

Abstract
The innate immune sensor NLRP3 assembles an inflammasome complex with NEK7 and ASC to activate caspase‐1 and drive the maturation of proinflammatory cytokines IL‐1β and IL‐18. NLRP3 inflammasome activity must be tightly controlled, as its over‐activation is involved in the pathogenesis of inflammatory diseases. Here, we show that NLRP3 inflammasome activation is suppressed by a centrosomal protein Spata2. Spata2 deficiency enhances NLRP3 inflammasome activity both in the macrophages and in an animal model of peritonitis. Mechanistically, Spata2 recruits the deubiquitinase CYLD to the centrosome for deubiquitination of polo‐like kinase 4 (PLK4), the master regulator of centrosome duplication. Deubiquitination of PLK4 facilitates its binding to and phosphorylation of NEK7 at Ser204. NEK7 phosphorylation in turn attenuates NEK7 and NLRP3 interaction, which is required for NLRP3 inflammasome activation. Pharmacological or shRNA‐mediated inhibition of PLK4, or mutation of the NEK7 Ser204 phosphorylation site, augments NEK7 interaction with NLRP3 and causes increased NLRP3 inflammasome activation. Our study unravels a novel centrosomal regulatory pathway of inflammasome activation and may provide new therapeutic targets for the treatment of NLRP3‐associated inflammatory diseases.
Keywords: CYLD, deubiquitination, NEK7, NLRP3, Spata2
Subject Categories: Immunology, Signal Transduction
The centrosomal protein Spata2 is as a novel regulator of NEK7 phosphorylation and NLRP3‐mediated inflammatory responses.
Introduction
Inflammasomes are innate immune sensors that activate inflammatory caspases, thereby mediating maturation and secretion of the proinflammatory cytokines IL‐1β and IL‐18 (Schroder & Tschopp, 2010; Davis et al, 2011; Latz et al, 2013). The NLRP3 inflammasome responses to diverse microbial products and damage‐associated signals and has been implicated in the pathogenesis of a number of inflammatory diseases (Guo et al, 2015; Broz & Dixit, 2016). Better understanding of the mechanisms that control NLRP3 inflammasome activation would be valuable for the design of new therapeutic strategies for the treatment of inflammatory diseases.
NLRP3 inflammasome activation requires two steps, the priming step that is mediated by TLR agonists, such as LPS, for the induction of the expression of NLRP3 and pro‐IL‐1β, and the inflammasome assembly step that is in response to multiple activating stimuli, including ATP, nigericin, and crystalline reagents, to trigger the assembly of the NLRP3‐ASC complex into a large single speck in a perinuclear region, which leads to the recruitment and activation of caspase‐1 and the subsequent production of active IL‐1β and IL‐18 (Schroder & Tschopp, 2010; Davis et al, 2011; Latz et al, 2013). Recent studies have demonstrated that NLRP3 inflammasome activation requires the association of NLRP3 with NEK7 (He et al, 2016; Schmid‐Burgk et al, 2016; Shi et al, 2016; Sharif et al, 2019), a serine/threonine kinase that localizes in centrosomes to regulate mitotic spindle formation and centrosome separation during mitosis (O'Regan et al, 2007) and centrosome duplication during interphase (Kim et al, 2011). NEK7 has also been shown recently to control telomere DNA replication via forming a multi‐protein complex with telomeric shelterin proteins (Tan et al, 2017). Interestingly, NEK7 kinase activity is dispensable for NLRP3 inflammasome activation (He et al, 2016; Shi et al, 2016). Notably, NEK7‐mediated NLRP3 inflammasome activation is induced in interphase and attenuated during mitosis, suggesting that functions of NEK7 in mitosis and inflammasome activation are mutually exclusive, likely due to insufficient quantity of NEK7 (Shi et al, 2016). Despite the importance of NEK7 in NLRP3 inflammasome activation, how NLRP3‐NEK7 association is regulated remains elusive.
A hallmark of inflammasome activation is the formation of one large speck structure in a perinuclear area per cell (Li et al, 2017; Chen & Chen, 2018). Little is known about what this cellular structure is and why solely one speck is formed per cell. A recent study demonstrates that in response to activating stimuli NLRP3 is transported through microtubules to the microtubule‐organizing center (MTOC), namely the centrosome in mammalian cells, for the assembly and activation of inflammasome, enabling the formation of a single inflammasome speck within the cell (Li et al, 2017). Importantly, the assembly of NLRP3 inflammasome in the centrosome is critical for its optimal activation since improper positioning of NLRP3 due to inhibition of the MARK4, a driver of NLRP3 transporting along microtubules, attenuates inflammasome activation (Li et al, 2017). These findings link centrosome to inflammasome activation and raise a possibility that inflammasome activity is subject to regulation by the centrosome or centrosome‐localized proteins.
Inflammasome activation is fine‐tuned by post‐translational modifications, among which protein ubiquitination has emerged as a critical mechanism of NLRP3 inflammasome regulation. An increasing number of E3 ligases and deubiquitinases (DUBs) have been identified as modifiers for different ubiquitin chain conjugation on or removal from components of NLRP3 inflammasome, such as NLRP3, ASC, and IL‐1β (Lopez‐Castejon & Edelmann, 2016). As a linear‐ and K63 linkage‐specific DUB, CYLD has long been known to function as a crucial regulator for inflammatory responses (Sun, 2010). However, it is still unclear if it is involved in the regulation of inflammasome activation. In a screen for CYLD‐binding proteins using proximity‐dependent biotin identification (BioID; Roux et al, 2013), we identify a CYLD‐binding adaptor protein Spata2. We show that Spata2 is specifically localized in centrosomes where it recruits CYLD for deubiquitination of Polo‐like kinase 4 (PLK4), a centrosome‐localized kinase that functions as a master regulator of centrosome duplication (Maniswami et al, 2018). Interestingly, we show further that deubiquitination of PLK4 leads to an augmented binding and phosphorylation of NEK7, which in turn inhibits the association of NEK7 with NLRP3, thereby preventing inflammasome activation. Our study reveals a novel signaling cascade on centrosomal activation of NLRP3 inflammasome and may provide potential therapeutic targets against inflammatory diseases.
Results
Spata2 negatively regulates inflammatory responses in vivo
CYLD, a DUB, has been shown to deconjugate both linear ubiquitin chains and lysine (K) 63‐linked ubiquitin chains, and plays a crucial role in negatively regulating inflammatory responses (Harhaj & Dixit, 2012; Lork et al, 2017). To understand the molecular mechanism underlying the signaling function of CYLD, we screened for CYLD‐binding proteins using BioID (Appendix Fig S1). Among the major CYLD‐binding proteins identified was an adaptor protein named Spata2, which was also identified as a CYLD‐binding protein by others using different approaches (Elliott et al, 2016; Kupka et al, 2016; Schlicher et al, 2016; Wagner et al, 2016). Co‐immunoprecipitation (co‐IP) assays confirmed the Spata2/CYLD interaction under both transfection and endogenous conditions (Appendix Fig S2A–C). This molecular interaction occurred constitutively and was not affected by TNFα stimulation (Appendix Fig S2B). Domain mapping revealed that the Spata2/CYLD interaction required the PUB domain of Spata2 and the B box of CYLD (Appendix Fig S2D–G).
Spata2 has been implicated in the regulation of TNFα‐induced cell death and NF‐κB activation, although the latter function has been controversial (Elliott et al, 2016; Kupka et al, 2016; Schlicher et al, 2016; Wagner et al, 2016; Wei et al, 2017). To investigate the in vivo function of Spata2, we generated Spata2 knockout (KO) mice. These mutant mice were born at expected Mendelian ratios and did not display obvious abnormalities in growth and survival when housed in specific pathogen‐free conditions. To examine the role of Spata2 in regulating inflammation, we employed a septic shock model of acute inflammation induced by LPS, known to be due to induction of proinflammatory cytokines, such as IL‐1β, IL‐18, and TNFα (Dinarello, 1997; Vanden Berghe et al, 2014). Compared with WT mice, Spata2 KO mice were profoundly more sensitive to LPS‐stimulated septic shock (Fig 1A). Consistently, the serum of LPS‐challenged Spata2 KO mice contained a significantly higher level of proinflammatory cytokines, IL‐1β, IL‐18, and TNFα, than that of WT mice (Fig 1B). These results demonstrate a crucial role for Spata2 in negatively regulating inflammatory responses in vivo.
Figure 1. Spata2 is a negative regulator of inflammation.
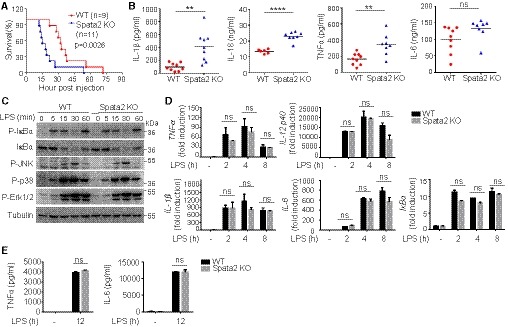
- Survival of age‐ and sex‐matched (8–10 week old) Spata2 +/+ (WT) and Spata2 −/− (KO) mice injected intraperitoneally (i.p.) with LPS (20 mg/kg bodyweight). Data pooled from three independent experiments are shown. Statistical analysis was performed using long‐rank test.
- ELISA of serum IL‐1β, IL‐18, TNFα, and IL‐6 at 12 h after injection of WT or Spata2‐KO mice with 20 mg/kg LPS. Circles and triangles represent individual mice. Horizontal bars indicate mean values. **P < 0.01; ****P < 0.0001. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
- Immunoblot analysis of the indicated phosphorylated (P) and total proteins in lysates of LPS‐stimulated BMDMs derived from WT or Spata2‐KO mice. Molecular weights in kDa are indicated to the right.
- qRT–PCR analysis of the indicated mRNAs in untreated or LPS‐stimulated WT or Spata2‐KO BMDMs. Bars and error bars represent the mean ± SD of triplicate experiments. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
- ELISA of TNFα and IL‐6 in the supernatants from untreated or LPS‐stimulated WT or Spata2‐KO BMDMs. Bars and error bars represent the mean ± SD of triplicate experiments. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
Source data are available online for this figure.
To examine the mechanism underlying the anti‐inflammatory function of Spata2, we analyzed the effect of Spata2 on cell signaling and gene induction using bone marrow‐derived macrophages (BMDMs) from WT and Spata2‐KO mice. Surprisingly, Spata2 deficiency had no effect on NF‐κB activation in BMDMs stimulated with the TLR4 ligand LPS (Fig 1C). The loss of Spata2 also had no effect on LPS‐stimulated expression of several NF‐κB target genes at the levels of both transcription (Fig 1D) and translation (Fig 1E). Thus, Spata2 is dispensable for NF‐κB and MAPK signaling and inflammatory gene induction in macrophages.
Spata2 specifically inhibits NLRP3 inflammasome activation
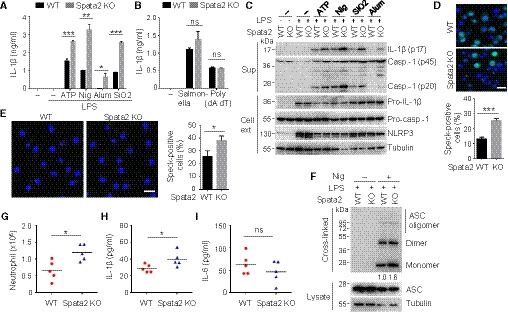
The results described above prompted us to examine whether Spata2 had a role in regulating proinflammatory cytokine secretion mediated by inflammasomes. Indeed, the Spata2‐deficient BMDMs secreted a significantly higher amount of IL‐1β in response to different inducers of NLRP3 inflammasome than WT BMDMs (Fig 2A). On the other hand, the Spata2 deficiency did not promote IL‐1β induction by the NLRC4 inflammasome inducer Salmonella, the AIM2 inflammasome inducer poly (dA:dT) (Fig 2B), or the non‐canonical NLRP3 inflammasome inducer cytoplasmic LPS (Appendix Fig S3A), suggesting a selective role of Spata2 in canonical NLRP3 inflammasome regulation. The negative role of Spata2 in regulating NLRP3 inflammasome activation was further confirmed by immunoblotting assays, demonstrating enhanced generation and secretion of mature IL‐1β p17 and cleaved form of caspase‐1 p20 in the Spata2 KO macrophages upon stimulation by several NLRP3 inducers (Fig 2C). NLRP3 inflammasome activation involves aggregation of the adaptor protein ASC to form microscopically visible complexes with NLRP3 (Fernandes‐Alnemri et al, 2007; Broz et al, 2010; Lu et al, 2014). Consistent with the negative role of Spata2 in regulating NLRP3 inflammasome activation, the Spata2 deficiency significantly promoted the formation of ASC specks (Fig 2D and E) and the level of ASC oligomerization (Fig 2F and Appendix Fig S4C) in both primary BMDMs and immortalized BMDMs (iBMDMs). In agreement with the enhanced canonical NLRP3 inflammasome activation in Spata2 KO cells, the loss of Spata2 resulted in more pyroptotic cell death indicated by higher levels of lactate dehydrogenase (LDH) release upon canonical but not noncononical NLRP3 inflammasome inducers (Appendix Figs S3C and S4B).
Figure 2. Spata2 suppresses NLRP3 inflammasome activation in vitro and in vivo .
-
A, BELISA of IL‐1β secretion by WT or Spata2‐KO BMDMs that were either untreated (−) or LPS‐primed and then treated with the indicated NLRP3 inducers (A) or stimulated with the NLRC4 inducer (Salmonella infection) or AIM2 inducer (Poly(dA:dT) transfection) (B). Bars and error bars represent the mean ± SD of triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
-
CImmunoblot analysis of the mature IL‐1β p17 and the active caspase‐1 p20 in cell supernatants (Sup) and the indicated proteins in the cell extracts (Cell ext) of WT or Spata2‐KO BMDMs stimulated as in (A).
-
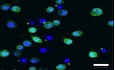
DWT and Spata2 CRISPR knockout (KO) iBMDMs stably expressing GFP‐ASC were primed with LPS and stimulated with Nigericin for 1 h. GFP‐ASC specks were imaged by confocal microscope, and percentage of ASC speck‐positive cells was quantified. Scale bar, 10 μm. Bars and error bars represent the mean ± SD of triplicate experiments. ****P < 0.0001. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
-
EBMDMs from Spata2 +/+ (WT) and Spata2 −/− (KO) mice were pretreated with caspase‐1 inhibitor, Z‐YVAD‐FMK, for 30 min, primed with LPS, and then stimulated with Nigericin for 1 h. ASC specks were assessed by immunofluorescence with antibody against ASC and imaged by confocal microscope, and percentage of ASC speck‐positive cells was quantified. Scale bar, 10 μm. Bars and error bars represent the mean ± SD of triplicate experiments. *P < 0.05. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
-
FImmunoblot analysis of ASC in soluble cell lysate and DSS cross‐linked insoluble fraction of WT or Spata2‐KO BMDMs primed with LPS and stimulated without or with nigericin (Nig) for 1 h. Relative levels of ASC in cross‐linked samples from LPS+ Nig treated cells were quantitated and shown below.
-
G‐ISpata2 +/+ (WT) and Spata2 −/− (KO) mice were injected intraperitoneally (i.p.) with alum (20 mg/kg bodyweight) and sacrificed 6 h latter to collect peritoneal lavages. Absolute numbers of neutrophils recruited to the peritoneum were counted by FACS (G); levels of IL‐1β (H) and IL‐6 (I) in the peritoneal fluids were measured by ELISA. Circles and triangles represent individual mice. Horizontal bars indicate mean values. *P < 0.05. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
To study the in vivo function of Spata2 in regulating NLRP3 inflammasome, we employed a mouse model of peritonitis. Intraperitoneal injection of alum into mice induces NLRP3‐dependent IL‐1β production and the recruitment of inflammatory cells, particularly neutrophils, to the peritoneal cavity (Guarda et al, 2011). When challenged with alum, Spata2 KO mice showed significantly increased recruitment of neutrophils to the peritoneal cavity and higher levels of IL‐1β in peritoneal fluid compared with WT mice (Fig 2G and H). In contrast, there was no difference in the level of peritoneal fluid IL‐6 between WT and Spata2 KO mice (Fig 2I). These results indicate that Spata2 inhibits NLRP3 inflammasome activation in vivo.
CYLD suppresses NLRP3 inflammasome activation
The DUB CYLD is important for preventing inflammation (Harhaj & Dixit, 2012; Lork et al, 2017). Although CYLD has been shown to negatively regulate NF‐κB activation in lymphocytes (Sun, 2010; Lork et al, 2017), its role in regulating inflammasome activation in innate immune cells has been undefined. Since Spata2 is a major partner protein of CYLD, we examined the role of CYLD in regulating inflammasome activation. As seen with Spata2 deficiency, the CYLD deficiency in macrophages profoundly promoted IL‐1β secretion, active caspase‐1 (p20) generation, ASC speck formation, and pyroptotic cell death in response to canonical activation of NLRP3 inflammasome (Fig 3A–D) but not the non‐canonical activation (Appendix Fig S3B and D). Consistent with these loss‐of‐function studies, overexpressed CYLD and Spata2 inhibited NLRP3 inflammasome activation in a HEK293 inflammasome reconstitution model (Appendix Fig S5). This function of CYLD was dependent on its DUB activity, since a catalytically inactive CYLD mutant (C601S) failed to inhibit NLRP3 activation (Appendix Fig S5).
Figure 3. CYLD suppresses NLRP3 inflammasome activation in a manner dependent on Spata2‐CYLD interaction.
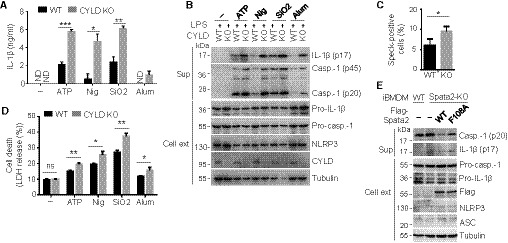
- ELISA of IL‐1β secretion by WT or CYLD KO BMDMs that were either untreated (−) or LPS‐primed and then treated with the indicated NLRP3 inducers. Bars and error bars represent the mean ± SD of triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001. Statistical analysis was performed using unpaired two‐tailed Student's t‐test. ND, not detected.
- Immunoblot analysis of IL‐1β and active caspase‐1 p20 in cell supernatants (Sup) and the indicated proteins in cell extracts (Cell ext) from WT and CYLD KO BMDMs, stimulated as in (A).
- BMDMs from WT and CYLD KO mice were pretreated with caspase‐1 inhibitor, Z‐YVAD‐FMK, for 30 min, primed with LPS, and then stimulated with Nigericin for 30 min. ASC specks were assessed by immunofluorescence with antibody against ASC, and the percentage of ASC speck‐positive cells was quantified. Bars and error bars represent the mean ± SD of triplicate experiments. *P < 0.05. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
- LDH release from WT and CYLD KO BMDMs primed with LPS and stimulated as indicated. Bars and error bars represent the mean ± SD of triplicate experiments. **P < 0.01. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
- Immunoblotting analysis of active caspase‐1 p20 and IL‐1β in cell supernatants (Sup) and the indicated proteins in cell extracts (Cell ext) of WT iBMDMs and Spata2 KO iBMDMs reconstituted with WT Spata2 or a Spata2 mutant (F108A) defective in CYLD binding that were primed with LPS and stimulated with Nigericin for 1 h.
To examine the role of Spata2‐CYLD physical interaction in inflammasome regulation, we knocked out Spata2 in iBMDMs and reconstituted the cells with either WT Spata2 or a Spata2 point mutant, F108A, known to be defective in CYLD binding (Schlicher et al, 2016). As expected, Spata2 knockout enhanced inflammasome activation, as revealed by the hyperproduction of cleaved caspase‐1 and IL‐1β (Fig 3E). Moreover, the expression of WT Spata2, but not Spata2 F108A, suppressed the inflammasome activation (Fig 3E). Collectively, these results provide genetic evidence that Spata2 and its partner protein CYLD serve as negative regulators of NLRP3 inflammasome activation.
Microtubule integrity and microtubule‐dependent transport have been shown to be critical for inflammasome activation (Misawa et al, 2013; Li et al, 2017). Specifically, inflammasome inducers stimulate α‐tubulin acetylation which mediates microtubule‐dependent transport and NLRP3 inflammasome activation (Misawa et al, 2013). Recent studies revealed that CYLD promotes acetylation of α‐tubulin and stabilization of microtubules in keratinocytes and MEFs (Wickstrom et al, 2010; Yang et al, 2014). To investigate whether CYLD and Spata2 are involved in regulation of α‐tubulin acetylation during inflammasome activation, we assessed the effect of CYLD or Spata2 deficiency on the inflammasome inducer‐stimulated acetylation of α‐tubulin in BMDMs. Interestingly, neither loss of Spata2 nor ablation of CYLD altered the basal level of nigericin‐stimulated acetylation of α‐tubulin (Appendix Fig S4A and D). These results suggest that Spata2 and CYLD may regulate inflammasome activation in a manner independent of α‐tubulin acetylation and microtubule stability.
Spata2 is localized to the centrosome where it recruits CYLD to inhibit NLRP3 inflammasome activation
To further delineate the mechanism by which Spata2 regulates NLRP3 inflammasome activation, we searched for Spata2‐associated proteins based on the BioGrid database (https://thebiogrid.org). Interestingly, in addition to CYLD, several centrosomal proteins are on the Spata2‐interacting protein list, including CEP72, CEP128, CEP135, CEP192, and CNTROB. This finding prompted us to examine whether Spata2 was associated with the centrosome. Confocal microscopy based on a well‐defined centrosomal marker, γ‐tubulin, indeed revealed the predominant localization of Spata2 in specks that were overlapping with the γ‐tubulin‐positive centrosomes in HEK293 cells (Fig 4A). The Spata2 staining was specific, as demonstrated by the negative staining in Spata2‐knockdown cells (Fig 4A). Since there is no commercial Spata2 antibody effective for immunofluorescence detection of murine Spata2, we infected the murine macrophage cell line Raw264.7 with a GFP‐tagged Spata2. As seen with endogenous Spata2, the GFP‐Spata2 was predominantly localized in the γ‐tubulin‐positive centrosomes (Fig 4B). This result was further confirmed using cells transfected with epitope‐tagged Spata2 and γ‐tubulin, demonstrating their colocalization in centrosomes (Fig 4C). Domain mapping studies revealed that the N‐terminal PUB domain of Spata2 was required for its centrosome localization since deletion of the entire PUB domain or a region (amino acids 51–81) within this domain abolished the centrosomal localization (Fig 4C).
Figure 4. Centrosomal localization of Spata2 is critical for recruiting CYLD to the centrosome and suppressing NLRP3 inflammasome activation.
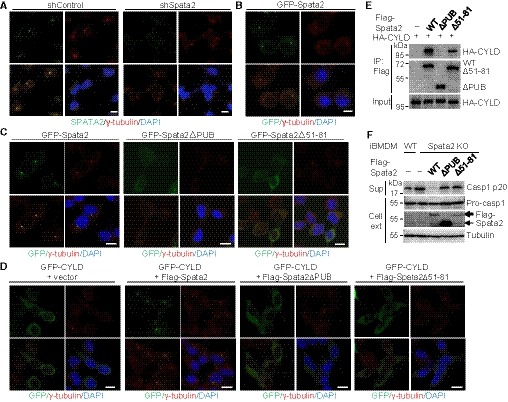
- Confocal immunofluorescence analysis of Spata2 centrosome localization in HEK293 cells stably expressing control shRNA or shRNA against Spata2, stained with Spata2 and γ‐tubulin antibodies and the nuclear dye DAPI. Scale bar, 10 μm.
- Confocal immunofluorescence analysis of Spata2 localization in RAW264.7 cells stably expressing GFP‐Spata2, stained with γ‐tubulin antibody and DAPI. Scale bar, 10 μm.
- Confocal microscopy of HEK293 cells stably expressing GFP‐tagged Spata2 or Spata2 mutants (ΔPUB or Δ51–81) that were stained with γ‐tubulin antibody and DAPI.
- Confocal microscopy of HEK293 cells stably expressing GFP‐tagged CYLD along with an empty vector or vectors encoding Flag‐tagged Spata2 or Spata2 mutants (ΔPUB or Δ51–81) stained as in (C). Scale bar, 10 μm.
- Co‐immunoprecipitation analysis of CYLD physical interaction with Spata2 or Spata2 mutants (ΔPUB or Δ51–81) in HEK293 cells transfected with the indicated expression vectors. CYLD and Spata2 proteins were detected by immunoblot using anti‐HA and anti‐Flag antibodies, respectively.
- Immunoblotting analysis of the active caspase‐1 p20 in cell supernatants (Sup) and pro‐caspase‐1 and Flag‐Spata2 proteins in cell extracts (Cell ext) from WT iBMDMs and Spata2 KO iBMDMs reconstituted with WT Spata2 or Spata2 mutant defective in centrosome localization (ΔPUB or Δ51–81). Cells were primed with LPS and stimulated with Nigericin for 1 h.
In contrast to the centrosome location of Spata2, CYLD was located in the cytoplasm (Fig 4D). However, when co‐expressed with Spata2, CYLD was re‐located to specks that were overlapping with the γ‐tubulin‐positive centrosomes (Fig 4D). The function of Spata2 to recruit CYLD to the centrosome depended on the PUB domain of Spata2 (Fig 4D). Since the PUB domain of Spata2 was required for both CYLD binding (Fig 4E) and centrosome localization (Fig 4D), we tested the function of Spata2 ∆51–81, which was defective in centrosome localization (Fig 4C) but retained a significant level of CYLD‐binding function (Fig 4E). Importantly, Spata2 ∆51–81 also failed to recruit CYLD to the centrosome specks (Fig 4D), suggesting the requirement of both centrosome localization and CYLD‐binding functions of Spata2 in mediating CYLD recruitment to the centrosome. These findings suggest that Spata2 is associated with centrosome and is able to recruit its partner protein CYLD into the centrosome.
To examine the functional significance of Spata2 centrosome location in inflammasome regulation, we reconstituted the Spata2‐deficient iBMDMs with either WT Spata2 or its mutants harboring deletions in the PUB domain, ∆PUB and ∆51–81, which were defective in centrosome localization (Fig 4C). The expression of WT Spata2 potently inhibited NLRP3 inflammasome activation stimulated by LPS plus nigericin, as demonstrated by the blockade of pro‐caspase‐1 processing to caspase‐1 p20 (Fig 4F). In contrast, the expression of Spata2 ∆PUB or Spata2 ∆51–81 failed to inhibit the NLRP3 inflammasome activation (Fig 4F). Thus, the centrosome‐association function of Spata2 appears to be required for inflammasome regulation.
Centrosomal kinase PLK4 binds Spata2 and CYLD and inhibits NLRP3 inflammasome activation
Recent studies have identified the centrosome‐associated protein NEK7 as an essential mediator of NLRP3 inflammasome activation (He et al, 2016; Schmid‐Burgk et al, 2016; Shi et al, 2016). Notably, the functions of NEK7 in regulating inflammasome activation and mitosis are mutually exclusive, although the underlying mechanism of NEK7 regulation is undefined (Shi et al, 2016). Our finding that Spata2 was located in the centrosome to negatively regulate inflammasome activation prompted us to examine whether Spata2 played a role in regulating NEK7 function. In agreement with previous reports (He et al, 2016; Shi et al, 2016), co‐IP assays detected NEK7/NLRP3 physical interaction in cells stimulated with LPS or LPS plus nigericin (Fig 5A). Importantly, Spata2 deficiency profoundly promoted the binding of NEK7 to NLRP3 (Fig 5A), which explains the enhanced inflammasome activation. Given that NLRP3 inflammasome is assembled and activated in the centrosome (Li et al, 2017), we searched for the proteins in the centrosome that might be associated with Spata2 function. We found that Spata2 and CYLD physically associated with protein kinase PLK4 (Fig 5B), a central regulator of centrosome formation and mitosis (Maniswami et al, 2018). Since the commercial antibodies to PLK4 we tested did not work for immunoprecipitation, virally transduced cells that stably expressed GFP‐tagged PLK4 were used for PLK4 immunoprecipitation assays. In HEK293 cells stably expressing GFP‐PLK4, PLK4 formed a stable complex with Spata2 and CYLD that was immunoprecipitated by the anti‐GFP antibody (Fig 5B). The physical association between Spata2 and PLK4 was also revealed by confocal microscopy analysis, showing that PLK4 and Spata2 were co‐localized in centrosomes in co‐transfected HEK293 cells (Appendix Fig S6A). We further confirmed the interaction of CYLD with PLK4 in iBMDMs stably expressing GFP‐PLK4. Similarly, CYLD was detected in anti‐GFP antibody immunoprecipitates from the cells stably expressing GFP‐PLK4 but not the parental cells without expressing GFP‐PLK4 (Fig 5C). Interestingly, the association of CYLD with PLK4 remained unchanged after cells were stimulated with LPS (Fig 5C), suggesting a constant binding of CYLD to PLK4.
Figure 5. Centrosomal kinase PLK4 suppresses NLRP3 inflammasome activation in a kinase activity‐dependent manner.
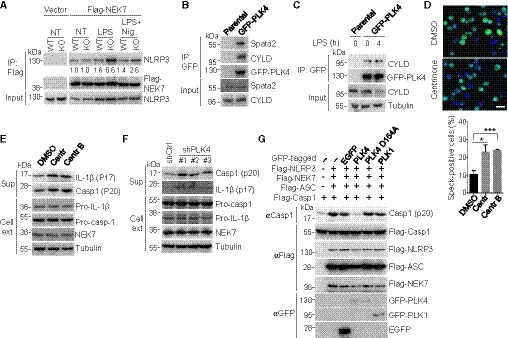
-
AWT and Spata2 KO iBMDMs stably expressing an empty vector or a vector encoding Flag‐NEK7 were untreated (NT), primed with LPS for 4 h, or primed with LPS for 4 h followed by stimulation with Nigericin for 30 min. Flag‐NEK7 was immunoprecipitated, and the association of endogenous NLRP3 was assessed by immunoblotting. Relative levels of NLRP3 in immunoprecipitates were normalized to Flag‐NEK7 and shown below.
-
BCo‐immunoprecipitation analysis of PLK4 interaction with endogenous Spata2 and CYLD in parental HEK293 cells and HEK293 cells stably expressing GFP‐PLK4.
-
CiBMDMs stably expressing GFP‐PLK4 and parental iBMDMs were unstimulated or stimulated with 0.5 μg/ml LPS for 4 h. The PLK4 interaction with endogenous CYLD was analyzed by co‐immunoprecipitation with anti‐GFP antibody.
-
DConfocal microscopy analysis of ASC specks in iBMDMs stably expressing GFP‐ASC, pretreated for 2 days with DMSO and two different PLK4 inhibitors, Centrinone (150 nM) and Centrinone‐B(500 nM), primed with LPS for 4 h, and stimulated with Nigericin for 30 min. GFP‐ASC specks were imaged and quantified (> 500 cells counted). Scale bar, 10 μm. Bars and error bars represent the mean ± SD of triplicate experiments. *P < 0.05; ***P < 0.001. Statistical analysis was performed using unpaired two‐tailed Student's t‐test.
-
E, FImmunoblot analysis of the indicated proteins in cell supernatants (Sup) and cell extracts (Cell ext) of LPS‐primed and Nigericin‐stimulated BMDMs that were pretreated for 2 days with PLK4 inhibitors, Centrinone (150 nM) and Centrinone‐B (500 nM) (E), or of LPS‐primed and Nigericin‐stimulated iBMDMs stably expressing a control shRNA or three different PLK4‐specific shRNAs (F).
-
GImmunoblot analysis of active caspase‐1 (p20) generation in HEK293 cells that were transfected with inflammasome components (Flag‐tagged NLRP3, NEK7, ASC, and caspase‐1) along with EGFP, GFP‐tagged PLK4, PLK4 kinase dead mutant (D154A), or PLK1 and stimulated with 10 μM Nigericin for 1 h 24 h post‐transfection.
The physical interaction of Spata2 and CYLD with PLK4 raised the possibility that PLK4 played a role in inflammasome regulation. To test this possibility, we investigated the effect of pharmacological inhibition of PLK4 on NLRP3 inflammasome activation by using two PLK4‐specific inhibitors, Centrinone and Centrinone‐B (Wong et al, 2015). It has been well documented that inhibition of PLK4 with Centrinone for sufficient length of time can reduce cell proliferation rate and block centrosome duplication in many types of cell lines, such as HeLa cells (Wong et al, 2015). For iBMDMs, treatment with 150 nM of Centrinone started to exhibit inhibitory effect on cell proliferation at day 3 (Appendix Fig S7A). However, 2‐day treatment changed neither cell proliferation rate (Appendix Fig S7A) nor centrosome number (Appendix Fig S8A and B). Interestingly, 2‐day treatment with the same concentration of Centrinone or 500 nM Centrinone‐B significantly enhanced LPS/nigericin‐stimulated formation of ASC specks in iBMDMs (Fig 5D) and activation of NLRP3 inflammasome in BMDMs (Fig 5E). As seen with Spata2 or CYLD deficiency, inhibition of PLK4 with Centrinone had little effect on non‐canonical NLRP3 inflammasome‐induced IL‐1β release and pyroptotic cell death (Appendix Fig S7B). Furthermore, much shorter PLK4 inhibitor treatments ranging from 3 h to 12 h could also markedly promote the LPS/nigericin‐stimulated activation of NLRP3 inflammasome in both BMDMs and iBMDMs (Appendix Fig S8C and D). These results suggest that PLK4 inhibits NLRP3 inflammasome activation in a kinase activity‐dependent manner but very likely acts independently of its function in controlling cell proliferation and centrosome duplication.
In addition to acute inhibition of PLK4 by pharmacological inhibitors, chronic silencing PLK4 using three different shRNAs also promoted NLRP3 inflammasome activation (Fig 5F and Appendix Fig S8F). In agreement with these loss‐of‐function studies, overexpression of PLK4 inhibited NLRP3 inflammasome activation in the HEK293 cell reconstitution model (Fig 5G). This function of PLK4 was specific, since the expression of another polo‐like kinase member, PLK1, failed to inhibit inflammasome activation (Fig 5G). Furthermore, the function of PLK4 in inflammasome regulation required its kinase activity, since a catalytically inactive PLK4 mutant, PLK4 D154A, was unable to inhibit inflammasome activation (Fig 5G). In line with these results, PLK4, like Spata2, inhibited the formation of ASC specks in HEK293 cells (Appendix Fig S6B–D). Collectively, these results demonstrated a crucial role of centrosomal protein kinase PLK4 in the negative regulation of NLRP3 inflammasome activation.
PLK4 phosphorylates NEK7 at Ser204
Our finding of Spata2‐PLK4 physical interaction suggested the possibility that Spata2 might function together with PLK4 to regulate inflammasome activation. To test this possibility, we treated the WT and Spata2‐KO iBMDMs with the PLK4 inhibitor Centrinone. PLK4 inhibition significantly enhanced LPS/nigericin‐induced ASC speck formation, mature IL1‐β production and pyroptotic cell death in WT but not Spata2‐KO iBMDMs (Fig 6A and Appendix Fig S9), suggesting that Spata2 and PLK4 may act in the same signaling pathway to inhibit NLRP3 inflammasome. Furthermore, like Spata2 deficiency, PLK4 knockdown markedly promoted the interaction of NLRP3 with NEK7 (Fig 6B), whereas its overexpression inhibited this interaction (Fig 6C), suggesting that PLK4 may negatively regulate the interaction of NEK7 with NLRP3. Moreover, the PLK4‐mediated inhibition of NEK7‐NLRP3 interaction could be partially overcome by treating cells with the PLK4 inhibitor, suggesting the dependence on its kinase activity (Fig 6C).
Figure 6. PLK4 inhibits NEK7‐NLRP3 interaction by mediating NEK7 phosphorylation at Ser204.
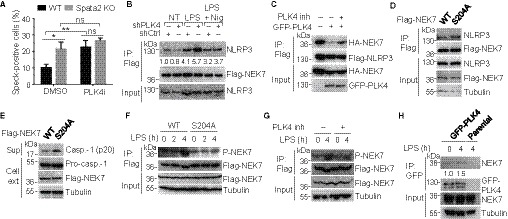
- WT and Spata2 CRISPR knockout (KO) iBMDMs stably expressing GFP‐ASC were pretreated with DMSO or PLK4 inhibitor Centrinone at a concentration of 150 nM for 2 days, primed with LPS, and stimulated with Nigericin for 1 h. GFP‐ASC specks were imaged by confocal microscope, and percentage of ASC speck‐positive cells was quantified. Bars and error bars represent the mean ± SD of triplicate experiments. *P < 0.05; **P < 0.01. Statistical analysis was performed using unpaired two‐tailed Student's t‐test. See Appendix Fig S9 for images of ASC specks and levels of IL‐1β and LDH.
- Co‐immunoprecipitation analysis of NEK7/NLRP3 interaction in iBMDMs stably expressing Flag‐NEK7 that were transduced with control or PLK4‐specific shRNA. The cells were untreated, primed with LPS, or primed with LPS and stimulated with Nigericin. Flag‐NEK7 was immunoprecipitated, and the association of endogenous NLRP3 was assessed by immunoblot analysis. Relative levels of NLRP3 in immunoprecipitates were normalized to Flag‐NEK7 and shown below.
- Co‐immunoprecipitation analysis of NEK7/NLRP3 interaction in HEK293 cells transfected with HA‐NEK7 and Flag‐NLRP3 along with (+) or without (−) GFP‐PLK4. The cells were either treated (+) or not treated (−) with the PLK4 inhibitor Centrinone.
- iBMDMs stably expressing Flag‐tagged NEK7 or NEK7 mutant S204A were treated with LPS. Anti‐Flag immunoprecipitates were prepared for immunoblot analysis for the interaction between NEK7 and endogenous NLRP3.
- Immunoblot analysis of active caspase‐1 (p20) in supernatants (Sup) and the indicated proteins in cell extracts (Cell ext) of iBMDMs stably expressing WT NEK7 or NEK7 mutant (S204A), primed with LPS and stimulated with nigericin.
- iBMDMs stably expressing Flag‐tagged WT NEK7 or NEK7 mutant S204A were left untreated or stimulated with LPS for indicated time. Anti‐Flag immunoprecipitates were prepared for immunoblot analysis for NEK7 phosphorylation at Ser204 (P‐NEK7).
- iBMDMs stably expressing Flag‐tagged WT NEK7 were left untreated or pretreated with PLK4 inhibitor Centrinone, and stimulated without or with LPS for 4 h. Anti‐Flag immunoprecipitates were prepared for immunoblot analysis for NEK7 phosphorylation at Ser204.
- iBMDMs stably expressing GFP‐tagged PLK4 and parental iBMDMs were left untreated or stimulated with LPS for 4 h. Anti‐GFP immunoprecipitates were prepared for immunoblot analysis for the interaction between GFP‐PLK4 and endogenous NEK7.
To understand the molecular mechanism by which PLK4 regulates NEK7, we expressed human NEK7 in HEK293 cells either in the absence or presence of PLK4 for mass spectrometry analysis of NEK7 phosphorylation. While several phosphorylation sites of NEK7 were identified, only two of them (Ser204 and Thr286) were induced by PLK4 (Appendix Fig S10A, Tables EV1 and EV2). Since Ser204, but not Thr286, was conserved between human and mouse NEK7, we generated a Ser204Ala point mutant of NEK7 to assess the functional significance of this phosphorylation in iBMDMs. Interestingly, the NEK7 Ser204Ala mutant displayed increased NLRP3‐binding ability (Fig 6D) and stronger inflammasome‐activation function (Fig 6E). Mutation of an adjacent phosphorylation site, Ser195, did not influence the NLRP3‐binding and inflammasome‐activation functions of NEK7 (Appendix Fig S10B and C). These results suggest that PLK4‐mediated suppression of inflammasome activation may involve phosphorylation of NEK7 at Ser204.
To confirm that NEK7 Ser204 was phosphorylated by PLK4, we generated a NEK7 Ser204 phospho‐specific antibody which detects the PLK4‐phosphorylated WT NEK7 but not the Ser204Ala mutant of NEK7 (Appendix Fig S10D). Next, we showed that LPS could stimulate phosphorylation of WT NEK7 but not that of Ser204Ala mutant in iBMDMs (Fig 6F), and that the LPS‐stimulated NEK7 Ser204 phosphorylation was blocked by PLK4 inhibition (Fig 6G). Finally, we found that PLK4 interacted with NEK7 in iBMDMs in a LPS stimulation‐dependent manner (Fig 6H). Together, these results reveal that LPS stimulates PLK4‐dependent phosphorylation of NEK7 at Ser204.
Spata2 and CYLD promote PLK4‐NEK7 interaction by deubiquitinating PLK4
Since Spata2 and PLK4 were shown to function in the same signaling axis, we then determined the epistasis between them. In transfected cells, NEK7‐NLRP3 interaction was inhibited by co‐expression of Spata2 (Fig 7A). However, PLK4 inhibitor blocked Spata2‐mediated inhibition of NEK7‐NLRP3 interaction, indicating that PLK4 might function downstream of Spata2 (Fig 7A). In support of this, the deficiency of Spata2 also caused a marked reduction of LPS‐stimulated phosphorylation of NEK7 at Ser204 (Fig 7B). As a major function of the Spata2/CYLD complex is to mediate deubiquitination, we examined the role of Spata2 and CYLD in regulating PLK4 ubiquitination. SiRNA‐mediated silencing of either Spata2 or CYLD resulted in marked accumulation of PLK4 conjugated with K63‐linked polyubiquitin chains but not the linear and K48‐linked ubiquitination of PLK4 (Fig 7C). Similarly, when using a K63‐specific tandem ubiquitin‐binding entity (TUBE; Hjerpe et al, 2009) to enrich and analyze K63‐linked polyubiquitinated proteins, we found that depletion of Spata2 or CYLD by shRNA also caused accumulation of K63‐linked polyubiquitin of PLK4 (Appendix Fig S11A). Furthermore, using an in vitro deubiquitination assay, we showed that Spata2/CYLD could directly catalyze the removal of K63 ubiquitin chains from PLK4 (Appendix Fig S11B). Consistent with no change in K48‐linked ubiquitination of PLK4 in the absence of Spata2 or CYLD, the knockdown of Spata2 or CYLD had no effect on either PLK4 protein level (Appendix Figs S11A and S12A) or the PLK4‐regulated duplication of centrioles (Appendix Fig S12B and C), while the Spata2 knockdown slightly reduced the kinase activity of PLK4, as measured by its activation loop autophosphorylation (Appendix Fig S12D). Interestingly, overexpression of Spata2 and CYLD, but not the catalytically inactive CYLD C601S mutant, augmented the binding of PLK4 with NEK7 (Fig 7D). Together, these data demonstrate that Spata2/CYLD regulates the binding of PLK4 to its downstream target NEK7.
Figure 7. Spata2 and CYLD regulate PLK4 ubiquitination and function in NLRP3 inflammasome inhibition.
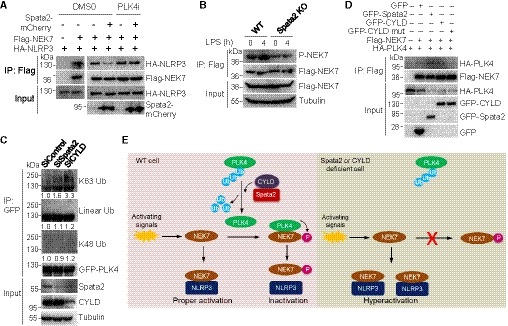
- Co‐immunoprecipitation analysis of NEK7/NLRP3 interaction in HEK293 cells transfected with the indicated expression vectors, treated with either DMSO or 150 nM PLK4 inhibitor Centrinone.
- WT and Spata2 KO iBMDMs stably expressing Flag‐tagged WT NEK7 were left untreated or pretreated with the PLK4 inhibitor Centrinone, and stimulated without or with LPS for 4 h. Anti‐Flag immunoprecipitates were prepared for immunoblot analysis for NEK7 phosphorylation at Ser204.
- HEK293 cells stably expressing GFP‐PLK4Δ24 were transfected with a control siRNA or siRNAs targeting Spata2 or CYLD. GFP‐PLK4Δ24 was immunoprecipitated under denaturing conditions and immunoblotted for K63‐linked, K48‐linked, and linear ubiquitination. Relative levels of PLK4 ubiquitination were normalized to GFP‐PLK4 and shown below.
- Co‐immunoprecipitation analysis of the PLK4‐NEK7 interaction in HEK293 cells co‐transfected with Flag‐NEK7 and HA‐PLK4 in the presence (+) or absence (−) of GFP, GFP‐Spata2, GFP‐CYLD, or GFP‐CYLD C601S, a catalytically inactive mutant.
- A model of NLRP3 inflammasome regulation by PLK4 and Spata2/CYLD. Polyubiquitination of PLK4 suppresses PLK4 binding to and phosphorylation of NEK7. This polyubiquitination‐mediated suppression can be reversed by Spata2 and CYLD‐mediated deubiquitination of PLK4, thereby allowing for PLK4 phosphorylation of NEK7, which attenuates NEK7 binding to NLRP3 and NLRP3 inflammasome activation. Spata2 or CYLD deficiency would cause accumulation of PLK4 polyubiquitination, reduction in NEK7 phosphorylation, increase in NEK7 binding to NLRP3, and eventually hyperactivation of NLRP3 inflammasome.
Discussion
Our BioID screen identified Spata2 as a CYLD‐interacting protein, and the Spata2‐CYLD interaction was also discovered by other groups using different strategies (Elliott et al, 2016; Kupka et al, 2016; Schlicher et al, 2016; Wagner et al, 2016). The best known function of Spata2 is to act as a CYLD partner protein that recruits CYLD to TNFSC complex for deubiquitination of RIP1 and initiation of necroptosis (Elliott et al, 2016; Kupka et al, 2016; Wagner et al, 2016; Wei et al, 2017). The results presented in this paper established Spata2 as a novel regulator of NLRP3 inflammasome activation and inflammatory responses. Our data suggest that Spata2 is located in and recruits its partner CYLD to the centrosome, which is in agreement with a recent work documenting the association of endogenous Spata2 and CYLD with well‐known centrosomal proteins (Gupta et al, 2015). Spata2 and CYLD physically interact with the centrosomal kinase PLK4 to deconjugate K63‐linked polyubiquitin chains from PLK4, thereby facilitating the physical interaction of PLK4 with NEK7. Based on these data, we propose a new model that PLK4 inhibits NLRP3 inflammasome activation through its direct phosphorylation of NEK7, which in turn inhibits the NEK7‐NLRP3 interaction (Fig 7E). Our data demonstrate a dynamic interplay between the centrosome and inflammasome and provide valuable insight into the molecular mechanism of the NEK7‐mediated NLRP3 inflammasome activation.
A number of E3 ligases have been discovered to regulate NLRP3 inflammasome by conjugating ubiquitin chains onto NLRP3, ASC, caspase‐1, or upstream regulators (Lopez‐Castejon & Edelmann, 2016). The E3‐mediated ubiquitination of NLRP3 inflammasome can be reversed by DUBs, but less is known about the DUBs that are involved in this process. Previously, BRCC3 was shown as a major DUB to promote inflammasome activation by directly removing ubiquitination of NLRP3 (Py et al, 2013; Ren et al, 2019). Another DUB capable of regulating inflammasome activation is A20. In contrast to BRCC3, A20 inhibits NLRP3 inflammasome activation by restricting ubiquitination of pro‐IL‐1β protein complexes (Vande Walle et al, 2014; Duong et al, 2015). Our study identified CYLD as a DUB that negatively regulates NLRP3 inflammasome activation by catalyzing the deubiquitination of an upstream regulator PLK4, instead of the inflammasome components themselves.
The centrosome is a non‐membrane‐bound organelle composed of a pair of centrioles surrounded by a large set of proteins termed pericentriolar material (PCM). The classic function of the centrosome is to serve as a major microtubule‐organizing center (MTOC) regulating cell cycle progression in animal cells. In addition, it has been shown that the centrosome is involved in immunological synapse formation between T cells and antigen‐presenting cells (Vertii et al, 2016a). Apart from the role in regulating adaptive immunity, a recent study indicates that the centrosome mediates the secretion of cytokines in response to proinflammatory stimuli which induce atypical interphase centrosome maturation in a manner independent of classic mitotic kinase PLK1(Vertii et al, 2016b). This links the interphase centrosome to innate immune response. Interestingly, upon NLRP3 activation, cells in interphase of cell cycle are capable of producing much higher levels of active caspase‐1 and IL‐1β than those produced by mitotic cells (Shi et al, 2016), suggesting that interphase is the major phase for NLRP3 inflammasome activation. Surprisingly, a more recent paper demonstrates that NLRP3 is positioned to and assembles with ASC in the centrosome upon inflammasome activation, and abnormal positioning of NLRP3 would compromise the inflammasome activity (Li et al, 2017). In agreement with this finding, our immunofluorescence results indicated that upon activation a substantial number of NLRP3 inflammasomes, represented by ASC specks, are associated with centrosomes during early stage of activation (Appendix Fig S13). Together, these findings reveal a cross‐talk between the centrosome and NLRP3 inflammasome. Additionally, our discovery that Spata2, CYLD, and PLK4 function in the same signaling axis in the centrosome to regulate inflammasome activation suggests a critical role for centrosomal proteins in controlling the activation of NLRP3 inflammasome.
PLK4 is a well‐known cell cycle regulator that controls centrosome duplication and some aspects of mitosis (Maniswami et al, 2018). In this study, we found a previously unknown function for PLK4. PLK4 suppressed NLRP3 inflammasome activation by specifically phosphorylating NEK7 during LPS‐mediated priming step, which in turn inhibited the interaction of NEK7 with NLRP3 and inflammasome activation. This study also defines the phosphorylation status of NEK7 as a previously uncharacterized factor fine‐tuning NLRP3 inflammasome activation. Recently, a cryo‐electron microscopy structure of inactive human NLRP3 in complex with NEK7 was solved to understand the mechanism of NEK7‐NLRP3 interaction (Sharif et al, 2019). This structure demonstrated that the NEK7 C‐lobe interacts with NLRP3 and the two halves of the NEK7 C‐lobe (AA120–259 and 260–302) form the two interfaces of NEK7‐NLRP3 interaction. While Ser204 is located in the first half of the NEK7 C‐lobe, it is not among the residues that directly interact with NLRP3. Therefore, it is reasonable to deduce that NEK7 Ser204 phosphorylation may inhibit NEK7‐NLRP3 interaction indirectly rather than directly, e.g., by favoring NEK7 binding to other proteins. In support of this notion, we found that Ser204 phosphorylation is also involved in a non‐NLRP3 inflammasome regulating function of NEK7 (Tan et al, 2017). The TRF1 stabilizing function of NEK7 was largely controlled by Ser204 phosphorylation (Tan et al, 2017; Appendix Fig S14).
How PLK4 and NEK7 are switched from cell cycle regulators to inflammasome regulators during inflammasome activation remains elusive. One possibility is that LPS priming alters the structure of the centrosome and changes the function of the proteins residing inside, including PLK4 and NEK7. It is reported that 4–6 h of LPS stimulation led to recruitment of more centrosome proteins to PCM and marked increase in the size of interphase centrosomes, resulting in enhanced microtubule nucleation, enriched recycling endosomes, and more secretion of cytokines (Vertii et al, 2016b). These data suggest that hours of LPS stimulation may drastically change the state of interphase centrosomes. Consistent with this notion, in a screen for new signaling components for TLR response in dendritic cells, PLK4 was found to be involved in LPS‐induced antiviral response in a cell cycle‐independent manner (Chevrier et al, 2011). In this case, PLK4 transcript was markedly enhanced after a few hours of LPS stimulation with concomitant NEK7 phosphorylation increase although the phosphorylation site(s) was not identified (Chevrier et al, 2011). This phosphorylation induction was suppressed with a pan PLK inhibitor (Chevrier et al, 2011), which is consistent with our finding that PLK4 phosphorylated NEK7 at S204 at the time point of 4 h of LPS priming. While the physiological significance of PLK4 in regulating inflammasome activation remains unclear, we speculate that LPS‐stimulated PLK4 expression/activation and subsequent NEK7 phosphorylation might serve as a break in the location where NLRP3 inflammasome is assembled and activated to tightly restraint inflammasome activity to a proper level so as to avoid deleterious effect of excessive activation.
As a key regulator of centrosome duplication, an appropriate level of PLK4 is critical for keeping the right number of centrosomes during cell divisions which would otherwise produce chromosomal instability and aneuploidy (Maniswami et al, 2018). Deregulated expression of PLK4 has been associated with multiple cancers, and PLK4 has become a drug target for cancer treatment (Maniswami et al, 2018). In this regard, its target NEK7 has recently been shown to control the integrity of telomere shelterin protein complex (Tan et al, 2017). It would be worthwhile to investigate whether aberrant activation of NLRP3 inflammasome contributes to tumorigenesis in cancers associated with deregulation of PLK4 and NEK7.
In summary, our findings define a centrosomal Spata2/CYLD‐PLK4 signaling axis that suppresses NLRP3 inflammasome activation by a PLK4‐mediated NEK7 phosphorylation, which in turn attenuates the association of NEK7 with NLRP3 leading to the inhibition of abnormally high inflammasome activation. Our data shed new light on the understanding of the centrosome as a platform for inflammasome assembly and activation and provide novel potential therapeutic targets for treatment of NLRP3 inflammasome‐mediated inflammatory diseases.
Materials and Methods
Mice
The Spata2 mouse strain Spata2tm1.1(KOMP)Vlcg (on C57BL/6NCrl genetic background) was created from ES cell clone 10289A‐F6, generated by Regeneron Pharmaceuticals, Inc. and obtained from the KOMP Repository (http://www.komp.org). Spata2 −/− (Spata2‐KO) and Spata2 +/+ (WT) mice were generated by intercrossing of heterozygous mice and genotyped with PCR primers (forward primer, AGATGAGGTAGCCCTTGGGTTTG, reverse primer 1, ACTCAGGCTGGAGCTGCTCAG, and reverse primer 2, GATAGGTCACGTTGGTGTAGATGG) producing a 274‐bp DNA fragment for Spata2 +/+ mice, a 351‐bp DNA fragment for Spata2 −/− mice, and both fragments for Spata2 +/− mice. Cyld −/− mice on a C57BL/DBA background were generated and genotyped as described previously (Reiley et al, 2006). All mice were housed under specific pathogen‐free conditions at Shanghai Jiao Tong University School of Medicine. All animal experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University School of Medicine.
Plasmids
Plasmids encoding human Spata2, mouse PLK4, and human PLK1, and lentiviral shRNA plasmids targeting mouse PLK4, were purchased from GE Dharmacon. Plasmids encoding human CYLD, human TRF1, and human NEK7 were described previously (Reiley et al, 2005; Tan et al, 2017). Plasmids encoding Flag‐tagged mouse NLRP3, mouse caspase‐1, and human ASC were from Addgene. Further cloning to add epitope tags for mammalian expression was done by PCR and conventional cloning approaches. Lentiviral expression vectors encoding Flag‐tagged NEK7 and Flag‐tagged Spata2 were constructed by subcloning human NEK7 and Spata2 into a modified lentiviral vector, pLVX. Lentiviral expression vectors encoding GFP‐tagged PLK4 and GFP‐tagged PLK4∆24 (a stabilized form of PLK4) were constructed by replacing the zsGREEN sequence of pLVX‐zsGREEN vector with GFP‐tagged PLK4 and GFP‐tagged PLK4∆24, respectively. Deletions and point mutations were generated by using the QuikChange Site‐Directed Mutagenesis Kit (Stratagene). All constructs were verified by DNA sequencing.
DNA and RNA transfections
DNA transfections of HEK293 cells were performed using the transfection reagent Neofect (Neofect Biotech, Beijing) according to the manufacturer's instructions. Transfected cells were either used for microscopy 16–24 h post‐transfection, or harvested 36–48 h post‐transfection for further experiments. siRNAs targeting human Spata2 (5′‐UGAAGAAGUACUAACGCAU‐3′; Wagner et al, 2016) and CYLD (5′‐GAACAGAUUCCACUCUUUA‐3′; Hrdinka et al, 2016) were transfected with Lipofectamine RNAiMax (Invitrogen) following the manufacturer's instructions.
Reagents
Nigericin (tlrl‐nlg), MSU (tlrl‐msu), Nano‐SiO2 (tlrl‐sio), Alum (tlrl‐alk), and poly(dA:dT) (tlrl‐patc) were purchased from Invivogen. LPS (derived from Escherichia coli O111:B4, L4391 for treatments of culture cells, and L4130 for injection of mice), ATP (A2383), and caspase‐1 inhibitor Z‐YVAD‐FMK (178603‐78‐6) were obtained from Sigma. SYBR Green Mix for real‐time PCR (A25918) was from Applied Biosystems. The PLK4 inhibitors, Centrinone (HY‐18682) and Centrinone‐B (HY‐18683), were purchased from MedChemExpress. All reagents were dissolved and used following the manufacturer's instructions.
LPS‐induced septic shock
Eight‐ to ten‐week‐old and gender‐matched mice were injected intraperitoneally with 20 mg kg−1 LPS and either killed at 12 h to collect blood for ELISA measurement of serum IL‐1β, IL‐18, IL‐6, and TNFα, or monitored for mortality every 2 h for 4 days.
Alum‐induced peritonitis
Eight‐ to ten‐week‐old and gender‐matched mice were injected intraperitoneally with 20 mg kg−1 alum and killed after 6 h to collect peritoneal lavages with 5 ml PBS. Peritoneal exudate cells (PECs) were analyzed by FACS to quantitate neutrophils (Ly6G+F4/80−). Peritoneal fluids were concentrated with Millipore Amicon Ultra 3K for ELISA analysis of IL‐1β and IL‐6.
Immunoblotting
Cells were lysed in NP‐40 lysis buffer (50 mM Tris–HCl pH 7.4, 250 mM NaCl, 1 mM EDTA, 1% NP‐40) supplemented with protease and phosphatase inhibitors (Roche 11873580001 and 04906845001), separated by SDS–PAGE, and transferred onto PVDF membrane (EMD Millipore) for blotting with the following antibodies: cleaved IL‐1β (R&D, AF‐401‐NA, 1:2,000 dilution), NLRP3 (clone Cryo‐2, AdipoGen, 1:2,000 dilution), ASC (Santa Cruz, sc‐22514, 1:2,000 dilution), PLK4 (Abcam, ab2642, 1:1,000 dilution), NEK7 (Abcam, ab133514, 1:2,000 dilution), CYLD (Santa Cruz, sc‐74435, 1:1,000 dilution), Tubulin (homemade, 1:100,000 dilution), Spata2 (Santa Cruz, sc‐515283, 1:3,000 dilution), V5 (HRP Conjugated, Thermo, MA5‐15253‐HRP, 1:2,000 dilution), HA (HRP Conjugated, Roche, 12013819001, 1:2,000 dilution), FLAG (M2, Sigma, F1804, 1:4,000 dilution), FLAG (Sigma, F7425, 1:4,000 dilution), c‐Myc (Santa Cruz, sc‐40, 1:2,000 dilution), IkBα (Santa Cruz, sc‐371, 1:2,000 dilution), phosphor‐IkBα (Cell Signaling, 2859S, 1:3,000 dilution), phosphor‐ERK1/2 (Santa Cruz, sc‐7383, 1:1,000 dilution), phosphor‐p38 (Cell Signaling, 9215S, 1:2,000 dilution), and phosphor‐JNK (Cell Signaling, 9251, 1:2,000 dilution). Polyclonal phospho‐NEK7 (S204) antibody was generated by Shanghai Guoyuan Biotechnology with a synthesized phosphor‐peptide corresponding to amino acids (aa) 201–212 of mouse NEK7 (YYMS204PERIHENG) as the antigen. All immunoblotting experiments were repeated at least two times but only a representative result for each experiment was shown. Quantification of immunoblotting results was done with Image‐Pro Plus 6.0.
Immunoprecipitation
For regular immunoprecipitations, cell lysates were incubated with antibody‐conjugated agarose beads at 4°C for 2 h, or with an unconjugated antibody for 2 h followed by 1.5 h of incubation with protein A/G beads. After three times of wash with lysis buffer, the beads were boiled in 2× SDS sample buffer, and eluted proteins were analyzed by immunoblotting. For protein ubiquitination assay, cells were lysed in 1% SDS containing NP‐40 lysis buffer and sonicated to denature proteins in lysates. Denatured cell lysates were diluted at 1:10 with regular NP‐40 lysis buffer, incubated with antibody‐conjugated agarose beads for 3 h, and washed three times before boiling in 2× SDS sample buffer. Beads used for immunoprecipitations included GFP Trap beads (gta‐20, Chromotek), Flag M2 agarose beads (A2220, Sigma), and protein A/G beads (sc‐2003, Santa Cruz). For K63‐specific tandem ubiquitin‐binding entity (TUBE) enrichment of K63‐linked polyubiquitinated proteins, FLAG K63‐TUBE 1 (UM604, LifeSensors) was used following the manufacturer's instructions. Briefly, 293 cells were lysed in the presence of protease inhibitors and the inhibitor of cysteine peptidases (5 mM). The whole cell lysates were incubated with FLAG K63‐TUBE 1 (200 nM) on ice for 2 h followed by pull‐down with Flag M2 agarose beads (A2220, Sigma) and stringent washes. Pull‐down beads were eventually boiled and analyzed by immunoblotting.
Immunofluorescence
Cells were seeded in 20‐mm glass‐bottom cell culture dishes (NEST, 801001), untreated or treated as indicated, fixed with −20°C methanol for 5 min, and stained with antibodies overnight at 4°C. After three washes in PBST, cells were stained with the indicated fluorescence‐labeled secondary antibodies and a DNA dye, 4′,6‐diamidino‐2‐phenylindole (DAPI). Microscopy of cells was performed with a Leica SP8 confocal microscope. Antibodies used for immunofluorescence included Spata2 (Santa Cruz, sc‐515283, 1:300 dilution,), γ‐tubulin (Abcam ab11317, 1:1,000 dilution), γ‐tubulin (Abcam, ab11316, 1:200 dilution,) ASC (Santa Cruz, sc‐22514, 1:200 dilution), and Centrin (Millipore, 04‐1624, 1:300 dilution). The following fluorescence‐labeled secondary antibodies were purchased from Abbkine Scientific: IFKine Green Donkey Anti‐Mouse IgG (A24211‐1, 1:500 dilution), IFKine Green Donkey Anti‐Rabbit IgG (A24221‐1, 1:500 dilution), IFKine Red Donkey Anti‐Mouse IgG (A24411‐1, 1:500 dilution), and IFKine Red Donkey anti‐Rabbit IgG (A24421‐1, 1:500 dilution). For quantitation of ASC speck‐positive cells, experiments were performed in triplicate. > 500 cells in 3 random areas were counted for each repeat, and the average percentage of ASC speck‐positive cells was calculated. For staining of centrioles, HeLa cells were treated with 100 μM Nocodazole for 19 h to arrest cells at G2/M phase and then released for 1 h to allow cells to proceed to M phage. Synchronized cells were fixed and stained as described above with Centrin and DAPI for confocal microscopy.
In vitro deubiquitination assay
In vitro deubiquitination of PLK4 was performed essentially as described previously (Schlicher et al, 2016). Briefly, V5‐Spata2 was co‐transfected with Flag‐CYLD, or Flag‐CYLD C601S into HEK293 cells. Anti‐Flag immunoprecipitation was prepared, and Flag‐CYLD/V5‐Spata2 complex was eluted with Flag peptide as a source of DUBs. To prepare ubiquitinated PLK4, CYLD siRNA and an ubiquitin‐encoding plasmid were co‐transfected into HEK293 cells that stably expressing GFP‐PLK4. Two days later, the accumulated ubiquitinated GFP‐PLK4 was affinity‐purified with anti‐GFP antibody beads and incubated with the purified DUBs at 37°C for 60 min. K63‐linked ubiquitination of PLK4 was assessed by immunoblot analysis as described above.
Inflammasome‐activation assays
Bone marrow cells were isolated from femurs of the indicated mice and differentiated for 6 days in DMEM medium supplemented with 10% fetal bovine serum and 20 μg ml−1 M‐CSF. Adherent BMDMs were split into 12‐well plates at a density of 5 × 105 per well. 12–16 h later, BMDMs were transfected with poly(dA:dT) for 16 h, infected with Salmonella for 16 h, or primed with 0.5 μg ml−1 LPS for 4–5 h and subsequently treated with ATP (5 mM for 30 min), nigericin (5 μM for 1 h), nano‐SiO2 (50 μM for 5 h), and Alum (5 mM for 5 h). Supernatants were collected for protein precipitation via methanol–chloroform extraction, and cells were lysed as whole cell lysates. Activation of inflammasomes was assessed based on immunoblotting analysis of active IL‐1β and cleaved caspase‐1 in supernatants and pro‐IL‐1β and pro‐caspase‐1 in whole cell lysates, ELISA of supernatants collected from cell culture, or ASC speck microcopy of cells that were immediately fixed after stimulation for immunofluorescence. NLRP3 inflammasome activation in iBMDMs was analyzed essentially as described above for BMDMs.
ASC oligomerization
Bone marrow‐derived macrophages or iBMDMs were stimulated as indicated and used for cross‐linking with disuccinimidyl suberate essentially as described previously (Shi et al, 2016).
Generation of CRISPR/Cas9 knockout cell lines
A CRISPR/Cas9 double nickase plasmid targeting mouse Spata2 was purchased from Santa Cruz (sc‐435216‐NIC) and transfected into iBMDMs by electroporation with Lonza Amaxa Nucleofector II Electroporator. GFP‐positive cells were sorted into single clones via flow cytometry (BD FACSAria III). Genomic DNA was purified from individual clones and PCR‐amplified using primers spanning the designed knockout region. Knockout was confirmed by sequencing the PCR product.
ELISA
Mouse sera and cell culture supernatants were assayed by ELISA (eBioscience) for IL‐1β, IL‐18, TNFα, and IL‐6 according to the manufacturer's instructions.
RT–PCR
Bone marrow‐derived macrophages were treated with LPS for the indicated time, and total RNA was extracted using RNeasy Mini Kit (Qiagen). cDNA was synthesized from RNA using PrimeScript RT Reagent Kit (TaKaRa, RR037A). Quantitative real‐time PCR was performed with SYBR Green Master Mix (Applied Biosystems, A25918). Gene expression was assessed in triplicate and normalized to that of GAPDH. Primers used for real‐time PCR are shown below.
Gapdh forward: 5′‐AGGTCGGTGTGAACGGATTTG‐3′
Gapdh reverse: 5′‐TGTAGACCATGTAGTTGAGGTCA‐3′
Il‐1β forward: 5′‐GAAATGCCACCTTTTGACAGTG‐3′
Il‐1β reverse: 5′‐TGGATGCTCTCATCAGGACAG‐3′
Il‐18 forward: 5′‐ATATCGACCGAACAGCCAA‐3′
Il‐18 reverse: 5′‐TAGGGTCACAGCCAGTCCTC‐3′
Il‐6 forward: 5′‐TCCAATGCTCTCCTAACAGATAAG‐3′
Il‐6 forward: 5′‐CAAGATGAATTGGATGGTCTTG‐3′
TNFα forward: 5′‐CTTCTGTCTACTGAACTTCGGG‐3′
TNFα reverse: 5′‐TGATCTGAGTGTGAGGGTCTG‐3′
IkBα forward: 5′‐ACGAGCAAATGGTGAAGGAG‐3′
IkBα reverse: 5′‐TTCTGGAAGTTGAGGAAGGC‐3′
Il‐12p40 forward: 5′‐CATTGAACTGGCGTTGGAAG‐3′
Il‐12p40 reverse: 5′‐TGAGGGAGAAGTAGGAATGGG‐3′
PLK4 forward: 5′‐AGGAGAAACTAATGAGCACCACA‐3′
PLK4 reverse: 5′‐TGGCTCTCGTGTCAGTCCAA‐3′.
Lentiviral infection
Lentivirus particles were generated from co‐transfection with lentiviral vectors and packaging plasmids in HEK293 cells, filtered through 0.45‐μm filters, and immediately used for infections. iBMDMs or HEK293 cells were incubated with lentivirus for 12 h. After recovery in regular medium for 1 day, cells were subjected to FACS sorting (for GFP‐expressing vectors) or antibiotic selection (for non‐GFP‐expressing vectors) to enrich the infected cells.
BioID
BioID identification of CYLD‐interacting proteins was performed as described (Roux et al, 2013). The retroviral expression vector encoding a modified bacterial biotin ligase (BirA*) was generated by transferring Myc‐tagged BirA* from pcDNA3.1‐myc‐BioID (obtained from Addgene) to the pCLXSN(GFP) retroviral vector. pCLXSN‐Myc‐BirA*‐CYLD was created by inserting human CYLD cDNA into the pCLSN‐Myc‐BirA* vector. HEK293 cells were infected with retroviruses expressing the BirA* or BirA*‐CYLD and selected by FACS sorting (based on GFP expression). The transduced cells were treated with 50 μM biotin for 20 h and lysed for purification of biotinylated proteins with streptavidin beads (M280, Invitrogen). The purified biotinylated proteins were subjected to SDS–PAGE visualized with Coomassie Brilliant Blue stain. Individual gel pieces were excised, destained, and subjected to in‐gel digestion with trypsin and protein identification by mass spectrometry in the Mass Spectrometry Proteomics Core at Baylor College of Medicine. Two independent experiments were performed, and three criteria were followed to determine highly reliable CYLD‐interacting candidates: (1) identified in both experiments, (2) no < 2 peptides were picked by mass spectrometry, and (3) either unique in CYLD BioID group or fivefold higher in relative abundance in the CYLD BioID group than in the control group.
Identification of NEK7 phosphorylation sites by mass spectrometry
HEK293 cells were transfected with Flag‐tagged NEK7 alone or together with GFP‐tagged PLK4∆24, and cells were lysed in NP‐40 lysis buffer supplemented with protease and phosphatase inhibitors for immunoprecipitation with anti‐Flag agarose beads. NEK7 was then eluted from agarose beads with Flag peptide, digested with trypsin, and subjected to LC/tandem MS (MS/MS) analysis in the National Center for Protein Science Shanghai in China. Briefly, the peptide mixture was first analyzed by a homemade 15‐cm‐long pulled‐tip analytical column (75 μm i.d.) which was then placed in‐line with an Easy‐nLC 1200 nano‐HPLC (Thermo Scientific, San Jose, CA) for mass spectrometry analysis. Data‐dependent MS/MS analysis and parallel reaction monitoring (PRM) analysis were performed with a Q‐exactive Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA) equipped with a nanoelectospray ionization source. The acquired MS/MS data were analyzed using PEAKS Studio 8 (Bioinformatics Solutions, Waterloo, Ontario, Canada). Oxidation on methionine and phosphorylation on serine\threonine\tyrosine were set as variable modifications during database search.
Cell proliferation assay
Cell Counting Kit‐8 (HY‐K0301‐500T, MedChemExpress) was employed to determine cell proliferation according to the manufacture's instruction. Briefly, 100 μl of cell suspension (1,500–2,000 cells/well) was dispensed in a 96‐well plate in triplicate. When assessing the effect of PLK4 inhibition on cell proliferation, DMSO or 150 nM of PLK4 inhibitor Centrinone was added into each well. The plate was incubated in a humidified incubator. CCK‐8 solution was added to each well of the plate at indicated time points for measurement of the absorbance at 450 nm.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 6.02. Statistical difference between two groups was determined by unpaired two‐tailed Student's t‐test except that the one between the survival curves of two groups of mice was determined by long‐rank test. A P value < 0.05 was considered significant, and different levels of significance were expressed as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Author contributions
X‐DY designed the study, supervised the work, performed experiments and data analysis, and wrote the manuscript; WL, SZ, DW, and XJ performed experiments; RT, XN, QW, XW, ZL, L‐FC, and JQ contributed to the performance of the experiments; BS supervised the work and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Table EV2
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Jun Zhou for the CYLD knockout mice. We thank Feng Shao for iBMDMs, Mutsuhiro Takekawa for PLK4 phosphor‐T170 antibody, Chao Peng and Min Huang for mass spec analysis of NEK7 phosphorylation, and all members of the Su laboratory for discussions. This work was supported by grants from the National Natural Science Foundation of China to X.D.Y. (31770818 and 31570770) and to B.S. (31470845 and 81430033) and the Shanghai Science and Technology Commission to B.S. (13JC1404700).
The EMBO Journal (2020) 39:e102201
§Correction added on 17 December 2019, after first online publication: affiliation 1 has been corrected.
Contributor Information
Xiao‐Dong Yang, Email: xdyang@shsmu.edu.cn.
Bing Su, Email: bingsu@sjtu.edu.cn.
References
- Broz P, von Moltke J, Jones JW, Vance RE, Monack DM (2010) Differential requirement for Caspase‐1 autoproteolysis in pathogen‐induced cell death and cytokine processing. Cell Host Microbe 8: 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16: 407–420 [DOI] [PubMed] [Google Scholar]
- Chen J, Chen ZJ (2018) PtdIns4P on dispersed trans‐Golgi network mediates NLRP3 inflammasome activation. Nature 564: 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, Gat‐Viks I, Tonti E, DeGrace MM, Clauser KR et al (2011) Systematic discovery of TLR signaling components delineates viral‐sensing circuits. Cell 147: 853–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BK, Wen H, Ting JP‐Y (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29: 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA (1997) Proinflammatory and anti‐inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest 112: 321S–329S [DOI] [PubMed] [Google Scholar]
- Duong BH, Onizawa M, Oses‐Prieto JA, Advincula R, Burlingame A, Malynn BA, Ma A (2015) A20 restricts ubiquitination of pro‐interleukin‐1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 42: 55–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott PR, Leske D, Hrdinka M, Bagola K, Fiil BK, McLaughlin SH, Wagstaff J, Volkmar N, Christianson JC, Kessler BM et al (2016) SPATA2 links CYLD to LUBAC, activates CYLD, and controls LUBAC signaling. Mol Cell 63: 990–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes‐Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase‐1 activation. Cell Death Differ 14: 1590–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P et al (2011) Type I interferon inhibits interleukin‐1 production and inflammasome activation. Immunity 34: 213–223 [DOI] [PubMed] [Google Scholar]
- Guo H, Callaway JB, Ting JPY (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GD, Coyaud E, Goncalves J, Mojarad BA, Liu Y, Wu Q, Gheiratmand L, Comartin D, Tkach JM, Cheung SW et al (2015) A dynamic protein interaction landscape of the human centrosome‐cilium interface. Cell 163: 1484–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhaj EW, Dixit VM (2012) Regulation of NF‐kappaB by deubiquitinases. Immunol Rev 246: 107–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zeng MY, Yang D, Motro B, Núñez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz‐Otsoa F, Lang V, England P, Rodriguez MS (2009) Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin‐binding entities. EMBO Rep 10: 1250–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdinka M, Fiil BK, Zucca M, Leske D, Bagola K, Yabal M, Elliott PR, Damgaard RB, Komander D, Jost PJ et al (2016) CYLD limits Lys63‐ and Met1‐linked ubiquitin at receptor complexes to regulate innate immune signaling. Cell Rep 14: 2846–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim S, Rhee K (2011) NEK7 is essential for centriole duplication and centrosomal accumulation of pericentriolar material proteins in interphase cells. J Cell Sci 124: 3760–3770 [DOI] [PubMed] [Google Scholar]
- Kupka S, De Miguel D, Draber P, Martino L, Surinova S, Rittinger K, Walczak H (2016) SPATA2‐mediated binding of CYLD to HOIP enables CYLD recruitment to signaling complexes. Cell Rep 16: 2271–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13: 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Thome S, Ma X, Amrute‐Nayak M, Finigan A, Kitt L, Masters L, James JR, Shi Y, Meng G et al (2017) MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule‐dependent mechanism. Nat Commun 8: 15986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Castejon G, Edelmann MJ (2016) Deubiquitinases: novel therapeutic targets in immune surveillance? Mediators Inflamm 2016: 3481371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lork M, Verhelst K, Beyaert R (2017) CYLD, A20 and OTULIN deubiquitinases in NF‐κB signaling and cell death: so similar, yet so different. Cell Death Differ 24: 1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, Egelman EH (2014) Unified polymerization mechanism for the assembly of ASC‐dependent inflammasomes. Cell 156: 1193–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniswami RR, Prashanth S, Karanth AV, Koushik S, Govindaraj H, Mullangi R, Rajagopal S, Jegatheesan SK (2018) PLK4: a link between centriole biogenesis and cancer. Expert Opin Ther Targets 22: 59–73 [DOI] [PubMed] [Google Scholar]
- Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S (2013) Microtubule‐driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14: 454–460 [DOI] [PubMed] [Google Scholar]
- O'Regan L, Blot J, Fry AM (2007) Mitotic regulation by NIMA‐related kinases. Cell Div 2: 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py BF, Kim MS, Vakifahmetoglu‐Norberg H, Yuan J (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49: 331–338 [DOI] [PubMed] [Google Scholar]
- Reiley W, Zhang M, Wu X, Granger E, Sun SC (2005) Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma‐dependent phosphorylation. Mol Cell Biol 25: 3886–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley WW, Zhang M, Jin W, Losiewicz M, Donohue KB, Norbury CC, Sun SC (2006) Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol 7: 411–417 [DOI] [PubMed] [Google Scholar]
- Ren G, Zhang X, Xiao Y, Zhang W, Wang Y, Ma W, Wang X, Song P, Lai L, Chen H et al (2019) ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J 38: e100376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Burke B (2013) BioID: a screen for protein‐protein interactions. Curr Protoc Protein Sci 74: Unit 19.23 [DOI] [PubMed] [Google Scholar]
- Schlicher L, Wissler M, Preiss F, Brauns‐Schubert P, Jakob C, Dumit V, Borner C, Dengjel J, Maurer U (2016) SPATA2 promotes CYLD activity and regulates TNF‐induced NF‐kappaB signaling and cell death. EMBO Rep 17: 1485–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid‐Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, Hornung V (2016) A Genome‐wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem 291: 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J (2010) The inflammasomes. Cell 140: 821–832 [DOI] [PubMed] [Google Scholar]
- Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Nunez G, Mao Y et al (2019) Structural mechanism for NEK7‐licensed activation of NLRP3 inflammasome. Nature 570: 338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S et al (2016) NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17: 250–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC (2010) CYLD: a tumor suppressor deubiquitinase regulating NF‐kappaB activation and diverse biological processes. Cell Death Differ 17: 25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan R, Nakajima S, Wang Q, Sun H, Xue J, Wu J, Hellwig S, Zeng X, Yates NA, Smithgall TE et al (2017) Nek7 protects telomeres from oxidative DNA damage by phosphorylation and stabilization of TRF1. Mol Cell 65: 818–831.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, Beyaert R, Elewaut D, Kanneganti TD, van Loo G et al (2014) Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512: 69–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Demon D, Bogaert P, Vandendriessche B, Goethals A, Depuydt B, Vuylsteke M, Roelandt R, Van Wonterghem E, Vandenbroecke J et al (2014) Simultaneous targeting of IL‐1 and IL‐18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med 189: 282–291 [DOI] [PubMed] [Google Scholar]
- Vertii A, Hehnly H, Doxsey S (2016a) The centrosome, a multitalented renaissance organelle. Cold Spring Harb Perspect Biol 8: a025049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertii A, Ivshina M, Zimmerman W, Hehnly H, Kant S, Doxsey S (2016b) The centrosome undergoes Plk1‐independent interphase maturation during inflammation and mediates cytokine release. Dev Cell 37: 377–386 [DOI] [PubMed] [Google Scholar]
- Wagner SA, Satpathy S, Beli P, Choudhary C (2016) SPATA2 links CYLD to the TNF‐alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J 35: 1868–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R, Xu LW, Liu J, Li Y, Zhang P, Shan B, Lu X, Qian L, Wu Z, Dong K et al (2017) SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev 31: 1162–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstrom SA, Masoumi KC, Khochbin S, Fassler R, Massoumi R (2010) CYLD negatively regulates cell‐cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J 29: 131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW et al (2015) Cell biology. Reversible centriole depletion with an inhibitor of Polo‐like kinase 4. Science 348: 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ran J, Liu M, Li D, Li Y, Shi X, Meng D, Pan J, Ou G, Aneja R et al (2014) CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell Res 24: 1342–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Table EV2
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7