

Abstract
Intrinsic genomic features of individual chromosomes can contribute to chromosome‐specific aneuploidy. Centromeres are key elements for the maintenance of chromosome segregation fidelity via a specialized chromatin marked by CENP‐A wrapped by repetitive DNA. These long stretches of repetitive DNA vary in length among human chromosomes. Using CENP‐A genetic inactivation in human cells, we directly interrogate if differences in the centromere length reflect the heterogeneity of centromeric DNA‐dependent features and whether this, in turn, affects the genesis of chromosome‐specific aneuploidy. Using three distinct approaches, we show that mis‐segregation rates vary among different chromosomes under conditions that compromise centromere function. Whole‐genome sequencing and centromere mapping combined with cytogenetic analysis, small molecule inhibitors, and genetic manipulation revealed that inter‐chromosomal heterogeneity of centromeric features, but not centromere length, influences chromosome segregation fidelity. We conclude that faithful chromosome segregation for most of human chromosomes is biased in favor of centromeres with high abundance of DNA‐dependent centromeric components. These inter‐chromosomal differences in centromere features can translate into non‐random aneuploidy, a hallmark of cancer and genetic diseases.
Keywords: aneuploidy, CENP, CENP‐B boxes, centromere, chromosome segregation
Subject Categories: Cell Cycle; Chromatin, Epigenetics, Genomics & Functional Genomics
Missegregation rates vary among different chromosomes, with biased based on abundance of DNA‐dependent centromeric components rather than centromere length.
Introduction
Defects during cell division can lead to loss or gain of chromosomes in the daughter cells, a phenomenon called aneuploidy. This alters gene copy number and cell homeostasis, leading to genomic instability and pathological conditions including genetic diseases and various types of cancers (Gordon et al, 2012; Santaguida & Amon, 2015). While it is known that selection is a key process in maintaining aneuploidy in cancer, a preceding mis‐segregation event is required. It was shown that chromosome‐specific aneuploidy occurs under conditions that compromise genome stability, such as treatments with microtubule poisons (Caria et al, 1996; Worrall et al, 2018), heterochromatin hypomethylation (Fauth & Scherthan, 1998), or following ionizing radiation (Balajee et al, 2014). This suggests that certain human chromosomes are more prone to mis‐segregate than others, indicating the existence of a heterogeneity between chromosomes that could be at the origin of chromosome‐specific aneuploidy.
Centromeres are key components in mediating equal distribution of genetic material. They are the chromosomal docking site for assembly of the kinetochore, the protein complex responsible for spindle attachment and chromosome separation during cell division. Centromere position is epigenetically defined by a specific chromatin enriched for the histone H3‐variant CENtromere Protein A (CENP‐A; Fukagawa & Earnshaw, 2014) via a two‐step mechanism (Fachinetti et al, 2013). Centromeres are built on centromeric DNA repeats of 171 base pairs (bp), named alpha‐satellites, that span several megabases (Miga, 2017). A fraction of these regions, called CENP‐B boxes, are bound by CENP‐B, the only DNA sequence‐dependent centromeric binding protein identified so far (Muro et al, 1992).
Differences in centromere features such as sequence variation (Alexandrov et al, 2001; Aldrup‐MacDonald et al, 2016; Contreras‐Galindo et al, 2017) and centromere length (Rudd & Willard, 2004; Contreras‐Galindo et al, 2017; Dumont & Fachinetti, 2017) could modulate the abundance of centromeric and kinetochore components, as shown for CENP‐A and Ndc80 (a subunit of the kinetochore; Irvine et al, 2004; Sullivan et al, 2011; Contreras‐Galindo et al, 2017; Drpic et al, 2017), and, thus, have a direct impact on chromosome segregation fidelity. A direct correlation between centromere size and bias in chromosome segregation was demonstrated in mouse asymmetric female meiosis, a phenomenon defined as centromere drive (Henikoff & Malik, 2002). Here it was shown that, between two homologous chromosomes, the chromosome that carries a centromere with a higher amount of centromeric DNA sequences and centromere proteins (a concept globally defined as “centromere strength”) was preferentially retained in the egg during the first meiotic division (Chmátal et al, 2017; Iwata‐Otsubo et al, 2017; Lampson & Black, 2017). This could explain part of the molecular mechanisms behind asymmetric division in female gametogenesis. However, if a similar phenomenon occurs also during mitotic division and between non‐homologous chromosomes remains untested.
We previously showed that CENP‐B plays an important role in chromosome segregation by reinforcing centromere function through its interaction with CENP‐C (Fachinetti et al, 2015; Hoffmann et al, 2016). CENP‐C is a key component of human centromeres recruited by CENP‐A (Guse et al, 2011; Hoffmann et al, 2016), and it is necessary to mediate the assembly of the kinetochore prior to mitosis (Fukagawa et al, 1999; Hoffmann et al, 2016; Weir et al, 2016). Interestingly, CENP‐B was revealed to be present in varying amounts among different chromosomes (Earnshaw et al, 1989). However, if these different amounts mirror the number of CENP‐B boxes within repetitive sequences is unknown. It is also noteworthy that CENP‐B binding to DNA might be regulated by DNA methylation (Tanaka et al, 2005), and DNA methylation patterns might be different from centromere to centromere. It is unclear as to whether a correlation exists between centromere length, the number of CENP‐B binding sites, and/or the amount of CENP‐B molecules at each human centromere. Furthermore, if variation of centromeric DNA translates into differing levels of other centromeric and kinetochore proteins that directly impact on the fidelity of chromosome segregation remains untested.
Here, we assessed the direct impact of centromeric DNA on providing strong connections between the chromosomes and the spindle microtubules and, consequently, on chromosome segregation fidelity. We show that in a non‐transformed diploid cell line context, chromosome‐specific aneuploidy occurs following centromere perturbations. We also show that human centromeres are intrinsically heterogeneous at the level of centromeric DNA and its binding components. Finally, we demonstrate that inter‐chromosomal differences in centromeres directly translate into non‐random aneuploidy during mitosis.
Results
Chromosome‐specific aneuploidy occurs in centromere perturbation conditions
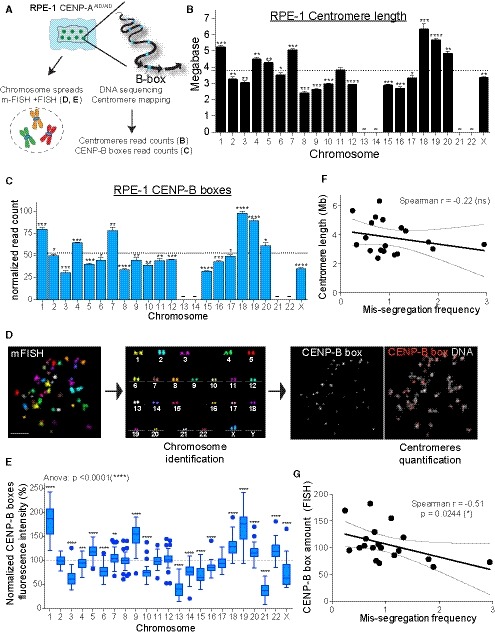
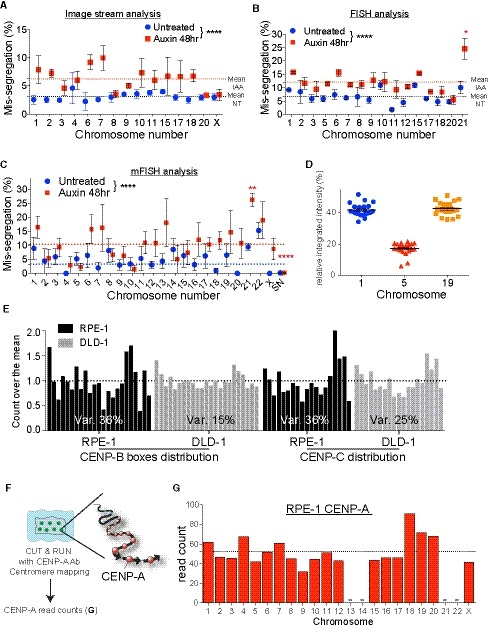
We first measured human chromosome‐specific aneuploidies in human female RPE‐1 cells with endogenously tagged CENP‐AAID/AID alleles (Hoffmann et al, 2016) as a model system. The use of this cell line provides several advantages. It is a non‐transformed cell line, thus we can exclude confounding effects due to mutations in genes that regulate cell cycle and transcription such as oncogene overexpression and cell checkpoint mutations. Moreover, it does not harbor chromosome rearrangements (with the exception of one known translocation on chromosome X). Additionally, RPE‐1 cells have a stable diploid karyotype with very low rates of spontaneous chromosome mis‐segregation, allowing us to explicitly test chromosome‐specific aneuploidy. To enhance the frequency of aneuploidy (necessary to generate enough data for statistical relevance) without perturbing mitosis with chemical inhibitors, we measured aneuploidy following removal of the epigenetic component of centromere function, CENP‐A, as recently described (Hoffmann et al, 2016). This also gives us the advantage of directly assessing the impact of centromeric DNA/CENP‐B on mediating chromosome segregation. Indeed, in CENP‐A‐deficient settings, centromere function and chromosome segregation fidelity depend mainly on CENP‐B bound to centromeric DNA as the sole source of centromere/kinetochore interaction (Fachinetti et al, 2015; Hoffmann et al, 2016; Fig 1A). Auxin (IAA) addition leads to rapid, complete, and uniform removal of CENP‐A molecules from all centromeres (Fig EV1A).
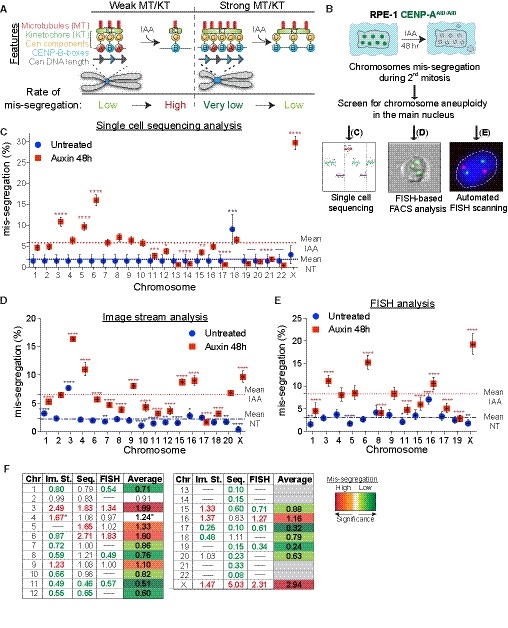
Figure 1. Chromosome‐specific aneuploidy arises following CENP‐A removal in RPE‐1 cells.
-
AModel of centromere strength via CENP‐C recruitment supported by DNA sequence and CENP‐B. KT = kinetochore, MT = microtubules. IAA = auxin. Cen = centromere. Blue arrows represent CENP‐B boxes. Upon IAA addition, AID‐tagged CENP‐A is degraded.
-
BSchematic of the experiments shown in (C–E).
-
C–ELogistic statistical model based on the (C) single‐cell sequencing, (D) ImageStream (E) analysis or automated FISH of RPE‐1 cells in untreated condition (blue circles) or treated with auxin for 48 h (red squares). Error bars represent the SEM based on the number of cells analyzed (see the statistical method section for details and Datasets EV1 and EV2). Dashed lines indicate the means of aneuploidy rates in untreated (blue line) or auxin‐treated (red line) condition. Red asterisks (IAA) and blue (Untreated) indicate significance over the respective mean using a binomial test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
-
FTable summarizing whole‐chromosome aneuploidy (fold over the mean) using the indicated methods to measure chromosome mis‐segregation rate. Bold numbers represent a statistically significant difference from the mean for each method. Orange to dark red gradient highlights chromosomes that mis‐segregate (with at least one method) at a significantly higher rate compared to the mean level (weak chromosomes). Light green to dark green gradient highlights chromosomes that mis‐segregate (with at least one method) at a significantly lower rate compared to the mean (stronger chromosomes). Data obtained with only one method were excluded. *centromere 4 probe with image stream was reported to lead to non‐specific signal (Worrall et al, 2018). Im. St. = image stream; Seq = single‐cell sequencing.
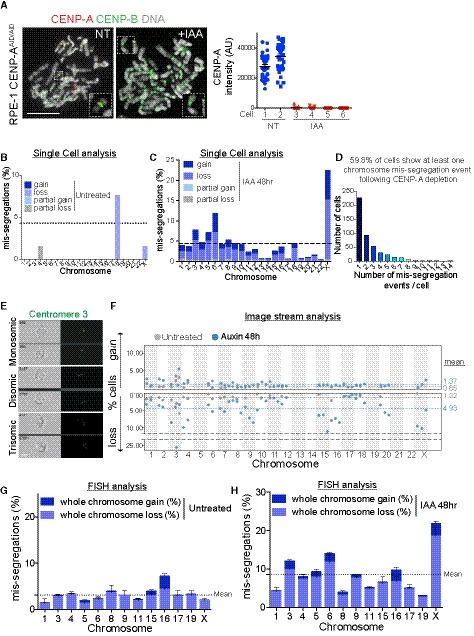
Figure EV1. Chromosome aneuploidy profile of RPE‐1 cells after 48 h of CENP‐A depletion (related to Fig 1).
-
A(Left) Representative image of a mitotic chromosome spread stained with CENP‐A and CENP‐B at the indicated conditions. Scale bar represents 10 μm. (Right) Dot plot showing the mean level of CENP‐A at the indicated conditions in different cells treated or not with auxin ± SEM. Each dot represents a centromere (n = 35).
-
B–DSingle‐cell sequencing analysis of the frequency of chromosome mis‐segregation rates determined (C) in untreated cells (n = 6 cells that show at least one event of chromosome mis‐segregation; total cells sequenced = 66), (D) after auxin treatment (n = 485 cells that show at least one event of chromosome mis‐segregation; total cells sequenced = 811), (D) number of mis‐segregated events per cells. Dashed lines in (B) and (C) show the expected 4.3% frequency of aneuploidy rate for a diploid chromosome.
-
ERepresentative images of ImageStream analyzed cells mono‐, di‐, or trisomic for chromosome 3 (green dots) labeled using a FISH centromeric probe. Scale bar represents 5 μm.
-
FImageStream analysis of RPE‐1 untreated (gray circles) or auxin‐treated cells (48 h, blue circles) cells. Dots represent independent experiments (between ˜ 600 cells to > 10,000 cells for each experiment for every single chromosome). Dashed lines indicate the mean of aneuploidy rate (blue IAA‐treated, gray untreated).
-
G, HBar graph represents aneuploidy profile with gain and loss frequencies of the chromosomes analyzed by automatic FISH scanning in untreated (G) and auxin‐treated (48 h) cells (H). Error bars represent the SEM of three to five independent experiments (between 600 cells and 2,400 cells for each experiment for every chromosome). Dashed lines indicate the mean of aneuploidy rate.
Whole‐chromosome mis‐segregation was measured following IAA treatment for 48 h, corresponding to approximately two full cell cycles. Within this short time frame, CENP‐A removal does not lead to cell death (Hoffmann et al, 2016), and therefore does not cause bias in the analysis due to loss of cells with a particular aneuploidy status. To measure aneuploidy, we used three different, complementary, and unbiased approaches (Fig 1B): (i) Single‐cell sequencing (Figs 1C and EV1B–D); (ii) ImageStream cytometry to quantify fluorescence in situ hybridization (FISH)‐marked individual centromeres in thousands of single cells on most human chromosomes (Figs 1D, and EV1E and F), as recently done (Worrall et al, 2018); and (iii) high‐throughput traditional centromeric FISH analysis on selected chromosomes with an automated scanning microscope (Figs 1E, and EV1G and H). Results from the three approaches were largely consistent among each other in detecting chromosome‐specific aneuploidy (with some exceptions mainly for the ImageStream data), particularly in identifying the chromosomes that show highest or lowest rates of aneuploidy. Altogether, our analysis of whole‐chromosome aneuploidy combined with statistical modeling (see Statistical data in Dataset EV1) revealed that, following CENP‐A depletion, specific chromosomes (mainly 3, 6, 16, and X) have a higher probability to mis‐segregate, while some others (e.g., 1, 11, 12, 17, and 19) show very low rates of mis‐segregation (Fig 1F and Dataset EV2). Although in the untreated condition we do not have sufficient aneuploidy events to draw strong conclusions, nonetheless our data indicate a similar trend (11 out of 18 chromosomes) of chromosome segregation fidelity as that of the CENP‐A depleted cells (with chromosomes 3, 16, and X always mis‐segregated in at least one method used to detect aneuploidy; Fig 1C–E).
Under this condition of centromere inactivation through CENP‐A depletion, mis‐segregation mainly involved chromosomes that failed to align to the metaphase plate during the second mitosis after IAA addition and were encapsulated into micronuclei (MNs), as observed by following specific chromosomes segregating during mitosis in real time (Fig 2A–E, Movies EV1 and EV2) or on fixed samples (Fig 2F–H). It is important to note that, with all three methods, we analyzed only the main nuclei and not the MNs, therefore explaining why we could detect more chromosome losses rather than gains (Fig EV1B–H).
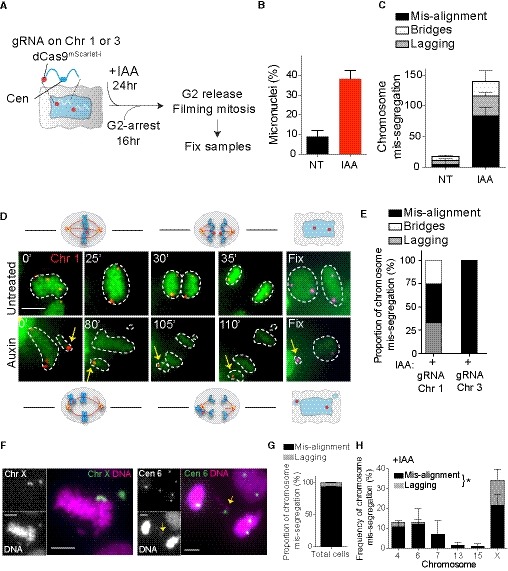
Figure 2. Analysis of chromosome‐specific mis‐segregation following CENP‐A removal.
-
ASchematic of the experimental procedure used in (B–E). Closer view of the labeled chromosome shows the targeted locus for the dCas9.
-
B, C(B) Micronuclei frequency formation by live cell imaging in untreated (NT) and auxin‐treated (IAA) condition. (C) Bar graph represents the type of chromosome mis‐segregation (independently of the dCas9 signal) observed in the indicated conditions, with (IAA) or without (NT) auxin, in the cell lines expressing dCas9 mScarlet‐I and sgRNA targeting chromosome 1 or 3. Error bars represent the SEM of four independent experiments in which cells labeled for chromosomes 1 and 3 were analyzed together (n = 45–149 cells).
-
DRepresentative live cell imaging of RPE‐1 cells dCas9 mScarlet‐I with sgRNA targeting chromosome 1 (red dots) starting from metaphase and showing example of correct (untreated) or mis‐aligned chromosome (auxin) leading to the formation of a micronucleus. Yellow arrows mark mis‐aligned‐ and micronucleus‐containing chromosome 1. Cells were imaged every 5 min. The numbers indicate the time point at which the presented images were taken. Cells were fixed at the end of the movie to detect Cas9 with an antibody. Scale bar represents 10 μm.
-
EBar graph representing the proportion of chromosome mis‐segregation observed in both cell lines after auxin addition. Only movies in which the dCas9 mScarlet‐I dots were clearly visible during the whole division were taken into consideration for the analysis (N = 6–10 cells).
-
FRepresentative images of mitotic errors leading to aneuploidy in fixed cells after CENP‐A depletion (28 h auxin). Chromosomes are stained using whole (X) or centromeric (6) FISH probes. Yellow arrows mark lagging chromosomes. Scale bar represents 5 μm.
-
G, HBars represent the (G) proportion of chromosome mis‐segregation and (H) the type of the different chromosome mis‐segregations observed. Error bars represent the SEM of two replicates. n > 66 mis‐segregation events. Paired t‐test lagging chromosomes versus mis‐alignment chromosomes, *P = 0.0275.
High‐order repeats vary in abundance among human chromosomes
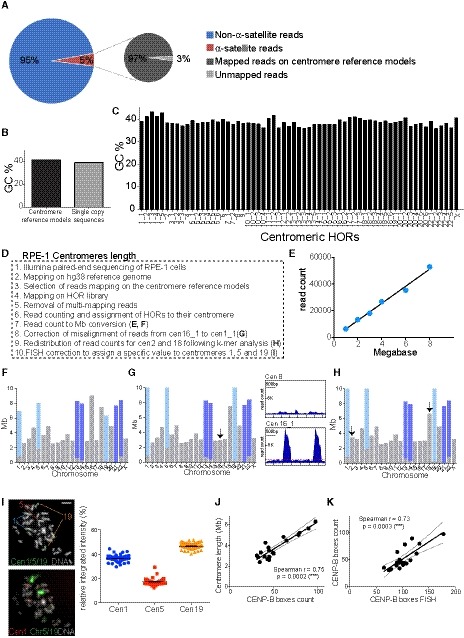
We then investigated if variations in centromere strength—a measure of microtubule binding capacity determined by centromere length and protein composition —could explain the observed variability in chromosome‐specific mis‐segregation. We first measured the length of centromeric DNA [defined as the sum of the lengths of all alpha‐satellite DNA organized into high‐order repeat (HOR) arrays present at each centromere] and the abundance of CENP‐B boxes in RPE‐1 cells using whole‐genome sequencing and mapping on centromere reference models (Fig 3A and Table EV1). Of the total reads obtained from whole‐genome sequencing, 5% were derived from alpha‐satellite DNA, as estimated using a comprehensive library of human alpha‐satellite k‐mers (Miga et al, 2014; Nechemia‐Arbely et al, 2017; see Materials and Methods). Our mapping on centromere reference models allows us to retrieve 97% of these alpha‐satellite containing reads, showing that there is no major loss of centromeric sequence information (Fig EV2A–C). Starting from these alignments, we reassigned mis‐mapped reads following a pipeline that includes k‐mers and FISH analysis to resolve possible ambiguity due to high sequence similarity between some of the centromeric alpha‐satellite arrays (Fig EV2D–I and Materials and Methods). It should be noted that the value assigned to each centromere represents the average between the two homologous chromosomes, whose centromeres features cannot be differentially assessed by sequencing.
Figure 3. Centromeres of individual chromosomes vary in DNA sequence and CENP‐B boxes abundance.
-
ASchematic of the experiments shown in (B–E).
-
B, CBar plot showing the mean of (B) centromere length (n = 4) and (C) CENP‐B boxes counts (n = 4) as determined by whole‐genome sequencing. Error bars represent the SEM of four independent experiments. Acrocentric chromosomes 13, 14, 21, and 22 were marked by a line as we could not assign the respective reads. Dashed lines indicate the mean. Bars were labeled with asterisks according to the significance of their difference from the mean (t‐test). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Bars represent the sum of the length or counts of different HOR arrays (see Table EV2). CENP‐B boxes counts are normalized to the average number of mapped reads in each replicate.
-
DSchematic using representative images of the mFISH labeling followed by CENP‐B box FISH method used to identify and quantify centromere specific CENP‐B boxes signal in (E). Scale bar represents 10 μm.
-
EBox and whisker plots of normalized CENP‐B boxes intensity over the mean on metaphase spread from three independent experiments (n > 50 cells) using the Tukey plot. One‐way ANOVA with post‐hoc Tukey's multiple comparison test shows high diversity between chromosomes. t‐Test against the mean was used to estimate the statistical significance for each chromosome. **P < 0.01; ***P < 0.001; ****P < 0.0001.
-
F, GScatter plot showing a non‐significant or significant negative correlation between the mean of (F) centromere length (n = 4) and mis‐segregation rate (from Fig 1F) or between the mean of (G) centromere CENP‐B boxes FISH signal (n > 50 cells) and mis‐segregation rate, respectively (r = Spearman rank coefficient). Lines represent the linear regression with a 95% confidence interval. Data from chr 13, 14, 21, and 22 were excluded from the analysis.
Figure EV2. Chromosome‐specific centromere length analysis in human cells (related to Fig 3).
-
APie charts representing the fraction of alpha‐satellite‐containing reads of the total read pool (left) and representing the fraction of alpha‐satellite‐containing reads that can be mapped on the centromere reference models by using our method (right). Both charts refer to the whole‐genome DNA sequencing of RPE‐1 cells.
-
BBarplot showing the average GC percentage of the centromere reference models versus the GC percentage of the single‐copy sequences that were used for centromere length determination with standard curve.
-
CBarplot reporting the GC content across all HOR array consensus sequences used as reference.
-
DStepwise procedure of RPE‐1 centromere length analysis.
-
EExample of standard curve used to convert whole‐genome sequencing read counts into megabases (Mb), for the determination of centromere length. Each point represents a randomly chosen single‐copy region of the genome.
-
FCentromere length after conversion to Mb using the standard curve shown in (E). The gray bars correspond to the length of centromere‐specific HOR arrays; the light and dark blue bars represent the length of the HOR array shared by chr1, 5, 19 (“cen1_1”) and chr13, 14, 21, 22 (“cen13_1”, “cen22_1”), respectively.
-
G(Left) Centromere length correction after reassignment of the reads from cen16_1 (chr16 specific) to cen1_1 (shared among chr1, 5, 19). The arrow marks the chromosome that changed compared to (F). (Right) Representative plots of read coverage along the HOR sequence of “cen 8”, not showing any misalignment, and “cen16_1”, showing misalignment.
-
HCentromere length correction after redistribution of read counts between centromere 2 and centromere 18 following k‐mer analysis. The arrows mark the chromosomes that changed compared to (G).
-
IRepresentative images of the sequential FISH using alpha‐satellite cen1/5/19 FISH probe (which recognizes the cen1_1 D1Z1 HOR) followed by chromosome 1, 5, 19 FISH labeling. The scatter plot represents signal quantification of alpha‐satellite cen1/5/19. Error bars represent the SEM of two independent experiments (n = 47 cells). Scale bar represents 5 μm.
-
J, KScatter plot showing a significant positive correlation between the mean of (J) centromere length (n = 4) and CENP‐B boxes count (n = 4) and (K) centromere CENP‐B boxes FISH data (n > 50 cells) and CENP‐B boxes count (r = Spearman rank coefficient). Data from chr 13, 14, 21 and 22 were excluded from the analysis. The lines represent linear regression with 95% confidence interval.
We were thus able to generate a comprehensive analysis of centromere length and abundance of CENP‐B boxes in the human model system RPE‐1 for all chromosomes, except for the acrocentrics 13, 14, 21, and 22, because they mainly share the same HOR arrays and thus become unassignable (Fig 3B and C, and Tables EV2 and EV3). As somewhat expected, centromere length and CENP‐B boxes abundance show a strong correlation profile (Fig EV2J). Sequencing data of CENP‐B box abundance was confirmed by a combination of multicolor FISH (mFISH) and CENP‐B boxes FISH (Fig 3A, D and E), which significantly correlated with the sequencing data (Fig EV2K). Interestingly, we revealed the existence of a statistically significant negative correlation between the rate of chromosome mis‐segregation and the abundance of CENP‐B boxes, while there was no significant correlation between chromosome mis‐segregation and centromere length (Fig 3F and G). A likely explanation for this difference in correlations can be that, even if longer centromeres allow more CENP‐B boxes to be present (Fig EV2J), not all HORs have equal CENP‐B box frequency, with some minor HORs that are almost devoid of CENP‐B boxes (Table EV2). Due to the lack of sequencing data, the acrocentric chromosomes 13, 14, 21, and 22 were excluded from this and all the following analyses.
CENP‐B box‐dependent features influence chromosome segregation fidelity
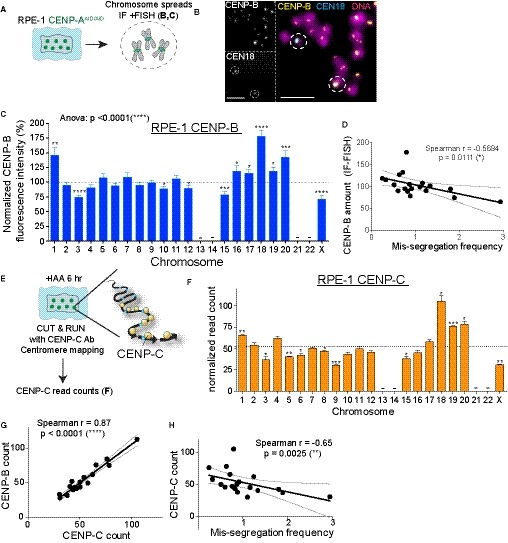
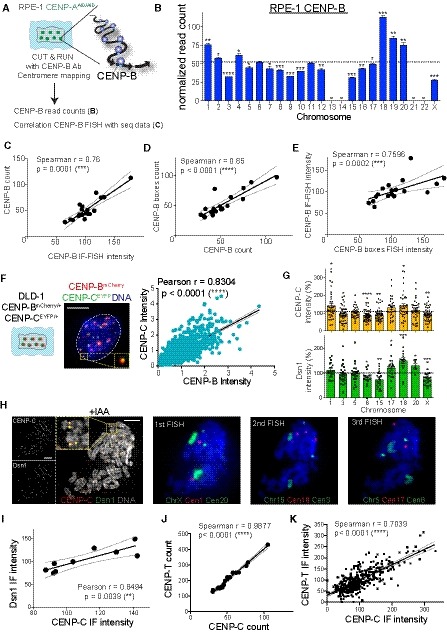
As CENP‐B boxes act as loading sites for the assembly of CENP‐B, the only known centromeric protein with DNA sequence‐specific binding (Earnshaw et al, 1987), we measured CENP‐B abundance across all human chromosomes in untreated conditions in RPE‐1 cells. To this end, we used both quantitative imaging approaches—immuno‐fluorescence‐FISH (IF‐FISH) (Fig 4A and B) and the Cleavage Under Targets & Release Using Nuclease (CUT&RUN) method (Skene & Henikoff, 2015)—using a CENP‐B antibody followed by next‐generation sequencing (Fig EV3A and B). Both of our analyses revealed variations in the abundance of CENP‐B within human chromosomes (Figs 4C and EV3B) that positively correlate with the abundance of CENP‐B boxes (Fig EV3C–E). As described for the CENP‐B boxes, we observed a significant negative correlation with chromosome mis‐segregation in CENP‐A‐depleted cells: chromosomes with high levels of CENP‐B (1, 18, 19, and 20) mis‐segregate less frequently compared to the ones that show low CENP‐B signals (3 and X; Fig 4D).
Figure 4. Faithful chromosome segregation is biased in favor of centromeres carrying a high frequency of CENP‐B/CENP‐C molecules.
-
ASchematic of RPE‐1 chromosome spreads used in experiment (B) and (C).
-
BRepresentative immunofluorescence FISH (IF‐FISH) images to measre CENP‐B intensity at specific chromosome in untreated cells. Scale bar represents 5 μm.
-
CBar plot showing the normalized CENP‐B fluorescence intensity at every chromosome over the mean on each metaphase spread (n > 37 per chromosome) ± SEM. Acrocentric chromosomes 13, 14, 21, and 22 were not analyzed and are marked by a line. One‐way ANOVA with post‐hoc Tukey's multiple comparison test shows high diversity between chromosomes. t‐Test against the mean was used to estimate the statistical significance for each chromosome. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
-
DScatter plot showing a significant negative correlation between the mean of CENP‐B IF‐FISH signal (n > 37) and the mis‐segregation rate (from Fig 1F; r = Spearman rank coefficient). The lines represent linear regression with 95% confidence band.
-
E, FBar graphs report the sum of the normalized read counts of different HOR arrays (see Table EV2) representing CENP‐C binding following CUT&RUN, sequencing, and centromere mapping. Cells were treated for 6 h with IAA to deplete CENP‐A. Error bars represent the SEM of three independent experiments and the dashed line represents the mean. Acrocentric chromosomes 13–14, 21, and 22 were marked by a line. t‐Test against the mean was used to estimate the statistical significance for each chromosome. *P < 0.05; **P < 0.01; ***P < 0.001.
-
GScatter plot showing a significant positive correlation between the mean of CENP‐C reads (n = 3) and CENP‐B reads (n = 3) (r = Spearman rank coefficient). Data from chr 1, 5, and 19 were excluded from the analysis in addition to chr 13, 14, 21, and 22 to better assess correlation without the FISH correction (as in sup Fig EV2I). The lines represent linear regression with a 95% confidence interval.
-
HScatter plot showing a significant negative correlation between the mean of CENP‐C reads (n = 3) and the mis‐segregation rate (from Fig 1F) (r = Spearman rank coefficient). The lines represent linear regression with 95% confidence interval. Data from chr 13, 14, 21, and 22 were excluded from the analysis. The lines represent linear regression with a 95% confidence interval.
Figure EV3. Centromeric and kinetochore proteins show a similar abundance profile (related to Fig 4).
-
ASchematic of the experimental procedure used in (B) and (C), Ab: Antibody.
-
BBarplot showing the sum of the normalized reads count of different HOR arrays (see Table EV2) representing CENP‐B binding following CUT&RUN, sequencing and centromere mapping. The dashed line represents the mean and error bars represent the SEM of three independent experiments. Acrocentric chromosomes 13, 14, 21, and 22 are missing and marked by a line. Bars are labeled with asterisks according to the significance of their difference from the mean (t‐test). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
-
C–EScatter plot showing significant positive correlation between the mean of (C) CENP‐B count (n = 3) and CENP‐B IF‐FISH intensity (n > 37), (D) CENP‐B count and CENP‐B boxes count (n = 4) and (E) CENP‐B IF‐FISH and CENP‐B boxes FISH intensity (n > 50 cells; r = Spearman rank coefficient). The lines represent linear regression with 95% confidence interval. In (D) data from chr 1, 5 and 19 were excluded from the analysis as chr 13, 14, 21 and 22 to better assess correlation without the FISH correction (as in Fig EV2I).
-
F(Left) Schematic and representative image of CENP‐BmCherry and CENP‐CEYFP cell and linear regression of signal quantification. Inset shows CENP‐BmCherry and CENP‐CEYFP signal colocalization. (Right) Scatter plot showing a significant positive correlation between centromere CENP‐C and CENP‐B intensity (r = Pearson rank coefficient). Scale bar represents 5 μm. N = 66. The lines represent linear regression with 95% confidence interval.
-
GBar graphs show CENP‐C and Dsn1 intensity quantification at the indicated chromosomes normalized over the mean on each metaphase spread. Error bars represent the SEM of three independent experiments and dashed line represents the mean of all the analyzed chromosomes. Each dot represents a cell. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
-
HRepresentative images of an immunofluorescence on CENP‐C and Dsn1 on metaphase‐arrested chromosome followed by sequential centromere or chromosome‐specific FISH probes staining. Cells were treated for 6 h with IAA to deplete CENP‐A. Scale bar represents 5 μm.
-
I–KScatter plot showing a significant positive correlation between the mean of (I) CENP‐C (n > 3) and Dsn1 (n > 3) intensity, CENP‐C (n = 3) and CENP‐T (n = 2) reads amount (J) and fluorescence intensity (K). r = Pearson or Spearman rank coefficient. The lines represent linear regression with 95% confidence interval.
We have previously shown that CENP‐B binding to alpha‐satellite DNA is sufficient to maintain chromosome segregation fidelity by preserving CENP‐C (directly) and CENP‐T (likely via CENP‐C) on CENP‐A‐depleted centromeres (Hoffmann et al, 2016). To determine the existence of a correlation between the number of CENP‐B binding sites, CENP‐C, and the fidelity of chromosome segregation, we measured the level of CENP‐C at human centromeres using the CUT&RUN method followed by next‐generation sequencing (Fig 4E and F). Since CENP‐C interacts with both CENP‐A and CENP‐B, we treated cells for 6 h with IAA to remove all CENP‐A/CENP‐C interaction (Fachinetti et al, 2015; Hoffmann et al, 2016). We found a strong positive correlation between CENP‐B and CENP‐C levels at individual centromeres (Fig 4G). The amount of CENP‐C obtained by sequencing data and the CENP‐B/C levels correlation were further confirmed using quantitative IF‐FISH and live cell imaging on endogenously tagged CENP‐CEYFP and CENP‐BmCherry (in the presence of CENP‐A), respectively (Fig EV3F and H). CENP‐C amount at individual centromeres also positively correlated with the amount of Dsn1 (Fig EV3G–I), member of the Mis12 kinetochore complex known to directly interact with CENP‐C (Weir et al, 2016), and with CENP‐T, another key centromeric component necessary for kinetochore assembly (Foltz et al, 2006; Huis in ‘t Veld et al, 2016; Fig EV3J and K). As observed for CENP‐B, CENP‐C negatively correlated with the chromosome mis‐segregation frequency (Fig 4H and Dataset EV3).
Putting all these data together, by removing CENP‐A, we unveiled the existence of a correlation between the levels of both centromere‐bound CENP‐B (via CENP‐B boxes) and CENP‐C and the rate of chromosome mis‐segregation for most human chromosomes: lower CENP‐B/C levels are concomitant with higher rates of mis‐segregation. This correlation is particularly strong for chromosomes carrying weak/very weak (6/3 and X, respectively) or strong/very strong (7, 11 and 17/1, 18, 19, and 20, respectively) centromeres (Dataset EV4).
To test if this bias in mis‐segregation is directly dependent on CENP‐B, we measured whole‐chromosome aneuploidy of few human chromosomes by high‐throughput centromeric FISH analysis in RPE‐1 CENP‐AAID CENP‐B WT versus CENP‐AAID CENP‐B KO cells (Hoffmann et al, 2016; Fig 5A and B). We depleted CENP‐A for 24 h by IAA addition, as this was shown to be an appropriate time to induce high rate of chromosome mis‐segregation in a CENP‐B‐defective background (Hoffmann et al, 2016). Our data show that, following CENP‐A depletion, chromosomes that did not show high levels of mis‐segregation in the presence of CENP‐B (1 and 11) later became highly mis‐segregated in the CENP‐B KO background, at rates similar to those of the X chromosome (comparing gray to red bars; Fig 5B). As in this condition the bias in chromosome aneuploidy was considerably reduced (with an increased fold change over CENP‐B WT/KO cells for chr 1 and 11 versus chr X), our data suggest a direct involvement of CENP‐B in mediating chromosome‐specific centromere strength and segregation fidelity. Our data also strengthen the notion that both CENP‐A and CENP‐B are somehow independently required for faithful chromosome segregation, since losing CENP‐A (gray bar) or losing CENP‐B (black bar) leads to the same mis‐segregation outcome, and depletion of both (red bar) reveals an additive effect (Fig 5B).
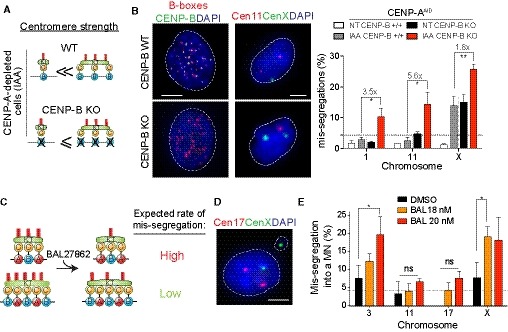
Figure 5. Chromosome‐specific aneuploidy arises following perturbation of centromere strength.
-
ASchematic of the centromere strength model in the RPE‐1 CENP‐AAID CENP‐B KO mutant. After CENP‐A depletion a reduced bias in chromosome mis‐segregation is expected.
-
BRepresentative image of centromere 11 (red) and centromere X (green) FISH on RPE‐1 CENP‐AAID CENP‐B KO mutant in control (top) or IAA‐treated condition (bottom). Scale bar represents 5 μm. Bar graphs show automatic FISH quantification of chromosome mis‐segregation in the indicated cell lines with and without IAA addition for 24 h. Error bars represent the SEM of three independent experiments (n > 200 cells per experiment). Dunnett's multiple comparisons test was used to compare conditions. Gray lines separate independent experiments. Fold changes between gray and red bars are also indicated.
-
CSchematic of the centromere strength model with the microtubule destabilizer BAL27862 compound.
-
DRepresentative image of centromere 17 (red) and centromere X (green) FISH on RPE‐1 cells treated with the BAL27862 compound. Scale bar represents 5 μm.
-
EBars represent the frequency of chromosome mis‐segregation into micronuclei after 28 h treatment with the BAL27862 drug at indicated concentration. Error bars represent the SEM of three independent experiments, dashed line represents the mean. n > 40 micronuclei were analyzed. Dunnett's multiple comparisons test, *P < 0.05. DMSO was used at the same concentration of BAL27862.
We further tested the existence of a direct correlation between the abundance of centromere components and chromosome/spindle fiber connections, as we know that the number of kinetochore microtubules can range between 12 and 24 (Wendell et al, 1993). To this end, we used the microtubule polymerization inhibitor BAL27862 (Basilea Pharmaceutica) that, when used at low concentration, leads to a reduction in the number of microtubules per kinetochore without generating completely unattached kinetochores (Dudka et al, 2018). As BAL27862 treatment will reveal weakening of centromere function, we expected that chromosomes with low levels of CENP‐B/C (e.g., chromosome 3 and X) would be more prone to mis‐segregate than others upon treatment, even in the presence of CENP‐A, due to a reduction of kinetochore‐interacting microtubules (Fig 5C). In agreement with this model, and similar to the CENP‐A auxin‐depleted condition, BAL27862 treatment over DMSO control increased in a dose‐dependent manner the frequency of micronuclei containing chromosomes 3 and X, but not chromosomes 11 and 17 (Fig 5D and E).
Different levels of CENP‐B among centromeres dictate different chromosome segregation outcomes
Homologous chromosomes may harbor different centromere lengths and/or abundance of CENP‐B boxes because they originate from different individuals. Our centromere sequencing analysis is not able to distinguish such differences, since it reports the average values between the two homologs. Using a FISH assay combined with mFISH in RPE‐1 cells, we found that the homologs of chromosome 3 harbor a twofold difference in the abundance of CENP‐B boxes at the centromere, while the overall centromere length remains the same (Figs 6A, and EV4A and B). Intriguingly, we found that chromosome 3‐containing micronuclei carried with higher frequency the homolog that harbors less CENP‐B boxes (Fig 6B and C). This was not due to any type of centromere erosion that may occur within the micronuclei, as this difference in the amount of CENP‐B boxes could also be observed within the main nucleus (Fig EV4B). Similarly, the same analysis on the two X chromosomes (easily distinguishable as one of them carried a fusion with a fragment of chromosome 10) showed that they harbored a slight difference in the number of CENP‐B boxes, which was reflected in a slight tendency of the chromosome with less CENP‐B to mis‐segregate at higher rates (Fig EV4C and D). Altogether, the finding that allelic differences in the amount of CENP‐B boxes observed for chromosomes 3 and X lead to allelic differences in the rate of mis‐segregation supports a direct role of DNA‐dependent features in influencing chromosome segregation fidelity. However, we cannot exclude that the chromosome X‐10 fusion may also have a negative impact on the segregation fidelity of this homolog possibly by increasing total chromosome length.
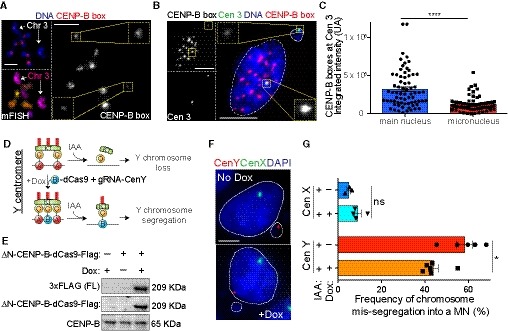
Figure 6. Different levels of CENP‐B across centromeres dictate chromosome segregation outcome.
-
ARepresentative image of CENP‐B box FISH and mFISH stained RPE‐1 metaphase spread cells. Chromosome 3 homologs are indicated by white arrows with single CENP‐B box chromosomes signal magnified in insets. Scale bar represents 5 μm.
-
BRepresentative image of CENP‐B box (red) and centromere 3 (green) FISH on RPE‐1 interphase nucleus after 48 h IAA treatment. CENP‐B box signal at individual homologs are magnified in insets. Scale bar represents 5 μm.
-
CBar plots represent the quantification of the CENP‐B box FISH intensity at the chromosome 3 homolog contained in the main nucleus versus the homolog present in the micronucleus as represented in (B). Error bars represent the SEM of three independent experiments. n = 69 cells. Unpaired t‐test, ****P < 0.0001.
-
DSchematic of the experiment shown in (E–G). ΔN‐CENP‐B protein was fused to dCas9‐doxycycline (Dox)‐inducible protein and was inserted at a single genomic locus in DLD‐1 CENP‐A−/EA male cell line expressing a gRNA targeting centromere Y.
-
EImmunoblot shows expression of the (ΔN)CENP‐B‐dCas9 after doxycycline induction.
-
FRepresentative images show a micronucleus or the main nucleus containing the Y chromosome (red) detected by FISH in no‐doxycycline or doxycycline treated cells, respectively. Chromosome X is shown in green. Scale bar represents 5 μm.
-
GBar plot shows the mean (n ≥ 3) frequency of micronuclei containing the chromosome Y or chromosome X ± doxycycline treatment for 72 h and IAA for 48 h ± SEM. n > 200 cells with a micronucleus. Mann–Whitney test. *P = 0.0173.
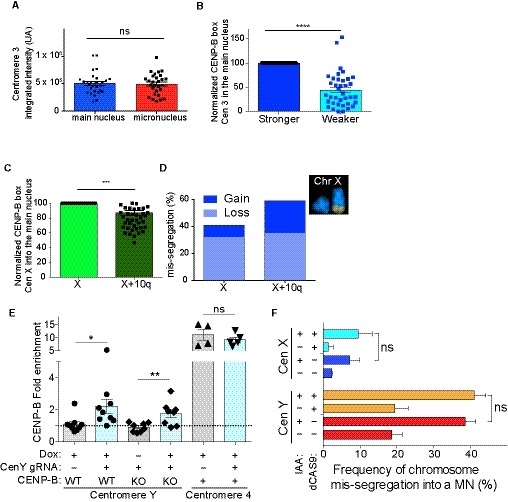
Figure EV4. Centromeric features at individual homologs and/or chromosomes show difference in their abundance (related to Fig 6).
-
ABars represent the quantification of centromere 3 FISH signal at the two chromosome 3 homologs in the main nucleus and in the micronuclei. Error bars represent SEM of at least three independent experiments. N = 62 cells, ns = not significant (Unpaired t‐test).
-
BBars represent the normalized quantification of CENP‐B box at the centromeres of the two homologs 3 in the main nucleus. N = 37 cells. Error bars graph represent the SEM. Unpaired t‐test, ****P < 0.0001.
-
CBars represent CENP‐B box FISH signal quantification on chromosome X versus chromosome X/q10 homologs. Error bars represent the SEM of three independent experiments. Unpaired t‐test, ***P = 0.0005.
-
DChromosome X homologs mis‐segregation as determined by single‐cell sequencing and representative mFISH imaging showing chromosome X and chromosome 10 q arm translocations (n = 811 cells).
-
EBars represent the mean of revealed qPCR CENP‐B enrichment normalized on IgG and with Alu repeats at centromere Y or 4 ± doxycycline induction of the (ΔN)CENP‐B‐dCas9‐Flag or dCas9‐Flag‐(ΔN)CENP‐B(ΔC)‐GST in CENP‐A−/AID or CENP‐A−/AID CENP‐B KO cells. Each dot represents an independent experiment (n = 4–9) ± SEM. Unpaired t‐test, *P = 0.0301 and **P = 0.0083.
-
FBar plot showing the mean (n = 3) of frequency of micronuclei containing the chromosome Y or chromosome X ± IAA for 48 h in the indicated cell lines. N > 50 cells for experiment with a micronucleus. ns = not significant (Unpaired t‐test).
We then tested whether increasing CENP‐B levels is enough to revert chromosome‐specific aneuploidy, by specifically rescuing in the pseudo‐diploid colorectal human male cell line (DLD‐1) the high level of chromosome mis‐segregation observed for the Y chromosome (Hoffmann et al, 2016), the only chromosome devoid of CENP‐B boxes and CENP‐B (Earnshaw et al, 1989; Miga et al, 2014). Indeed, we previously demonstrated that, following CENP‐A depletion by IAA or removal of the CENP‐A/CENP‐C interaction site, the Y chromosome undergoes high rates of mis‐segregation (~40% to ~80%) due to the loss of centromeric CENP‐C (Hoffmann et al, 2016; Ly et al, 2017). To this end, we generated a cell line harboring a doxycycline (Dox)‐inducible fusion protein of CENP‐B (lacking the DNA binding domain) with dCas9 and expressing a stably integrated gRNA specific for the CENP‐A binding site on the Y centromere (DYZ3) (Henikoff et al, 2015) in male DLD‐1 CENP‐AAID cells (Fig 6D and E). Following dox (to express CENP‐B‐dCas9) and IAA (to deplete CENP‐A) addition, we observed a partial but significant rescue of the rate of micronuclei containing the Y chromosome (Fig 6F and G). This partial rescue was presumably caused by a transient recruitment of CENP‐B to the DYZ3 or by a small subset of cells correctly localizing the CENP‐B‐dCas9, as we could detect just a minor enrichment of CENP‐B compared to the control on the Y centromere by CUT&RUN (Fig EV4E). Stable expression of dCas9 only at DYZ3 was insufficient to rescue the Y chromosome mis‐segregation (Fig EV4F). Altogether, these results strongly support a model in which different levels of CENP‐B across centromeres dictate chromosome segregation outcome.
CENP‐B box‐dependent features and aneuploidy in mouse and cancer cell model systems
We next tested the positive role of CENP‐B in determining centromere strength in two different model systems. We first analyzed chromosome‐specific aneuploidy in the pseudo‐diploid colorectal cancer DLD‐1 cells, in which we can induce rapid degradation of the endogenous CENP‐A, similarly to what we have done for RPE‐1 (Fig 7A; Hoffmann et al, 2016). As expected, following CENP‐A depletion for 48 h we could observe a strong increase in chromosome mis‐segregation compared to control (Fig EV5A–C). Nevertheless, with the exception of chromosome 21, we failed to detect any significant chromosome‐specific aneuploidy over the mean (Fig 7B) [the Y chromosome was not analyzed here since we have already reported its very high rate of mis‐segregation under this condition; Fig EV4F (Hoffmann et al, 2016)]. This could be explained by the observed higher heterogeneity in the number of chromosomes between cells in DLD‐1 compared to RPE‐1 (Fig 7C) and/or by the presence of driver mutations and/or rearrangements that govern segregation fidelity. In support of this last hypothesis, the supernumerary chromosome—a centromeric fusion between acrocentric chromosome 13–14 with the addition of chromosome 10 (Fig 7B–E)—present in all DLD‐1 cells was never observed to mis‐segregate in untreated or even auxin‐treated cells (Fig EV5C). Interestingly, chromosomes 1, 6, and 7 that carry intra‐ and/or inter‐chromosome translocations (Ghandi et al, 2019) show a slighter, although not significant, increase in the mis‐segregation rate (Fig 7B). We then tested if reduced variability in DNA‐dependent centromeric features could also partially explain the lack of chromosome‐specific aneuploidy. To this aim, we measured centromere length, abundance of CENP‐B boxes, and CENP‐C in DLD‐1 cells using a combination of whole‐genome sequencing, CUT&RUN, mapping on centromere reference models (Tables EV2 and EV3, and Fig EV5D and E), and mFISH+FISH (Fig 7E and F), as done for RPE‐1. Interestingly, centromeres of DLD‐1 cells show reduced heterogeneity in the abundance of CENP‐B boxes and CENP‐C among the different chromosomes compared to RPE‐1 (Figs 7F and EV5E). Nevertheless, similar to RPE‐1 cells, we could observe a very strong correlation between CENP‐B boxes and CENP‐C, but a weaker, although significant, correlation between CENP‐B boxes and centromere length (Fig 7G and H). The lack of chromosome‐specific aneuploidy in the DLD‐1 cell line could at least in part be explained by the low inter‐chromosomal variation in centromere features. Furthermore, the complex and instable karyotype of the DLD‐1 cancer cell line possibly makes it harder to extricate the effect of centromeric features on mis‐segregation in this system, which is very likely driven by other factors as well.
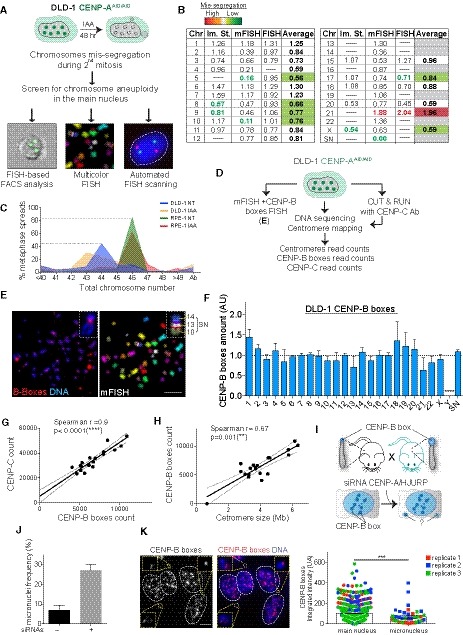
Figure 7. Abundance of CENP‐B boxes influences segregation fidelity in mouse but has a weaker impact in a cancer model system.
-
ASchematic of the experiment shown in (B).
-
BTable summarizing whole‐chromosome aneuploidy (fold over the mean) using the indicated methods to measure chromosome mis‐segregation rate. Bold numbers represent significant statistical difference from the mean for each method. Orange to dark red gradient highlights chromosomes that mis‐segregate (with at least one method) at a significantly higher rate compared to the mean level (weak chromosomes). Light green to dark green gradient highlights chromosomes that mis‐segregate (with at least one method) at a significantly lower rate compared to the mean (stronger chromosomes). Data obtained with only one method were excluded. Im. St. = image stream.
-
CPlot shows the karyotype distribution of DLD‐1 and RPE‐1 cells in untreated or auxin‐treated condition.
-
DSchematic of the experiment shown in (E).
-
ERepresentative image of chromosome spreads in DLD‐1 cells following mFISH and subsequently CENP‐B boxes FISH. Inset shows the supernumerary (SN) chromosome. Scale bar represents 10 μm.
-
FBar plot shows the mean variation in CENP‐B boxes in DLD‐1 cells from data from (E) (n = 10 cells) and whole‐genome sequencing and centromere mapping ± SD.
-
G, HScatter plot showing a significant positive correlation between the indicated features in DLD‐1 cells (n = 1). The lines represent linear regression with 95% confidence band. Data from chr 13, 14, 21, 22, Y, and SN were excluded from the analysis.
-
ISchematic of the experiment described in (J) and (K).
-
JBars show frequency of micronuclei with or without siRNA treatment. Error bars represent the SEM of three independent experiments (n > 170 cells per replicate).
-
K(Left) Representative image of CENP‐B box (red) staining after siRNA against CENP‐A/HJURP for 48 h. CENP‐B box signals at individual micronuclei are magnified in insets. Scale bar represent 5 μm. (Right) Bar graph represents the mean of the CENP‐B box FISH intensity contained in the main nucleus versus the one present in the micronucleus ± SEM. Color dots represent independent experiments (n > 43 micronuclei and 840 main nucleus) Mann–Whitney U test: ***P = 0.0001.
Figure EV5. Chromosome aneuploidy profile of DLD‐1 cells after 48 h of CENP‐A depletion (related to Fig 7).
-
A–CImageStream analysis (A), automated FISH (B) or multicolor FISH (C) of DLD‐1 cells in untreated condition (blue circles) or treated with Auxin for 48 h (red squares). Error bars represent the SEM of 2–4 independent experiments. (see the Dataset EV2 for details). Dashed lines indicate the means of aneuploidy rates in untreated (blue line) or Auxin‐treated (red line) condition. One‐way ANOVA with post‐hoc Tukey's multiple comparison test shows high diversity between untreated and IAA‐treated). Red asterisks (IAA) indicated significant over the respective mean. *P < 0.05; **P < 0.01; ****P < 0.0001.
-
DThe scatter plot represents signal quantification of alpha‐satellite cen1/5/19. Error bars represent the SEM of 22 cells.
-
EHistograms show the normalized distribution of CENP‐B boxes (RPE‐1 n = 4; DLD‐1 from Fig 7F) and CENP‐C (RPE‐1 n = 3; DLD‐1 n = 1) in the indicated cell lines. The coefficient of variation between individual chromosomes is indicated.
-
FSchematic of the experimental procedure used in (G). Ab: Antibody.
-
GBar graph represents the quantification of RPE‐1 CENP‐A counts at each chromosome following CUT&RUN, sequencing and centromere mapping. Dashed line represents the mean (n = 1). Acrocentric chromosomes 13, 14, 21 and 22 are missing and marked by a line.
Finally, we studied mitosis in a mouse embryonic fibroblast cell line derived from a hybrid mouse model system (CF‐1 × CHPO) where the two sets of chromosomes harbor a ~ 6 fold difference in the length of minor satellite sequence on all chromosomes (Iwata‐Otsubo et al, 2017; Fig 7I). In accordance with the difference in centromeric length, centromeres of CF‐1 were shown to have a higher amount of CENP‐B (~ 5 fold) compared to CHPO, but also a mild increase in CENP‐A and Hec1 (a subunit of the kinetochore) (Chmátal et al, 2014; Iwata‐Otsubo et al, 2017). We then assessed if chromosomes containing a reduced number of minor satellites were mis‐segregated more frequently within the hybrid cell line under conditions of compromised mitosis via simultaneous CENP‐A and HJURP [the CENP‐A chaperon (Dunleavy et al, 2009; Foltz et al, 2009)] reduction by RNAi (Fig 7J). In agreement with the data on human RPE‐1 cells, we observed that micronuclei preferentially contained chromosomes with low intensity of CENP‐B boxes (as a read out of a shorter minor satellite arrays) (Fig 7K). These data indicate that the impact of centromeric DNA‐dependent features on chromosome segregation fidelity during mitotic divisions exists also in mouse cells.
Discussion
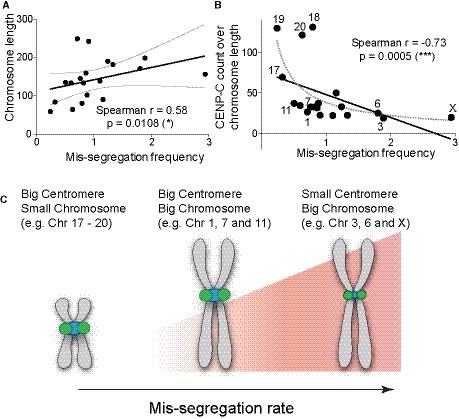
Our work demonstrates that, at least in compromised mitosis conditions, the amount of centromere components of individual chromosomes—measured by CENP‐B and kinetochore proteins levels and dependent on CENP‐B boxes—influences chromosome mis‐segregation. Surprisingly, despite these features being mainly dependent on centromere length, the size of HOR arrays per se does not seem to directly influence segregation fidelity. It has been previously observed that larger chromosomes tend to mis‐segregate more frequently in cancer cells (Bochtler et al, 2018) or under conditions that compromise the function of CENP‐E (Tovini & McClelland, 2019), a key component of chromosome congression (Kapoor et al, 2006). Here, we also found that larger chromosomes have a slightly higher tendency to mis‐segregate, but this correlation is strongly enhanced by the diversity in the centromere features (centromere strength) over the whole chromosome length (Fig 8A and B). This correlation is particularly evident for large chromosomes characterized by a low amount of centromere components (CENP‐B boxes, CENP‐B, CENP‐C), such as chromosome X (particularly the X + 10 fusion), 3, and 6. Vice versa, small chromosomes harboring strong centromeres (e.g., chromosome 17, 19, and 20) have very low chances to mis‐segregate (Fig 8B and C).
Figure 8. DNA‐dependent centromeric feature and chromosome size influence chromosome‐specific aneuploidy.
-
A, BScatter plot showing (A) a significant positive or (B) a negative correlation between chromosome length and the mis‐segregation rate (from Fig 1F) or CENP‐C (n = 3) reads over chromosome length and mis‐segregation rate (r = Spearman rank coefficient). The lines represent linear regression with 95% confidence interval. Data from chr 13, 14, 21, and 22 were excluded from the analysis. Numbers indicate chromosomes with low or high mis‐segregation frequency.
-
CModel of how chromosome segregation is influenced by centromere size and chromosome length.
Recently, it has been shown that the unusually large centromeres of the Indian muntjac deer chromosomes [covering up to 26% of whole chromosome length (Levy et al, 1993)] display proportional kinetochore size which, however, negatively impacts on the segregation fidelity due to a higher chance of incorrect attachment (Drpic et al, 2017). Human chromosomes do not carry this extremely high diversity in centromere length and kinetochore size and, differently from what happens in the Indian muntjac deer, we demonstrated that lower levels of centromere components lead to increased chromosome mis‐segregation. Interestingly, in the RPE‐1 cell line, chromosome 1, that carries a large centromere, tends to lag (likely due to merotelic attachment) at a higher frequency than chromosomes with a smaller centromere, such as chromosome 3, even in unperturbed conditions (Fig 2E and data not shown). Accordingly, chromosome 1 was recently found to be lost at higher frequency compared to others following nocodazole release (Worrall et al, 2018), a treatment known to promote merotelic attachment (Crasta et al, 2012). This suggests that stronger centromeres, as the ones from chromosome 1, are less affected in conditions that compromise centromere function, but more vulnerable to incorrect attachment. This could explain the reasonably moderate range of variation in centromere strength between human chromosomes.
Although we had limited data on the rate of chromosome mis‐segregation and centromere features for the acrocentric chromosomes (except chromosome 15), our results in RPE‐1 do not show the correlation described above for this category of chromosomes. This is probably due to the fact that human acrocentric chromosomes, all of which carry ribosomal DNA (rDNA) close to their centromeres, cluster around the nucleolus, which may give them special adhesion properties through their rDNA (Ferguson‐Smith & Handmaker, 1961) that limits non‐disjunction during mitosis/meiosis, independently of their centromeres. Indeed, a recent report demonstrated that UBF‐dependent transcribed rDNA can form inter‐ and intra‐chromosomal connections that link acrocentric chromosomes together, and that are naturally resolved just prior to anaphase onset (Potapova et al, 2019). Surprisingly, while RPE‐1 showed remarkably no evidence of acrocentric chromosomes mis‐segregation even after centromere perturbation (Fig 1D), acrocentric chromosomes 13, 21, and 22 mis‐segregated at higher, although variable, rates compared to the overall mean in DLD‐1 cells (Figs 7B and EV5C). Notably, within the small range of centromere features seen in DLD‐1 cells, chromosomes 13, 21, and 22 have the lowest number of CENP‐B boxes (Figs 7F and EV5E). In agreement with our data, cell lines derived from patients harboring trisomy 21 were found to carry shorter D21Z1 arrays and less CENP‐B amounts on their chromosome 21 compared to cell lines derived from healthy individuals (Contreras‐Galindo et al, 2017).
Our findings do not exclude that, besides the capacity to form a stronger kinetochore in the absence of CENP‐A, other factor(s) directly or indirectly related to centromere length could potentially control this chromosome mis‐segregation bias. Indeed, it is well‐known that not all CENP‐B is in complex with CENP‐A and CENP‐C. This might explain why CENP‐C level has a higher correlation with chromosome mis‐segregation compared to CENP‐B boxes or CENP‐B. In any case, CENP‐A has a similar centromere enrichment profile to that of CENP‐B (Fig EV5F and G), in accordance to the fact that CENP‐A binding is proportional to centromere length [it tends to occupy ~ 25–30% of the higher order repeats array (Sullivan et al, 2011); Table EV2]. Other (peri‐)centromeric features could impact on chromosome‐specific aneuploidy in a kinetochore‐independent manner, among which we can mention variations within HOR arrays such as SNPs and indels (Sullivan et al, 2017), changes in the heterochromatin surrounding functional centromeres (Pezer & Ugarković, 2008), fluctuations in centromere transcripts (Smurova & De Wulf, 2018), DNA methylation (Scelfo & Fachinetti, 2019), and variation in centromeric cohesion (Kitajima et al, 2006) and/or in microtubule‐destabilizing activity (Akera et al, 2019). All these factors can potentially promote different patterns of chromosome‐specific aneuploidy. For example, following mitotic delay, certain chromosomes were shown to be more prone to cohesion fatigue and consequently to mis‐segregation (Worrall et al, 2018).
Our results reveal the existence of biological differences between different chromosomes and might help understand certain types of whole‐chromosome aneuploidy known to cause human genetic diseases such as Down (as discussed above) and Turner syndromes, as well as various types of cancer, such as multiple myeloma, adenocarcinoma, and leukemia (Gordon et al, 2012). Indeed, while tumor aneuploidy is largely modeled by selection during evolution, as our results on the supernumerary chromosome of DLD‐1 suggested, centromere strength can still have an impact at the initial stage of mis‐segregation, before clonal selection. As an example, it has been shown that the CENP‐B‐free Y chromosome mis‐segregates at higher rates in an age‐ and smoke‐dependent manner, and it was recently observed that its loss (LOY) is associated with shorter survival and high risk of blood cancer (Forsberg et al, 2014; Dumanski et al, 2015; Forsberg, 2017). It is tempting to speculate that this event may also be related to reduced level of CENP‐A expression during aging, as observed in certain cell types (Lee et al, 2010; McGregor et al, 2013). Interestingly, loss of chromosome 3 has been frequently observed in uveal melanoma and can be used as prognostic predictor (Prescher et al, 1996; Scholes et al, 2001). Furthermore, loss of heterozygosity at chromosome X is a common feature of breast cancer, with the inactivated X being more likely to be fully lost (Loupart et al, 1995; Sirchia et al, 2005). A recent work pointed out that specific heterozygous genetic variations flanking the centromere X (DXZ1) negatively affect segregation fidelity in hematopoietic cells (Loh et al, 2018). Remarkably, according to the karyotypic analyses of astrocytomas at different stages, loss of X occurs almost exclusively at early stages during tumor development (Duijf et al, 2013), thus in a phase where the effects of selective pressure may be less prevalent.
Whether centromere strength could also control aneuploidy during meiosis, particularly in association to aging, still needs to be tested. Indeed, besides other well‐studied causes of age‐dependent aneuploidy such as the maternal age effect [increase in chromosome non‐disjunction due to defective cohesion and altered recombination (Nagaoka et al, 2012)], it is possible that reduced levels of CENP‐A may also be a direct cause of this phenomenon. We could envision that, in these conditions of impaired CENP‐A levels, the underlying frequency of CENP‐B boxes and CENP‐B would have a strong impact on age‐related oocyte aneuploidy. Certainly, it is known that mammalian oogenesis meiosis is naturally biased, because homologous chromosomes have unequal chances of being inherited by the offspring (Henikoff & Malik, 2002). In these conditions of female asymmetric meiosis, “stronger” centromeres are known to preferentially segregate to the egg due to higher amounts of centromere proteins (Lampson & Black, 2017). This could explain the positive evolution of the centromere toward the presence of a higher number of CENP‐B boxes and the absence of CENP‐B boxes at the Y chromosome which does not undergo this female‐specific selection.
Materials and Methods
Constructs
For tetracycline‐inducible expression, the siRNA‐resistant delta N‐terminus (ΔN)CENP‐B‐dCas9‐Flag or dCas9‐Flag‐(ΔN)CENP‐B(ΔC)‐GST was cloned into a pcDNA5/FRT/TO‐based vector (Invitrogen), and the sgRNA used to target the chromosome Y centromere (targeted sequence 5′AAATGATAGGTTGAACTCC3′) was cloned into a pSB700‐H2B‐ECFP plasmid.
H2B‐mTurquoise2 was cloned by replacing the mNeon in pLV‐H2B‐mNeon‐IRES‐Blast for mTurquoise2 via Gibson reaction, using a PCR product obtained from the mTq2 D13 plasmid (a gift from Bas Ponsioen). The lentiviral vector pHAGE‐UbC‐dCas9‐3XmScarlet‐I was generated from pHAGE‐TO‐dCas9‐3XGFP, a gift from Thoru Pederson (Addgene plasmid # 64107). The Chromosome (Chr.) 1 and Chr. 3 sgRNAs were selected as in Ma et al (2015; sequence GATGCTCACCT for Chr. 1 and sequence TGATATCACAG for Chr. 3, both located in sub‐telomeric repeats) and cloned into lentiviral vector pLH‐spsgRNA2 (gift from Thoru Pederson, Addgene plasmid # 64114) as in Ma et al (2015).
The baculoviral sgRNA expression plasmid was created by inserting the sgRNA cassette from pLH‐spsgRNA1‐2xPP7, a gift from Thoru Pederson [Addgene plasmid # 75390 (Ma et al, 2016)] into the baculovirus donor plasmid pAcebac1 [gift from Dr. Imre Berger, EMBL, Grenoble, France (Berger et al, 2004)]. After cloning the sgRNA sequences into the plasmids, bacmids were generated and Baculovirus was produced as in Hindriksen et al (2017).
Cell culture conditions
Cells were maintained at 37°C in a 5% CO2 atmosphere. hTERT‐immortalized RPE‐1 cells CENP‐AAID/AID or CENP‐AAID/AID CENP‐B KO (Hoffmann et al, 2016) were grown in DMEM:F12 medium containing 10% fetal bovine serum (FBS, BioSera), 0.123% sodium bicarbonate, 100 U/ml penicillin, 100 U/ml streptomycin, and 2 mM l‐glutamine.
Flp‐In TRex‐DLD‐1 CENP‐AAID/− (Hoffmann et al, 2016) and CENP‐AAID/− CENP‐B KO were maintained in DMEM containing 10% tetracycline‐free fetal bovine serum (Pan Biotech). IAA (I5148; Sigma) was used at 500 μM, colcemid (Roche) at 100 ng/ml, the microtubule destabilizer BAL27862 (Basilea Pharmaceutica International) at 18 or 20 nM (dissolved in DMSO), and doxycycline (Sigma) at 0.2 μg/ml.
Immortalized mouse embryonic fibroblast cells (MEF) (a generous gift from M. Lampson) were grown in DMEM supplemented with 0.123% sodium bicarbonate, 1 mM sodium pyruvate, and 10% FBS at 37°C in a 3% O2 atmosphere.
Generation of stable cell lines
Stable, isogenic cell lines expressing (ΔN)CENP‐B‐dCas9‐Flag or dCas9‐Flag‐(ΔN)CENP‐B(ΔC)‐GST were generated using the FRT/Flp‐mediated recombination system as described previously (Fachinetti et al, 2013). Stable integration was selected with 400 μg/ml hygromycin, and the gRNA was introduced by lentiviral infection. Isolation of positive population was performed using fluorescence‐activated cell sorting (FACS Aria, BD Biosciences). Doxycyclin‐induced ΔN‐CENP‐B‐dCas9 expression was validated by immunostaining and immunoblotting assays. Control DLD‐1 cell lines were infected with a lentivirus containing pHAGE‐EFS‐dCas9‐GFP plasmid (Addgene 64104) and the same lentivirus as above containing the gRNA. Double‐positive cells were isolated using fluorescence‐activated cell sorting technique (S3™ sorter, Bio Rad).
To generate the RPE1 CENP‐AEA/EA, H2B‐mTq2, dCas9‐3xmScarlet‐I cell line, cells were first transduced with pLV‐H2B‐mTurquoise2‐IRES‐BLAST, and fluorescent H2B‐mTurquoise2 cells were isolated by fluorescence‐activated cell sorting (BD Biosciences Aria II). This population was subsequently transduced with pHAGE‐UbC‐dCas9‐3xmScarlet‐I lentivirus, and fluorescent dCas9‐3xmScarlet‐I cells were isolated by fluorescence‐activated cell sorting (BD Aria III). Cell clones were subsequently screened for levels of dCas9‐3xmScarlett optimal for genomic locus visualization by live imaging. This was done by baculoviral transduction with spsgRNA2‐C1‐A.1s, as described in Hindriksen et al (2017). Forty hours after transduction, the infected clones were screened for the presence of two nuclear fluorescent foci by live cell imaging. The clones with uniform mScarlet‐I expression and optimal signal to noise ratio of the foci were selected for further experiments.
MEF siRNA transfection
siRNAs against CENP‐A and HJURP (gift from G. Almouzni) were introduced using Lipofectamine RNAiMax (Invitrogen) following manufacturer's instructions. About 6 pmol of each siRNA was co‐transfected twice at 24‐h interval. Cells were fixed 48 h after the first transfection.
Single‐cell sequencing
Single‐cell karyotype sequencing (scKaryo‐seq) was performed as described previously with some modifications (Bolhaqueiro et al, 2019). Briefly, nuclei were isolated from cells using a nuclear staining buffer (Bakker et al, 2016). We used 10 μg/ml Hoechst 34580 (Sigma‐Aldrich) to determine DNA content. The mixture of nuclear staining buffer and cells was kept on ice for 1 h before G1 cells were sorted by a BD FACSJazz in Hard‐Shell 384 wells PCR plates (Bio‐Rad) containing 5 μl of mineral oil (Sigma‐Aldrich) in each well. Plates were stored at −20°C until further processing.
Library preparation started by performing a cell lysis overnight at 50°C using 0.05 units of Qiagen Protease in 1× NEBuffer 4 (NEB) followed by heat inactivation at 75°C for 20 min and 80°C for 5 min. The genomic DNA was subsequently fragmented with 100 nl 1 U NlaIII (NEB) in 1× CutSmart (NEB) for 60 min at 37°C followed by heat inactivation at 80°C for 20 min. About 100 nl of 1 μM barcoded double‐stranded NlaIII adapters and 100 nl of 40 U T4 DNA ligase (NEB) in 1× T4 DNA ligase buffer (NEB) supplemented with 3 mM ATP (Invitrogen) was added to each well and ligation proceeded overnight at 16°C. Then, ligation samples were pooled and library preparation was performed as described previously (Muraro et al, 2016).
Libraries were sequenced 1 × 75 bp single end on an Illumina Nextseq 500. Fastq files were mapped to GRCh38 using the Burrows‐Wheeler Aligner. The mapped data were analyzed using custom scripts in Python, which parsed for library barcodes, removed reads that start without a NlaIII sequence, and removed PCR‐duplicated reads. Single‐cell sequencing data of lymphocytes harvested from two healthy donors were used as a diploid reference to determine variable bin sizes of average 8 Mb in size. Copy numbers were called by DNAcopy, and quality control was performed as described previously in Aneufinder (Bakker et al, 2016). Copy number variations were manually scored.
Image stream
FISH in suspension for ImageStream analysis was performed as previously described (Worrall et al, 2018) with the following modifications: Cells were fixed by adding freshly prepared 3:1 methanol‐glacial acetic acid dropwise to a pellet of PBS‐washed cells (1.5 × 106 cells per sample). Cells were washed two times in 4× SSC with 1% BSA, pelleted, and resuspended in 0.1% Tween20, 4× SSC. About 1.5 × 106 cells were pelleted and resuspended with 28 μl of hybridization buffer, 10 μl of H2O (nuclease‐free water), and 2 μl of centromere probe (Cytocell, UK). Denaturation and probe hybridization were performed in a thermocycler under the following conditions: 2 h at 65°C, 5 min at 80°C, and 16 h 37°C. About 200 μl 0.3% Tween20 in 4× SSC prewarmed at 73°C was added to the reaction mixture which was then incubated at 75°C for 2 min, and 200 μl of ice‐cold FBS was added to reduce the temperature. Cells were pelleted and resuspended in 100 μl of ice‐cold FBS before ImageStream analysis.
All samples were analyzed on an ImageStream®X Mark II cytometer as previously described (Worrall et al, 2018). Briefly, samples were excited with the blue laser with a power of 100 mW at a “high” flow speed and using the Extended Depth of Field (EDF) function to capture the full depth of each cell. Data obtained by the ImageStream were analyzed in IDEAS 6.2 (Merck Millipore). Raw data files were opened in the IDEAS software package and the built‐in compensation matrix was applied. Single, in‐focus, hybridized cells were then analyzed for centromere copy number using the built‐in spot‐counting wizard on the ImageStream analysis software. To correct for overlapping centromeric signals, centromere signal intensity was plotted as a histogram. Disomic cells had a medium (M) intensity of hybridization signal intensity, representing two spots. Cells with one centromere will fall below two standard deviations below the mean fluorescent intensity; cells that had gained a chromosome will fall above two standard deviations of the mean of the hybridization signal intensity. Cells designated as one spot that fell outside the 2 standard deviation windows were deemed to be true monosomies. Cells designated as 2n + 1 by the spot‐counting wizard were manually verified by visual inspection of each image and correlating it with the 2‐standard deviation cut‐off above the mean diploid fluorescence intensity.
Immunofluorescence and chromosome spreads
For chromosome spreads, cells grown to ~ 75–80% confluency on a 4‐well glass slide (Millipore) were treated with colcemid for 3 h. Growth medium was replaced by a hypotonic medium (60% growth medium, 40% ddH2O) for 5 min. After centrifugation (3 min, 800 g) in a humid chamber, cells were pre‐extracted 1 min in blocking buffer (0.2 M glycine, 2.5% FBS, 0.1% Triton X‐100 in 1× PBS) and fixed in 4% formaldehyde at room temperature for 10 min. Incubation with primary antibodies was conducted in blocking buffer for 1 h at room temperature using the following antibodies: CENP‐A (Enzo Life sciences, ADI‐KAM‐CC006‐E 1:1,000), CENP‐C (MBL, 1:1,000), CENP‐B (Abcam ab25734, 1:1,000), ACA (Antibodies Inc 15‐235‐0001, 1:500), Dsn1 (1:1,000, a gift from A. Desai, Ludwig, San Diego), Flag (Sigma F3165, 1:1,000), DM1A (alpha‐tubulin, 1:2,000), and CENP‐T (MBL, A302‐313A, 1:1,000). Immunofluorescence on chromosome spreads was done as described previously (Fachinetti et al, 2015). Immunofluorescence imaging was performed using a Deltavision Core system (Applied Precision). For IF‐FISH, we followed the IF protocol with additional post‐fixation step (2% formaldehyde, PBS 1× for 10 min) followed by the FISH protocol (see below). When applicable, alternatively, point coordinates were recorded for sequential FISH hybridizations.
Fluorescence in situ hybridization
Chromosome painting and centromere enumeration probes were purchased from MetaSystems probes and fluorescence in situ hybridization (FISH) was performed as previously described (Hoffmann et al, 2016), with the exception of the probes used to label the DNA binding site of CENP‐B (PNA Bio), the chromosome 16 alpha‐satellite (AmpliTech), and to target the common alpha‐satellite repeats of the chromosomes 1, 5, and 19 (denominated here as ar34TG, a kindly gift from C. Escudé). Manufacturer's instructions were used for the CENP‐B probe. Probe ar34TG was used at a final 0.1 μM working concentration diluted in a commercial hybridization buffer (HB1000L, AmpliTech). Detailed table of FISH probes and conditions is available in the Supplemental material (Table EV4).
Enumeration of chromosome 19 was done by combining chromosome painting and ar34TG probes. The deletion probe XL Del(5)(q31) (MetaSystems probes) was used to assess chromosome 5 aneuploidy by counting the green control signal located on 5p15 locus.
Metafer imaging platform (MetaSystems) and the metacyte spot‐counting software were used for the automatic FISH signal detection on interphase cells with additional manual validation.
Metaphase spread preparation and mFISH karyotyping
Cells grown to ~ 75–80% confluency were treated with colcemid for 3 h and prepared as described in Trott et al (2017) for the mFISH karyotyping. The Metafer imaging platform (MetaSystems) and the Isis software were used for automated acquisition of the chromosome spread and mFISH image analysis.
Sequential FISH
In order to combine mFISH karyotyping and FISH signal quantification, the first mFISH hybridization was stripped. After coverslip removal, the slide was washed in ethanol 70% for 1 min. Prewarmed denaturation solution (70% formamide, 2× SSC, pH 7.0) was applied and the slide was placed on a hotplate at 75°C for 2 min. The slide was then washed in 70% ethanol for 1 min and subsequently dehydrated in 90% and 100% ethanol for 1 min. The sample was air‐dried and hybridization with new probes was performed.
Live cell imaging
For live imaging of dCas9 tagged chromosome mis‐segregation, RPE‐1 CENP‐AEA/EA, H2B‐mTq2, and dCas9‐3xmScarlet‐I cells were transduced with lentivirus carrying sgRNA for either Chr1 or Chr3 for 24 h. Cells were transferred to a high optical quality plastic 8‐well slide (IBIDI, cat. no. 80826) and 500 μM IAA was added. After 24 h, cells were synchronized for 16 h in 6.25 μM of the Cdk1 inhibitor, RO‐3306 (Merck Millipore, Billerica, MA). Cells were subsequently washed three times and imaged on a Zeiss AIM System—Cell Observer microscope equipped with an AxioImager Z1 stand, a Hamamatsu ORCA‐flash 4.0 camera, and a Colibri 7 LED module using a 40×/1.4 oil PLAN Apochromat lens. Images were acquired every 5 min. Directly after live cell imaging, cells were fixed using 4% PFA for 8 min and subsequently permeabilized with ice‐cold methanol. Slides were incubated with anti‐Cas9 (Diagenode, Liège, Belgium, cat., mouse, monoclonal, 1:1,000) diluted in PBST (1× PBS, 0.1% Triton X‐100) with 3% BSA for 2 h, washed three times with PBST, followed by a 1‐h incubation with secondary antibody (goat anti‐mouse IgG Alexa Fluor 568 conjugate, Thermo Fisher Scientific, Waltham, 1:500) in PBST with 3% BSA. Cells were relocated using the live cell imaging positions and imaged. Images acquisition and processing was done using the Zeiss Zen 2.3 (Blue edition). Representative movies are orthogonal maximum intensity Z projections.
Live cell imaging on DLD‐1 CENP‐BmCherry CENP‐CEYFP cells was performed at 37°C on a DeltaVision microscope using Softworx software (Applied Precision) equipped with a CoolSnap HQ2 camera (Photometrics) at 1 × 1 binning using a ×60 planApo objective. Cells were incubated in CO2‐independent media (ThermoFisher) in 8‐channel μ‐slides (Ibidi) during image acquisition.
Image analysis
Quantification of centromere signal intensity on metaphase cells was done manually as described previously (Fachinetti et al, 2013). Deconvolved 2D maximum intensity projections were saved as un‐scaled 16‐bit TIFF images and signal intensities determined using MetaMorph (Molecular Devices). A 25 × 25 pixel circle was drawn around the two sister centromeres and an identical circle drawn adjacent to the structure to determine the background. The integrated signal intensity of each individual centromere was calculated by subtracting the fluorescence intensity of the background from the intensity of the adjacent centromere. Twenty‐five centromeres were averaged to provide the average fluorescence intensity for each individual cell and the average intensity of five identical circles drawn on the background was subtracted. Quantification of the CENP‐B boxes intensity in MEF was done following the same method with some modifications. A 20 × 20 pixel circle was drawn around ten high CENP‐B box signal intensity spots and ten around low CENP‐B box signal intensity spots in the main nucleus. Five adjacent circles were drawn to subtract the background. A similar circle was drawn around the CENP‐B box signal contained into the micronucleus and one circle was drawn adjacent to the structure (background). Each individual signal was normalized to the average fluorescence intensity quantified in the main nucleus.
For DLD‐1 CENP‐BmCherry CENP‐CEYFP cells, fluorescence intensities were measured separately by first subtracting nucleoplasmic background in individual slices using an automated approach (available upon request). Background subtracted stacks were then projected using a maximum intensity projection, and centromeres were segmented using a probabilistic segmentation approach on CENP‐BmCherry spots (preprint: Boudreau et al, 2019). Segmented centromeres were then used to measure fluorescence raw integrated densities of both CENP‐BmCherry and CENP‐CEYFP spots. All image analyses were performed using Fiji (Schindelin et al, 2012).
Immunoblotting
For immunoblot analysis, protein samples were separated by SDS–PAGE, transferred onto PVDF membranes (GE Healthcare, Amersham), and then probed with the following antibodies: CENP‐B (Abcam ab25734, 1:1,000) and Flag (Sigma F3165, 1:1,000).
CUT&RUN and next‐generation sequencing
CUT&RUN was performed according to the procedure reported by Skene & Henikoff (2017) starting from 1 million cells and using anti‐CENP‐A (Ozyme, 2186S), ‐CENP‐B (Abcam, ab25734), ‐CENP‐T (MBL, A302‐313A), or ‐CENP‐C (Abcam, ab33034) antibodies. Rabbit IgG isotype control antibodies (ThermoFisher, 10500C) were used for background detection.
Illumina sequencing library was prepared using the Illumina TruSeq ChIP Library Preparation Kit according to the manufacturer's instructions. Sequencing was performed with an Illumina Hi‐seq 2500 system.
Bioinformatic analysis
For mapping reads to HOR arrays, after barcodes removal, reads were merged using the PEAR software (v 0.9.11) (Zhang et al, 2014) using default parameters. Reads were mapped using the bwa‐mem algorithm of the BWA software package (preprint: Li, 2013; Li & Durbin, 2009) on the human reference genome GRCh38.p12, which includes the reference models for the α‐satellite DNA sequences of the X and Y chromosome (Miga et al, 2014) as well as the reference models of the centromeres of the 22 autosomes (Nechemia‐Arbely et al, 2017; Schneider et al, 2017). These centromere models include the observed variation in centromeric arrays of higher order repeat (HOR) sequences contained in the HuRef genome (Levy et al, 2007), and are deposited under the accession numbers listed in Table EV5. The reads mapped on the centromere reference models were extracted using samtools (Li et al, 2009) (according to the centromeric coordinate reported in Table EV5) and remapped with bwa‐mem on a reference composed of 64 centromeric HOR array consensus sequences [in which each array is repeated twice in tandem (see Table EV1)] (Nechemia‐Arbely et al, 2019). Multi‐mapping reads were removed by maintaining only the alignment with the lowest edit distance, corresponding to the NM tag of the bam alignment file. Read counts per each HOR array are reported in Table EV2. When read mapping on a HOR is unambiguous, read distribution along the HOR array consensus is homogeneous, leading to an approximately flat profile (see as example the cen8 HOR array coverage plot in Fig EV2G, top right panel). As previously reported, a portion of the cen16_1 HOR consensus (corresponding to D16Z2) has a high similarity to the cen1_1 HOR consensus (corresponding to D1Z7/D5Z2/D19Z3), leading to mis‐mapping of reads originating from the cen1_1 onto the cen16_1 reference. This is proven by the very heterogeneous distribution of reads on the cen16_1, where the presence of two clear peaks on the cen16_1 consensus is indicative of mis‐mapping (see Fig EV2G, bottom right panel). As expected, the sequence underlying these peaks corresponds to the region of high similarity between cen1_1 and cen16_1. To correct this misalignment, the average read count in the out‐of‐peak region of cen16_1 was calculated and subtracted from the in‐peak read count. The resulting value was assigned to the total cen1_1 and removed from the total cen16_1 read count. Given the known similarity between the cen2 and cen18_1 HOR (corresponding to D2Z1 and D18Z1), a more detailed analysis was performed to correct possible alignment inaccuracies. We used the software jellyfish [version 2.2.10; (Marçais & Kingsford, 2011)] to identify all the 18‐, 24‐, and 40‐mers that are specific to the cen2 and cen18_1 consensus sequences. Then, we performed the same k‐mer analysis on the pool of reads mapping on the cen2 or cen18_1 and calculated the proportion between cen2‐ and cen18_1‐specific reads. This ratio was used to distribute all the reads between the two HOR consensus references. Overall, this correction led to a ~10% decrease of read counts on cen18_1 and a ~30% increase on cen2. The subsequent correction steps are recapitulated in Table EV3 and in Fig EV2D, F, G and H.
To convert read counts into megabases, a standard curve was produced by counting the read mapping on several single‐copy genomic regions of known size (Fig EV2E). To avoid potential GC bias, the GC content of these single‐copy regions was tested to be very similar to that of the centromere reference models; similarly, the GC content of different HOR consensus sequences was shown not to be extremely variable, thus decreasing the risk of GC content bias affecting our analysis (Fig EV2B and C). The occurrences of the CENP‐B box minimal sequence (TTCGNNNNANNCGGG) and its reverse complement were counted on the reads mapping on each HOR array. CENP‐A, CENP‐B, CENP‐C, and CENP‐T levels are derived from the read counts of the respective CUT&RUN‐seq. In order to make the replicates comparable, CENP‐B boxes and read counts of CENP‐A, ‐B, ‐C, ‐T, and CUT&RUN‐seq were normalized to the total number of mapped reads. The values from HORs located on the same chromosomes were summed to give a single value per each chromosome, except for the cen1_1 HOR. A quantitative FISH approach was used to precisely assign the centromere length and CENP‐B box values of the cen1_1 HOR to chr1, 5, and 19 (Fig EV2I). Since we have proven a correlation between length, amount of CENP‐B boxes, and amounts of CENP‐B and CENP‐C, we distributed the reads from CUT&RUN‐seq according to the same proportion among chr1, 5, and 19.
Statistical analysis
To estimate chromosome mis‐segregation rates for Image stream, FISH and single‐cell data, a logistic model was fitted to assess the proportion of events (gains, losses, or gains and losses pooled) by chromosome. For each estimated proportion, 95% confidence interval was computed and the difference to the mean value was assessed by a Wald test with a chi‐squared statistic.
For single‐cell data, a multinomial model was fitted considering only the cells with at least one mis‐segregation event to estimate the proportion for each chromosome. The fitted proportions were tested against the mean value with a binomial test. The expected frequency of mis‐segregation per each chromosome is 4.3% only when we consider cells with at least one mis‐segregation event. We chose to calculate the frequency as a percentage of all the analyzed cells, including the ones not showing any mis‐segregation, therefore the mean value can vary. All analyses were performed with R software version 3.5.1. For all statistical results, see the statistical dataset info.
Author contributions
PG performed statistical analysis of all the three methods used to detect aneuploidy. SK performed and analyzed single‐cell sequencing and he was supervised by GJPLK; JTW executed and analyzed all the image stream experiments under the supervision of SEM; SGDV performed live cell imaging on dCas9‐targeted chromosomes under the supervision of SMAL; VB acquired and analyzed live centromere quantification of CENP‐B/C double‐tagged cell lines and he was supervised by PSM; KHM gave key input to analyze centromere sequences using her unpublished reference model. CS‐L performed and analyzed certain experiments. MD performed and analyzed all FISH, mFISH, and IF experiments. RG implemented centromere read mapping and performed chromatin–protein interaction experiments and analysis. DF, MD, and RG made figures, wrote the manuscript, and all authors contributed to manuscript editing. DF conceived the experimental design.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Movie EV1
Movie EV2
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Acknowledgements
The authors would like to thank: (from I. Curie, Paris) Cristina Bartocci, Oumou Goundiam, Sara Rivas Lopez, Amandine Grelier, and the Fachinetti, Basto and Drinnenberg team members for helpful suggestions and technical help. We would also like to thank L. Tovini (BCI, UK) for technical help; Christophe Escudé (Museum National d'Histoire Naturelle), Genevieve Almouzni (I. Curie), Peter Ly and Don Cleveland (UCSD, San Diego), Steve Henikoff (Fred Hutchinson Cancer Research Center), Arshad Desai (UCSD, San Diego), Mike Lampson (UPENN, US) for providing reagents; and Heidi Lane and Felix Bachmann (Basilea Pharmaceutica International, Switzerland) for providing the BAL27862 compound. We also thank the flow cytometry platform, the cell and tissue imaging facility (PICT‐IBiSA, member of the French National Research Infrastructure France‐BioImaging ANR10‐INBS‐04), and the sequencing platform at Institut Curie. High‐throughput sequencing has been performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR‐10‐EQPX‐03 (Equipex) and ANR‐10‐INBS‐09‐08 (France Génomique Consortium) from the Agence Nationale de la Recherche (“Investissements d'Avenir” program), by the Canceropole Ile‐de‐France and by the SiRIC‐Curie program—SiRIC Grant « INCa‐DGOS‐ 4654. D.F. receives salary support from the CNRS. D.F. has received support for this project from Labex « CelTisPhyBio », the Institut Curie, the ATIP‐Avenir 2015 program, the program «Investissements d'Avenir» launched by the French Government and implemented by ANR with the references ANR‐10‐LABX‐0038 and ANR‐10‐IDEX‐0001‐02 PSL. D.F. is also supported by the City of Paris, Emergence(s) 2018, an annual call for proposals that aim to help young researchers. R.G. was supported by AIRC post‐doctoral fellowship for abroad research. P.S.M. is supported by National Science Foundation CAREER Award 1652512 and is William Burwell Harrison Fellow. S.M.A.L. is a member of Oncode Institute which is partly financed by the Dutch Cancer Society.
The EMBO Journal (2020) 39: e102924
Data availability
Whole‐genome and CUT&RUN sequencing data have been deposited at GEO (accession #GSE132193; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132193).
References
- Akera T, Trimm E, Lampson MA (2019) Molecular strategies of meiotic cheating by selfish centromeres. Cell 178: 1132–1144.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrup‐MacDonald ME, Kuo ME, Sullivan LL, Chew K, Sullivan BA (2016) Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles. Genome Res 26: 1301–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov I, Kazakov A, Tumeneva I, Shepelev V, Yurov Y (2001) Alpha‐satellite DNA of primates: old and new families. Chromosoma 110: 253–266 [DOI] [PubMed] [Google Scholar]
- Bakker B, Taudt A, Belderbos ME, Porubsky D, Spierings DCJ, de Jong TV, Halsema N, Kazemier HG, Hoekstra‐Wakker K, Bradley A et al (2016) Single‐cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol 17: 115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balajee AS, Bertucci A, Taveras M, Brenner DJ (2014) Multicolour FISH analysis of ionising radiation induced micronucleus formation in human lymphocytes. Mutagenesis 29: 447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger I, Fitzgerald DJ, Richmond TJ (2004) Baculovirus expression system for heterologous multiprotein complexes. Nat Biotechnol 22: 1583–1587 [DOI] [PubMed] [Google Scholar]
- Bochtler T, Kartal‐Kaess M, Granzow M, Hielscher T, Cosenza MR, Herold‐Mende C, Jauch A, Krämer A (2018) Micronucleus formation in human cancer cells is biased by chromosome size. Genes Chromosom Cancer 34: 369 [DOI] [PubMed] [Google Scholar]
- Bolhaqueiro ACF, Ponsioen B, Bakker B, Klaasen SJ, Kucukkose E, van Jaarsveld RH, Vivié J, Verlaan‐Klink I, Hami N, Spierings DCJ et al (2019) Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat Genet 51: 824–834 [DOI] [PubMed] [Google Scholar]
- Boudreau V, Hazel J, Sellinger J, Chen P, Manakova K, Radzyminski R, Garcia H, Allard J, Gatlin J, Maddox P (2019) Nucleo‐cytoplasmic trafficking regulates nuclear surface area during nuclear organogenesis. bioRxiv 10.1101/326140 [PREPRINT] [DOI] [Google Scholar]
- Caria HL, Chaveca T, Rueff J (1996) Preferential sensitivity of acrocentric chromosomes to the aneugenic effect of colchicine. Teratog Carcinog Mutagen 16: 243–252 [DOI] [PubMed] [Google Scholar]
- Chmátal L, Gabriel SI, Mitsainas GP, Martínez‐Vargas J, Ventura J, Searle JB, Schultz RM, Lampson MA (2014) Centromere strength provides the cell biological basis for meiotic drive and karyotype evolution in mice. Curr Biol 24: 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmátal L, Schultz RM, Black BE, Lampson MA (2017) Cell biology of cheating‐transmission of centromeres and other selfish elements through asymmetric meiosis. Prog Mol Subcell Biol 56: 377–396 [DOI] [PubMed] [Google Scholar]
- Contreras‐Galindo R, Fischer S, Saha AK, Lundy JD, Cervantes PW, Mourad M, Wang C, Qian B, Dai M, Meng F et al (2017) Rapid molecular assays to study human centromere genomics. Genome Res 27: 2040–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D (2012) DNA breaks and chromosome pulverization from errors in mitosis. Nature 482: 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drpic D, Almeida A, Aguiar P, Maiato H (2017) Chromosome (mis)segregation is biased by kinetochore size. Curr Biol 28: 1344–1356.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudka D, Noatynska A, Smith CA, Liaudet N, McAinsh AD, Meraldi P (2018) Complete microtubule‐kinetochore occupancy favours the segregation of merotelic attachments. Nat Commun 9: 2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duijf P, Schultz N, Benezra R (2013) Cancer cells preferentially lose small chromosomes. Int J Cancer 132: 2316–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanski JP, Rasi C, Lönn M, Davies H, Ingelsson M, Giedraitis V, Lannfelt L, Magnusson PKE, Lindgren CM, Morris AP et al (2015) Mutagenesis. Smoking is associated with mosaic loss of chromosome Y. Science 347: 81–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont M, Fachinetti D (2017) DNA sequences in centromere formation and function. Prog Mol Subcell Biol 56: 305–336 [DOI] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray‐Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni‐Pettinotti G (2009) HJURP is a cell‐cycle‐dependent maintenance and deposition factor of CENP‐A at centromeres. Cell 137: 485–497 [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Sullivan KF, Machlin PS, Cooke CA, Kaiser DA, Pollard TD, Rothfield NF, Cleveland DW (1987) Molecular cloning of cDNA for CENP‐B, the major human centromere autoantigen. J Cell Biol 104: 817–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Ratrie H, Stetten G (1989) Visualization of centromere proteins CENP‐B and CENP‐C on a stable dicentric chromosome in cytological spreads. Chromosoma 98: 1–12 [DOI] [PubMed] [Google Scholar]
- Fachinetti D, Folco HD, Nechemia‐Arbely Y, Valente LP, Nguyen K, Wong AJ, Zhu Q, Holland AJ, Desai A, Jansen LET et al (2013) A two‐step mechanism for epigenetic specification of centromere identity and function. Nat Cell Biol 15: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, Cleveland DW (2015) DNA sequence‐specific binding of CENP‐B enhances the fidelity of human centromere function. Dev Cell 33: 314–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauth E, Scherthan H (1998) Frequencies of occurence of all human chromosomes in micronuclei from normal and 5‐azacytidine‐treated lymphocytes as revealed by chromosome painting. Mutagenesis 13: 235–241 [DOI] [PubMed] [Google Scholar]
- Ferguson‐Smith MA, Handmaker SD (1961) Observations on the satellited human chromosomes. Lancet 1: 638–640 [DOI] [PubMed] [Google Scholar]
- Foltz DR, Jansen LET, Black BE, Bailey AO, Yates JR, Cleveland DW (2006) The human CENP‐A centromeric nucleosome‐associated complex. Nat Cell Biol 8: 458–469 [DOI] [PubMed] [Google Scholar]
- Foltz DR, Jansen LET, Bailey AO, Yates JR III, Bassett EA, Wood S, Black BE, Cleveland DW (2009) Centromere‐specific assembly of CENP‐A nucleosomes is mediated by HJURP. Cell 137: 472–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA, Rasi C, Malmqvist N, Davies H, Pasupulati S, Pakalapati G, Sandgren J, Diaz de Ståhl T, Zaghlool A, Giedraitis V et al (2014) Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat Genet 46: 624–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA (2017) Loss of chromosome Y (LOY) in blood cells is associated with increased risk for disease and mortality in aging men. Hum Genet 136: 657–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukagawa T, Pendon C, Morris J, Brown W (1999) CENP‐C is necessary but not sufficient to induce formation of a functional centromere. EMBO J 18: 4196–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukagawa T, Earnshaw WC (2014) The centromere: chromatin foundation for the kinetochore machinery. Dev Cell 30: 496–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané‐Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H et al (2019) Next‐generation characterization of the cancer cell line encyclopedia. Nature 569: 503–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DJ, Resio B, Pellman D (2012) Causes and consequences of aneuploidy in cancer. Nat Rev Genet 13: 189–203 [DOI] [PubMed] [Google Scholar]
- Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF (2011) In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 477: 354–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Malik HS (2002) Centromeres: selfish drivers. Nature 417: 227 [DOI] [PubMed] [Google Scholar]
- Henikoff JG, Thakur J, Kasinathan S, Henikoff S (2015) A unique chromatin complex occupies young ‐satellite arrays of human centromeres. Sci Adv 1: e1400234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindriksen S, Bramer AJ, Truong MA, Vromans MJM, Post JB, Verlaan‐Klink I, Snippert HJ, Lens SMA, Hadders MA (2017) Baculoviral delivery of CRISPR/Cas9 facilitates efficient genome editing in human cells. PLoS One 12: e0179514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Dumont M, Barra V, Ly P, Nechemia‐Arbely Y, McMahon MA, Hervé S, Cleveland DW, Fachinetti D (2016) CENP‐A is dispensable for mitotic centromere function after initial centromere/kinetochore assembly. Cell Rep 17: 2394–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huis in ‘t Veld PJ, Jeganathan S, Petrovic A, Singh P, John J, Krenn V, Weissmann F, Bange T, Musacchio A (2016) Molecular basis of outer kinetochore assembly on CENP‐T. Elife 5: 576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine DV, Amor DJ, Perry J, Sirvent N, Pedeutour F, Choo KHA, Saffery R (2004) Chromosome size and origin as determinants of the level of CENP‐A incorporation into human centromeres. Chromosome Res 12: 805–815 [DOI] [PubMed] [Google Scholar]
- Iwata‐Otsubo A, Dawicki‐McKenna JM, Akera T, Falk SJ, Chmátal L, Yang K, Sullivan BA, Schultz RM, Lampson MA, Black BE (2017) Expanded satellite repeats amplify a discrete CENP‐A nucleosome assembly site on chromosomes that drive in female meiosis. Curr Biol 27: 2365–2373.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, Lampson MA, Hergert P, Cameron L, Cimini D, Salmon ED, McEwen BF, Khodjakov A (2006) Chromosomes can congress to the metaphase plate before biorientation. Science 311: 388–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima TS, Sakuno T, Ishiguro K‐I, Iemura S‐I, Natsume T, Kawashima SA, Watanabe Y (2006) Shugoshin collaborates with protein phosphatase 2A to protect cohesin. Nature 441: 46–52 [DOI] [PubMed] [Google Scholar]
- Lampson MA, Black BE (2017) Cellular and molecular mechanisms of centromere drive. Cold Spring Harb Symp Quant Biol 82: 249–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S‐H, Itkin‐Ansari P, Levine F (2010) CENP‐A, a protein required for chromosome segregation in mitosis, declines with age in islet but not exocrine cells. Aging 2: 785–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy HP, Schultz RA, Ordonez JV, Cohen MM (1993) DNA content measurements and an improved idiogram for the Indian muntjac. Cytometry 14: 362–368 [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G et al (2007) The diploid genome sequence of an individual human. PLoS Biol 5: e254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv 1303.3997v2 [PREPRINT] [Google Scholar]
- Loh P‐R, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, Birmann BM, Talkowski ME, Bakhoum SF, McCarroll SA et al (2018) Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559: 350–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loupart ML, Adams S, Armour JA, Walker R, Brammar W, Varley J (1995) Loss of heterozygosity on the X chromosome in human breast cancer. Genes Chromosom Cancer 13: 229–238 [DOI] [PubMed] [Google Scholar]
- Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, Cleveland DW (2017) Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non‐homologous end joining. Nat Cell Biol 19: 68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Naseri A, Reyes‐Gutierrez P, Wolfe SA, Zhang S, Pederson T (2015) Multicolor CRISPR labeling of chromosomal loci in human cells. Proc Natl Acad Sci USA 12: 3002–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Tu L‐C, Naseri A, Huisman M, Zhang S, Grunwald D, Pederson T (2016) Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol 34: 528–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Kingsford C (2011) A fast, lock‐free approach for efficient parallel counting of occurrences of k‐mers. Bioinformatics 27: 764–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor M, Hariharan N, Joyo A, Margolis RL, Sussman M (2013) CENP‐A is essential for cardiac progenitor cell proliferation. Cell Cycle 13: 739–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga KH, Newton Y, Jain M, Altemose N, Willard HF, Kent WJ (2014) Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res 24: 697–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga KH (2017) The promises and challenges of genomic studies of human centromeres. Prog Mol Subcell Biol 56: 285–304 [DOI] [PubMed] [Google Scholar]
- Muraro MJ, Dharmadhikari G, Grün D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJP et al (2016) A single‐cell transcriptome atlas of the human pancreas. Cell Syst 3: 385–394.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro Y, Masumoto H, Yoda K, Nozaki N, Ohashi M, Okazaki T (1992) Centromere protein B assembles human centromeric alpha‐satellite DNA at the 17‐bp sequence, CENP‐B box. J Cell Biol 116: 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka SI, Hassold TJ, Hunt PA (2012) Human aneuploidy: mechanisms and new insights into an age‐old problem. Nat Rev Genet 13: 493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechemia‐Arbely Y, Fachinetti D, Miga KH, Sekulic N, Soni GV, Kim DH, Wong AK, Lee AY, Nguyen K, Dekker C et al (2017) Human centromeric CENP‐A chromatin is a homotypic, octameric nucleosome at all cell cycle points. J Cell Biol 216: 607–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechemia‐Arbely Y, Miga KH, Shoshani O, Aslanian A, McMahon MA, Lee AY, Fachinetti D, Yates JR, Ren B, Cleveland DW (2019) DNA replication acts as an error correction mechanism to maintain centromere identity by restricting CENP‐A to centromeres. Nat Cell Biol 21: 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezer Z, Ugarković D (2008) Role of non‐coding RNA and heterochromatin in aneuploidy and cancer. Semin Cancer Biol 18: 123–130 [DOI] [PubMed] [Google Scholar]
- Potapova TA, Unruh JR, Yu Z, Rancati G, Li H, Stampfer MR, Gerton JL (2019) Superresolution microscopy reveals linkages between ribosomal DNA on heterologous chromosomes. J Cell Biol 218: 2492–2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jöckel KH, Becher R (1996) Prognostic implications of monosomy 3 in uveal melanoma. Lancet 347: 1222–1225 [DOI] [PubMed] [Google Scholar]
- Rudd MK, Willard HF (2004) Analysis of the centromeric regions of the human genome assembly. Trends Genet 20: 529–533 [DOI] [PubMed] [Google Scholar]
- Santaguida S, Amon A (2015) Short‐ and long‐term effects of chromosome mis‐segregation and aneuploidy. Nat Rev Mol Cell Biol 16: 473–485 [DOI] [PubMed] [Google Scholar]
- Scelfo A, Fachinetti D (2019) Keeping the centromere under control: a promising role for DNA methylation. Cells 8: 912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider VA, Graves‐Lindsay T, Howe K, Bouk N, Chen H‐C, Kitts PA, Murphy TD, Pruitt KD, Thibaud‐Nissen F, Albracht D et al (2017) Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27: 849–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholes AG, Liloglou T, Maloney P, Hagan S, Nunn J, Hiscott P, Damato BE, Grierson I, Field JK (2001) Loss of heterozygosity on chromosomes 3, 9, 13, and 17, including the retinoblastoma locus, in uveal melanoma. Invest Ophthalmol Vis Sci 42: 2472–2477 [PubMed] [Google Scholar]
- Sirchia SM, Ramoscelli L, Grati FR, Barbera F, Coradini D, Rossella F, Porta G, Lesma E, Ruggeri A, Radice P et al (2005) Loss of the inactive X chromosome and replication of the active X in BRCA1‐defective and wild‐type breast cancer cells. Cancer Res 65: 2139–2146 [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S (2015) A simple method for generating high‐resolution maps of genome‐wide protein binding. Elife 4: e09225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S (2017) An efficient targeted nuclease strategy for high‐resolution mapping of DNA binding sites. Elife 6: 576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smurova K, De Wulf P (2018) Centromere and pericentromere transcription: roles and regulation … in sickness and in health. Front Genet 9: 674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LL, Boivin CD, Mravinac B, Song IY, Sullivan BA (2011) Genomic size of CENP‐A domain is proportional to total alpha satellite array size at human centromeres and expands in cancer cells. Chromosome Res 19: 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LL, Chew K, Sullivan BA (2017) α satellite DNA variation and function of the human centromere. Nucleus 8: 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kurumizaka H, Yokoyama S (2005) CpG methylation of the CENP‐B box reduces human CENP‐B binding. FEBS J 272: 282–289 [DOI] [PubMed] [Google Scholar]
- Tovini L, McClelland SE (2019) Impaired CENP‐E function renders large chromosomes more vulnerable to congression failure. Biomolecules 9: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott J, Tan EK, Ong S, Titmarsh DM, Denil SLIJ, Giam M, Wong CK, Wang J, Shboul M, Eio M et al (2017) Long‐term culture of self‐renewing pancreatic progenitors derived from human pluripotent stem cells. Stem Cell Rep 8: 1675–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir JR, Faesen AC, Klare K, Petrovic A, Basilico F, Fischböck J, Pentakota S, Keller J, Pesenti ME, Pan D et al (2016) Insights from biochemical reconstitution into the architecture of human kinetochores. Nature 537: 249–253 [DOI] [PubMed] [Google Scholar]
- Wendell KL, Wilson L, Jordan MA (1993) Mitotic block in HeLa cells by vinblastine: ultrastructural changes in kinetochore‐microtubule attachment and in centrosomes. J Cell Sci 104(Pt 2): 261–274 [DOI] [PubMed] [Google Scholar]
- Worrall JT, Tamura N, Mazzagatti A, Shaikh N, van Lingen T, Bakker B, Spierings DCJ, Vladimirou E, Foijer F, McClelland SE (2018) Non‐random Mis‐segregation of human chromosomes. Cell Rep 23: 3366–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kobert K, Flouri T, Stamatakis A (2014) PEAR: a fast and accurate Illumina Paired‐End reAd mergeR. Bioinformatics 30: 614–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Movie EV1
Movie EV2
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Data Availability Statement
Whole‐genome and CUT&RUN sequencing data have been deposited at GEO (accession #GSE132193; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132193).