

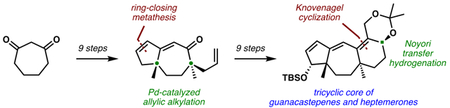
Abstract
For nearly two decades, synthetic chemists have been fascinated by the structural complexity and synthetic challenges afforded by the guanacastepene and heptemerone diterpenoids.Numerous synthetic approaches to these compounds have been reported, but to date the application of enantioselective catalysis to this problem has not been realized. Herein we report an enantioselective synthesis of an advanced intermediate corresponding to the tricyclic core common to the guanacastepenes and heptemerones. Highlights of this work include sequential Pd-catalyzed decarboxylative allylic alkylation reactions to generate the two all carbon quaternary stereocenters, the use of ring-closing metathesis to close the A ring in the presence of a distal allyl sidechain, and a region and diastereoselective oxidation of an trienol ether to introduce oxygenation on the A ring.
Keywords: allylic alkylation, Pd catalysis, olefin metathesis, stereoselectivity, natural products
Graphical Abstract
1. Introduction
The guanacastepenes are a family of diterpenoids (Figure 1) originally isolated by Clardy and coworkers from an unidentified fungus that was found growing on a tree, Daphnopsis americana, in Costa Rica.1 Various members of the family have displayed interesting anticancer2 and antibacterial3 activity. In 2005, Sterner and co-workers reported the isolation of structurally similar diterpenoids, the heptemerones, from the mushroom Coprinus heptemerus.4 Structurally, the guanacastepenes and heptemerones are interesting due to their unusual 5–7-6 ring system with a highly oxidized upper portion and fully saturated lower half. Of particular note are the two quaternary carbon stereocenters that have proven to be the one of the biggest synthetic challenges posed by these targets.
Figure 1.

Representative members of the guanacastepene and heptemerone diterpenoids.
Although initial excitement over this family of natural products has tempered, due to their hemolytic activity,5 the synthetic community continues to demonstrate interest in these molecules.6,7 While there have been several nonasymmetric and asymmetric total syntheses of these molecules, no catalytic enantioselective routes have been described in the literature. When our work on these targets started, our group had recently disclosed a series of asymmetric Pd-catalyzed decarboxylative allylic alkylation reactions capable of constructing all-carbon quaternary stereocenters with high levels of stereocontrol.8 At the same time, we had a burgeoning interest in deploying this technology in natural product synthesis,9 and believed these targets would be an ideal challenge for this newly developed technology. Herein, we describe our enantioselective synthesis of the tricyclic core common to the guanacastepenes and heptemerones.
2. Results and Discussion
2.1. Synthetic plan
We planned to use keto-acetonide 1 (Scheme 1) as our initial synthetic target owing to Danishefsky and co-workers’ success in accessing (±)-guanacastepene A from this intermediate.10 The acetonide-protected diol would arise through reduction of the corresponding ketoester. We envisoned that the six-membered ring of 1 could be constructed through a Knoevenagel condensation, after chain elongation of the allyl moiety of 2, while the isopropyl-substituted cyclopentanone moiety would be constructed from the cyclopentene portion of 2. The α-quaternary stereocenter in 2 would be installed by a Pd-catalyzed decarboxylative allylic alkylation, while the five-membered ring could be closed by ring-closing metathesis (RCM) of 3. The styryl moiety in cycloheptenone 3 would be installed by performing Stork–Danheiser chemistry on vinylogous ester 4.11 Ultimately, an asymmetric decarboxylative allylic alkylation of s β-ketoester (±)-5 would serve as the source of asymmetry in our synthesis.
Scheme 1.

2.2. Installation of first stereocenter
Reaction of 1,3-cycloheptanedione (6)12 with isobutanol under Dean–Stark conditions in the presence of PPTS produced vinylogous ester 7 (Scheme 2). Although vinylogous esterification proceeded smoothly, a significant amount of the retro-Dieckmann product was obtained if the generated water was not efficiently removed. This side product could be eliminated with two simple procedural modifications. First, the isobutanol should be distilled from CaO. Second, the heating bath should be preheated before addition of the reaction mixture. Acylation of 7 with allyl cyanoformate, and subsequent methylation, gave the required β-ketoester (±)-5. It should be noted at this point that the absolute configuration of the natural products requires the use of (R)-t-BuPHOX, which is derived from the unnatural enantiomer of tert-leucine. Consequently, much of the subsequent work was carried out with racemic material obtained by using PPh3 as the ligand. We also used the more readily available (S)-t-BuPHOX to demonstrate that the planned enantioselective decarboxylative allylic alkylation could proceed in good yield and acceptable enantioselectivity [(±)-5→(S)-4].
Scheme 2.

Addition of β-lithiosytrene to vinylogous ester 4 afforded cyclohepteneone 3 (Scheme 2). In contrast to what is observed when performing Stork–Danheiser chemistry on six-membered rings, the intermediate β-hydroxy cycloheptanone was found to be particularly stable.13 Fortunately, warming with aqueous HCl was sufficient to push the elimination toward 3. Attempts were made to derivatize 3 (e.g. oxime, semicarbazone, various hydrazones) in order to affect enantioenrichment through recrystallization; unfortunately none were successful in forming suitably crystalline products. Fortunately, however, further transformations would make such enrichment unnecessary.
2.3. Installation of second stereocenter
Ring-closing metathesis (RCM) with the second-generation Grubbs catalyst afforded bicyclic dieneone 8, which was acylated and methylated to give alkylation substrate 9. When screening ligands to promote the Pd-catalyzed alkylation reaction, we found that the use of an achiral phosphinooxazoline (PHOX) ligand or either antipode of t-BuPHOX resulted in the formation of a single diastereomer of 2 in all cases. Unfortunately, comparison with later synthetic intermediates revealed this to be the undesired syn-methyl diastereomer (syn-2).
Molecular modeling of the enolate intermediate derived from ketone 9 (enolate A) was performed in order to understand the high degree of diastereoselectivity observed in this reaction (Figure 2). This effort revealed that the methyl group of the previously installed quaternary stereocenter sits in a pseudoaxial position and effectively blocks the Re face of the enolate. This is in excellent agreement with related computational work by Houk that was reported after our experimental work was completed.14,15 Houk’s work also revealed that steric effects, rather than torsional effects,16 were primarily responsible for the observed stereoselectivity.
Figure 2.

Calculated (B3LYP/6–31+G(d), SMDTHF) structure of enolate A.
Clearly the large amount of substrate control imposed by enolate A would be difficult to overcome using a bicyclic scaffold. However, we believed diminished substrate control might be experienced with a monocyclic enolate.17 To test this hypothesis, ketone 3 was converted to β-ketoester 10 (Scheme 4). Subjecting ketoester 10 to Pd-catalyzed decarboxylative allylic alkylation, in the presence of an achiral PHOX ligand, afforded ketone 11 with a diastereomeric ratio of 2.6:1. Using either antipode of t-BuPHOX resulted in a dr of 1.3:1. It should be emphasized that these results were obtained using material derived from racemic ketone 3. This means that if we were to observe complete catalyst control, the best possible dr we could see with (S)- or (R)-t-BuPHOX is 1:1. With this in mind, the diastereoselectivity afforded by (S)- and (R)-t-BuPHOX was actually quite encouraging.18
Scheme 4.

With this result in hand, ketone (R)-4, of 83% ee, was prepared using (R)-t-BuPHOX and advanced to β-ketoester (R,R/S)-10. Performing the second Pd-catalyzed allylic alkylation with (R)-t-BuPHOX furnished ketone (R,R)-11 with 9:1 diastereoselectivity. Furthermore, the major diastereomer was formed with improved levels of enantiopurity (96% ee), in accordance with the Horeau principle.19 Because both stereocenters of 11 were formed through catalyst control using the same antipode of the ligand, we are confident that the major diastereomer has the desired anti-methyl relationship. Additionally, subsequent RCM furnished a bicyclic compound that was diastereometic to 2, vide infra.
2.4. Elaboration of the allyl group and Knoevenagel cyclization
With the two quaternary stereocenters in place, our attention turned to completing the tricyclic ring system (Scheme 5). First, treating compound 11 with the second-generation Grubbs catalyst affected the closure of the A ring. Once the initial RCM reaction was complete, the indicated propenyl boronic ester20 was added to the reaction mixture in order to carry out a cross metathesis with the remaining allyl group. Undesired cross metathesis with the liberated styrene was minimized by carefully monitoring (TLC) the initial RCM. The resulting boronic ester (12) was not isolated. Instead it was immediately oxidized to aldehyde 13 with anhydrous21 Me3NO.22 A number of other cross metathesis partners were also examined, but none proved to be as useful or successful as the coupling with the vinyl boronate.
Scheme 5.

Aldehyde 13 was then coupled to ethyl diazoacetate23 to furnish β-ketoester 14. To our delight, heating 14 with NaOEt affected the Knoevenagel cyclization10 needed to produce tricycle 15 representing the complete guanacastepene ring skeleton. It should be noted the reactions presented in Scheme 3 were not optimized at this point. Nevertheless, they serve as a useful guide for how the tricyclic ring system can be constructed.
Scheme 3.

2.5. Oxygenation of the A-ring.
Having identified a serviceable route to the tricyclic ring system, we then concerned ourselves with oxidizing the cyclopentene ring. Considerable effort was expended on this particularly troublesome task. We investigated a number of conditions including regioselective epoxidations, dihydroxylations, and allyic oxidations, but all resulted in either no reaction or general decomposition of the starting material. Our prospects for carrying out this necessary transformation seemed bleak until we located a useful procedure from the literature. Kirk and Wiles found that α,β-unsaturated ketones could be converted into γ-hydroxylated ketones by utilizing a two-step procedure involving extended enol ether formation, followed by m-CPBA oxidation and hydrolysis.24,25,26 The authors found that solvent choice was critical to the regioselectivity, as use of CH2Cl2 or other anhydrous organic solvents resulted in oxidation at the α-position. Conversely, the use of aqueous organic solvents (e.g. THF, dioxane, EtOH) along with slow addition of the oxidant provided the γ-hydroxy enone in good yields.24
Applying these conditions to the present case proved to be particularly rewarding (Scheme 6). First, an RCM was used to convert anti-11 into anti-2. Careful monitoring of the reaction was needed in order to minimize competitive cross metathesis reactions (homodimer and styrene cross product) involving the allyl sidechain present in 2. Performing the reaction under an atmosphere of ethylene further minimized these pathways. Treating ketone 2 with TBSOTf furnished silyl enol ether 16. To our delight, oxidation of 16 with m-CPBA in 95% EtOH smoothly formed alcohol 17 in high yield and as the only observed isomer. Through experimentation it was found that magnesium monoperoxyphthalate (MMPP) performed better than m-CPBA in this oxidation. The secondary alcohol was then converted to TBS ether 18.
Scheme 6.

2.6. Multigram synthetic route
Having finally succeeded in identifying conditions to oxygenate the cyclopentene ring, we then scaled up the route with enantioenriched material. Our final optimized route to compound 18 is shown in Scheme 7. Gratifyingly, many of the steps could be scaled with little problem, but there were a few last minute optimizations made along the way. The solvent of the initial decarboxylative allylic alkylation was changed from THF to toluene. This allowed for the relatively small increase in selectivity from 83% to 87% ee. Other notable details include the use of freshly prepared LHMDS for the acylation of 3 and using Cs2CO3, rather than NaH, for the subsequent methylation in order to improve the impurity profile of these particular transformations.
Scheme 7.

The conversion of 4 to 3 was the one step that did need some more optimization. While the lithium-halogen exchange of βbromostyrene proceeded readily on small scale in THF, the large-scale reaction proceeded to give products of phenylacetylene addition (e.g. deprotonation of α-hydrogen, elimination of bromide, deprotonation of phenylacetylene). Presumably, this was due to inefficient cooling of the exothermic lithium-halogen exchange reaction when performed in large volumes of THF. Changing the solvent to Et2O alleviated this problem. However, attempts to heat the mixture of Et2O with aqueous HCl to affect elimination of the hydroxyl group were unsuccessful, presumably due to the two-phase nature of the system. This could be overcome by first quenching the reaction with 10% HCl followed by removal of the volatiles under rotary evaporation. THF was then added and the mixture warmed to 50 °C. By employing this procedure, 3 was formed in high yield on multigram scale. By following this 12-step route, and starting with 5.0 g of cycloheptanedione, we were able to synthesize 4.5 g of 18 as a pure, colorless oil.
2.7. Generation of tricyclic core
With multigram quantities of 18 in hand, we then had a difficult choice to make. Either we could take the time to elaborate the five membered ring into the required isopropyl-containing cyclopentanone, or investigate that problem after forming the six-membered ring. For better or worse, we chose the latter.
Starting with intermediate 18, a cross metathesis reaction was performed between the allyl group and the indicated vinyl boronate (Scheme 8). The resulting boronic ester was not isolated. Instead it was immediately oxidized to aldehyde 19 with anhydrous Me3NO. The aldehyde was then coupled to ethyl diazoacetate23 to furnish β-ketoester 20. Preliminary attempts at using an alkoxide base (NaOEt) to affect the desired Knoevenagel ring closure were successful,10 but we found that using KF as the base27 provided higher yields. Notably, the use of a protic solvent prevented cleavage of the TBS ether. Finally, stereoselective reduction of the β-ketoester, to give alcohol 22, was accomplished using a Noyori transfer hydrogenation.28 By relying on reagent control of the newly formed stereocenter, we were able to address the low diastereoselectivity (~4:1) observed by Danishefsky,10 Snider,29 and Wicha.7h Ester 22 was then converted into acetonide 23 using standard methods.
Scheme 8.

2.8. Attempts to functionalize the A ring
With acetonide 23 in hand, we turned our attention to functionalizing the A ring (Scheme 9). We thought that the allylic ether already present in the A ring (A) contained enough functionality to allow for installation of the C12 isopropyl group (B). Following that, we planned to access the C14 ketone through isomerization of a C13-C14 epoxide (B→C→D). In this manner, and by starting with acetonide 23, we would generate the same intermediate ketone (1) used by Danishefsky and co-workers in their synthesis of guanacastepene A.10 This same intermediate would also be directly applicable to heptemerone G.7h
Scheme 9.

As the silyl ether at C12 was positioned on the α-face, we first considered using a Cu-catalyzed coupling reaction with i-PrMgCl. Similar coupling reactions, albeit not as sterically crowded as the present example, have been shown to proceed with net inversion of the initial stereocenter.30,31 If successful, this would install the C12 isopropyl group with the correct relative configuration. Conversion of silyl ether 23 into allyl pivalate 24 proceeded smoothly (Scheme 10), but all cross coupling attempts with this intermediate failed. This is likely due to the presence of the adjacent quaternary carbon.
Scheme 10.

In an effort to relieve steric crowding, we decided to convert silyl ether 23 into a vinyl triflate. First, the silyl ether was cleaved using TBAF. The more sterically accessible alkene (A ring) was then hydrogenated under palladium catalysis. Oxidation with TPAP/NMO32 afforded cyclopentanone 25 in high yield for the sequence. Formation of the requisite vinyl triflate proceeded smoothly. To our delight, coupling between the vinyl triflate and i-PrMgCl proceeded to give triene 26 in high yield, using catalytic conditions reported by Bäckvall and co-workers.33
Installing oxygenation on the A ring from compound 26, once again proved taxing. All attempts to isomerize the C12-C13 double bond failed. Preliminary molecular modeling suggested that the tricyclic ring system adopted a twisted structure, which does not allow for efficient conjugation in the desired triene. Consequently, the trisubsubstituted A ring alkene is more thermodynamically favored.
In a final attempt to functionalize the A ring, we planned to employ a Grignard addition/oxidative transposition34,35 sequence (Scheme 11, box) in order to install the C12 isopropyl and C14 ketone. Our prospects were bolstered by precedent from the Phillips36 and Mander37 labs, who performed an analogous transformation with cyclopentenones. This approach was evaluated by first converting silyl ether 18 into cyclopentenone 27. Performing nOe experiments on compound 27 confirmed the site of A-ring oxidation. Treating ketone 27 with i-PrMgBr provided a product, whose 1H NMR spectrum was deficient by two alkene protons. This was tentatively assigned as conjugate addition product 28. A similar result was obtained when the Grignard addition was performed in the presence of CeCl3.38 After our work was completed, Wicha and co-workers successfully performed a similar 1,2-addition/oxidative transposition en route to the A ring of guacastepene.7h,39 Notably, their bicyclic intermediate contained a trans ring fusion between the A and B rings. This likely enforces a different conformation that provides a more open approach to the C12 ketone.
Scheme 11.

3. Conclusion
We have developed the first catalytic enantioselective route to the tricyclic core common to the guanacastepene and heptemerone diterpenoids. Our route relies on sequential Pdcatalyzed decarboxylative allylic alkylation reactions to generate the two all carbon quaternary stereocenters with high fidelity. We have also identified conditions through which oxygenation and the C12 isopropyl group can be introduced to the A ring. Although there is still work to be done, particularly on the A ring, the advanced intermediates we have generated may yet prove useful; especially when one considers the potential utility of biocatalysis as a means to perform regioselective C–H oxidation reactions.40
4. Experimental section
Unless otherwise stated, reactions were performed in flame-dried glassware under an Ar or N2 atmosphere using dry, deoxygenated solvents. Solvents were dried by passage through an activated alumina column under argon. Triethylamine, pyridine, and diisopropylamine were distilled from calcium hydride immediately prior to use. Isobutanol was distilled from CaO prior to use. β-bromostyrene was distilled (110 °C, 20 mmHg) and storred under Ar in a Schlenk flask. Allyl cyanoformate,41 TBSOTf,42 and Ru[(S,S)-Ts-DPEN](pcymene)28a were prepared by known methods. (S)- and (R)-t-Bu-PHOX was prepared by known methods.43 (R)-t-Leucinol was resolved using a known procedure.44 Reaction temperatures were controlled by an IKAmag temperature modulator. Thin-layer chromatography (TLC) was performed using E. Merck silica gel 60 F254 precoated plates (0.25 mm) and visualized by UV fluorescence quenching, anisaldehyde, or KMnO4 staining. ICN Silica gel (particle size 0.032–0.063 mm) was used for flash chromatography. Analytical chiral HPLC was performed with an Agilent 1100 Series HPLC utilizing chiralcel AD, OD-H, or OJ columns (4.6 mm · 25 cm) obtained from Daicel Chemical Industries, Ltd with visualization at 254 nm. Analytical achiral GC was performed with an Agilent 6850 GC utilizing a DBWAX (30m · 0.25 mm) column (1.0 mL/min carrier gas flow). Optical rotations were measured with a Jasco P-1010 polarimeter at 589 nm. 1H and 13C NMR spectra were recorded on a Varian Mercury 300 (at 300 MHz and 75 MHz respectively) or Varian Unity Inova 500 (at 500 MHz and 125 MHz respectively), and are reported relative to CDCl3/CHCl3(1 H NMR δ 7.26, 13C NMR δ 77.0). Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). The following abbreviations are used to report NMR data: s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, br = broad, obsc = obscured, app = apparent. IR spectra were recorded on a Perkin Elmer Paragon 1000 spectrometer and are reported in frequency of absorption (cm−1). High resolution mass spectra were obtained from the Caltech Mass Spectral Facility.
4.1. 3-Isobutoxycyclohept-2-enone (7)
A 250 mL round-bottom flask fixed with a Dean–Stark trap was charged with 1,3-cycloheptanedione (6, 5.0054 g, 39.7 mmol) and toluene (40 mL). Isobutanol (30 mL, 325 mmol, 8.2 equiv) and PPTS (149.6 mg, 0.595 mmol, 1.5 mol%) were added and the mixture was then placed in an oil bath pre-heated to 130 °C. After two hours TLC indicated complete consumption of starting material. The reaction was cooled and concentrated in vacuo. The residue was then distilled at 0.6 mmHg, collecting the portion that distilled at 91–96 °C, to afford the title compound (5.3025 g, 73% yield) as a yellow oil. RF = 0.06 (10:1 Hexane:EtOAc); 1H NMR (500 MHz, CDCl3) δ5.37 (s, 1H), 3.49 (d, J = 6.6 Hz, 2H), 2.60–2.56 (m, 4H), 2.00 (app sept, J = 6.6 Hz, 1H), 1.88-1.77 (m, 4H), 0.96 (d, J = 6.8 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 202.5, 176.6, 106.0, 75.0, 41.9, 33.1, 27.9, 23.7, 21.5, 19.3; IR (Neat Film NaCl) 2958, 2872, 1646, 1607, 1469, 1237, 1190, 1174 cm−1; HRMS (EI) m/z calc’d for C11 H18 O2 [M]+•: 182.1307; found 182.1310.
4.2. Allyl 4-isobutoxy-1-methyl-2-oxocyclohept-3-enecarboxylate (5)
n-Butyllithium (2.12 M in hexane, 15 mL, 31.8 mmol) was added to a solution of N,N-diisopropylamine (4.4 mL, 31.4 mmol) in 100 mL THF at −78 °C and the solution stirred for 30 min. A solution of 3-isobutoxycyclohept-2-enone (7, 4.9996 g, 27.43 mmol) in 10 mL THF was added via cannula with a 5 mL THF rinse. After stirring for 30 min, allyl cyanoformate (3.3677 g, 30.3 mmol) was added. The reaction was kept at −78 °C for 3 hours and then quenched with 50 mL of half saturated aq. NH4Cl and allowed to thaw. The mixture was diluted with 50 mL Et2O and the aqueous layer washed 3 × 50 mL Et2O. The combined organic layers were washed sequentially with water and brine, dried over MgSO4, and evaporated in vacuo. The residue was then dissolved in 60 mL THF and cooled to 0 °C. NaH (60% dispersion in mineral oil, 1.2218 g, 30.5 mmol) was added in portions over 10 min. The mixture was stirred cold 20 min, and then MeI (5 mL, 80.3 mmol) was added. The reaction was then heated to 50 °C for 1 hour. The reaction was then quenched by the careful addition of 50 mL of half saturated aq. NH4Cl. The mixture was diluted with 50 mL Et2O and the aqueous layer washed 3 × 25 mL Et2O. The combined organic layers were washed with brine, dried over MgSO4, and evaporated in vacuo. Silica gel chromatography (5 × 21 cm, 10:1 hexanes:EtOAc) afforded the title compound as a pale yellow oil (5.9924 g, 78% yield). RF = 0.20 (10:1 Hexane:EtOAc); 1H NMR (300 MHz, CDCl3) δ 5.86 (dddd, J = 5.4, 5.4, 10.5, 16.8 Hz, 1H), 5.40 (s, 1H), 5.29 (dddd, J = 1.5, 1.5, 1.5, 17.1 Hz, 1H), 5.20 (dddd, J = 1.2, 1.2, 1.2, 10.5 Hz, 1H), 4.63 (dddd, J = 1.2, 1.2, 5.7, 13.2 Hz, 1H), 4.56 (dddd, J = 1.2, 1.2, 5.1, 12.9 Hz, 1H), 3.51 (dd, J = 6.6, 9.6 Hz, 1H), 3.46 (dd, J = 6.6, 9.3 Hz, 1H), 2.59 (ddd, J = 4.2, 9.3, 18 Hz, 1H), 2.47–2.36 (m, 2H), 2.04–1.92 (obsc m, 1H), 1.98 (app sept, J = 6.8 Hz, 1H), 1.81 (ddd, J = 4.2, 9.3, 18.9 Hz, 1H), 1.70 (ddd, 4.5, 7.5, 14.4 Hz, 1H), 1.43 (s, 3H), 0.95 (app d, J = 6.6 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 198.8, 173.7, 173.3, 131.7, 118.2, 104.9, 74.6, 65.5, 58.8, 34.1, 33.7, 27.7, 24.0, 21.1, 19.0; IR (Neat Film NaCl) 3084, 2959, 2935, 1733, 1652, 1612, 1456, 1383, 1233, 1197, 1170, 1114, 994 cm−1; HRMS (EI) m/z calc’d for C16 H24 O4 [M]+•: 280.1675; found 280.1686.
4.3. (R)-7-Allyl-3-isobutoxy-7-methylcyclohept-2-enone ((R)-4)
In a nitrogen filled glove box, a flask was charged with Pd(dmdba)2 (568.4 mg, 0.697 mmol, 2 mol%), (R)-tBu-PHOX (334.8 mg, 0.864 mmol, 2.5 mol%), and 200 mL toluene. The mixture was stirred 30 min, at which time allyl 4-isobutoxy-1-methyl-2-oxocyclohept-3-enecarboxylate (5, 9.8032 g, 34.97 mmol) was added with a total of 150 mL toluene. The reaction was then taken out of the govebox, placed under a stream of argon, and stirred 60 hrs. The mixture was then evaporated in vacuo. Silica gel chromatography (5 × 18 cm, 25:1 hexanes:EtOAc) afforded the title compound as a colorless oil (7.8401 g, 95% yield). RF = 0.36 (10:1 Hexane:EtOAc); 1H NMR (300 MHz, CDCl3) δ 5.72 (dddd, J = 7.2, 7.2, 10.8, 15.9 Hz, 1H), 5.31 (s, 1H), 5.05 (br s, 1H), 5.01 (dddd, J = 1.5, 1.5, 2.7, 5.1 Hz, 1H), 3.51 (dd, J = 6.6, 9.3 Hz, 1H), 3.45 (dd, J = 6.6, 9.3 Hz, 1H), 2.50–2.45 (m, 2H), 2.38 (app dd, J = 7.2, 13.5 Hz, 1H), 2.20 (dddd, J = 1.2, 1.2, 7.5, 8.7 Hz, 1H), 1.98 (app sept, J = 6.6 Hz, 1H), 1.89–1.70 (m, 3H), 1.63–1.55 (m, 1H), 1.14 (s, 3H), 0.95 (app d, J = 6.9 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 206.5, 171.1, 134.4, 117.7, 104.8, 74.3, 51.3, 45.2, 35.9, 35.0, 27.8, 25.0, 19.7, 19.1; IR (Neat Film NaCl) 3075, 2959, 2932, 1614, 1470, 1387, 1213, 1192, 1172, 998, 912 cm−1; HRMS m/z calc’d for C15 H24O2 [M]+: 236.1776, found 236.1775; [α] 24.9D +61.39 (c 1.055, CH2Cl2, 87% ee).
4.4. (R)-4-Allyl-4-methyl-3-styrylcyclohept-2-enone ((R)-3)
β-Bromostyrene (7 mL, 54.57 mmol) was dissolved in 64 mL Et2O and cooled to −78 °C. t-BuLi (1.6 M in pentane, 64 mL, 102.4 mmol) was added over 40 min. The mixture was stirred at −78 °C for 1 hr, at which time (R)-7-allyl-3-isobutoxy-7-methylcyclohept-2-enone (4, 7.8335 g, 33.14 mmol) in 15 mL Et2O was added by cannula with a 5 mL rinse. After 90 min, the reaction was warmed with an ice bath and stirred for 1 hr. The reaction was quenched with 10% HCl (100 mL), and the volatiles removed in vacuo. To the residue was added 100 mL THF. The mixture was then heated to 50 °C for 12 hrs. After cooling to room temperature, the mixture was extracted 4 × 100 mL Et2O. The combined organic extracts were dried over MgSO4, and evaporated in vacuo. Silica gel chromatography (5 × 17 cm, ~700 mL 20:1 hexanes:EtOAc then ~1.6 L 15:1 hexanes:EtOAc) afforded the title compound as a viscous yellow oil (8.0542 g, 91% yield). RF = 0.23 (10:1 Hexane:EtOAc); 1H NMR (300 MHz, CDCl3) δ 7.45–7.27 (m, 5H), 6.90 (s, 2H), 6.31 (s, 1H), 5.68 (dddd, J = 6.6, 8.1, 10.5, 17.1 Hz, 1H), 5.09–5.01 (m, 2H), 2.66–2.61 (m, 2H), 2.47 (dd, J = 6.6, 14.1 Hz, 1H), 2.14 (dd, J = 8.1, 14.1 Hz, 1H), 1.93–1.80 (m, 3H), 1.70–1.60 (m, 1H), 1.27 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 204.5, 158.3, 136.5, 133.7, 133.1, 129.0, 128.7, 128.3, 126.9, 126.9, 118.3, 46.0, 44.3, 44.2, 38.5, 26.6, 17.4; IR (Neat Film NaCl) 3075, 3026, 2925, 1640, 1582, 1450, 1344, 1250, 1217, 963, 916, 752, 694 cm−1; HRMS m/z calc’d for C19H22O [M]+: 266.1671, found 266.1668; [α]D24.8 +33.70 (c 1.19, CHCl3, 88% ee).
4.5. (±)-8a-methyl-6,7,8,8a-tetrahydroazulen-5(1H)-one (8)
To a solution of (±)-4-allyl-4-methyl-3-styrylcyclohept-2-enone (3, 634.g mg, 2.38 mmol) in 80 mL degassed (argon bubbling) CH2Cl2 was added the second generation Grubbs catalyst (5.2 mg, 0.00612 mmol, 0.25 mol%). The mixture was then heated to 50 °C for 50 min and then cooled to ambient temperature. Ethyl vinyl ether (5 mL) was added and the mixture stirred 30 min. Evaporation in vacuo, followed by silica gel chromatography (2 cm · 16 cm, 15:1 hexane:EtOAc) afforded the title compound as a colorless oil (351.3 mg, 91% yield). RF = 0.17 (10:1 7 Hexane:EtOAc); 1H NMR (300 MHz, CDCl3) δ 6.34 (ddd, J = 0.6, 3, 5.4 Hz, 1H), 6.14 (ddd, 1.5, 2.1, 5.4 Hz, 1H), 5.87 (s, 1H), 2.71 (ddddd, J = 0.9, 0.9, 3.6, 6.3, 15 Hz, 1H), 2.59–2.49 (m, 2H), 2.41 (ddd, J = 1.5, 2.7, 18 Hz, 1H), 2.14–1.80 (m, 4H), 1.23 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 204.6, 169.0, 142.6, 134.2, 121.5, 51.4, 46.5, 44.9, 37.7, 29.5, 21.0; IR (Neat Film NaCl) 3059, 2930, 1650, 1615, 1449, 1352, 1261, 968 cm−1; HRMS m/z calc’d for C11H 14O [M]+: 162.1045, found 162.1040.
4.6. (±)-(6R,8aR)-6-Allyl-6,8a-dimethyl-6,7,8,8atetrahydroazulen-5(1H)-one (syn-2)
To s solution of (±)-8a-methyl-6,7,8,8a-tetrahydroazulen-5(1H)-one (8, 132.9 mg, 0.819 mmol) in 5 mL THF cooled to −78 °C was added a solution of LHMDS (1.0M in THF, 0.9 mL, 0.9 mmol). The mixture was stirred 30 min and then allyl cyanoformate (106 mg, 0.954 mmol) was added. After 30 min, 8 mL 50% sat. NH4Cl was added and the mixture allowed to warm to ambient temperature. The aqueous layer was extracted with 3 × 10 mL Et2O, and the combined organic layers dried with MgSO4 and evaporated in vacuo. The crude residue was then dissolved in 5 mL THF and cooled to 0 °C. NaH (60% dispersion in mineral oil, 34.8 mg, 0.87 mmol) was added and the mixture stirred 10 min. MeI (140 μL, 2.25 mmol) was added and the reaction warmed to ambient temperature. After 1 hour, the reaction was quenched by the careful addition of ~5 mL 10% HCl. The mixture was then diluted with 10 mL H2O and 10 mL Et2O. The aqueous layer was awashed 3 × 10 mL Et2O. The combined organic layers were dried with MgSO4 and evaporated in vacuo. Flash chromatography (2 × 14 cm, 10:1 Hex/EtOAc) afforded βketoester 9 (213.3 mg, 74% yield, 1 diastereomer) as a yellow oil. RF = 0.36 (5:1 Hexane:EtOAc); 1H NMR (300 MHz, CDCl3)δ 6.38 (ddd, J = 2.7, 2.7, 5.4 Hz, 1H), 6.15–6.10 (m, 1H), 5.95 (dddd, J = 5.4, 5.4, 10.8, 16.8 Hz, 1H), 5.76 (s, 1H), 5.34 (dddd, J = 1.5, 1.5, 1.5, 17.4 Hz, 1H), 5.22 (dddd, J = 1.5, 1.5, 1.5, 10.5 Hz, 1H), 4.74–4.62 (m, 2H), 2.81 (ddd, J = 2.7, 14.7, 14.7 Hz, 1H), 2.55 (bd, J = 18 Hz, 1H), 2.41 (ddd, J = 1.5, 3, 18 Hz, 1H), 2.11 (ddd, J = 2.1, 14.1, 14.1 Hz, 1H), 1.81 (ddd, J = 2.7, 6, 14.7 Hz, 1H), 1.70 (ddd, J = 1.8, 5.7, 14.4 Hz, 1H), 1.48 (s, 3H), 1.25 (s, 3H).
A flame-dried vial was charged with Pd(dm-dba)2 (4.5 mg, 0.0.00552 mmol, 5 mol%), (S)-tBu-PHOX (2.5 mg, 0.00645 mmol, 5.8 mol%), and 3 mL THF. The mixture is stirred at 25 °C for 30 min at which time β-ketoester 9 (29 mg, 0.111 mmol) prepared above was added by syringe. The reaction was stirred at 25 °C for 5.5 hrs. Evaporation in vacuo followed by silica gel chromatography (3 × 3 cm, 20:1 hexanes:EtOAc) afforded the title compound as a light yellow oil (20.8 mg, 86% yield, >10:1 mixture of diastereomers). RF = 0.40 (10:1 Hexane:EtOAc); 1H NMR (300 MHz, CDCl3) Major diastereomer: δ 6.26 (ddd, J = 3, 3, 5 Hz, 1H), 6.09 (ddd, J = 1.8, 1.8, 5.4 Hz, 1H), 5.71 (s, 1H), 5.61 (dddd, J = 6.9, 7.5, 10.2, 18 Hz, 1H), 5.06–4.97 (m, 2H), 2.55–2.41 (m, 2H), 2.35 (ddd, J = 1.8, 2.7, 18 Hz, 1H), 2.26–1.93 (m, 3H), 1.70 (app d, J = 5.1 Hz, 1H), 1.64 (app d, J = 5.7 Hz, 1H), 1.12 (s, 3H), 1.07 (s, 3H); Diagnostic peaks of minor diastereomer: δ5.74 (s), 1.15 (s), 1.11 (s); 13C NMR (75 MHz, CDCl3) δ 208.9, 164.5, 141.4, 133.6, 133.3, 119.5, 118.0, 50.9, 50.7, 45.2, 42.2, 34.1, 33.2, 28.8, 23.4; IR (Neat Film NaCl) 3075, 3059, 2961, 2929, 1656, 1625, 1450, 1375, 1220, 1204, 1122, 913 cm−1.
4.7. (5R)-Allyl 5-allyl-1,5-dimethyl-2-oxo-4-styrylcyclohept-3-enecarboxylate (10)
To a solution of hexamethyldisilazane (10 mL, 47.71 mmol) in 155 mL THF at −78 °C, was added n-butyllithium (2.4 M in hexane, 19 mL, 45.6 mmol) over 5 min. The mixture was stirred 30 min and then (R)-4-allyl-4-methyl-3-styrylcyclohept-2-enone (3, 8.0542 g, 30.24 mmol) in 15 mL THF (precooled to −78 °C) was added via cannula with a 5 mL rinse. After 30 min, allyl cyanoformate (4.4772 g, 40.30 mmol) was added quickly. After 10 min, 100 mL of half saturated aq. NH4Cl was added to the cold reaction. The layers were separated and the aqueous layer washed with 3 × 100 mL EtOAc. The combined organic layers were dried with MgSO4 and evaporated in vacuo. The residue was then dissolved in 60 mL CH3CN, and Cs2CO3 (14.68 g, 45.06 mmol) and MeI (10 mL, 160.28 mmol) were added. The mixture was then heated to 80 °C. After 4 hrs and 20 min the reaction was cooled to room temperature and filtered through a plug of silica gel (5 × 1 cm) which was then rinsed with 3 × 50 mL EtOAc. The filtrate was then evaporated in vacuo. Silica gel chromatography (5 × 17 cm, 20:1 hexanes:EtOAc) afforded the title compound as an orange-yellow oil (9.9142 g, 90% yield). 1H NMR indicated an ~ 3:1 mixture of diasteromers. RF = 0.26 (10:1 Hexane:EtOAc); 1H NMR (500 MHz, CDCl3) Major diastereomer:δ 7.44–7.41 (m, 2H), 7.37–7.33 (m, 2H), 7.30–7.26 (m, 1H), 6.91 (d, J = 15.5 Hz, 1H), 6.85 (d, J = 15.5 Hz, 1H), 6.28 (s, 1H), 5.90 (dddd, J = 5.5, 5.5, 11, 22.5 Hz, 1H), 5.68 (dddd, J = 7.5, 7.5, 10.5, 17.5 Hz, 1H), 5.33 (dddd, J = 1.5, 1.5, 1.5, 17.5 Hz, 1H), 5.26–5.21 (m, 1H), 5.09–5.03 (m, 2H), 4.70–4.56 (m, 2H), 2.44 (dd, J = 7.5, 14.5 Hz, 1H), 2.24 (app dd, J = 8.5, 15 Hz, 1H), 2.18–2.13 (m, 2H), 1.80 (dd, J = 9.5 14 Hz, 1H), 1.54 (dd, J = 8.5 Hz, 15 Hz, 1H), 1.44 (s, 3H), 1.23 (s, 3H), Minor diastereomer: δ 7.44–7.41 (m, 2H), 7.37–7.33 (m, 2H), 7.30–7.26 (m, 1H), 6.89 (d, J = 16 Hz, 1H), 6.82 (d, J = 15.5 Hz, 1H), 6.31 (s, 1H), 5.89 (dddd, J = 5.5, 5.5, 11.5, 22.5 Hz, 1H), 5.61 (dddd, J = 7, 8.5, 10.5, 17 Hz, 1H), 5.31 (J = 1.5, 1.5, 1.5, 17 Hz, 1H), 5.26–5.21 (m, 1H), 5.06–4.99 (m, 2H), 4.70–4.56 (m, 2H), 2.41–2.34 (obsc m, 1H), 2.11 (obsc dd, J = 8, 14 Hz, 1H), 1.79 (obsc ddd, 2H), 1.66 (ddd, J = 1, 9, 10 Hz, 2H), 1.47 (s, 3H), 1.26 (s, 3H); 13C NMR (125 MHz, CDCl3) Major diastereomer: δ 202.6, 173.1, 156.9, 136.5, 133.5, 133.3, 131.8, 128.7, 128.4, 128.1, 126.9, 124.6, 118.4, 118.0, 65.6, 60.2, 44.9, 44.5, 34.5, 29.3, 26.3, 23.4, Minor diastereomer: δ 202.2, 172.8, 154.9, 136.5, 133.4, 133.1, 131.7, 128.7, 128.3, 128.0, 126.8, 125,2, 118.4, 118.2, 65.7, 59.8, 46.1, 44.3, 34.1, 28.9, 25.5, 21.8, ; IR (Neat Film NaCl) 3078, 3025, 2974, 2934, 1735, 1653, 1648, 1584, 1448, 1375, 1221, 1196, 1103, 965, 918 cm−1; FAB+ HRMS m/z calc’d for C24 H29 O3 [M+H] +: 365.2117, found 365.2117.
4.8. (4R,7R)-4,7-Diallyl-4,7-dimethyl-3-styrylcyclohept-2-enone (anti-11)
In a nitrogen filled glove box, a flask was charged with Pd(dmdba)2 (440.2 mg, 0.540 mmol, 2 mol%) and (R)-tBu-PHOX (253.0 mg, 0.653 mmol, 2.4 mol%) and 2500 mL THF. The mixture is stirred 30 min, at which time (5R)-allyl 5-allyl-1,5-dimethyl-2-oxo-4-styrylcyclohept-3-enecarboxylate (10, 9.9052 g, 27.18 mmol) was added with a total of 50 mL THF. The reaction was taken out of the glovebox and stirred at 25 °C for 24 hrs. Evaporation in vacuo followed by silica gel chromatography (5 × 17 cm, 25:1 hexanes:EtOAc) afforded the title compound as a light yellow oil (8.163 g, 94% yield, 10:1 mixture of diastereomers). RF = 0.52 (10:1 Hexane:EtOAc); 1H NMR (300MHz, CDCl3) Major diastereomer: δ 7.45–7.27 (m, 5H), 6.90 (d, J = 15.3 Hz, 1H), 6.82 (d, J = 15.6 Hz, 1H), 6.19 (s, 1H), 5.79–5.54 (m, 2H), 5.08–4.97 (m, 4H), 2.34 (app dd, J = 6.3, 14.1 Hz, 1H), 2.24 (app dd, J = 6.9, 12.9 Hz, 1H), 2.16 (app dd, J = 8.1, 14.1 Hz, 1H), 2.08 (app dd, J = 8.1, 13.8 Hz, 1H), 1.93–1.84 (m, 1H), 1.75–1.66 (m, 1H), 1.56–1.45 (m, 2H), 1.25 (s, 3H), 1.16 (s, 3H); Diagnostic peaks of minor diastereomer: δ 6.20 (s), 1.20 (s), 1.11 (s); 13C NMR (75 MHz, CDCl3) 209.4, 154.8, 136.7, 133.7, 133.7, 132.8, 128.7, 128.3, 128.2, 126.8, 125.2, 118.1, 118.0, 52.9, 46.2, 45.3, 44.4, 34.3, 29.8, 25.6, 22.8; IR (Neat Film NaCl) 3076, 3027, 2967, 2931, 1659, 1587, 1449, 1373, 1194, 1147, 1120, 962, 915, 753, 694 cm−1; FAB+ HRMS m/z calc’d for C 23H29O [M+H]+: 321.2218, found 321.2225; 24.6D +87.18 (c 1.27, CHCl3, 10:1 dr, anti-Me diastereomer 97% ee).
4.9. (6R,8aR)-6-Allyl-6,8a-dimethyl-6,7,8,8a-tetrahydroazulen-5(1H)-one (anti-2)
A 3000 mL flask was charged with (4R,7R)-4,7-diallyl-4,7-dimethyl-3-styrylcyclohept-2-enone (11, 8.1630 g, 25.47 mmol) and 2000 mL CH2Cl2. Argon was bubbled through the solution using a glass gas dispersion tube for 20 min, at which time ethylene was bubbled through the mixture for 5 min. The second generation Grubbs catalyst (434.1 mg, 0.511 mmol, 2 mol%) was added and ethylene was bubbled through the mixture for 15 min, followed by flushing the headspace for 15 min. The flask was then sealed. After 17 hrs, a second portion of the second generation Grubbs catalyst (112.7 mg, 0.133 mmol, 0.5 mol%) was added. After 3 hrs, the reaction was quenched by adding 90 mL ethyl vinyl ether and allowed to stir 1.5 hrs. Evaporation in vacuo, followed by silica gel chromatography (5 cm · 17 cm, 4% Et2O in hexane) afforded the title compound as a yellow oil (5.3386 g, 89% yield with 6% starting material). RF = 0.40 (10:1 Hexane:EtOAc); 1H NMR (500 MHz, CDCl3) Major diastereomer: δ 6.28 (ddd, J = 3, 3, 5 Hz, 1H), 6.10–6.08 (m, 1H), 5.83 (dddd, J = 6.5, 7.5, 11, 16 Hz, 1H), 5.73 (s, 1H), 5.06–5.04 (m, 1H), 5.04–5.00 (m, 1H), 2.53–2.46 (m, 2H), 2.35 (ddd, J = 1.5, 3, 18 Hz, 1H), 2.15 (app dd, J = 8, 13.5 Hz, 1H), 2.13–2.06 (m, 2H), 1.76–1.67 (m, 1H), 1.55–1.47 (m, 1H), 1.15 (s, 3H), 1.11 (s, 3H); Diagnostic peaks of minor diastereomer: δ5.71 (s), 1.12 (s), 1.07 (s); 13C NMR (125 MHz, CDCl3) δ 208.5, 165.3, 141.7, 135.7, 133.8, 119.9, 117.4, 50.6, 50.0, 45.4, 44.2, 33.3, 32.7, 28.8, 24.7; IR (Neat Film NaCl) 3072, 2966, 2914, 1648, 1625, 1448, 1378, 1225, 1122, 1079, 912 cm−1; HRMS m/z calc’d for C +15H20O [M]: 216.1514, found 216.1505; [α]D 24.9 +12.59 (c 1.165, CHCl3).
4.10. (1R,6R,8aS,Z)-6-allyl-1-hydroxy-6,8a-dimethyl-6,7,8,8atetrahydroazulen-5(1H)-one (17)
A flask was charged with (6R,8aR)-6-allyl-6,8a-dimethyl-6,7,8,8a-tetrahydroazulen-5(1H)-one (2, 4.4093 g, 20.38 mmol) and 60 mL CH2Cl2. To the ice cooled mixture was added Et3N (12 mL, 86.10 mmol) followed by TBSOTf (9.5 mL, 41.36 mmol) over 10 min. The reaction was stirred cold for 10 min and then allowed to warm to ambient temperature. After 3 hrs the reaction was cooled with an ice bath and 50 mL sat. NaHCO3 was added. The mixture was extracted with hexane (3 × 50 mL) and the combined organic layers dried with Na2SO4. Evaporation in vacuo afforded a yellow residue that was dissolved in 100 mL 95% EtOH and cooled with an ice bath. MMPP (12.6 g, 80%, 20.38 mmol) was added in portions over 1 hour. After stirring a further 15 min the mixture was evaporated in vacuo to a volume of 20–30 mL. Water (100 mL) was added and the mixture extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried with MgSO4 and evaporated in vacuo. Silica gel chromatography (3 cm · 18 cm, ~330 mL 5:1 Hex/EtOAc, then ~600 mL 3:1 Hex/EtOAc) afforded the title compound as a pale yellow oil (3.9786 g, 84% yield). RF = 0.10 (5:1 Hexane:EtOAc); 1H NMR (500 MHz, CDCl3) δ 6.35 (dd, J = 2, 5.5 Hz, 1H), 6.31 (d, J = 5 Hz, 1H), 5.83 (dddd, J = 7, 8, 11.5, 11.5 Hz, 1H), 5.82 (s, 1H), 5.07–5.03 (m, 2H), 4.24 (d, J = 2.5 Hz, 1H), 2.49 (dd, J = 6.5, 14 Hz, 1H), 2.29 (m, 1H), 2.20 (dd, J = 8, 14 Hz, 1H), 2.10 (m, 1H), 1.93 (br s, 1H), 1.64 (app ddd, J = 2, 6.5, 6.5 Hz, 1H), 1.60 (app ddd, J = 2, 6, 6 Hz, 1H), 1.15 (s, 3H), 1.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 208.1, 161.6, 141.1, 137.4, 135.4, 122.6, 117.6, 84.8, 49.7, 48.1, 43.8, 30.9, 27.6, 27.1, 24.4; IR (Neat Film NaCl) 3401, 3073, 2963, 2915, 1631, 1450, 1226, 1089, 1037, 915, 861 cm−1; HRMS m/z calc’d for C15 H20 +O2 [M]+: 232.1463, found 232.1467; [α]D 23.1 −64.33 (c 1.49, CHCl3).
4.11. (1R,6R,8aS)-6-Allyl-1-(tert-butyldimethylsilyloxy)-6,8adimethyl-6,7,8,8a-tetrahydroazulen-5(1H)-one (18)
A solution of (1R,6R,8aS,Z)-6-allyl-1-hydroxy-6,8a-dimethyl-6,7,8,8a-tetrahydroazulen-5(1H)-one (17, 3.9786 g, 17.13 mmol) in 84 mL CH2Cl2 was cooled to −78 °C. To this mixture was added 2,6-lutidine (8 mL, 68.68 mmol, 4 equiv.), followed by TBSOTf (5.9 mL, 25.69 mmol, 1.5 equiv.) over ~5 min. The reaction was stirred cold for 45 min and then quenched by adding 100 mL sat. NaHCO3. The mixture was allowed to thaw and the layers separated. The aqueous layer was washed with EtOAc (3 × 100 mL) and the combined organic layers dried with MgSO4. Evaporation in vacuo, followed by silica gel chromatography (5 cm · 15 cm, 35:1 pet. ether:EtOAc) afforded the title compound as a colorless oil (4.5484 g, 77% yield). RF = 0.17 (20:1 Hexane:EtOAc); 1H NMR (500 MHz, CDCl3) δ 6.23 (d, J = 5.5 Hz, 1H), 6.19 (dd, J = 2.5, 5 Hz, 1H), 5.84 (dddd, J = 7, 8, 12.5, 12.5 Hz, 1H), 5.78 (s, 1H), 5.06–5.01 (m, 2H), 4.23 (d, J = 2.5 Hz, 1H), 2.48 (dd, J = 7, 13.5 Hz, 1H), 2.31 (ddd, J = 2.5, 14.5, 14.5 Hz, 1H), 2.20 (dd, J = 8, 14 Hz, 1H), 2.04 (ddd, J = 2, 15, 15 Hz, 1H), 1.59 (ddd, J = 2, 5.5, 14.5 Hz, 1H), 1.46 (ddd, J = 2, 5.5, 14.5 Hz, 1H), 1.15 (s, 3H), 1.06 (s, 3H), 0.90 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 208.3, 162.4, 141.6, 136.2, 135.6, 122.0, 117.4, 85.1, 49.7, 48.4, 43.8, 31.0, 27.9, 27.6, 25.8, 24.4, 18.3, −4.3, −4.7; IR (Neat Film NaCl) 3073, 2957, 2929, 1656, 1637, 1472, 1258, 1096, 1067, 872, 837, 775 cm−1; HRMS m/z calc’d for C21 H34 O2Si [M]+: 346.2328, found 346.2326; [α]D 23.1 −149.36 (c 1.56, CHCl3).
4.12. (6R,8aS)-6-Allyl-6,8a-dimethyl-6,7,8,8atetrahydroazulene-1,5-dione (27)
A vial was charged with a mixture of (1R,6R,8aS)-6-allyl-1-(tertbutyldimethylsilyloxy)-6,8a-dimethyl-6,7,8,8a-tetrahydroazulen-5(1H)-one and (6R,8aR)-6-allyl-6,8a-dimethyl-6,7,8,8atetrahydroazulen-5(1H)-one (20.3 mg) and 0.3 mL THF. TBAF (1.0M in THF, 50 μL, 0.050 mmol) was added and the mixture stirred 15 min. The volatiles were removed in vacuo and the residue taken up in EtOAc and filtered through a plug of silica gel, rinsing with EtOAc. Evaporation in vacuo gave a residue that was dissolved in 0.3 mL CH2Cl2. Dess–Martin periodinane (19.6 mg, 0.0462 mmol) was added. After 40 min, isopropanol was added and the mixture evaporated in vacuo. Flash chromatography (0.7 cm · 7 cm, 10:1 Hex/EtOAc) afforded the title compound as a colorless oil (8 mg): RF = 0.11 (10:1 Hexane:EtOAc); 1H NMR (300 MHz, CDCl3) δ 7.69 (d, J = 5.7 Hz, 1H), 6.37 (d, J = 5.4 Hz, 1H), 6.00 (s, 1H), 5.83 (dddd, J = 7.2, 7.2, 10.5, 14.7 Hz, 1H), 5.12–5.02 (m, 2H), 2.40 (app dd, J = 7.2, 14.1 Hz, 1H), 2.29 (app dd, J = 7.2, 13.5 Hz, 1H), 2.05 (ddd, J = 2.4, 14.7, 14.7 Hz, 1H), 1.92 (ddd, J = 3.3, 5.1, 14.7 Hz, 1H), 1.77 (obsc ddd, J = 2.4, 18.3 Hz, 1H), 1.67 (ddd, J = 2.7, 5.1, 14.7 Hz, 1H), 1.13 (s, 3H), 1.09 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 209.4, 207.8, 155.8, 152.8, 134.7, 134.1, 125.2, 118.2, 50.8, 48.9, 43.3, 32.2, 26.8, 22.6, 22.0; IR (Neat Film NaCl) 3071, 2973, 2932, 1713, 1675, 1550, 1448, 1379, 1345, 1227, 1090, 1073, 917, 866, 627 cm−1; HRMS m/z calc’d for C15H18O2 [M]+: 230.1307, found 230.1302; [α]D 23.5 +46.28 (c 0.40, CH2Cl2).
4.13. 3-((1R,6R,8aS,Z)-1-(tert-Butyldimethylsilyloxy)-6,8adimethyl-5-oxo-1,5,6,7,8,8a-hexahydroazulen-6-yl)propanal (19)
To a solution of (1R,6R,8aS)-6-allyl-1-(tertbutyldimethylsilyloxy)-6,8a-dimethyl-6,7,8,8a-tetrahydroazulen-5(1H)-one (18, 4.5484 g, 13.12 mmol) in 130 mL degassed CH2Cl2 was added 4,4,5,5-tetramethyl-2-vinyl-1,3,2-dioxaborolane20 (10.3126 g, 66.96 mmol, 5 equiv.) followed by the second generation Hoveyda–Grubbs catalyst (412.5 mg, 0.658 mmol, 5 mol%). The mixture was then heated to reflux for 12 hrs at which time it was cooled and 10 mL ethyl vinyl ether was added. After stirring for 30 min at ambient temperature, the reaction mixture was then concentrated in vacuo. The residue was applied to the top of a 6.5 × 1.5 cm pad of silica gel and eluted with a total of 600 mL 10:1 Hex/EtOAc. The filtrate was evaporated in vacuo and the residue dissolved in 130 mL THF. Anhydrous Me3 NO21(5.03 g, 66.97 mmol) was added and the mixture heated to reflux for 10 hrs. To the cooled reaction mixture was added 50 mL H2O and the mixture was allowed to stir ~15 min. Brine (50 mL) was added and the layers separated. The organic layer was washed with 50 mL brine, and the combined aqueous layers washed with EtOAc (3 × 100 mL). The combined organic layers were dried with MgSO4 and concentrated in vacuo. Silica gel chromatography (5 × 15 cm, ~600 mL 7:1 Hex/EtOAc then ~450 mL 6:1 Hex/EtOAc) afforded the title compound (3.7336 g, 78%) as a yellow oil. RF = 0.3 (5:1 Hexane:EtOAc); 1H NMR (500 MHz, CDCl3) δ 9.79 (dd, J = 1.5, 1.5 Hz, 1H), 6.22 (d, J = 5.5 Hz, 1H), 6.20 (dd, J = 2.5, 5.5 Hz, 1H), 5.76 (s, 1H), 4.23 (d, J = 2.5 Hz, 1H), 2.53 (m, 1H), 2.47 (m, 1H), 2.36 (ddd, J = 2, 14.5, 14.5 Hz, 1H), 2.10 (ddd, J = 2.5, 14.5, 14.5 Hz, 1H), 2.02 (ddd, J = 6.5, 9.5, 14.5 Hz, 1H), 1.72 (ddd, J = 6, 9.5, 14 Hz, 1H), 1.54 (ddd, J = 2, 6, 14.5 Hz, 1H), 1.46 (ddd, J = 2, 5.5, 14.5 Hz, 1H), 1.17 (s, 3H), 1.06 (s, 3H), 0.89 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 207.9, 202.7, 162.6, 141.9, 136.1, 121.8, 85.0, 49.5, 48.3, 39.8, 31.6, 31.2, 27.8, 27.4, 25.8, 24.8, 18.2, −4.3, −4.8; IR (Neat Film NaCl) 3059, 2956, 2928, 1725, 1654, 1636, 1472, 1451, 1382, 1361, 1250, 1095, 1062, 936, 870, 837, 775 cm−1; HRMS m/z calc’d for C21 H34O3Si [M]+: 362.2277, found 362.2260; [α]D 23.3 −127.87 (c 2.305, CHCl3).
4.14. Ethyl 5-((1R,6R,8aS,Z)-1-(tert-butyldimethylsilyloxy)-6,8adimethyl-5-oxo-1,5,6,7,8,8a-hexahydroazulen-6-yl)-3-oxopentanoate (20)
A 250 mL round-bottom flask was charged with 3-((1R,6R,8aS,Z)-1-(tert-butyldimethylsilyloxy)-6,8a-dimethyl-5-oxo-1,5,6,7,8,8a-hexahydroazulen-6-yl)propanal (19, 3.7336 g, 10.30 mmol) and 100 mL CH2Cl2. Anhydrous SnCl2 (195.4 mg, 1.031 mmol) was added, followed by ethyl diazoacetate (1.1946 g, 10.47 mmol) over ~ 5 min. The reaction was stirred 1.5 hrs at ambient temperature and a further portion of ethyl diazoacetate (210 μL, 2.00 mmol) was added. After stirring another 1.5 hrs, the reaction was concentrated in vacuo, and the residue subjected to silica gel chromatography (3 cm · 31 cm, 7:1 Hex:EtOAc) to afford the title compound as a viscous yellow oil (4.0558 g, 88% yield). RF = 0.28 (5:1 Hexane:EtOAc); Major keto tautomer: 1H NMR (500 MHz, CDCl3) δ 6.23 (d, J = 6 Hz, 1H), 6.20 (dd, J = 2.5, 5.5 Hz, 1H), 5.75 (s, 1H), 4.23 (d, J = 2.5 Hz, 1H), 4.19 (app q, J = 7.5 Hz, 2H), 3.48 (s, 1.6H), 2.64 (m, 1H), 2.62 (m, 1H), 2.36 (ddd, J = 2, 14.5, 14.5 Hz, 1H), 2.09 (ddd, J = 2, 14.5, 14.5 Hz, 1H), 1.97 (ddd, J = 6.5, 8.5, 14 Hz, 1H), 1.71 (ddd, J = 6, 9.5, 14 Hz, 1H), 1.54 (ddd, J = 2.5, 6, 15 Hz, 1H), 1.46 (ddd, J = 2, 6, 15 Hz, 1H), 1.27 (dd, J = 7, 7 Hz, 3H), 1.17 (s, 3H), 1.06 (s, 3H), 0.89 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 208.0, 203.1, 167.4, 162.5, 141.9, 136.1, 121.9, 85.0, 61.3, 49.5, 49.2, 48.3, 38.9, 33.1, 31.2, 27.8, 27.4, 25.8, 24.8, 18.2, 14.1, −4.3, −4.7; IR (Neat Film NaCl) 3054, 2956, 2929, 1744, 1718, 1653, 1636, 1472, 1449, 1367, 1318, 1258, 1165, 1095, 1071, 936, 869, 837, 802, 775 cm−1; HRMS m/z calc’d for C25H40O5Si [M]+: 448.2645, found 448.2631; [α]D 23.4 – 122.05 (c 0.870, CHCl3).
4.15. Knoevenagel cyclization (preparation of compound 21)
To a solution of ethyl 5-((1R,6R,8aS,Z)-1-(tertbutyldimethylsilyloxy)-6,8a-dimethyl-5-oxo-1,5,6,7,8,8ahexahydroazulen-6-yl)-3-oxopentanoate (20, 2.3213 g, 5.17 mmol, 1 equiv) in 52 mL EtOH was added KF (337.9 mg, 0.280 mmol, 1.1 equiv). The mixture was then heated to 80 °C for 10 hrs, at which time TLC analysis (twice developed in 10:1 Hex/EtOAc) indicated no SM was present. The reaction was then cooled and evaporated in vacuo. The residue was directly applied to a column of silica (3 × 22 cm) and eluted with 10:1 Hex/EtOAc. Unclean fractions were chromatographed again (2 × 18 cm silica gel) to afford compound 21 (1.6580 g, 74%) as a yellow oil that solidified on standing. RF = 0.29 (5:1 Hexane:EtOAc); 1H NMR (500 MHz, CDCl3) δ 6.19 (d, J = 5.5 Hz, 1H), 6.13 (dd, J = 2, 5 Hz, 1H), 6.00 (s, 1H), 4.30 (dq, J = 7, 13.5 Hz, 1H), 4.23 (dq, J = 7, 13.5 Hz, 1H), 4.22 (obsc d, J = 3.5 Hz, 1H), 2.64 (m, 1H), 2.62 (m, 1H), 2.27 (app br t, J = 13.5 Hz, 1H), 2.10 (br s, 1H), 2.01 (m, 1H), 1.81 (ddd, J = 5.5, 5.5, 11 Hz, 1H), 1.62 (ddd, J = 2, 7, 15 Hz, 1H), 1.37 (ddd, J = 2, 7, 15.5 Hz, 1H), 1.27 (dd, J = 7, 7 Hz, 3H), 1.22 (s, 3H), 1.05 (s, 3H), 0.89 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 195.2, 167.3, 162.1 (br), 161.4 (br), 140.1, 136.6, 132.6, 117.6 (br), 85.2, 61.0, 48.2, 37.6 (br), 36.5, 33.6, 27.2, 27.1 (br), 25.8, 25.8, 24.6 (br), 18.2, 14.1, −4.3, −4.7; IR (Neat Film NaCl) 3054, 2955, 2928, 1734, 1671, 1617, 1472, 1451, 1367, 1321, 1229, 1136, 1093, 1071, 1031, 872, 836, 774 cm−1; FAB+ HRMS m/z calc’d for C25H39O4Si [M+H]+: 431.2618, found 431.2614; [α]D 23.4 −909.69 (c 1.945, CHCl3).
4.16. Diastereoselective reduction of β-ketoester 21
A solution of Knoevenagel product 21 (1.6173 g, 3.76 mmol) in 40 mL isopropanol, was degassed by bubbling argon through the mixture for 30 min. Ru[(S,S)-Ts-DPEN](p-cymene) (112.9 mg, 0.188 mmol, 5 mol%) was then added. The mixture was stirred at ambient temperature for 24 hrs and then evaporated in vacuo. After silica gel chromatography (3 × 24 cm, 7:1 Hex/EtOAc), any fractions containing unreacted starting material were pooled and collected away from the reaction product. This residue (461.8 mg) containing ketoester 21 was then dissolved in 11 mL isopropanol and degassed as above. Ru[(S,S)-Ts-DPEN](pcymene) (7.2 mg, 0.0120 mmol) was then added. The mixture was stirred at ambient temperature for 36 hrs and then evaporated in vacuo. After silica gel chromatography (2 × 17 cm, 7:1 Hex/EtOAc), any fractions containing unreacted starting material were pooled and subjected to one further silica gel column (2 × 17 cm, 7:1 Hex/EtOAc). The final product (22) was isolated as a brown viscous oil (1.3934 g, 86%). RF = 0.26 (5:1 Hexane:EtOAc); 1H NMR (500 MHz, CDCl3, 50 °C) δ 6.21 (s, 1H), 6.20 (d, J = 5.5 Hz, 1H), 5.95 (dd, J = 2.5, 5.5 Hz, 1H), 4.54 (br d, J = 3.5 Hz, 1H), 4.24 (dq, J = 7, 13.5 Hz, 1H), 4.20 (obsc d, J = 2.5 Hz, 1H), 4.19 (dq, J = 7, 13.5 Hz, 1H), 2.59 (bs, 1H), 2.12 (app t, J = 13 Hz, 1H), 1.95–1.85 (m, 4 H), 1.58 (ddd, J = 2, 7.5, 14.5 Hz, 1H), 1.39–1.37 (obsc m, 1H), 1.33 (ddd, J = 2, 7.5, 15 Hz, 1H), 1.27 (dd, J = 7, 7 Hz, 3H), 1.05 (s, 3H), 1.00 (s, 3H), 0.90 (s, 9H), 0.082 (s, 3H), 0.076 (s, 3H); IR (Neat Film NaCl) 3435, 3054, 2951, 2929, 1700, 1472, 1366, 1257, 1216, 1089, 1073, 873, 835, 773 cm−1; HRMS m/z calc’d for C25H O+40 4Si [M]+: 432.2696, found 432.2716; [α]D 23.4 −707.61 (c 0.620, CHCl3). Note: due to conformational instability, we were unable to obtain a suitable 13C NMR spectrum.
4.17. Conversion to acetonide 23
Ester 22 (1.3934 g, 3.22 mmol) was dissolved in 30 mL THF and cooled to 0 °C. Red-Al (~3.5 M solution in toluene, 3.6 mL, 12.6 mmol) was added slowly over 5 min. The mixture was allowed to stir cold for 1 hr. The reaction was quenched by the careful addition of EtOH (15 mL). The mixture was then warmed to ambient temperature and Na2SO4•10 H2O (15 g) was added. After stirring vigorously for 1 hour, the mixture was filtered through a fritted glass funnel and the salts rinsed with EtOAc(3 × 50 mL). Evaporation in vacuo gave a residue that was subjected to flash chromatography (3 × 18 cm, 2:1 Hex/EtOAc) to afford a diol product (960.2 mg, 76% yield). RF = 0.16 (2:1 Hexane:EtOAc); 1H NMR (300 MHz, CDCl3) δ 6.18 (d, J = 5.4 Hz, 1H), 5.95 (s, 1H), 5.92 (dd, J = 2.1, 5.4 Hz, 1H), 4.40–4.32 (m, 2H), 4.28–4.20 (m, 2H), 2.40 (bs, 1H), 2.15–1.65 (m, 5H), 1.60 (bs, 1H), 1.55–1.22 (m, 3H), 0.99 (s, 3H), 0.97 (s, 3H), 0.89 (s, 9H), 0.07 (s, 6H); IR (Neat Film NaCl) 3352, 3054, 2953, 2929, 1650, 1472, 1463, 1450, 1362, 1256, 1092, 1059, 1005, 870, 836, 774 cm−1; FAB+ HRMS m/z calc’d for C23 H37 O3Si [(M+H)-H2]+: 389.2512, found 389.2509. Note: due to conformational instability, we were unable to obtain a suitable 13C NMR spectrum.
The isolated diol was dissolved in 25 mL CH2Cl2 and cooled to 0 °C. PPTS (30.3 mg, 0.121 mmol, 5 mol%) was added, followed by 2,2-dimethoxypropane (6 mL, 48.80 mmol, 20 equiv). After stirring cold for 1 hour, the reaction mixture was evaporated in vacuo and the residue subjected to flash chromatography (3 × 25 cm, 25:1 Hex/EtOAc) to afford acetonide 23 (552.8 mg, 52% yield) as a white solid. RF = 0.32 (20:1 Hexane:EtOAc); 1H NMR (500 MHz, CDCl3) δ 6.15 (d, J = 5.5 Hz, 1H), 5.88 (dd, J = 2.5, 5.5 Hz, 1H), 5.69 (s, 1H), 4.35 (dddd, J = 1.5, 3.5, 3.5, 7.5 Hz, 1H), 4.29 (d, J = 15.5 Hz, 1H), 4.26 (d, J = 2.5 Hz, 1H), 4.13 (ddd, J = 2, 2, 16 Hz, 1H), 2.24 (ddd, J = 3, 14.5, 14.5 Hz, 1H), 2.00 (ddd, J = 3, 14, 14 Hz, 1H), 1.84 (dddd, J = 3.5, 3.5, 7, 12.5 Hz, 1H), 1.71 (dddd, J = 6, 10.5, 10.5, 10.5 Hz, 1H), 1.60–1.52 (m, 2H), 1.43 (s, 3H), 1.38 (obsc ddd, J = 3, 4.5, 13.5 Hz, 1H), 1.35 (s, 3H), 1.29 (ddd, J = 3, 5, 15 Hz, 1H), 0.97 (s, 3H), 0.96 (s, 3H), 0.89 (s, 9H), 0.07 (s, 6H); 13C NMR (125 MHz, CDCl3) 153.5, 135.6, 135.5, 134.9, 133.1, 117.8, 99.5, 86.4, 67.8, 60.5, 46.7, 38.6, 36.9, 36.0, 26.9, 25.9, 25.9, 25.3, 25.0, 24.4, 23.8, 18.3, −4.4, −4.7; IR (Neat Film NaCl) 3054, 2951, 2930, 1472, 1378, 1250, 1224, 1092, 1064, 866, 835, 774 cm−1; FAB+ HRMS m/z calc’d for C26 H41O3Si [(M+H)-H2]+: 429.2825, found 429.2805; [α]D 24.9 −514.23 (c 1.54, CHCl3).
4.18. Conversion to pivalate 24
Compound 23 (30.5 mg, 0.708 mmol) was dissolved in 0.2 mL THF and TBAF (1.0 M in THF, 150 μL, 0.150 mmol) was added at ambient temperature. The reaction was allowed to stir at ambient temperature 2.5 hrs at which time it was filtered through a plug of silica gel, eluted with EtOAc, and evaporated in vacuo. The residue was then dissolved in 0.3 mL pyridine and trimethylacetyl chloride (50 L, 0.406 mmol) was added. The mixture was then heated to 50 °C for 30 min. The cooled reaction mixture was then applied to a column of silica (3 × 2 cm) and eluted with 20:1 Hex/EtOAc to afford pivalate ester 24 (24.7 mg, 87%) as a white solid. RF = 0.33 (10:1 Hexane:EtOAc); 1 H NMR (500 MHz, CDCl3) δ 6.29 (d, J = 6 Hz, 1H), 5.90 (dd, J = 2.5, 5 Hz, 1H), 5.77 (s, 1H), 5.33 (d, J = 2.5 Hz, 1H), 4.35 (ddd, J = 1, 5.5, 9 Hz, 1H), 4.29 (d, J = 15.5 Hz, 1H), 4.13 (ddd, J = 2, 2, 16 Hz, 1H), 2.24 (ddd, J = 2.5, 13.5, 13.5 Hz, 1H), 1.95 (ddd, J = 3, 14, 14 Hz, 1H), 1.85 (dddd, J = 3.5, 3.5, 6.5, 13 Hz, 1H), 1.70 (m, 1H), 1.6–1.55 (m, 2H), 1.43 (s, 3H), 1.39–1.23 (m, 2H), 1.35 (s, 3H), 1.19 (s, 9H), 1.08 (s, 3H), 0.98 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.2, 152.6, 138.5, 134.2, 134.1, 131.4, 118.8, 99.7, 86.8, 67.7, 60.4, 46.2, 39.0, 38.5, 36.7, 36.0, 27.2, 26.8, 26.1, 25.2, 24.9, 23.9, 23.8; IR (Neat Film NaCl) 3049, 2980, 2938, 1724, 1455, 1379, 1279, 1224, 1152, 1093, 1028, 976, 865 cm−1; HRMS m/z calc’d for C25H36O4 [M]+: 400.2614, found 400.2610; [α]D 23.7 −533.13 (c 1.185, CHCl3).
4.19. Conversion to cyclopentanone 25
Compound 23 (262 mg, 0.608 mmol) was dissolved in 5 mL THF and cooled with an ice bath. TBAF (1.0M in THF, 2.4 mL, 2.4 mmol) was added dropwise and the ice bath was removed. After stirring at ambient temperature for 2 hours, the volatiles were evaporated in vacuo to leave a viscous orange residue which was applied to a 3 × 2 cm plug of silica gel and eluted with 3:1 Hex/EtOAc (~350 mL total). Evaporation in vacuo provided a residue that was dissolved in 3 mL EtOAc and cooled with an ice bath. Pd/C was added (5 wt% Pd, 65.4 mg, 0.0307 mmol Pd, 5 mol%). The atmosphere was evacuated three times, filling with H2 (balloon) each time. The reaction was stirred at 0 °C for 2.5 hours, with monitoring by TLC (AgNO3 treated silica plates, 5:1 Hex/EtOAc). Once complete, the reaction was filtered through a 3 × 1 cm plug of silica gel and rinsed with ~65 mL EtOAc. Evaporation in vacuo afforded a residue that was dissolved in 3 mL CH2Cl2. To the solution was added 413.2 mg 4Å molecular sieves, NMO (110.8 mg, 0.946 mg, 1.5 equiv), and then TPAP (10.6 mg, 0.0302 mmol, 5 mol%). The reaction was allowed to stir 40 min and was then filtered through a 3 × 1 cm plug of silica gel which was rinsed with ~50 mL EtOAc. Evaporation in vacuo, followed by silica gel chromatography (2 × 12 cm, 10% Et2O in petroleum ether) afforded cyclopentatnone 25 (174 mg, 90% over three steps) as a viscous colorless oil. RF = 0.16 (10:1 Hexane:EtOAc), 0.36 (5:1 Hex:EtOAc); 1H NMR (500 MHz, CDCl3) 5.78 (s, 1H), 4.40–4.30 (m, 1H), 4.27 (d, J = 15 Hz, 1H), 4.14 (ddd, J = 2, 2, 15 Hz, 1H), 2.71–2.58 (m, 2H), 2.52 (ddd, J = 4.5, 9.5, 18 Hz, 1H), 2.42 (ddd, J = 10, 10, 18 Hz, 1H), 2.24 (ddd, J = 2.5, 13.5, 13.5 Hz, 1H), 1.89–1.83 (m, 1H), 1.74–1.66 (m, 2H), 1.61 (ddd, J = 3.5, 5, 14 Hz, 1 H), 1.57–1.50 (m, 2H), 1.43 (s, 3H), 1.39–1.34 (obsc m, 1H), 1.36 (s, 3H), 1.07 (s, 3H), 0.98 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 222.2, 146.3, 133.8, 132.4, 120.5, 99.5, 67.4, 60.5, 51.5, 37.9, 36.7, 36.7, 36.1, 28.4, 26.9, 26.5, 25.3, 25.2, 23.5, 20.0; IR (Neat Film NaCl) 2982, 2937, 1743, 1445, 1378, 1224, 1090, 1026, 863 cm−1; HRMS m/z calc’d for C20H28O3 [M]+: 316.2039, found 316.2030; [α]D 25.2 −91.95 (c 0.695, CHCl3).
4.20. Installation of isopropyl (preparation of compound 26)
A solution of ketone 25 (42.9 mg, 0.136 mmol, evaporated twice from benzene) dissolved in 0.5 mL THF and cooled to −78 °C was added via teflon cannula to a solution of KHMDS (37.2 mg, 0.186 mmol) in 0.6 mL THF at −78 °C with a 0.5 mL rinse. The mixture was stirred 30 min and then added, via Teflon cannula, to a solution of PhNTf2 (70 mg, 0.196 mmol, evaporated twice from benzene) in 1 mL THF at −78 °C. After 50 min, silica gel was added and the suspension allowed to warm to ambient temperature. After evaporation in vacuo, the solid mixture was subjected to flash chromatography (2 × 12 cm, 5% Et2O in petroleum ether) to afford a vinyl triflate (36 mg, 59% yield) as a colorless oil. RF = 0.2 (20:1 Hexane:EtOAc); 1H NMR (300 MHz, C6D6) δ 5.33 (bs, 1H), 5.22 (dd, J = 2.4, 2.4 Hz, 1H), 4.31–4.21 (m, 1H), 4.18–4.06 (m, 2H), 2.59 (ddd, J = 2.4, 2.4, 20.7 Hz, 1H), 2.49 (ddd, 2.4, 2.4, 20.7 Hz, 1H), 1.96 (ddd, J = 3.3. 13.8, 13.8 Hz, 1H), 1.80–1.68 (m, 2H), 1.62 (ddd, J = 3, 13.8, 13.8 Hz, 1H), 1.45–1.12 (m, XH), 1.40 (s, 3H), 1.36 (s, 3H), 1.05 (s, 3H), 0.96 (ddd, J = 3, 4.2, 14.4 Hz, 1H), 0.81 (s, 3H).
The vinyl triflate prepared above was evaporated twice from benzene. CuI (1.8 mg, 0.00945 mg, 12 mol%) and THF (0.65 mL) were added under an atmosphere of argon and the suspension cooled to −15 °C. i-PrMgCl (1.91 M in THF, 0.13 mL, 0.248 mmol, 3.1 equiv) was added quickly and the reaction turned from blue to green to yellow brown. The reaction mixture was kept between −20 and −15 °C for 2.5 hrs and then warmed with an ice bath and silica gel added. After evaporation in vacuo, the solid mixture was subjected to flash chromatography (3 × 2 cm, 5% Et2O in petroleum ether) to afford compound 26 (23.5 mg, 85%, contaminated with ~10% reduction product) as a colorless oil. RF = 0.43 (20:1 Hexane:EtOAc); 1H NMR (300 MHz, CDCl3) 5.62 (bs, 1H), 5.37 (dd, J = 2.4, 2.4 Hz, 1H), 4.39–4.31 (m, 1H), 4.26 (d, J = 15 Hz, 1H), 4.16 (ddd, J = 1.5, 1.5, 15 Hz, 1H), 2.59 (ddd, J = 2.1, 2.1, 21.6 Hz, 1H), 2.49 (ddd, 2,1, 2.1, 21 Hz, 1H), 2.28 (ddd, J = 3.3, 13.2, 13.2 Hz, 1H), 2.14 (app pent, J = 6.9 Hz, 1H), 1.90–1.79 (m, 1H), 1.79–1.57 (m, 3H), 1.57–1.50 (m, 2H), 1.44 (s, 3H), 1.37 (s, 3H), 1.09 (s, 3H), 1.06 (d, J = 6.6 Hz, 3H), 1.04 (d, J = 6.6 Hz, 3H), 0.99 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.4, 150.9, 134.9, 130.9, 117.9, 117.6, 99.4, 67.7, 60.5, 52.8, 38.3, 37.8, 37.3, 35.9, 30.6, 26.8, 25.5, 25.4, 25.3, 25.0, 24.9, 23.6, 21.4; IR (Neat Film NaCl)3044, 2956, 2935, 1651, 1455, 1377, 1223, 1091, 863, 755 cm−1; HRMS m/z calc’d for C23H33O2 [(M+H)-H2]+: 341.2481, found 341.2489; [α]D 24.4 −169.01 (c 0.075, CHCl3).
Supplementary Material
Acknowledgments
We thank the NIH-NIGMS (R01GM080269 and postdoctoral fellowship F32GM073332 to A.M.H.), Amgen, the Gordon and Betty Moore Foundation, and Caltech for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary material includes selected spectra of new synthetic compounds.
This paper is dedicated to Professor Ryan A. Shenvi on receipt of the Tetrahedron Young Investigator Award.
References and notes
- 1.(a) Brady SF; Singh MP; Janso JE; Clardy JJ Am. Chem. Soc 2000, 122, 2116–2117. [Google Scholar]; (b) Brady SF; Bondi SM; Clardy JJ Am. Chem. Soc 2001, 123, 9900–9901. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y; Li Y; Ou Y; Xiao S; Lu C; Zheng Z; Shen Y Bioorg. Med. Chem. Lett 2012, 22, 5059–5062. [DOI] [PubMed] [Google Scholar]
- 3.Singh MP; Janso JE; Luckman SW; Brady SF; Clardy J; Greenstein M; Maiese WM J. Antibiot 2000, 53, 256–261. [DOI] [PubMed] [Google Scholar]
- 4.(a) Valdivia C; Kettering M; Anke H; Thines E; Sterner O Tetrahedron 2005, 61, 9527–9532. [Google Scholar]; (b) Kettering M; Valdivia C; Sterner O; Anke H; Thines EJ Antibiot. 2005, 58, 390–396. [DOI] [PubMed] [Google Scholar]
- 5.Singh MP; Janso JE; Luckman SW; Brady SF; Clardy J; Greenstein M; Maiese WM J. Antibiot 2000, 53, 256–261. [DOI] [PubMed] [Google Scholar]
- 6.(a) Reviews: Mischne M Curr. Org. Synth 2005, 2, 261–279. [Google Scholar]; (b) Maifeld SV; Lee D Synlett 2006, 1623–1644. [Google Scholar]
- 7.(a) Cheong PH-Y; Yun H; Danishefsky SJ; Houk KN Org. Lett 2006, 8, 1513–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shipe WD; Sorensen EJ J. Am. Chem. Soc 2006, 128, 7025–7035. [DOI] [PubMed] [Google Scholar]; (c) Li C-C; Wang C-H; Liang B; Zhang X-H; Deng L-J; Liang S; Chen J-H; Wu Y-D; Yang ZJ Org. Chem 2006, 71, 6892–6897. [DOI] [PubMed] [Google Scholar]; (d) Iimura S; Overman LE; Paulini R; Zakarian AJ Am. Chem. Soc 2006, 128, 13095–13101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Miller AK; Hughes CC; Kennedy-Smith JJ; Gradl SN; Trauner DJ Am. Chem. Soc 2006, 128, 17057–17062. [DOI] [PubMed] [Google Scholar]; (f) McGowan CA; Schmieder A-K; Roberts L; Greaney MF Org. Biomol. Chem 2007, 5, 1522−1524. [DOI] [PubMed] [Google Scholar]; (g) Oonishi Y; Taniuchi A; Sato Y Synthesis 2010, 2884–2892. [Google Scholar]; (h) Michalak K; Michalak M; Wicha JJ Org. Chem 2010, 75, 8337–8350. [DOI] [PubMed] [Google Scholar]; (i) Gampe CM; Carreira EM Angew. Chem. Int. Ed 2011, 50, 2962–2965. [DOI] [PubMed] [Google Scholar]; (j) Gampe CM; Carreira EM Chem.—Eur. J 2012, 18, 15761–15771. [DOI] [PubMed] [Google Scholar]; (k) Michalak K; Wicha J Synlett 2013, 24, 1387–1390. [Google Scholar]; (l) Peng S-Z; Sha C-K Org. Lett 2015, 17, 3486–3489. [DOI] [PubMed] [Google Scholar]; (m) Zhurakovsky O; Ellis SR; Thompson AL; Robertson J Org. Lett 2017, 19, 2174–2177. [DOI] [PubMed] [Google Scholar]
- 8.(a) Behenna DC; Stoltz BM J. Am. Chem. Soc 2004, 126, 15044–15045. [DOI] [PubMed] [Google Scholar]; (b) Mohr JT; Behenna DC; Harned AM; Stoltz BM Angew. Chem. Int. Ed 2005, 44, 6924–6927. [DOI] [PubMed] [Google Scholar]; (c) Behenna DC; Mohr JT; Sherden NH; Marinescu SC; Harned AM; Tani K; Seto M; Ma S; Novák Z; Krout MR; McFadden RM; Roizen JL; Enquist JA Jr.; White DE; Levine SR; Petrova KV; Iwashita A; Virgil SC; Stoltz BM Chem.—Eur. J 2011, 17, 14199–14223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) McFadden RM; Stoltz BM J. Am. Chem. Soc 2006, 128, 7738–7739. [DOI] [PubMed] [Google Scholar]; (b) Behenna DC; Stockdill JL; Stoltz BM Angew. Chem. Int. Ed 2007, 46, 4077–4080. [DOI] [PubMed] [Google Scholar]; (c) White DE; Stewart IC; Grubbs RH; Stoltz BM J. Am. Chem. Soc 2008, 130, 810–811. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Enquist JA Jr.; Stoltz BM Nature 2008, 453, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandal M; Yun H; Dudley GB; Lin S; Tan DS; Danishefsky SJ J. Org. Chem 2005, 70, 10619–10637. [DOI] [PubMed] [Google Scholar]
- 11.(a) Bennett NB; Hong AY; Harned AM; Stoltz BM Org. Biomol. Chem 2012, 10, 56–59. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hong AY; Bennett NB; Krout MR; Jensen T; Harned AM; Stoltz BM Tetrahedron 2011, 67, 10234–10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Ragan JA; Makowski TW; am Ende DJ; Clifford PJ; Young GR; Conrad AK; Eisenbeis SA Org. Proc. Res. Dev 1998, 2, 379–381. [Google Scholar]; (b) Ragan JA; Murry JA; Castaldi MJ; Conrad AK; Jones BP; Li B; Makowski TW; McDermott R; Sitter BJ; White TD; Young GR Org. Proc. Res. Dev 2001, 5, 498–507. [Google Scholar]; (c) Do N; McDermott RE; Ragan JA Org. Synth 2008, 85, 138–146. [Google Scholar]
- 13.Hong AY; Krout MR; Jensen T; Bennett NB; Harned AM; Stoltz BM Angew. Chem. Int. Ed 2011, 50, 2756–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H; Michalak K; Michalak M; Jiménez-Osés G; Wicha J; Houk KN J. Org. Chem 2010, 75, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danishefky and co-workers also reported extremely high diastereoselectivities when working with similar bicyclic cycloheptenones. See:Dudley GB; Tan DS; Kim G; Tanski JM; Danishefsky SJ Tetrahedron Lett 2001, 42, 6789–6791.
- 16.Wang H; Houk KN Chem. Sci 2014, 5, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearson AJ; Bansal HS Tetrahedron Lett 1986, 27, 287–290. [Google Scholar]
- 18.Similar observations were made by our group after the current work was completed. See:Ma S; Reeves CM; Craig RA II; Stoltz BM Tetrahedron 2014, 70, 4208–4212.
- 19.Review: Harned AM Tetrahedron 2018, 74, 3797–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrill C; Grubbs RH J. Org. Chem 2003, 68, 6031–6034. [DOI] [PubMed] [Google Scholar]
- 21.Soderquist JA; Anderson CL Tetrahedron Lett 1986, 27, 3961–3962. [Google Scholar]
- 22.Njardarson JT; Biswas K; Danishefsky SJ Chem. Commun 2002, 2759–2761. [DOI] [PubMed] [Google Scholar]
- 23.Holmquist CR; Roskamp EJ J. Org. Chem 1989, 54, 3258–3260. [Google Scholar]
- 24.Kirk DN; Wiles JM J. Chem. Soc. D 1970, 1015–1016. [Google Scholar]
- 25.Ireland RE; Beslin P; Giger R; Hengartner U; Kirst HA; Maag HJ Org. Chem 1977, 42, 1267–1276. [DOI] [PubMed] [Google Scholar]
- 26.For a related oxidation with Oxone®, see : Suryawanshi SN; Fuchs PL Tetrahedron Lett 1981, 22, 4201–4204.
- 27.Arai H; Ohno A; Tani Y.-i.; Imachi S; Mukaiyama T Chem. Lett 2002, 92–93. [Google Scholar]
- 28.(a) Haack K-J; Hashiguchi S; Fujii A; Ikariya T; Noyori R Angew. Chem. Int. Ed., Engl 1997, 36, 285–288. [Google Scholar]; (b) Matsumura K; Hashiguchi S; Ikariya T; Noyori RJ Am. Chem. Soc 1997, 119, 8738–8739. [Google Scholar]
- 29.Shi B; Hawryluk NA; Snider BB J. Org. Chem 2003, 68, 1030–1042. [DOI] [PubMed] [Google Scholar]
- 30.Underiner TL; Goering HL J. Org. Chem 1988, 53, 1140–1146. [Google Scholar]
- 31.Bäckvall J-E; Sellén M; Grant BJ Am. Chem. Soc 1990, 112, 6615–6621. [Google Scholar]
- 32.Ley SV; Norman J; Griffith WP; Marsden SP Synthesis 1994, 639–666. [Google Scholar]
- 33.Karlström ASE; Rönn M; Thorarensen A; Bäckvall J-EJ Org. Chem 1998, 63, 2517–2522. [DOI] [PubMed] [Google Scholar]
- 34.Luzzio FA Tetrahedron 2012, 68, 5323–5339. [Google Scholar]
- 35.(a) Dauben WG; Michno DM J. Org. Chem 1977, 42, 682–685. [Google Scholar]; (b) Shibuya M; Ito S; Takahashi M; Iwabuchi Y Org. Lett 2004, 6, 4303–4306. [DOI] [PubMed] [Google Scholar]
- 36.Pfeiffer MWB; Phillips AJ J. Am. Chem. Soc 2005, 127, 5334–5335. [DOI] [PubMed] [Google Scholar]
- 37.Mander LN; Thomson RJ J. Org. Chem 2005, 70, 1654–1670. [DOI] [PubMed] [Google Scholar]
- 38.Imamoto T; Takiyama N; Nakamura K; Hatajima T; Kamiya YJ Am. Chem. Soc 1989, 111, 4392–4398. [Google Scholar]
- 39.Michalak K; Michalak M; Wicha J Tetrahedron Lett 2008, 49, 6807–6809. [Google Scholar]
- 40.Loskot SA; Romney DK; Arnold FH; Stoltz BM J. Am. Chem. Soc 2017, 139, 10196–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donnelly DMX, Finet J-P, Rattigan BA, J. Chem. Soc. Perkin Trans 1 1993, 1729–1735. [Google Scholar]
- 42.Corey EJ; Cho H; Rücker C; Hua DH Tetrahedron Lett 1981, 22, 3455–3458. [Google Scholar]
- 43.Tani K; Behenna DC; McFadden RM; Stoltz BM Org. Lett 2007, 9, 2529–2531. [DOI] [PubMed] [Google Scholar]
- 44.Drauz K; Jahn W; Schwarm M Chem.—Eur. J 1995, 1, 538–540. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.