isolate ID numbers are derived from the Neisseria PubMLST database.
(XLSX)
Table shows strain name, date and country of isolation, serogroup, as well as sequence type and clonal complex designation. Colored cells indicate the presence of ORF–disrupting SNVs or CREE insertions (see Fig 6 for details). ID numbers are derived from the Neisseria PubMLST database.
(XLSX)
Table shows strain name, date and country of isolation, serogroup, as well as sequence type and clonal complex designation. Colored cells indicate the presence of ORF–disrupting SNVs or CREE insertions (see Fig 6 for details). ID numbers are derived from the Neisseria PubMLST database.
(XLSX)
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
CH, JHA, NH, RV, ÅV and MK were supported in part by Research Council of Norway grant 214442 (https://www.forskningsradet.no/) and by the Center for Integrative Microbial Evolution at the Department of Biosciences, University of Oslo (https://www.mn.uio.no/ibv/english/research/groups/cime/, https://www.mn.uio.no/forskning/sentre-satsinger/endringsmiljoer/). YHG was supported by the Richard and Susan Smith Family Foundation (https://www.smithfamilyfoundation.net/) and NIH R01 AI132606 (https://grants.nih.gov/grants/funding/r01.htm). KCM was supported by the National Science Foundation Graduate Research Fellowship Program (https://www.nsfgrfp.org/). This publication made use of the Neisseria Multi Locus Sequence Typing website (http://pubmlst.org/neisseria/) developed by Keith Jolley and sited at the University of Oxford. The development of this site has been funded by the Wellcome Trust and European Union. This study also made use of the Meningitis Research Foundation Meningococcus Genome Library (http://www.meningitis.org/research/genome) developed by Public Health England, the Wellcome Trust Sanger Institute, and the University of Oxford as a collaboration. That project is funded by the Meningitis Research Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Mostowy RJ, Holt KE. Diversity-Generating Machines: Genetics of Bacterial Sugar-Coating. Trends Microbiol. 2018;26(12):1008–21. 10.1016/j.tim.2018.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bentley SD, Aanensen DM, Mavroidi A, Saunders D, Rabbinowitsch E, Collins M, et al. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2006;2(3):e31 10.1371/journal.pgen.0020031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L, Wang Q, Reeves PR. The variation of O antigens in gram-negative bacteria. Subcell Biochem. 2010;53:123–52. 10.1007/978-90-481-9078-2_6 . [DOI] [PubMed] [Google Scholar]
- 4.Eichler J, Koomey M. Sweet New Roles for Protein Glycosylation in Prokaryotes. Trends Microbiol. 2017;25(8):662–72. 10.1016/j.tim.2017.03.001 . [DOI] [PubMed] [Google Scholar]
- 5.Borud B, Anonsen JH, Viburiene R, Cohen EH, Samuelsen AB, Koomey M. Extended glycan diversity in a bacterial protein glycosylation system linked to allelic polymorphisms and minimal genetic alterations in a glycosyltransferase gene. Molecular microbiology. 2014;94(3):688–99. Epub 2014/09/13. 10.1111/mmi.12789 . [DOI] [PubMed] [Google Scholar]
- 6.Nothaft H, Scott NE, Vinogradov E, Liu X, Hu R, Beadle B, et al. Diversity in the protein N-glycosylation pathways within the Campylobacter genus. Molecular & cellular proteomics: MCP. 2012;11(11):1203–19. Epub 2012/08/04. 10.1074/mcp.M112.021519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott NE, Kinsella RL, Edwards AV, Larsen MR, Dutta S, Saba J, et al. Diversity within the O-linked protein glycosylation systems of acinetobacter species. Molecular & cellular proteomics: MCP. 2014;13(9):2354–70. 10.1074/mcp.M114.038315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coyne MJ, Fletcher CM, Chatzidaki-Livanis M, Posch G, Schaffer C, Comstock LE. Phylum-wide general protein O-glycosylation system of the Bacteroidetes. Molecular microbiology. 2013;88(4):772–83. 10.1111/mmi.12220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett JS, Jolley KA, Earle SG, Corton C, Bentley SD, Parkhill J, et al. A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology (Reading, England). 2012;158(Pt 6):1570–80. Epub 2012/03/17. 10.1099/mic.0.056077-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tønjum T. Genus I. Neisseria. In: Garrity G. M. BDJ, Krieg N. R, Staley J. R, editor. Bergey’s Manual of Systematic Bacteriology. New York: Springer-Verlag; 2005. p. 777–98. [Google Scholar]
- 11.Marri PR, Paniscus M, Weyand NJ, Rendon MA, Calton CM, Hernandez DR, et al. Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PloS one. 2010;5(7):e11835 Epub 2010/08/03. 10.1371/journal.pone.0011835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snyder LA, Saunders NJ. The majority of genes in the pathogenic Neisseria species are present in non-pathogenic Neisseria lactamica, including those designated as 'virulence genes'. BMC Genomics. 2006;7:128 10.1186/1471-2164-7-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stabler RA, Marsden GL, Witney AA, Li Y, Bentley SD, Tang CM, et al. Identification of pathogen-specific genes through microarray analysis of pathogenic and commensal Neisseria species. Microbiology (Reading, England). 2005;151(Pt 9):2907–22. 10.1099/mic.0.28099-0 . [DOI] [PubMed] [Google Scholar]
- 14.Veyrier FJ, Biais N, Morales P, Belkacem N, Guilhen C, Ranjeva S, et al. Common Cell Shape Evolution of Two Nasopharyngeal Pathogens. PLoS Genet. 2015;11(7):e1005338 10.1371/journal.pgen.1005338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borud B, Viburiene R, Hartley MD, Paulsen BS, Egge-Jacobsen W, Imperiali B, et al. Genetic and molecular analyses reveal an evolutionary trajectory for glycan synthesis in a bacterial protein glycosylation system. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(23):9643–8. Epub 2011/05/25. 10.1073/pnas.1103321108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aas FE, Li X, Edwards J, Hongro Solbakken M, Deeudom M, Vik A, et al. Cytochrome c-based domain modularity governs genus-level diversification of electron transfer to dissimilatory nitrite reduction. Environmental microbiology. 2015;17(6):2114–32. Epub 2014/10/21. 10.1111/1462-2920.12661 . [DOI] [PubMed] [Google Scholar]
- 17.Clemence MEA, Maiden MCJ, Harrison OB. Characterization of capsule genes in non-pathogenic Neisseria species. Microbial genomics. 2018;4(9). 10.1099/mgen.0.000208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borud B, Barnes GK, Brynildsrud OB, Fritzsonn E, Caugant DA. Genotypic and Phenotypic Characterization of the O-Linked Protein Glycosylation System Reveals High Glycan Diversity in Paired Meningococcal Carriage Isolates. Journal of bacteriology. 2018;200(16). Epub 2018/03/21. 10.1128/jb.00794-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borud B, Aas FE, Vik A, Winther-Larsen HC, Egge-Jacobsen W, Koomey M. Genetic, structural, and antigenic analyses of glycan diversity in the O-linked protein glycosylation systems of human Neisseria species. Journal of bacteriology. 2010;192(11):2816–29. Epub 2010/04/07. 10.1128/JB.00101-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartley MD, Morrison MJ, Aas FE, Borud B, Koomey M, Imperiali B. Biochemical characterization of the O-linked glycosylation pathway in Neisseria gonorrhoeae responsible for biosynthesis of protein glycans containing N,N'-diacetylbacillosamine. Biochemistry. 2011;50(22):4936–48. Epub 2011/05/06. 10.1021/bi2003372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aas FE, Vik A, Vedde J, Koomey M, Egge-Jacobsen W. Neisseria gonorrhoeae O-linked pilin glycosylation: functional analyses define both the biosynthetic pathway and glycan structure. Molecular microbiology. 2007;65(3):607–24. Epub 2007/07/05. 10.1111/j.1365-2958.2007.05806.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chamot-Rooke J, Rousseau B, Lanternier F, Mikaty G, Mairey E, Malosse C, et al. Alternative Neisseria spp. type IV pilin glycosylation with a glyceramido acetamido trideoxyhexose residue. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(37):14783–8. Epub 2007/09/07. 10.1073/pnas.0705335104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Power PM, Roddam LF, Rutter K, Fitzpatrick SZ, Srikhanta YN, Jennings MP. Genetic characterization of pilin glycosylation and phase variation in Neisseria meningitidis. Molecular microbiology. 2003;49(3):833–47. Epub 2003/07/17. 10.1046/j.1365-2958.2003.03602.x . [DOI] [PubMed] [Google Scholar]
- 24.Johannessen C, Koomey M, Borud B. Hypomorphic glycosyltransferase alleles and recoding at contingency loci influence glycan microheterogeneity in the protein glycosylation system of Neisseria species. Journal of bacteriology. 2012;194(18):5034–43. Epub 2012/07/17. 10.1128/JB.00950-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anonsen JH, Vik A, Borud B, Viburiene R, Aas FE, Kidd SW, et al. Characterization of a Unique Tetrasaccharide and Distinct Glycoproteome in the O-Linked Protein Glycosylation System of Neisseria elongata subsp. glycolytica. Journal of bacteriology. 2016;198(2):256–67. Epub 2015/10/21. 10.1128/JB.00620-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang N, Anonsen JH, Viburiene R, Lam JS, Vik A, Koomey M. Disrupted Synthesis of a Di-N-acetylated Sugar Perturbs Mature Glycoform Structure and Microheterogeneity in the O-Linked Protein Glycosylation System of Neisseria elongata subsp. glycolytica. Journal of bacteriology. 2019;201(1). Epub 2018/10/17. 10.1128/jb.00522-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kahler CM, Martin LE, Tzeng YL, Miller YK, Sharkey K, Stephens DS, et al. Polymorphisms in pilin glycosylation Locus of Neisseria meningitidis expressing class II pili. Infection and immunity. 2001;69(6):3597–604. Epub 2001/05/12. 10.1128/IAI.69.6.3597-3604.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Power PM, Roddam LF, Dieckelmann M, Srikhanta YN, Tan YC, Berrington AW, et al. Genetic characterization of pilin glycosylation in Neisseria meningitidis. Microbiology (Reading, England). 2000;146 (Pt 4):967–79. Epub 2000/04/28. 10.1099/00221287-146-4-967 . [DOI] [PubMed] [Google Scholar]
- 29.Vik A, Aas FE, Anonsen JH, Bilsborough S, Schneider A, Egge-Jacobsen W, et al. Broad spectrum O-linked protein glycosylation in the human pathogen Neisseria gonorrhoeae. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(11):4447–52. Epub 2009/03/03. 10.1073/pnas.0809504106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamelas A, Harris SR, Roltgen K, Dangy JP, Hauser J, Kingsley RA, et al. Emergence of a new epidemic Neisseria meningitidis serogroup A Clone in the African meningitis belt: high-resolution picture of genomic changes that mediate immune evasion. mBio. 2014;5(5):e01974–14. Epub 2014/10/23. 10.1128/mBio.01974-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viburiene R, Vik A, Koomey M, Borud B. Allelic variation in a simple sequence repeat element of neisserial pglB2 and its consequences for protein expression and protein glycosylation. Journal of bacteriology. 2013;195(15):3476–85. Epub 2013/06/05. 10.1128/JB.00276-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gault J, Ferber M, Machata S, Imhaus AF, Malosse C, Charles-Orszag A, et al. Neisseria meningitidis Type IV Pili Composed of Sequence Invariable Pilins Are Masked by Multisite Glycosylation. PLoS pathogens. 2015;11(9):e1005162 Epub 2015/09/15. 10.1371/journal.ppat.1005162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anonsen JH, Vik A, Egge-Jacobsen W, Koomey M. An extended spectrum of target proteins and modification sites in the general O-linked protein glycosylation system in Neisseria gonorrhoeae. Journal of proteome research. 2012;11(12):5781–93. Epub 2012/10/04. 10.1021/pr300584x . [DOI] [PubMed] [Google Scholar]
- 34.Ku SC, Schulz BL, Power PM, Jennings MP. The pilin O-glycosylation pathway of pathogenic Neisseria is a general system that glycosylates AniA, an outer membrane nitrite reductase. Biochemical and biophysical research communications. 2009;378(1):84–9. Epub 2008/11/18. 10.1016/j.bbrc.2008.11.025 . [DOI] [PubMed] [Google Scholar]
- 35.Swanson J. Studies on gonococcus infection. IV. Pili: their role in attachment of gonococci to tissue culture cells. J Exp Med. 1973;137(3):571–89. 10.1084/jem.137.3.571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Virji M, Saunders JR, Sims G, Makepeace K, Maskell D, Ferguson DJ. Pilus-facilitated adherence of Neisseria meningitidis to human epithelial and endothelial cells: modulation of adherence phenotype occurs concurrently with changes in primary amino acid sequence and the glycosylation status of pilin. Molecular microbiology. 1993;10(5):1013–28. Epub 1993/12/01. 10.1111/j.1365-2958.1993.tb00972.x . [DOI] [PubMed] [Google Scholar]
- 37.Hobbs MM, Sparling PF, Cohen MS, Shafer WM, Deal CD, Jerse AE. Experimental Gonococcal Infection in Male Volunteers: Cumulative Experience with Neisseria gonorrhoeae Strains FA1090 and MS11mkC. Front Microbiol. 2011;2:123 10.3389/fmicb.2011.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jen FE, Warren MJ, Schulz BL, Power PM, Swords WE, Weiser JN, et al. Dual pili post-translational modifications synergize to mediate meningococcal adherence to platelet activating factor receptor on human airway cells. PLoS pathogens. 2013;9(5):e1003377 10.1371/journal.ppat.1003377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jennings MP, Jen FE, Roddam LF, Apicella MA, Edwards JL. Neisseria gonorrhoeae pilin glycan contributes to CR3 activation during challenge of primary cervical epithelial cells. Cellular microbiology. 2011;13(6):885–96. Epub 2011/03/05. 10.1111/j.1462-5822.2011.01586.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vik A, Aspholm M, Anonsen JH, Borud B, Roos N, Koomey M. Insights into type IV pilus biogenesis and dynamics from genetic analysis of a C-terminally tagged pilin: a role for O-linked glycosylation. Molecular microbiology. 2012;85(6):1166–78. Epub 2012/08/14. 10.1111/j.1365-2958.2012.08166.x . [DOI] [PubMed] [Google Scholar]
- 41.Zhang QY, DeRyckere D, Lauer P, Koomey M. Gene conversion in Neisseria gonorrhoeae: evidence for its role in pilus antigenic variation. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(12):5366–70. 10.1073/pnas.89.12.5366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Power PM, Seib KL, Jennings MP. Pilin glycosylation in Neisseria meningitidis occurs by a similar pathway to wzy-dependent O-antigen biosynthesis in Escherichia coli. Biochemical and biophysical research communications. 2006;347(4):904–8. Epub 2006/07/28. 10.1016/j.bbrc.2006.06.182 . [DOI] [PubMed] [Google Scholar]
- 43.Siddique A, Buisine N, Chalmers R. The transposon-like Correia elements encode numerous strong promoters and provide a potential new mechanism for phase variation in the meningococcus. PLoS Genet. 2011;7(1):e1001277 Epub 2011/02/02. 10.1371/journal.pgen.1001277 ; PubMed Central PMCID: PMC3024310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robertson BD, Frosch M, van Putten JP. The role of galE in the biosynthesis and function of gonococcal lipopolysaccharide. Molecular microbiology. 1993;8(5):891–901. Epub 1993/05/01. 10.1111/j.1365-2958.1993.tb01635.x . [DOI] [PubMed] [Google Scholar]
- 45.Ison CA, Golparian D, Saunders P, Chisholm S, Unemo M. Evolution of Neisseria gonorrhoeae is a continuing challenge for molecular detection of gonorrhoea: false negative gonococcal porA mutants are spreading internationally. Sexually transmitted infections. 2013;89(3):197–201. Epub 2012/12/18. 10.1136/sextrans-2012-050829 . [DOI] [PubMed] [Google Scholar]
- 46.Kuo CH, Ochman H. The extinction dynamics of bacterial pseudogenes. PLoS Genet. 2010;6(8). Epub 2010/08/12. 10.1371/journal.pgen.1001050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heidrich N, Bauriedl S, Barquist L, Li L, Schoen C, Vogel J. The primary transcriptome of Neisseria meningitidis and its interaction with the RNA chaperone Hfq. Nucleic Acids Res. 2017;45(10):6147–67. 10.1093/nar/gkx168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swartley JS, Marfin AA, Edupuganti S, Liu LJ, Cieslak P, Perkins B, et al. Capsule switching of Neisseria meningitidis. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(1):271–6. Epub 1997/01/07. 10.1073/pnas.94.1.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill DM, Lucidarme J, Gray SJ, Newbold LS, Ure R, Brehony C, et al. Genomic epidemiology of age-associated meningococcal lineages in national surveillance: an observational cohort study. Lancet Infect Dis. 2015;15(12):1420–8. 10.1016/S1473-3099(15)00267-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tonjum T, Freitag NE, Namork E, Koomey M. Identification and characterization of pilG, a highly conserved pilus-assembly gene in pathogenic Neisseria. Molecular microbiology. 1995;16(3):451–64. Epub 1995/05/01. 10.1111/j.1365-2958.1995.tb02410.x . [DOI] [PubMed] [Google Scholar]
- 51.Johnston DM, Cannon JG. Construction of mutant strains of Neisseria gonorrhoeae lacking new antibiotic resistance markers using a two gene cassette with positive and negative selection. Gene. 1999;236(1):179–84. 10.1016/s0378-1119(99)00238-3 . [DOI] [PubMed] [Google Scholar]
- 52.Wattam AR, Davis JJ, Assaf R, Boisvert S, Brettin T, Bun C, et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017;45(D1):D535–D42. 10.1093/nar/gkw1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44(D1):D457–62. 10.1093/nar/gkv1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS one. 2010;5(6):e11147 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution. 2013;30(4):772–80. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thingholm TE, Larsen MR. Sequential Elution from IMAC (SIMAC): An Efficient Method for Enrichment and Separation of Mono- and Multi-phosphorylated Peptides. In: vS L., editor. Phospho-Proteomics Methods in Molecular Biology. 1355 New York, NY 2016. [DOI] [PubMed] [Google Scholar]
- 58.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular biology and evolution. 2016;33(7):1870–4. 10.1093/molbev/msw054 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics (Oxford, England). 2014;30(14):2068–9. Epub 2014/03/20. 10.1093/bioinformatics/btu153 . [DOI] [PubMed] [Google Scholar]
- 60.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PloS One. 2010;5(3):e9490 Epub 2010/03/13. 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics (Oxford, England). 2015;31(22):3691–3. Epub 2015/07/23. 10.1093/bioinformatics/btv421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bovre K, Holten E. Neisseria elongata sp.nov., a rod-shaped member of the genus Neisseria. Re-evaluation of cell shape as a criterion in classification. J Gen Microbiol. 1970;60(1):67–75. 10.1099/00221287-60-1-67 . [DOI] [PubMed] [Google Scholar]
- 63.Muzzi A, Mora M, Pizza M, Rappuoli R, Donati C. Conservation of meningococcal antigens in the genus Neisseria. mBio. 2013;4(3):e00163–13. Epub 2013/06/14. 10.1128/mBio.00163-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A, B). Detection of the NirK-His glycoprotein in N. elongata subsp. glycolytica pgl mutant / variant backgrounds by immunoblotting with polyHis-epitope recognizing mAb (C). WT: KS944; pglC: KS994, pglPS74-R148: NW180; pglP::kan/rpsL+: NW154; pglP rescue: NW254; pglPN. oralis: NW182 and pglPN. cinerea: NW212. Multiple isoforms of NirK-His are the result of macrohereogeneity (variable glycan site occupancy) as NirK has five sites of glycan occupancy.
(TIF)
Genomes shown as lacking pglG and pglH retain the canonical pglG3´/H5´ spanning deletion. The asterisks for pglP denote a pseudogene. Other genes are annotated as follows: diagonal line fill = O-antigen ligase like (UNIPROT D7N379 in N. oralis); horizontal line fill = HAD hydrolase (UNIPROT D7N385 in N. oralis); vertical line fill = formyl transferase (UNIPROT D7N386 in N. oralis) and blank fill = three unannotated ORFs (NELON_10550, NELON_10555 and NELON_10560 in N. elongata subspecies glycolytica).
(TIF)
% values are percentage of strains in the species group with that configuration. Other genes shown are annotated as encoding 3-oxoacyl-[acyl-carrier-protein] synthase 2 (KASII in Ngo—UNIPROT Q5F603), a transposase (IS in Nme and Npo—IS110) and a potassium transporter (kefC in Npo—UNIPROT E2PBV4).
(TIF)
% values are percentage of strains in the species group with that configuration. Other genes shown are annotated as encoding a putrescine-binding periplasmic protein (potF in Ngo—UNIPROT Q5FA28) and an uncharacterized protein (blue in Nlact—UNIPROT E4ZEM6)
(TIF)
% values are percentage of strains in the species group with that configuration. Other genes shown are annotated as encoding an uncharacterized protein (grey in Ngo–UNIPROT Q5F9H4), a metalloprotease (pmbA in Nsub -UNIPROT C0EPK5), an NADH-dependent flavin oxidoreductase (nox in Nmuc–UNIPROT F9EUJ4) and an uncharacterized protein (brown in Nmuc—UNIPROT F9EUJ7).
(TIF)
Selected PglP alleles/ORFs were aligned with MAFFT using Geneious and subsequently used to generate an identity and a similarity—based matrix. The two tables were imported into Excel and combined into a single panel (top). Alleles of pglP were selected from across the genus as representatives of each species group, aligned using MAFFT and visualized using Jalview (bottom). Strains in which the allele is located within the core pgl locus begin with an asterix (*) and protein regions with >90% conservation are highlighted in red. The locations of the two glycosyltransferase domains (as predicted by NCBI) are underlined in the alignment (blue line = pfam13439 and green line = pfam00534).
(TIF)
A maximum likelihood phylogenetic tree of aligned pglP nucleotide sequences was generated using MEGA (Molecular Evolutionary Genetics Analysis) V7 using the Tamura-Nei model [58]. A total of 500 bootstrap iterations were undertaken allowing a confidence interval for each node to be determined. The resulting consensus tree was then annotated for each Neisseria species. The analysis involved 204 nucleotide sequences.
(TIF)
De novo assemblies from PubMLST were annotated using Prokka (version 1.14) [59]. A genus-level phylogeny was constructed with FastTree (version 2.1.11) [60] using a core genome alignment generated by Roary (version 3.12.0) [61] with a minimum BLASTP identity of 90%.
(TIF)
See Fig 7 and text for further details.
(TIF)
(PDF)
(PDF)
(PDF)
(PDF)
Gonococcal isolates used originate from those used in a prior study of protein antigen distribution across the genus [63]. Meningococcal isolates originate from a public database which represents global meningococcal diversity in the 20th century. The remaining species include all respective isolates with complete genome sequences available at the Neisseria PubMLST database at the time of this work. Colored cells indicate presence of a gene, crossed cells (X) denote out-of-frame alleles (exclusive of those resulting from phase variable mutational hotspots, the presence of transposon elements is marked with TN, diagonal lines (/) indicate that gene is partially present and isolates with two alleles are marked with 2. Isolate ID numbers are derived from the Neisseria PubMLST database.
(XLSX)
Table shows strain name, date and country of isolation and multi-locus sequence type (MLST). Isolate ID numbers are derived from the Neisseria PubMLST database.
(XLSX)
Table shows strain name, date and country of isolation, serogroup, as well as sequence type and clonal complex designation. Colored cells indicate the presence of ORF–disrupting SNVs or CREE insertions (see Fig 6 for details). ID numbers are derived from the Neisseria PubMLST database.
(XLSX)
Table shows strain name, date and country of isolation, serogroup, as well as sequence type and clonal complex designation. Colored cells indicate the presence of ORF–disrupting SNVs or CREE insertions (see Fig 6 for details). ID numbers are derived from the Neisseria PubMLST database.
(XLSX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.