

Abstract
As a ubiquitous second messenger, cyclic adenosine monophosphate (cAMP) mediates diverse biological processes such as cell growth, inflammation, and metabolism. The ability to probe these pathways would be significantly enhanced if we had a DNA-based sensor for cAMP. Herein, we describe a new, 31-base long single stranded DNA aptamer for cAMP, denoted caDNApt-1, isolated by in vitro selection using Systemic Evolution of Ligands after Exponential Enrichment (SELEX). caDNApt-1 has ~3-fold higher affinity for cAMP compared to ATP, ADP and AMP. Using non-denaturing gel electrophoresis and fluorescence spectroscopy, we characterize the structural changes caDNApt-1 undergoes upon binding to cAMP and reveal its potential as a cAMP sensor.
Graphical abstract:
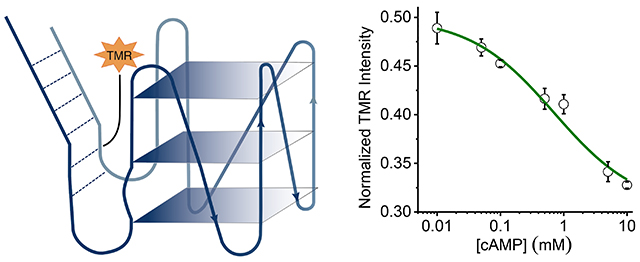
Characterizing conformational changes of a DNA aptamer: A DNA aptamer that binds cAMP was isolated using SELEX. Diverse spectroscopic and electrophoretic analyses reveal its parallel G-quadruplex conformation, as well as its adaptive recognition of high concentrations of cAMP.
Introduction:
Cyclic adenosine monophosphate (cAMP) is a well-known intracellular second messenger in eukaryotes and prokaryotes. A wide variety of processes are regulated by cAMP gradients that are governed by cAMP degrading enzymes, phosphodiesterases, and cAMP effector proteins within the cell.[1] In order to study cAMP dynamics and localization in live cells, genetically encodable sensors have been developed based on endogenous eukaryotic cAMP-binding proteins such as protein kinase A (PKA) and exchange protein directly activated by cAMP (Epac).[2,3] For example, FRET-based sensors have been developed that sandwich Epac between donor and acceptor fluorescent proteins.[4] A single-wavelength sensor was recently developed that fuses circularly permutated GFP in between two PKA subunits.[5] While valuable, these sensors have the following limitations: (1) they need to be expressed in cells and the overexpression of cAMP effector molecules can modulate cell behavior; (2) they cannot measure cAMP levels ≥ 100 μM, and cAMP levels exceed this threshold in certain cellular contexts[6–9]; and (3) they have limited or poorly characterized signal fold change.
Aptamers are ssDNA and ssRNA sequences that specifically bind to a target molecule.[10] Aptamers can be isolated using a method of in vitro evolution called SELEX from a library of randomized nucleic acid sequences.[11,12] Aptamers have applications in the fields of therapeutics,[13,14] targeted delivery,[15–17] and analyte sensing.[11,18] Sensors based on nucleic acid aptamers offer a solution to the aforementioned drawbacks by having tuneable target affinities and significantly higher changes in signal.[19,20] This is especially necessary for quantitative and dynamic imaging with improved spatiotemporal resolution. For example, our group has established that nucleic acid-based sensors can be deployed to quantitatively image analytes such as pH, Cl−, and Ca2+ in cells and in living organisms.[21–26]
Previously, we have developed an RNA-based sensor for cAMP that can be used to quantitatively image cAMP inside membrane-bound compartments.[27] It is suitable for imaging elevated cAMP concentrations, but its application is limited due to the instability of RNA inside cells. An ssDNA aptamer would greatly improve the stability of a multipurpose cAMP sensor.
Therefore, we performed an in vitro isolation of an ssDNA aptamer that binds to cAMP with a Kd of 1 mM. As a result, we isolated a 31 base-long aptamer that has ~3-fold higher affinity for cAMP over ATP, ADP and AMP. This aptamer thus adopts a scaffold structure that optimally binds to nucleotides having a cyclic phosphodiester bond, rather than a phosphate chain at the 5 position of the ribose sugar. Further, we observed that this aptamer undergoes conformational changes upon binding to cAMP, leaving it well-placed for sensor development.
Results and Discussion:
SELEX for in vitro isolation of an ssDNA aptamer for cAMP:
We selected cAMP aptamers by incubating an ssDNA library with cAMP-agarose beads in selection buffer (SB). The unbound DNA in solution was separated from the bound DNA by spinning down the beads and removing the supernatant. The bound DNA was eluted by incubating the beads in elution buffer (EB) which is simply SB containing 10 mM cAMP. The eluted DNA was amplified and subjected to further rounds of selection. After five rounds of selection and amplification, the cAMP-agarose beads were washed three times with SB containing ATP, ADP, AMP and cGMP, prior to elution with EB, to enrich for sequences that bind specifically to cAMP. The sequences obtained after different SELEX experiments were pooled (Supplementary Table 1). All the sequences were G-rich and shared significant similarity. On the basis of multiple sequence alignment of the randomized regions, the sequences were divided into four groups. Sequences within a group could be aligned without any insertion or deletion (Figure 1a).
Figure 1:

In vitro isolation and characterization of an ssDNA aptamer for cAMP. a) Multiple sequence alignment of the randomized region of sequences obtained after SELEX. b) Schematic illustrating the truncated sequences, obtained from the sequence that was represented most often in isolated sequences. Grey sequence shows middle randomized region and black sequences show the fixed region. c) Fraction of the indicated 5’-32P labelled constructs bound to cAMP-agarose beads in selection buffer containing Tween-20 (SBT) in presence (black bars) or absence of cAMP (white bars). d) Percentage reduction in fraction of 5’-32P –caDNApt-1 bound to cAMP-agarose in SBT containing indicated amounts of cAMP (cyan), cGMP (blue), ATP (black), ADP (red) and AMP (green). Error bars: mean ± sem, n=3.
Characterization of the isolated aptamers:
In order to characterize the binding properties of the aptamers, we selected a specific sequence from Group B, which is most highly represented in sequence space. The sequences in Group B show high similarity at their 5’ termini, indicating its potential importance in cAMP binding. On the other hand, high variability in the randomized region after the 41st base (Figure 1b) indicates the potential redundancy of these bases in binding to cAMP. Therefore, we evaluated two fragments: one of length 34 bases from the 3’ end denoted Apt-A and one of length 41 bases from the 5’ end denoted Apt-B.
In order to quantify the cAMP binding of these truncated sequences, we compared the radioactivity of cAMP bound 32P-labelled Apt-A and Apt-B. After incubating cAMP-agarose beads with radiolabelled Apt-A and Apt-B in SB containing Tween-20 (SBT), we washed the beads with SBT and eluted bound DNA with EB containing Tween-20 (EBT). The cAMP-bound fraction is revealed by the percentage of total DNA radioactivity in the eluate. As expected, Apt-B to cAMP significantly better than Apt-A (Figure 1c).
Because shorter aptamer sequences are easier to synthesize and also provide the potential to integrate fluorophores or organelle-targeting motifs, we sought to determine the minimal motif in Apt-B that is necessary for cAMP binding. Removing ten bases from the 5’ terminus of Apt-B yielded the 31mer Apt-C, which showed improved binding to cAMP-agarose beads. However, removal of six bases from the 5’ terminus of Apt-C to form the 25mer Apt-D completely abrogated cAMP binding. These six bases correspond to fixed sequences at the 5´ terminus of the original library and indicate a role for the fixed sequence in cAMP binding. The phenomenon of a fixed sequence in a SELEX library participating in target binding has been observed previously.[28] Removal of six bases from the 3’ terminus of the Apt-C to form the 25mer Apt-E significantly reduced binding to cAMP. The fraction bound to cAMP-agarose beads was also calculated in the presence of 5 mM free cAMP. A reduction in the fraction bound in the presence of free cAMP confirms that these DNA sequences specifically bind cAMP. Apt-C shows the highest bound fraction, and longer sequences show less binding, presumably due to their potential to adopt unfavourable conformations. We therefore selected the Apt-C, and hereafter denote it as cAMP DNA Aptamer-1, (caDNApt-1) for future study (Supplementary Table 2).
In order to compare the binding affinity of caDNApt-1 for cAMP to structurally similar molecules such as ATP, ADP, AMP and cGMP, the fraction bound was also calculated in the presence of varying concentration of these analytes (Figure 1d). The percentage reduction in fraction bound was plotted as a function of corresponding analyte concentration. Concentrations at which a 50% reduction in fraction bound was obtained are reported as IC50 values. A comparison of the IC50 values revealed that caDNApt-1 binds to cAMP and cGMP with similar affinities but with a ~3-fold lower affinity for ATP, ADP and AMP as compared to cAMP.
caDNApt-1 adopts a G-quadruplex containing structure:
Since the caDNApt-1 sequence is G-rich, we wondered if it could potentially adopt a G-quadruplex containing structure as seen in many other aptamers (Supplementary Figure 2a).[29] A circular dichroism (CD) spectrum of caDNApt-1 in SBT showed a positive maximum at 260 nm and a minimum at 240 nm (Figure 2a). This is a signature of a parallel G-quadruplex and is consistent with the reported CD spectra of other G-quadruplex forming oligonucleotides.[30–32] A CD spectrum of caDNApt-1 in a solution of Tris.Cl (20 mM, PH=7.5) and Tween-20 (0.2 %) showed a G-quadruplex signature only in the presence of 5 mM KCl but not in the presence of 100 mM NaCl (Supplementary Figure 1a). Interestingly, the solution containing KCl was enough to bind cAMP-agarose, whereas no binding was observed in a solution containing 100 mM NaCl (Supplementary Figure 1b). These results indicate that caDNAapt-1 likely adopts a G-quadruplex conformation in the presence of K+ that is essential in order to bind cAMP.
Figure 2:

caDNApt-1 adopts a G-quartet containing structure. a) CD spectra of caDNApt-1 showing the signature parallel G-quadruplex structure in SBT. b) Products of DMS protection assay performed with 5’-32P -caDNApt-1 electrophoresed on 20 % denaturing PAGE. Lane 1: no DMS and piperidine treatment. Lane 2-5: DMS protection assay in Tris.Cl (20 mM, pH 7.5) and Tween-20 (0.2%) with either no added salt (lane 2), NaCl (5mM, lane 3), LiCl (5mM, lane 4), or KCl (5mM, lane 5). c) Initial proposed secondary structure of an ATP aptamer. d) Expected secondary structure of caDNApt-1. Arrows indicate the position of TMR in caDNAptT-2 (red) and caDNAptT-3 (green).
In order to confirm the formation of a G-quadruplex structure by caDNApt-1, we performed dimethyl sulfate (DMS) footprinting studies. 5’-32P-labelled caDNApt-1 was incubated in solutions containing Tris.Cl (20 mM, PH 7.5) and Tween-20 (0.2 %) containing either LiCl (5 mM) or NaCl (5 mM) or KCl (5 mM). caDNApt-1 was then treated with DMS followed by cleavage with piperidine. The reaction products were then analysed by denaturing gel electrophoresis and visualized by auto-radiography. The efficacy for stabilization of G-quadruplex by metal ions is in the following order: K+ > Na+ > Li+ [23]. caDNApt-1 underwent cleavage at each of its 13 guanine residues in the DMS protection assay in the presence of NaCl and LiCl. However, in the presence of KCl only G-28 showed cleavage (Figure 2b), indicating that the remaining 12 guanine residues are involved in G-quartet base pairing. This increased protection of guanines in the presence of K+ is consistent with G-quartet base pairing in caDNApt-1. Combined, this evidence supports caDNApt-1 forming a parallel G-quadruplex, which resembles the second structure initially proposed for a DNA aptamer for ATP, with important distinctions discussed later (Figure 2c,d).[33]
Structural changes upon binding to cAMP:
Aptamers can undergo conformational changes upon ligand binding.[34] We therefore studied whether caDNApt-1 underwent conformational changes upon cAMP binding, using gel electrophoresis. 5´-32P labelled caDNApt-1 was incubated in SBT containing varying concentrations of cAMP and subjected to non-denaturing PAGE. The band with slowest mobility, representing the conformation caDNApt-1 assumes upon binding, had increased intensity with increasing cAMP (Figure 3a). The ratio of upper band intensity to total intensity per lane was plotted as a function of cAMP concentration which gave a binding curve (Figure 3b). Fitting the Hill equation (Materials and methods Eq. 5) to this curve yielded a Kd of 1 mM. As expected from the specificity of caDNApt-1 observed in Figure 1d, the conformation represented by the slower mobility band is assumed upon binding to cAMP or cGMP but not by ATP, ADP or AMP (Supplementary Figure 4). Interestingly, in both the cAMP bound and unbound states, caDNApt-1 maintains a G-quadruplex conformation (Supplementary Figure 3).
Figure 3:

Adaptive recognition of caDNApt-1 by cAMP. a) Non denaturing PAGE showing the increased intensity of the slowest migrating band with increasing cAMP concentrations in SBT containing 9 mM MgCl2. b) Fraction of the intensity of slower moving band as a function of cAMP concentration. c) Percent decrease in fluorescence intensity of TMR relative to Alexa647 in the presence of 5 mM cAMP in SBT containing 9 mM MgCl2. Bar graph color corresponds to position of TMR label (Figure 2d and Supplementary Table 2). d) Decrease in normalized fluorescence intensity of caDNAptT-3 (5 nM) as a function of cAMP in SBT containing 9 mM MgCl2.
It is well known that tetramethyl rhodamine (TMR) fluorescence intensity is sensitive to local DNA conformation.[35–37] Therefore, in order to monitor the conformational changes, we labelled caDNApt-1 with TMR at three different positions (Figure 2d) and added Alexa647 in solution as a normalizing dye. We observed that all three showed a decrease in TMR fluorescence intensity relative to added Alexa647 in the presence of 5 mM cAMP (Figure 3c), indicating a conformational change in caDNApt-1 upon cAMP binding. We also monitored the change in fluorescence intensity of caDNAptT-3, which showed the largest change in intensity, as a function of cAMP concentration (Figure 3d). Fitting the Hill equation to this curve yielded a Kd of ~1 mM.
When an aptamer undergoes a large structural change upon binding its cognate ligand, resulting in an overall conformation change, this is referred to as adaptive recognition.[34,38,39] Previously isolated nucleic acid aptamers for adenosine containing ligands reveal two types of changes upon ligand binding, namely “induced-fit” and “conformation-selection.”[40,41] The induced-fit model describes a conformation change as a consequence of ligand binding, whereas the conformation-selection model describes ligand stabilization of one of multiple conformations. Our experiments show that two major species are present in the absence of cAMP, and that cAMP increases the concentration of one of these conformations. This reveals that the adaptive recognition of cAMP by caDNApt-1 occurs through a process of conformation selection.
Conclusions:
In vitro isolation of aptamers that have high specificity for adenosine-based biomolecules has been challenging. Independent attempts made in three different laboratories to isolate RNA aptamers for ATP,[42] SAM,[43] and NAD+, [44] resulted in the repeated isolation of a similar binding motif.[45] Interestingly, a recently isolated RNA aptamer for cAMP has a 2.5-fold higher affinity for cAMP over ATP.[27,28] An ssDNA aptamer for cAMP has ~2-fold higher affinity for cAMP over ATP.[46]
Using mutational analysis, a previously described adenosine-recognizing DNA aptamer for ATP was suggested as adopting a G-quadruplex conformation (Figure 2c).[33] In this aptamer, the 5’ and 3’ termini were complementary to each other, forming a duplex, while the central region was hypothesized to form two stacked G-quartets. Further, two highly conserved adenines stacked between the duplex and top G-quartet were proposed to form the adenosine binding site. More rigorous structure determination revealed a different architecture not containing a G-quadruplex, with the invariant adenines instead forming G-A mismatches adjacent to the binding site.[47]
It is therefore interesting that our spectroscopic and DMS footprinting at single nucleotide resolution, provides compelling evidence of a G-quadruplex in caDNApt-1 (Figure 2d). Its complementary 5´ and 3´ termini form a duplex, while the central G-rich sequence exists as three stacked G-quartets. We find a conserved adenine at position 11 present in all members of Group B. The significant difference between the two aptamers is that caDNApt-1 has a single nucleotide between each stretch of guanines, which likely enforces a parallel G-quadruplex conformation.[32] The increased rigidity in this binding site may be optimal to accommodate a cyclic phosphodiester, as compared to a triphosphate.
The ssDNA aptamer caDNApt-1 described in our study shows a comparable specificity for cAMP with the added advantage that cAMP induces a conformational change upon binding. This is potentially important for future design of cAMP reporters, as it minimizes the potential reduction of affinity to the ligand upon converting a binder to sensor. Further, performing an in vitro selection on a library based on partial randomization of this aptamer could potentially yield either cAMP or cGMP aptamers with even higher affinities.
Experimental Section:
Equipment and Reagents:
Fluorescence and absorbance measurements were carried out in fluoromax-4 (Horiba Scientific, Japan) and Cary-300-Bio (Agilent technologies, USA) respectively. Gels with 32P labelled samples were visualized by exposing them to Kodak storage phosphor screen (S02330) and scanned in the phosphorimager (Typhoon Trio+™, GE Healthcare Life Sciences, USA). Cherenkov counting was done in scintillation counter (MicroBeta2® LumiJET® Microplate Counter 2460, Perkin Elmer, USA). CD spectra were acquired in JASCO-J-720 spectrophotometer (JASCO, Tokyo, Japan). Unmodified DNA oligonucleotides were purchased from Sigma and modified oligonucleotides and library for SELEX was purchased from IBA GmbH (Germany). cAMP (A9501-1G), ATP(A2383-1G), ADP(A2754-100MG), AMP(A9501-1G) and cGMP (G6129-25MG) were purchased from Sigma-Aldrich (USA) and their stocks (~100 mM) were prepared by dissolving the respective salts in Tris.Cl (pH=7.5, 20 mM) and pH was adjusted to 7-7.5 with NaOH. Concentration of stocks were measured using absorbance, taking extinction coefficient of ATP, ADP, AMP and cAMP to be 15000 M−1cm−1 and that of cGMP to be 12320 M−1cm−1 in water at 260 nm. Other reagents used are C8-cAMP-agarose (A0144 Sigma Aldrich, USA). Band intensities of gels were calculated using ImageJ ver 1.47 (NIH, USA), Multiples sequence alignment was done using ClustalW2 (EMBL-EBI), JalView Desktop (EMBL-EBI) was used to make representative images of alignments and for rest of the data analysis OriginPro 8.5 (OriginLab, USA) was used.
SELEX:
The ssDNA library was suspended in Tris.Cl (20 mM, pH=7.5), NaCl (100 mM), KCl (5 mM), MgCl2 (1 mM) and CaCl2 (1 mM) (selection buffer, SB) heated at 90 ºC for 10 min and kept at RT (5 min) and incubated for 2 h at RT with cAMP-agarose beads. Post incubation the cAMP-agarose beads were washed 6 times with selection buffer by spinning the solution at 13000 RPM (2 min). The DNA bound to cAMP-agarose beads was eluted by re-suspending the cAMP-agarose beads in SB containing 10 mM cAMP (elution buffer, EB) for 5 min. The eluted DNA from each round was ethanol precipitated in the presence of glycogen (8 μg), and sodium acetate (1/10 V, 3 M, pH=5.2). The purified eluted ssDNA was amplified to get PCR product with strands of unequal lengths as described. The strand of interest was separated from complementary strand on denaturing PAGE and purified using crush and soak method followed by desalting using 10 kDa cut off centrifugal filtration device and ethanol precipitation. In the first round of selection 1-5 nmol of DNA was used for binding and the volume was adjusted so that concentration of DNA does not exceed 1 μM. For subsequent rounds of selections, maximum of 200 pmol of selected and amplified DNA was used. After last round of selection eluted DNA was PCR amplified in 10 μL PCR reaction using unmodified primers FPL and RPL. The PCR product was cloned in TopoTA cloning vector (PCR® 2.1 TOPO®, Life technologies, USA) and sequenced.
Bead Binding Assay:
The DNA was labelled at 5´ terminus with 32P using T4 PNK and γ-32P-ATP. The amount of DNA used for bead binding assay was adjusted so as to never exceed a concentration of 1nM. The 5´−32P labelled DNA in SBT (Selection buffer containing 0.2 % Tween-20, 100 μL) was heated at 90 °C (10 min) and kept at RT (5 min) and added to C8-cAMP-agarose beads suspension (100 μL) in SBT either not containing any molecule or containing cAMP/ATP/ADP/AMP at twice the concentration than required. Resulting solution was incubated at 25 °C (3 h) with constant shaking. After incubation beads were washed 3 times in ice cold SBT by spinning at 13000 RCF for 1 min at 4 °C. In order to get the DNA bound to cAMP on C8-cAMP-agarose beads, they were resuspended in elution buffer containing Tween 20 (EBT) at RT and mixed gently for 1 min and spun (13000 RCF, 1 min). The elution was repeated two more times. The CPM of 32P in all the washes (w) and eluates (e) were estimated using Cherenkov counting. Fraction bound and percentage reduction in fraction bound was calculated as follows.
Here, FBno cAMP and FBx mM cAMP means fraction bound of 5´−32P labelled to cAMP agarose-beads when the binding was done in absence of cAMP and in the presence of cAMP (x mM) respectively.
The mean percentage reduction in the fraction bound obtained from three independent experiments was plotted as function of cAMP concentration or relevant structural variant concentration. The logistic sigmoid given below was fitted through the plots using Origin.
The parameter A2 is the maximum percentage reduction in fraction bound possible. We fixed A2 to be 100. The parameter A1 is the % reduction in fraction bound in the absence of any structural mimics, this parameter was not fixed while fitting and as expected it reached a value of 0 after fitting. The x0 is the concentration of cAMP or structural variant where 50 % reduction in fraction bound was observed and this is the value that was reported as IC50.
Gel electrophoresis:
5´-32P DNA was added to a solution containing SBT (40 μL), MgCl2 (9 mM) and unlabelled DNA and indicated concentration of cAMP or structural variant. The solution was heated at 90 °C (10 min) and incubated at 25 °C (3 h). Ice cold loading dye was added to this solution and was immediately loaded on a pre-chilled 10 % poly-acrylamide gel made in TBEK. Electrophoresis was done in 1X TBEK (1X TBE + KCl (5 mM)) at 10 Vcm−1 inside a cold cabinet. The gels were dried in a vacuum drier and exposed to IP plate and scanned in phosphorimager. After appropriate background subtraction the total intensities corresponding to the fast mobility band (FB) and slow mobility band (SB) band were quantified using ImageJ. The size of ROI for all the bands of a gel was kept constant. Fraction of slower band was calculated as below.
Where, ISB and IFB are the intensities corresponding to slower band and faster band respectively. Mean fraction bound from three different gels was plotted as a function of cAMP concentration and a modified Hill equation as shown below was fitted through the curve using OriginPro8.1.
Here, yo and y∞ are fraction of upper band in the absence of cAMP and the highest concentration of cAMP used, k is Kd.
CD Spectroscopy:
CD spectra were acquired in 210-400 nm range with a data pitch of 1 nm in continuous scanning mode using 100 mdeg sensitivity, scanning speed of 100 nm/min, response time 0.5 s and band width 2 nm. Spectra reported were an average of 5 scans.
Fluorescence Spectroscopy:
The fluorescently labelled oligonucleotides (5 nM) were heated at 90 °C for 5 min in a solution containing SBT and 9 mM MgCl2 in the presence of the indicated concentrations of cAMP. The labelled DNA was always added from a stock solution (1 μM) that also contained Alexa-647 (1 μM). After heating, the solutions were incubated at RT for 3h and fluorescence spectra of TMR and Alexa-647 were acquired using excitation wavelengths of 555 nm and 647 nm, respectively. The fluorescent emission intensity of TMR at 580 nm was divided by florescence emission intensity of Alexa-647 at 670 nm to obtain the TMR/A647 ratio. Percentage decrease in TMR intensity represents the change in TMR/A647 in the presence of cAMP.
DMS assay:
The 5´ 32P labelled cADNApt-1 was suspended in solutions of desired ionic composition and heated at 90 °C for 5 min followed by incubation at RT for 3 h. For, obtaining a G ladder the incubation was done in Tris.Cl (1 mM, PH= 7.5). After the incubation DMS (Dimethyl Sulphate (DMS, Spectrochem, India) was added (0.8 %) and incubated at RT for 5 min, the DMS reaction was stopped by addition of one fourth of reaction volume of stop solution (1.5 M Sodium acetate pH=7.0 and 1 M β-Mercaptoethanol). This mixture was ethanol precipitated and the precipitate was resuspended in sodium acetate (50 μL of 300 mM and pH=5.2). This mixture was once more ethanol precipitated and the precipitate was washed with 70 % ethanol. The precipitate was now suspended in piperidine (10 % piperidine in water) and heated at 90 °C for 30 min followed by vacuum drying in a centrivap (Refrigerated Centrivap Concentrator, Labconco, USA). After drying it was again resuspended in MQ water (500 μL) and dried again. Finally, the precipitate was resuspended in 20 uL of MQ water and loaded on 20 % polyacrylamide gel containing 8 M Urea and electrophoresed in 1 X TBE at RT, 40 V cm−1 for 2 h and scanned in a phosphorimager preceded by gel fixation and drying.
Supplementary Material
Acknowledgments:
This work was supported by The Wellcome Trust DBT India Alliance. We thank V. Noyola for assistance in preparing graphics and schematics. MZ thanks the NIH Chemistry-Biology Interface (CBI) Predoctoral Training Program for funding.
References:
- [1].Baillie GS, FEBS J. 2009, 276, 1790–1799. [DOI] [PubMed] [Google Scholar]
- [2].Willoughby D, Cooper DMF, Nat. Methods 2008, 5, 29–36. [DOI] [PubMed] [Google Scholar]
- [3].Sprenger JU, Nikolaev VO, Int. J. Mol. Sci 2013, 14, 8025–8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, Moolenaar WH, Bos JL, Jalink K, EMBO Rep. 2004, 5, 1176–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hackley CR, Mazzoni EO, Blau J, Sci. Signal 2018, 11, DOI 10.1126/scisignal.aah3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bai G, Schaak DD, McDonough KA, FEMS Immunol. Med. Microbiol. 2009, 55, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dass BKM, Sharma R, Shenoy AR, Mattoo R, Visweswariah SS, J. Bacteriol 2008, 190, 3824–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Buettner MJ, Spitz E, Rickenberg HV, J. Bacteriol 1973, 114, 1068–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lowrie DB, Jackett PS, Ratcliffe NA, Nature 1975, 254, 600–602. [DOI] [PubMed] [Google Scholar]
- [10].Ellington AD, Szostak JW, Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- [11].Cho EJ, Lee J-W, Ellington AD, Annu. Rev. Anal. Chem. (Palo Alto, Calif.) 2009, 2, 241–264. [DOI] [PubMed] [Google Scholar]
- [12].Pollard J, Bell SD, Ellington AD, Curr. Protoc. Nucleic Acid Chem. 2001, Chapter 9, Unit 9.2. [DOI] [PubMed] [Google Scholar]
- [13].Bruno JG, Pharmaceuticals (Basel) 2013, 6, 340–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jani MS, Veetil AT, Krishnan Y, Nat. Rev. Mater 2019, DOI 10.1038/s41578-019-0105-4. [DOI] [Google Scholar]
- [15].Dua P, Kim S, Lee D-K, Methods 2011, 54, 215–225. [DOI] [PubMed] [Google Scholar]
- [16].Banerjee A, Bhatia D, Saminathan A, Chakraborty S, Kar S, Krishnan Y, Angew. Chem. Int. Ed. Engl 2013, 52, 6854–6857. [DOI] [PubMed] [Google Scholar]
- [17].Saha S, Prakash V, Halder S, Chakraborty K, Krishnan Y, Nat. Nanotechnol 2015, 10, 645–651. [DOI] [PubMed] [Google Scholar]
- [18].Chakraborty K, Veetil AT, Jaffrey SR, Krishnan Y, Annu. Rev. Biochem 2016, 85, 349–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kellenberger CA, Wilson SC, Sales-Lee J, Hammond MC, J. Am. Chem. Soc 2013, 135, 4906–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krishnan Y, Simmel FC, Angew. Chem. Int. Ed. Engl 2011, 50, 3124–3156. [DOI] [PubMed] [Google Scholar]
- [21].Modi S, M G S, Goswami D, Gupta GD, Mayor S, Krishnan Y, Nat. Nanotechnol 2009, 4, 325–330. [DOI] [PubMed] [Google Scholar]
- [22].Modi S, Halder S, Nizak C, Krishnan Y, Nanoscale 2014, 6, 1144–1152. [DOI] [PubMed] [Google Scholar]
- [23].Surana S, Bhat JM, Koushika SP, Krishnan Y, Nat. Commun 2011, 2, 340. [DOI] [PubMed] [Google Scholar]
- [24].Modi S, Nizak C, Surana S, Halder S, Krishnan Y, Nat. Nanotechnol 2013, 8, 459–467. [DOI] [PubMed] [Google Scholar]
- [25].Narayanaswamy N, Chakraborty K, Saminathan A, Zeichner E, Leung K, Devany J, Krishnan Y, Nat. Methods 2019, 16, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Leung K, Chakraborty K, Saminathan A, Krishnan Y, Nat. Nanotechnol 2019, 14, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sharma S, Zaveri A, Visweswariah SS, Krishnan Y, Small 2014, 10, 4276–4280. [DOI] [PubMed] [Google Scholar]
- [28].Koizumi M, Breaker RR, Biochemistry 2000, 39, 8983–8992. [DOI] [PubMed] [Google Scholar]
- [29].Huppert JL, Chem. Soc. Rev 2008, 37, 1375–1384. [DOI] [PubMed] [Google Scholar]
- [30].Bugaut A, Balasubramanian S, Biochemistry 2008, 47, 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Paramasivan S, Rujan I, Bolton PH, Methods 2007, 43, 324–331. [DOI] [PubMed] [Google Scholar]
- [32].Hazel P, Huppert J, Balasubramanian S, Neidle S, J. Am. Chem. Soc 2004, 126, 16405–16415. [DOI] [PubMed] [Google Scholar]
- [33].Huizenga DE, Szostak JW, Biochemistry 1995, 34, 656–665. [DOI] [PubMed] [Google Scholar]
- [34].Li T, Li B, Dong S, Chem. Eur. J 2007, 13, 6718–6723. [DOI] [PubMed] [Google Scholar]
- [35].Vámosi G, Gohlke C, Clegg RM, Biophys. J 1996, 71, 972–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Qu P, Chen X, Zhou X, Li X, Zhao X, Sci. China Ser. B-Chem 2009, 52, 1653–1659. [Google Scholar]
- [37].Edman L, Mets U, Rigler R, Proc. Natl. Acad. Sci. USA 1996, 93, 6710–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Patel DJ, Suri AK, Jiang F, Jiang L, Fan P, Kumar RA, Nonin S, J. Mol. Biol 1997, 272, 645–664. [DOI] [PubMed] [Google Scholar]
- [39].Hermann T, Patel DJ, Science 2000, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- [40].Monod J, Wyman J, Changeux JP, J. Mol. Biol 1965, 12, 88–118. [DOI] [PubMed] [Google Scholar]
- [41].Gianni S, Dogan J, Jemth P, Biophys Chem 2014, 189, 33–39. [DOI] [PubMed] [Google Scholar]
- [42].Sassanfar M, Szostak JW, Nature 1993, 364, 550–553. [DOI] [PubMed] [Google Scholar]
- [43].Burke DH, Gold L, Nucleic Acids Res. 1997, 25, 2020–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Burgstaller P, Famulok M, Angew. Chem. Int. Ed. Engl 1994, 33, 1084–1087. [Google Scholar]
- [45].Saran D, Frank J, Burke DH, BMC Evol. Biol. 2003, 3, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Barbu M, Stojanovic MN, Chembiochem 2012, 13, 658–660. [DOI] [PubMed] [Google Scholar]
- [47].Lin CH, Patel DJ, Chem. Biol 1997, 4, 817–832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.