

Abstract
Developmental cell fate specification is a unidirectional process that can be reverted in response to injury or experimental reprogramming. Whether differentiation and de‐differentiation trajectories intersect mechanistically is unclear. Here, we performed comparative screening in lineage‐related mouse naïve embryonic stem cells (ESCs) and primed epiblast stem cells (EpiSCs), and identified the constitutively expressed zinc finger transcription factor (TF) Zfp281 as a bidirectional regulator of cell state interconversion. We showed that subtle chromatin binding changes in differentiated cells translate into activation of the histone H3 lysine 9 (H3K9) methyltransferase Ehmt1 and stabilization of the zinc finger TF Zic2 at enhancers and promoters. Genetic gain‐of‐function and loss‐of‐function experiments confirmed a critical role of Ehmt1 and Zic2 downstream of Zfp281 both in driving exit from the ESC state and in restricting reprogramming of EpiSCs. Our study reveals that cell type‐invariant chromatin association of Zfp281 provides an interaction platform for remodeling the cis‐regulatory network underlying cellular plasticity.
Keywords: cell state transition, cellular plasticity, differentiation, pluripotency, reprogramming
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Regenerative Medicine; Transcription
Cell type‐invariant chromatin association of zinc finger transcription factor Zfp281 facilitates bidirectional regulation of mouse naïve pluripotency.
Introduction
Mammalian development is a hierarchical process that coordinates organismal growth with increasing cellular differentiation. The lineage progression of the few pluripotent cells in the blastocyst toward the many specialized cell types in the mature embryo is by and large unidirectional. However, fully differentiated cells can be de‐differentiated into induced pluripotent stem cells (iPSCs) by ectopic expression of the transcription factors (TFs) Oct4, Sox2, Klf4, and c‐Myc (Takahashi & Yamanaka, 2006). Reprogramming of somatic cells into iPSCs requires erasure of the entire developmental history of a somatic cell, but whether this depends on the reversal of developmental hierarchies is unclear (Ladewig et al, 2013; Takahashi & Yamanaka, 2015).
Transcriptional and epigenomic profiling of the reprogramming process has revealed an ordered series of events that include the transient and sequential activation of late and early developmental genes (Takahashi et al, 2014; Cacchiarelli et al, 2015; Amlani et al, 2018). Although the specific trajectory is dictated by the identity of the starting somatic cell type (Jackson et al, 2016; Nefzger et al, 2017) and the experimental regime (Chantzoura et al, 2015; Stuart et al, 2019), iPSC formation may involve the reversion of natural developmental mechanisms (Takahashi & Yamanaka, 2015). Consistent with this possibility, a mesenchymal‐to‐epithelial transition is necessary for iPSC formation (Li et al, 2010; Samavarchi‐Tehrani et al, 2010), while the converse epithelial‐to‐mesenchymal transition is crucial for embryogenesis, e.g., during gastrulation and neural crest formation (Acloque et al, 2009). Although it is debated whether these observations reflect a shared developmental intermediate (Raab et al, 2017), they suggest that de‐differentiation and differentiation employ common mechanisms in opposite directions. Here, we systematically and functionally examine this concept using naïve pluripotent embryonic stem cells (ESCs) and primed pluripotent epiblast stem cells (EpiSCs; Smith, 2017).
Embryonic stem cells and EpiSCs are developmentally related derivatives of mouse embryonic day (E) 3.75–4.5 blastocysts (Boroviak et al, 2014) and E5.5–8.0 embryos (Brons et al, 2007; Tesar et al, 2007; Osorno et al, 2012), respectively. ESCs cultured in the presence of two inhibitors (2i) resemble naïve pluripotent cells of the preimplantation epiblast (Boroviak et al, 2014), while primed pluripotent EpiSCs cultured in the presence of FGF2 and activin A (FA) resemble cells of the late gastrula (Osorno et al, 2012; Kojima et al, 2014; Tsakiridis et al, 2014). Upon in vitro differentiation, ESCs progress through a transient post‐implantation epiblast‐like (EpiLC) cell state that is amenable to EpiSC derivation (Zhang et al, 2010; Hayashi et al, 2011). Conversely, activation of just one TF, such as Stat3, Klf4, or Esrrb, is sufficient to reprogram EpiSCs into naïve pluripotent EpiSC‐derived iPSCs (Epi‐iPSCs) in the presence of 2i (Guo et al, 2009; Yang et al, 2010; Festuccia et al, 2012). The interconvertibility of ESCs and EpiSCs thus provides an experimental system to explore whether de‐differentiation includes the reversion of differentiation mechanisms.
Using a large‐scale loss‐of‐function reprogramming screen in sensitized EpiSCs, we identify the zinc finger TF Zfp281 as a prominent bidirectional ESC‐EpiSC transition regulator. We show that Zfp281 exhibits stable chromatin association and drives ESC progression through differentiation‐specific interaction with Ehmt1 and Zic2. Genomic analysis revealed activation of Ehmt1 and enrichment of Zic2 at Zfp281‐bound cis‐regulatory elements (CREs) that are associated with developmental transcription in EpiLCs and EpiSCs. Zfp281 therefore establishes and stabilizes cell fate commitment to safeguard the unidirectionality of pluripotent state transitions.
Results
Zfp281 is a bidirectional ESC‐EpiSC transition regulator
We hypothesized that mechanisms common to differentiation and de‐differentiation may be encoded in genes that both promote exit from the naïve ESC state and impair reprogramming of EpiSCs. ESC differentiation drivers have been determined in several genetic loss‐of‐function screens (Guo et al, 2011; Westerman et al, 2011; Betschinger et al, 2013; Leeb et al, 2014; Li et al, 2018), but it is unknown whether those also inhibit reprogramming of EpiSCs into naïve pluripotency. We therefore set out to systematically identify reprogramming roadblocks using a large‐scale endoribonuclease‐prepared small interfering RNA (esiRNA) loss‐of‐function screen (Ding et al, 2009). We made use of O4GIPGY118F EpiSCs expressing green fluorescent protein (GFP) and puromycin N‐acetyltransferase under the regulatory sequences of the Oct4 gene (Guo et al, 2009), and a Stat3‐activating receptor (GY118F) responsive to granulocyte colony‐stimulating factor (Gcsf) driven by a constitutive promoter (Yang et al, 2010). Upon exposure to Gcsf and 2i for 4 days (d), O4GIPGY118F EpiSCs gave rise to self‐renewing Epi‐iPSCs at an efficiency of roughly 0.1% (Fig EV1A), thus providing a sensitized setup to identify reprogramming inhibitors. O4GIPGY118F EpiSCs were transfected with esiRNAs targeting 9,540 transcripts and control esiRNAs targeting Luciferase (Luc) and the GY118F downstream effector Stat3 in 384‐well plates (Fig 1A). The next day, reprogramming was induced by changing to 2i and Gcsf. After 4 days, we selected Epi‐iPSCs in the presence of puromycin and quantified viability with a fluorescent assay after 3–4 days. The screen was performed in duplicate, and Z scores were calculated per plate (Table EV1). Positive (Stat3 esiRNA), but not negative (non‐targeting Luc esiRNA and no esiRNA), controls induced negative Z scores (Fig EV1B). Screen hits with average Z scores < −2 included ribosome and proteasome subunits, Stat3 and Oct4 (Fig 1B), and were strongly enriched for functions associated with RNA maturation and translation using gene ontology (GO) analysis (Fig EV1C). These therefore contain genes required for reprogramming and/or cell survival. Screen hits with positive Z scores, conversely, are expected to inhibit reprogramming and/or proliferation. Among the 146 hits with an average Z score > 2, the zinc finger TF Zfp281 and the E3 ubiquitin ligase Fbxw7 scored highest. Zfp281 and Fbxw7 have previously been shown to restrict iPSC generation from somatic cells (Buckley et al, 2012; Fidalgo et al, 2012, 2016; Okita et al, 2012), thus suggesting successful identification of reprogramming roadblocks.
Figure EV1. Enhanced reprogramming of EpiSCs in the absence of Zfp281.
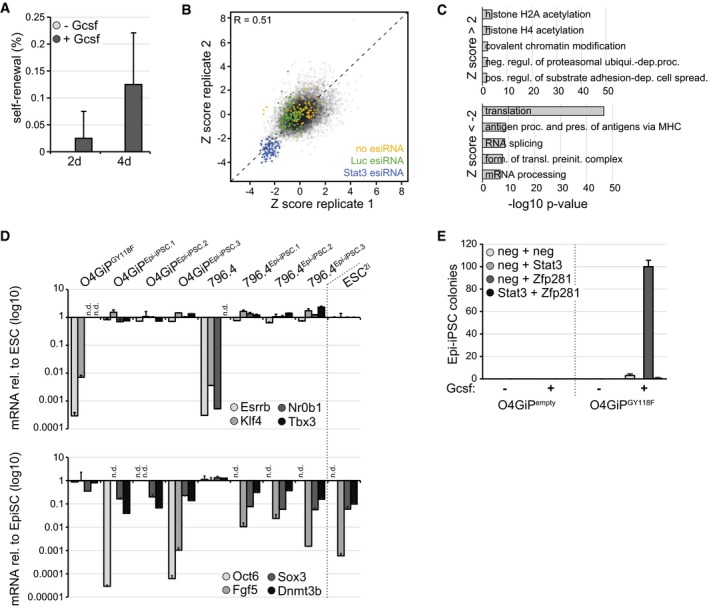
- Self‐renewal of O4GIPGY118F reprogramming intermediates after 2 or 4 days in 2i in the presence or absence of Gcsf. Average and SD of 2 experiments performed in duplicates.
- Scatter plot of Z scores between screen replicates. Negative controls (no esiRNA and non‐targeting Luc esiRNA) are marked in yellow and green, respectively, and positive controls (Stat3 esiRNA) in blue. Pearson's correlation coefficient (R).
- Top 5 GO terms enriched in screen hits with Z scores > 2 (top) and < −2 (bottom).
- Induction of naïve (top) and repression of primed (bottom) pluripotency markers in Epi‐iPSCs derived from Zfp281‐depleted and Gcsf‐stimulated O4GIPGY118F and 796.4 EpiSCs. mRNA fold changes relative to ESCs (top) and EpiSCs (bottom) are shown on a log(10)‐scaled axis. Average and SD of two technical replicates. Not detected (n.d.).
- Epi‐iPSC colonies derived from O4GIPempty and O4GiPGY118F EpiSCs transfected with indicated siRNAs, incubated for 4 days in 2i in the presence or absence of Gcsf, and selected with puromycin. Average and SD of two technical replicates.
Figure 1. Zfp281 inhibits reprogramming of EpiSCs.
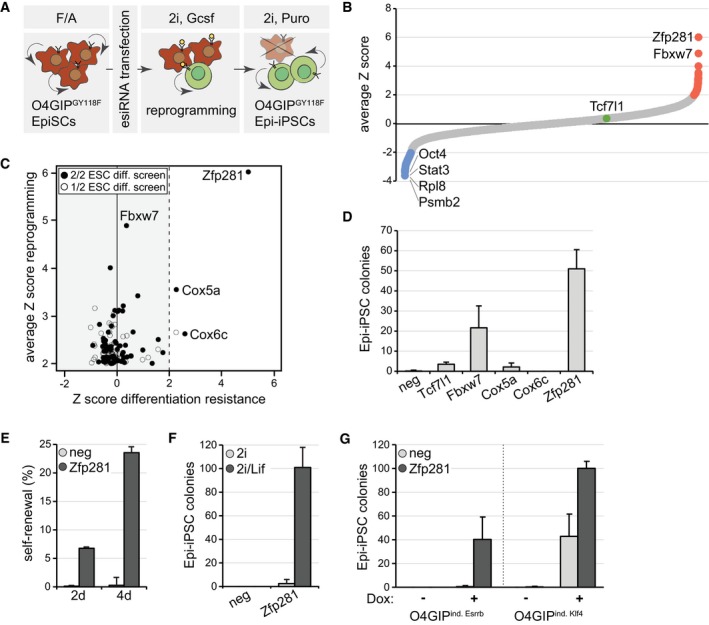
- Schematic outline of the reprogramming screen. Red indicates O4GiPGY118F EpiSCs and green O4GIPGY118F Epi‐iPSCs.
- Average Z scores of the two screen replicates. Note that esiRNAs targeting Mll1 (Zhang et al, 2016a) and Mbd3 (Rais et al, 2013) were not included in our library and that Otx2 (Acampora et al, 2013) scored below the significance threshold. Screen hits with negative (blue) and positive (red) Z scores (red), and Tcf7l1 (green) are highlighted.
- Number of Epi‐iPSC colonies derived from 796.4 EpiSCs transfected with indicated siRNAs, stimulated with Gcsf and 2i for 4 days, and selected with puromycin. Average and standard deviation (SD) of three experiments performed in duplicates. Negative siRNA (neg).
- Self‐renewal of O4GIPGY118F reprogramming intermediates after 2 days or 4 days of stimulation with Gcsf and 2i following transfection with indicated siRNAs. Average and SD of two experiments performed in duplicates.
- Number of Epi‐iPSC colonies derived from OEC2 EpiSCs transfected with indicated siRNAs, treated for 4 days in 2i or 2i/Lif medium, and selected with puromycin. Average and SD of two experiments performed in duplicates.
- Number of Epi‐iPSC colonies derived from O4GIP EpiSCs carrying Dox‐inducible Esrrb or Klf4 transgenes after transfection with indicated siRNAs, stimulation with or without Dox for 2 days, and selection with puromycin. Average and SD of two experiments performed in duplicates.
To determine whether any of the 146 genes also drive ESC differentiation, we compared our screen hits with those from two previous large‐scale ESC differentiation loss‐of‐function studies (Betschinger et al, 2013; Li et al, 2018; Fig 1C). Zfp281 and the cytochrome c oxidase subunits Cox5a and Cox6c scored strongest in all screens. For validation, we depleted each of them by siRNA transfection in independent GY118F‐expressing Oct4 reporter 796.4 EpiSCs (Yang et al, 2010) and also included siRNAs targeting Fbxw7 and Tcf7l1 as controls (Fig 1B and C). Knockdown of Fbxw7 and Zfp281, but not of Cox5a, Cox6c, or Tcf7l1, increased reprogramming (Fig 1D and Appendix Fig S1A). Therefore, Cox5a and Cox6c are false‐positive or cell line‐dependent screen hits, and we focused our further efforts on Zfp281. Consistent with previous findings (Fidalgo et al, 2016), Epi‐iPSCs derived by Zfp281 depletion expressed the naïve TFs Esrrb, Klf4, Nr0b1, and Tbx3, and reduced levels of the primed markers Oct6, Fgf5, Sox3, and Dnmt3b (Fig EV1D), suggesting successful reversion to the pluripotent ground state.
To quantify the dynamics of this process, we used self‐renewal in 2i as a proxy for acquisition of Epi‐iPSC identity. Compared to controls, Zfp281 depletion dramatically increased the colony‐forming capacity of single cells after 2 and 4 days of Gcsf addition (Fig 1E), indicating accelerated and more efficient reprogramming of EpiSCs. Gcsf supplementation was essential, and co‐depletion of Stat3 abolished Epi‐iPSC formation from O4GIPGY118F EpiSCs in the presence of Gcsf (Fig EV1E). Similarly, Leukemia inhibitory factor (Lif), which activates Stat3 in EpiSCs (Yang et al, 2010), was required for reprogramming of Zfp281‐depleted OEC2 EpiSCs (Fig 1F). To test whether Zfp281 acts only in the context of active Stat3, we used conditional expression of Esrrb or Klf4 in O4GIP EpiSCs through addition of doxycycline (Dox) to induce reprogramming. In the absence of extrinsic Lif, we observed an increase in Dox‐induced Epi‐iPSC colonies upon knockdown of Zfp281 (Fig 1G), suggesting that Zfp281 functions independent of the specific reprogramming regime. Taken together, these findings demonstrate that the vast majority of cell state transition regulators act unidirectionally. Zfp281, in contrast, acts bidirectionally as it drives ESC differentiation and inhibits reprogramming of EpiSCs. Notably, this is inverse to the activity of reprogramming TFs, e.g., Klf4 and Esrrb, that induce and consolidate the naïve ESC state (Guo et al, 2009; Niwa et al, 2009; Festuccia et al, 2012; Martello et al, 2012; Yamane et al, 2018).
Zfp281 promotes exit from naïve pluripotency independent of Tet1 and Tet2
To characterize the function of Zfp281 in ESC differentiation, we inactivated the gene in naïve RGd2 ESCs that contain a destabilized GFP protein downstream of the Rex1 (Zfp42) promoter (Appendix Fig S2A and B), which allows near real‐time tracking of cell state transition (Kalkan et al, 2017): GFP is homogeneously expressed in 2i and up to 16 hours (h) after 2i withdrawal (GFPhigh; Kalkan et al, 2017) before becoming progressively downregulated (GFPlow) as ESCs exit from self‐renewal (Fig EV2A). In 2i, reporter expression in two independent Zfp281 knockout (KO) clones was similar to the parental wild‐type cell line (WT) and an untargeted wild‐type sibling clone (Zfp281 WT) (Fig EV2B). In contrast, 32 and 72 h after 2i withdrawal, 30 and < 1% of WT cells were GFPhigh, while 75 and 10% of Zfp281 KO cells maintained high GFP expression, respectively. Consistent with impaired exit from the ESC state, 10% of Zfp281 KO cells formed colonies in 2i after 72 h of differentiation (Fig 2A). This phenotype was reverted by transgenic Zfp281 expression (Fig 2B). Resistance to exit self‐renewal was also observed in KO cells generated in a different ESC lines (Appendix Fig S2A and B, Fig EV2C), and in EpiLC (Hayashi et al, 2011) and embryoid body (EB) differentiation regimes (Figs 2A and EV2B). Zfp281 mutant cells maintained Rex1 reporter expression and self‐renewal even after lengthy periods in the absence of 2i (Figs 2A and EV2B), demonstrating that differentiation resistance is persistent.
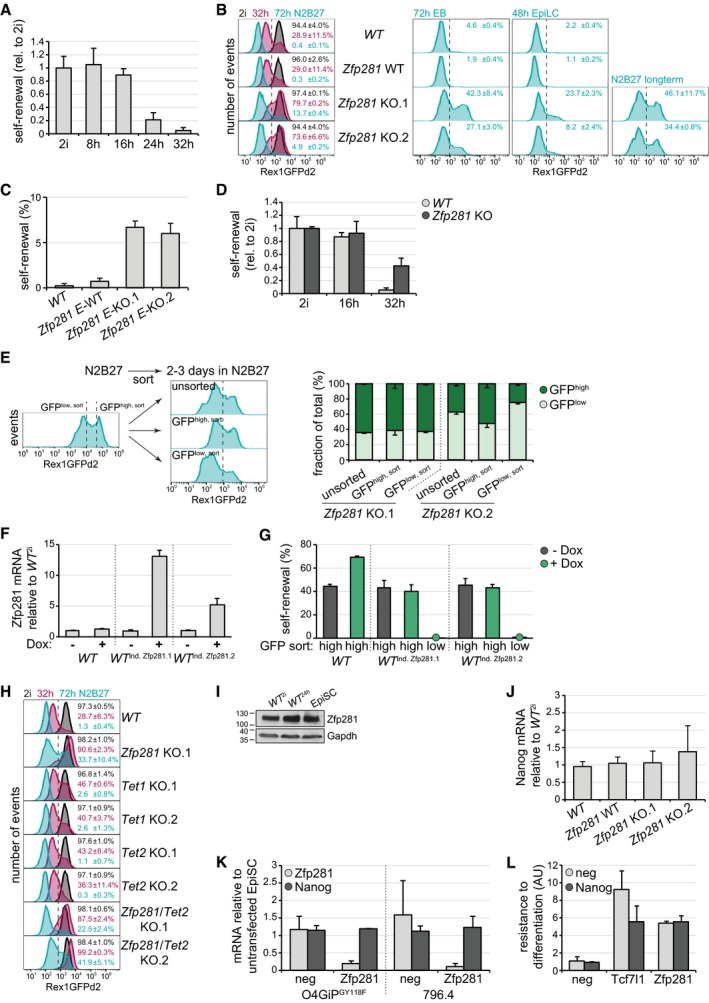
Figure EV2. Characterization of Zfp281 and Tet enzymes in ESC differentiation.
-
ASelf‐renewal in RGd2 cells at indicated time points of 2i withdrawal. Average and SD of two experiments performed in duplicates.
-
BRepresentative flow cytometry profiles of RGd2 ESCs of specified genotypes, at indicated time points and in indicated conditions. Numbers are average and SD of GFPhigh cells in two experiments.
-
C, DSelf‐renewal in RGd2 cells of indicated genotypes after 3 days (C) or indicated time points (D) of 2i withdrawal. Average and SD of two experiments performed in duplicates. E denotes E14 parental cell line origin.
-
EFlow cytometry profiles (left panel) of long‐term differentiated Zfp281 KO.2 cells in N2B27 and indicating GFP sorting gates (left), and of unsorted or sorted GFPlow,sort and GFPhigh,sort cells after an additional 2–3 days of culture in N2B27 and indicating gates used for quantification of GFP distribution (right). Please note that profiles shown on the right were recorded on a different instrument than the profile presented on the left. Quantification of GFP distribution (right panel) in N2B27 cultures derived from indicated sorted cells of specified genotypes. Average and SD of 2 experiments.
-
FZfp281 transcription relative to untreated WT 2i cells in Zfp281‐inducible ESC clones after 48 h in 2i and in the presence or absence of Dox. Average and SD of two technical replicates.
-
GSelf‐renewal of sorted GFPhigh,sort and GFPlow,sort populations of indicated genotypes after exposure to Dox (green) or control conditions (black) for 48 h. Green circles on x‐axis mark Dox‐treated non‐self‐renewing samples. Average and SD of two technical replicates.
-
HRepresentative flow cytometry profiles of RGd2 ESCs of specified genotypes at indicated time points and in indicated conditions. Numbers are average and SD of GFPhigh cells in two experiments.
-
IWestern blot showing Zfp281 protein levels during ESC progression.
-
J, KNanog (J, K) and Zfp281 (K) mRNA levels relative to WT 2i cells in ESCs of specified genotypes (J) and in indicated EpiSCs 24 h after transfection of neg and Zfp281 siRNAs (K). Average and SD of 5 (J) and 2 (K) experiments performed in duplicates.
-
LResistance to differentiation in O4GIP ESCs transfected with indicated siRNA combinations after 3 days of 2i withdrawal relative to untransfected cells. Average and SD of two experiments performed in duplicates.
Figure 2. Zfp281 drives exit from naïve pluripotency independent of Tet enzymes.

-
A, BSelf‐renewal in RGd2 ESCs of specified genotypes expressing indicated transgenes (B) after differentiation in indicated conditions (A) or 72 h in N2B27 (B). Average and SD of two experiments performed in duplicates. Note that control cells were lost during continuous passaging in N2B27 (A). Not determined (n.d.).
-
CRepresentative flow cytometry profiles of WT and Zfp281 KO.1 cells after 32 h of 2i withdrawal before (unsorted) and after purification of cells with indicated GFP expression (top panel). Self‐renewal of undifferentiated (2i) and sorted GFPhigh,sort and GFPlow,sort cells of indicated genotypes (bottom panel). Average and SD of three experiments performed in duplicates.
-
DRepresentative flow cytometry profiles of control and Zfp281‐inducible ESCs (top panel) and quantification of GFPlow cells (bottom panel) after 2 days in 2i and in the presence (green) or absence (black) of Dox. Average and SD of two experiments.
-
ESelf‐renewal in RGd2 ESCs of specified genotypes after 72 h in N2B27. Average and SD of two experiments performed in duplicates.
- F
Differentiating Zfp281 KO cells expressed varied levels of the Rex1 reporter (Fig EV2B) and formed colonies in 2i less efficiently than naïve pluripotent ESCs. For example, 32 h after 2i withdrawal, mutant cells displayed only 40% of the self‐renewal capacity of ESCs (Fig EV2D). To test whether this reduction is linked to population heterogeneity, we purified GFPhigh and GFPlow cells at 32 h using fluorescence‐activated cell sorting. As expected (Kalkan et al, 2017), sorted WT GFPlow cells were largely committed to differentiation and unable to generate clones in 2i (Fig 2C). In contrast, Zfp281 KO GFPlow cells formed colonies almost as efficiently as GFPhigh cells. Rex1 downregulation and exit from the ESC state is, thus, disconnected in Zfp281 mutants. However, the efficiency with which GFPhigh cells formed colonies after 32 h of 2i withdrawal was lower than of GFPhigh cells after 24 h (Kalkan et al, 2017) and of ESCs (Fig 2C). This was irrespective of genotype, suggesting a gradual decline in self‐renewal during differentiation both in Zfp281 mutant and in WT GFPhigh cells. The reduced clonogenicity of Zfp281 KO populations compared to ESCs may therefore be consequential to impaired progression of an advanced cell state with limited self‐renewal capacity and independent of population heterogeneity. In fact, GFPlow cells in long‐term differentiated Zfp281 mutants re‐established GFPhigh expression within a few days (Fig EV2E), revealing reversibility of the GFPlow state in the absence of Zfp281. To test sufficiency, we generated naïve RGd2 cells conditionally overexpressing Zfp281 under Dox regulation (Fig EV2F). Dox treatment in the presence of 2i induced silencing of the Rex1 reporter and loss of self‐renewal in a subset of cells (Figs 2D and EV2G). Zfp281 is therefore required and sufficient for exit from naïve pluripotency.
A previous study showed that differentiation of Serum/Lif‐cultured ESCs is accompanied by upregulation of Zfp281, which in turn destabilizes metastable pluripotency by binding to the methylcytosine hydroxylase Tet1 and transcriptionally suppressing Tet2 (Fidalgo et al, 2016). If the same mechanisms were to regulate exit from naïve pluripotency downstream of Zfp281, loss of Tet1 should induce the same phenotype as loss of Zfp281, and loss of Tet2, the opposite. We therefore generated Tet1, Tet2, and Zfp281/Tet2 KO alleles in naïve RGd2 ESCs (Appendix Fig S2C and D). Strikingly, the extinction of the Rex1 reporter and self‐renewal was similar in differentiating WT cells and Tet1 and Tet2 mutants, while the absence of Tet2 in Zfp281/Tet2 KO cells did not revert resistance to differentiation caused by absence of Zfp281 alone (Figs 2E and EV2H). We furthermore noted only modest changes in Zfp281 mRNA or protein during ESC differentiation, and across existing RNA sequencing (RNA‐seq) datasets of EpiLC and EpiSC differentiation (Buecker et al, 2014; Factor et al, 2014; Bao et al, 2018) and epiblast development (Boroviak et al, 2015; Figs 2F and EV2I). Zfp281 has also been reported to repress Nanog transcription through interacting with the NuRD complex in Serum/Lif‐cultured ESCs (Fidalgo et al, 2012). However, Nanog mRNA was unchanged in naïve Zfp281 KO ESCs or EpiSCs depleted of Zfp281 by siRNA transfection (Fig EV2J and K). Furthermore, knockdown of Nanog did not restore differentiation in ESCs depleted of Zfp281 while it partially did so, as expected (Pereira et al, 2006), in ESCs depleted of Tcf7l1 (Fig EV2L). Taken together, these results suggest that Zfp281 is expressed fairly constantly during exit from naïve pluripotency and drives differentiation independent of Tet1, Tet2, and Nanog.
Zfp281 acts independent of cell state‐exclusive chromatin association
To identify the transcriptional defects causing differentiation resistance, we performed RNA‐seq of WT and Zfp281 KO cells in 2i, and 16 and 32 h after 2i withdrawal (WT 2i,16 h,32 h and Zfp281 2i,16 h,32 h; Table EV2). The expression of several naïve and primed pluripotency markers was perturbed in Zfp281 16 h and Zfp281 32 h cells (Fig EV3A), confirming impaired silencing of naïve identity in Zfp281 mutants. k‐means clustering of mRNAs that significantly changed during WT differentiation or in Zfp281 KO cells (2,495 genes) identified six gene clusters (Fig 3A and B): Clusters 1–4 (1,898 genes) contain the majority of genes that were differentially transcribed in WT 32 h cells and of which a subset was already regulated in WT 16 h cells. Comparison with external EpiLC (Buecker et al, 2014) and EpiSC (Factor et al, 2014; Bao et al, 2018) expression datasets, which were not employed in the clustering analysis, revealed persistence of the bulk transcriptional changes established in WT 32 h cells, suggesting that clusters 1–4 contribute to pluripotent cell state progression. Clusters 5 and 6 (597 genes), in contrast, contain genes that were mostly unchanged in WT 32 h cells, but transiently regulated in WT 16 h cells and differentially expressed in EpiSCs. Clusters 5 and 6 may therefore act in gastrulation stage epiblast development and/or EpiSCs.
Figure EV3. Genomics of Zfp281.
-
AmRNA log2FC relative to WT 2i cells of selected core, naïve, and primed pluripotency markers in differentiating between WT and Zfp281 KO cells at indicated time points.
-
BTop 5 enriched GO terms in clusters 1–6.
-
CSequence logo from the de novo identified binding motif in 82.4% of 23,756 Zfp281 peaks.
-
DScatter plot comparing log2 Zfp281 peak ChIP enrichment over matched inputs between replicates in WT 2i (top) and WT 32 h (bottom) cells.
- E, F
-
GHeatmap of Zfp281, H3K27ac, histone H3K4 tri‐methylation (H3K4me3), H3K4 mono‐methylation (H3K4me1), and histone H3K27 tri‐methylation (H3K27me3) ChIP‐seq, DNase‐hypersensitive sites (DHS), and global run‐on sequencing (GRO‐seq) read densities across all proximal (± 2 kb of TSS) Zfp281 (top) and distal Zfp281 (bottom) peaks. Each row represents a 10‐kb window centered on the peak mid of Zfp281. Rows are sorted for H3K27ac ChIP read densities in ESCs. Reads per million (RPM).
Figure 3. Zfp281 directs sequential gene expression despite stable occupancy of target sites.
-
AmRNA log2 fold changes (log2FC) in WT 16 h, WT 32 h, Zfp281 16 h, and Zfp281 32 h samples relative to WT 2i cells, and in EpiSCs relative to WT 2i/Lif cells (Factor et al, 2014; Bao et al, 2018). Zfp281 2i, Zfp281 16 h , and Zfp281 32 h and WT 16 h and WT 32 h samples were used for k‐means clustering.
-
B, CQuantification of (A) including mRNA log2FC in EpiLCs relative to WT 2i/Lif (Buecker et al, 2014) and as indicated (C). (B) Dots represent the median, and shades, the lower and upper quartile. (C) Middle line represents median; notches, 95% confidence interval of the median; box, interquartile range; and whiskers, 1.5 times the interquartile range. Number of datapoints: 477 (cluster 1), 397 (cluster 2), 534 (cluster 3), 490 (cluster 4), 337 (cluster 5), and 260 (cluster 6).
-
DRepresentative immunofluorescence staining of spheroids in Matrigel derived from WT or Zfp281 KO.1 ESCs grown in 2i or N2B27 for 3 days. Blue: DNA. Red: F‐actin. Scale bar is 10 μm.
-
EScatter plot comparing Zfp281 log2 ChIP enrichment relative to matched inputs in WT 2i and WT 32 h cells.
-
FSame as in (E) with dots colored according to H3K27ac ChIP log2FC at the same peaks (top left), and to gene expression log2FC associated with peaks by nearest distance to TSS (bottom left) in WT 32 h relative to WT 2i cells. Quantification of H3K27ac ChIP (top right) and mRNA (bottom right) log2FC at top 1,000 Zfp281 peaks with increased (red) or decreased (blue) Zfp281 binding during ESC differentiation. Boxes as in Fig 3C for 1,000 datapoints each.
-
GQuantification of Zfp281 (left) and H3K27ac (right) ChIP log2FC in WT 32 h compared to WT 2i cells at Zfp281 peaks assigned to gene clusters 1–6. Boxes and number of datapoints as in Fig 3C.
Clusters 1 and 2 were largely unaffected in differentiating Zfp281 KO cells, whereas the repression and induction, respectively, of cluster 3 and 4 genes were blunted in both Zfp281 16 h and Zfp281 32 h cells (Fig 3A and B). Cell state‐specific comparison revealed that this was predominantly due to deregulation during differentiation (Fig 3C). Although we cannot exclude that the transcriptional defects in Zfp281 32 h cells were influenced by cell state heterogeneity (Fig EV2D), perturbed expression of cluster 3 and 4 genes in Zfp281 16 h cells, a time point at which downregulation of Rex1 reporter expression (Kalkan et al, 2017) and exit from self‐renewal (Fig EV2A and D) has not yet commenced, suggests a direct role of Zfp281 in regulating these genes. Zfp281 may therefore drive exit from naïve pluripotency through controlling gene clusters 3 and 4, which contain the naïve pluripotency TFs Klf4, Klf5, and Nr0b1, and the primed markers Sox3 and Dnmt3b, respectively (Table EV2), and are enriched for generic developmental terms using GO analysis (Fig EV3B). Conversely, cluster 5 and 6 genes were similarly mis‐expressed in Zfp281 2i, Zfp281 16 h, and Zfp281 32 h cells, notably with directionalities that are inverse to the changes observed in EpiSCs. GO analysis revealed significant enrichment of regulators of cell adhesion, which is critical for cell polarization (Ebnet et al, 2018) that initiates lumenogenesis after exit from naïve pluripotency (Shahbazi et al, 2017). To test whether Zfp281 controls polarization, we generated spheroids in Matrigel as described before (Shahbazi et al, 2017). WT ESCs formed polarized spheroids with expanded lumens that were encircled by apical F‐actin, while Zfp281 KO cells grew as unpolarized and disorganized cellular aggregates that were morphologically similar to ESCs (Fig 3D). Although we cannot exclude that this is consequential to impaired exit from self‐renewal, regulation of cluster 5 and 6 genes by Zfp281 may therefore contribute to cell polarization and cavity formation during ESC differentiation.
Oct4, similar to Zfp281, is expressed at equal levels in ESCs and EpiLCs, but occupies distinct CREs in the two cell states (Buecker et al, 2014). To determine whether Zfp281 acts through cell state‐specific chromatin association, we profiled its genome localization in WT 2i and WT 32 h cells using chromatin immunoprecipitation (ChIP) coupled to deep sequencing (ChIP‐seq) (Table EV2). De novo motif finding identified the consensus CCCCTCCCCC motif in 82.4% of 23,756 peaks (Fig EV3C), which is similar to results obtained in Serum/Lif ESCs (Fidalgo et al, 2016). Surprisingly, Zfp281 occupancy in WT 2i and WT 32 h cells was as highly correlated (Pearson's correlation coefficient R = 0.81) as between replicates (R = 0.84 and R = 0.85, respectively), with only few peaks exclusively detected in any of the two cell states (Figs 3E and EV3D). A lower correlation was observed between WT 32 h cells and published data for EpiSCs (Huang et al, 2017; R = 0.69) and trophoblast stem cells (TSCs; Ishiuchi et al, 2019; R = 0.55), but binding at peaks associated with cluster 1–6 genes was largely unchanged (Fig EV3E and F), suggesting stable chromatin association also during later pluripotency progression and in lineage‐unrelated TSCs. To determine whether Zfp281 binds to CREs, we profiled histone H3 lysine 27 acetylation (H3K27ac), a chromatin mark associated with active promoters and enhancers. Comparison of our Zfp281 and H3K27ac with published histone mark ChIP‐seq data (Kurimoto et al, 2015; Buecker et al, 2014; Fig EV3G) identified 7,697 Zfp281 peaks proximal to transcriptional start sites (TSSs), of which 54% were at active promoters (co‐localization with H3K27ac and H3K4 tri‐methylation), and 16,059 distal Zfp281 peaks of which 62% were at putative enhancers (co‐localization with H3K4 mono‐methylation). 38% of the latter were also enriched for H3K27ac, qualifying them as active enhancers. Notably, peaks with slightly increased Zfp281 binding in WT 32 h cells gained H3K27ac and expression of associated genes during differentiation, while decreased binding was associated with reduced H3K27ac and transcription (Fig 3F). Despite stable occupancy of target sites, quantitative binding changes in Zfp281 at these sites therefore parallel differences in CRE activity. However, this was similar at peaks linked to Zfp281‐insensitive cluster 1/2 and Zfp281‐sensitive cluster 3/4 genes (Fig 3G), showing that differential binding strength at CREs correlates with differentiation‐specific gene expression, but only partially with transcriptionally regulated targets.
Zfp281 interacts with Ehmt1 and Zic2 during ESC differentiation
Since chromatin occupancy was largely unchanged, we reasoned that Zfp281 may control transcription through cell state‐specific protein interaction partners. To test this, we performed Zfp281 immunoprecipitations (IPs) coupled to semi‐quantitative mass spectrometry (MS) in nuclear extracts of WT 2i and WT 40 h cells, including Zfp281 KO lysates to control for antibody specificity (Table EV3). Stringent selection criteria identified the previously reported interactor Nanog (Fidalgo et al, 2012) in WT 2i cells and several proteins specifically enriched in WT 40 h cells (Fig 4A). Strikingly, the latter were transcriptionally induced and the former repressed during differentiation (Fig 4B), suggesting that differential binding to Zfp281 may, at least in part, be driven by protein abundance. To determine functional downstream effectors, we decided to probe genetic interaction of differentiation‐specific interactors with Zfp281 in our conditionally overexpressing ESCs (Fig 2D). To this end, we depleted selected binding partners using siRNA transfection, induced Zfp281 by Dox treatment, and quantified Rex1 reporter distribution after 32 h in 2i (Fig 4C). As controls, we included siRNAs targeting Zfp281 itself and Tcf7l1. Transfection of Zfp281 but not Tcf7l1 siRNAs blocked emergence of GFPlow cells (76% reduction) (Figs 4C and EV4A), thus confirming suitability for identifying genetic Zfp281 interactors. Of all candidates tested individually, only knockdown of Ehmt1 and Zic2 reduced the fraction of GFPlow cells (34 and 32%, respectively), an effect enhanced by simultaneous depletion of both (63%). Conversely, conditional overexpression of Ehmt1 and Zic2 in RGd2 ESCs (Fig EV4B and C), similar to Zfp281, induced downregulation of the Rex1 reporter in a subset of cells (Fig EV4D). The de novo DNA methyltransferases Dnmt3a and Dnmt3b have overlapping functions during embryogenesis (Okano et al, 1999) and, hence, may act redundantly. However, simultaneous depletion of Dnmt3a and Dnmt3b by siRNAs or in compound Dnmt3a/3b KO cells (Appendix Fig S2E and Fig EV4E) did not impair Zfp281‐induced reporter repression (Figs 4C, and EV4A and F), demonstrating that Zfp281 drives differentiation independent of Dnmt3a and Dnmt3b.
Figure 4. Zfp281 acts by associating with Ehmt1 and Zic2.

- Cell state‐specific Zfp281 interactors in WT 2i and WT 40 h cells. Pink and cyan mark Zfp281 and selected binding partners, respectively. Quantification is based on three biological replicates.
- Same as (A) with mRNA log2FC of differential binding partners during ESC differentiation instead of P‐values.
- Procedure to identify Zfp281 effectors in naïve Zfp281‐inducible RGd2 cells (left). Quantification of GFPlow WT ind. Zfp281 cells transfected with indicated siRNAs and incubated for 32 h in 2i in the presence (green) or absence (black) of Dox (right). Dashed line marks fraction of GFPlow cells in control cells exposed to Dox. Significance was determined using a Wilcoxon–Mann–Whitney rank sum test compared to neg control sample. *P < 0.05; not significant (n.s.). Average and SD of four experiments.
Figure EV4. Characterization of Ehmt1 and Zic2 downstream of Zfp281.
-
ARepresentative flow cytometry profiles of Zfp281‐inducible RGd2 ESCs transfected with indicated siRNAs after 32 h in 2i and in the presence (green) or absence (black) of Dox.
-
B, CFC Ehmt1 (B) and Zic2 (C) transcription relative to untreated WT 2i cells in indicated ESC clones after 32 h in 2i in the presence or absence of Dox. Average and SD of two technical replicates.
-
DRepresentative flow cytometry profiles of RGd2 ESCs with conditional Ehmt1 and Zic2 expression after 32 h in 2i and in the presence (green) or absence (black) of Dox. Numbers are the average and SD of GFPlow cells in two experiments.
-
EZfp281 transcription relative to untreated WT 2i cells in indicated ESC clones after 32 h in 2i in the presence or absence of Dox. Average and SD of two technical replicates.
-
FRepresentative flow cytometry profiles of Dnmt3a/3b compound KO RGd2 ESCs with conditional Zfp281 expression after 32 h in 2i and in the presence (green) or absence (black) of Dox. Significance was determined using a Wilcoxon–Mann–Whitney rank sum test compared to WT ind. Zfp281.1 Dox‐treated cells. Not significant (n.s.) ≥ 0.05. Numbers are the average and SD of GFPlow cells in four experiments.
-
GWhole‐cell lysate Zfp281 IPs in WT and Zfp281 KO cells in 2i or 40 h after 2i withdrawal, and probed for indicated proteins. Input (left) and Zfp281 IP (right). (*) Ig heavy chain.
-
H–JCell morphologies (H), growth curves (I), and cell cycle analyses using propidium iodide staining (J) of indicated genotypes in 2i. Average and SD of three experiments (I, J). Scale bar is 50 μm (H).
-
KRepresentative flow cytometry profiles of indicated genotypes in 2i, and after 32 and 72 h of 2i withdrawal. Numbers are the average and SD of GFPhigh cells in two experiments.
-
LQuantification and hierarchical clustering of normalized F‐actin intensity in 20 concentric rings (from center to circumference) in spheroids derived from ESCs with indicated genotypes in 2i or N2B27 for 4 days. Intensity is color‐coded and illustrates central F‐actin accumulation and, hence, polarization of WT and Zic2 KO cells during differentiation.
-
MRepresentative immunofluorescence staining of WT or Ehmt1 KO ESCs expressing the indicated transgenes. Top: H3K9me2 and DAPI. Bottom: Ehmt1. Co‐localization of H3K9me2 with DAPI‐rich speckles in Ehmt1 2i cells expressing no transgene, the ∆NHHC, or NH‐LE alleles is indicated by arrowheads. Please note the absence of nuclear Ehmt1 staining in Ehmt1 2i cells and restoration by Ehmt1 transgenes. Scale bar is 10 μm.
-
NRepresentative flow cytometry profiles of indicated genotypes in 2i and 32 h after 2i withdrawal.
Zic2 is a zinc finger TF that represses poised developmental enhancers in Serum/Lif ESCs (Luo et al, 2015). Ehmt1 (GLP) is a methyltransferase that can be found in a complex with Ehmt2 (G9a), which both mediate mono‐ and di‐methylation of histone H3 lysine 9 (H3K9me1 and H3K9me2; Tachibana et al, 2005). The genetic interactions in naïve ESCs (Fig 4C) together with the preferential binding during differentiation observed in both nuclear extracts using IP‐MS (Fig 4A) and in whole‐cell lysates using IP–Western blot (Fig EV4G) suggest that Ehmt1 and Zic2 are functional downstream effectors of Zfp281.
Ehmt1 and Zic2 regulate ESC differentiation and reprogramming of EpiSCs
We therefore generated individual and compound Ehmt1 and Zic2 KO RGd2 ESCs (Appendix Fig S2F and G). In contrast to wild‐type or Zic2 KO cells, Ehmt1 and Ehmt1/Zic2 KO cells were spindle‐shaped (Fig EV4H) and proliferated slowly (Fig EV4I). They were not arrested at a specific cell cycle stage (Fig EV4J) and did not exhibit downregulation of the Rex1 reporter in 2i (Fig EV4K). After 2i withdrawal for 32 h (or 72 h), 75% (9%) of Ehmt1 and 55% (7%) of Zic2 KO cells maintained GFP expression, increasing to 90% (35%) in Ehmt1/Zic2 compound KO cells (Fig EV4K), while 30% (1%) of cells from untargeted sibling clones (Zic2 WT and Ehmt1 WT) were GFPhigh. Correspondingly, 5, 4, and 12% of Zic2, Ehmt1, and Ehmt1/Zic2 KO cells retained self‐renewal after 72 h of differentiation (Fig 5A). Ehmt1 and Ehmt1/Zic2, but not Zic2 KO ESCs, were unable to form polarized spheroids in Matrigel (Fig 5B). Quantification of this effect was similar to Zfp281 mutants (Fig EV4L). We therefore conclude that Ehmt1 is required for polarization and that Ehmt1 and Zic2 promote exit from self‐renewal independently of each other.
Figure 5. Ehmt1 and Zic2 drive exit from the ESC state and restrict reprogramming of EpiSCs.
- Self‐renewal in cells with indicated genotypes 3 days after 2i withdrawal. Significance was determined using a Wilcoxon–Mann–Whitney rank sum test comparing the specified genotype groups. *P < 0.05. Average and SD of two experiments performed in duplicates.
- Representative immunofluorescence staining of spheroids in Matrigel derived from indicated genotypes in 2i or N2B27 for 4 days. Blue: DNA. Red: F‐actin. Scale bar is 10 μm.
- Quantification of GFPhigh cells in WT cells or Ehmt1 KO clones expressing indicated transgenes in 2i (black) or 32 h after 2i withdrawal (pink). Average and SD of two experiments.
- Number of Epi‐iPSC colonies derived from 796.4 (gray) and O4GIPGY118F (black) EpiSCs transfected with indicated siRNAs, stimulated with Gcsf and 2i for 4 days, and selected with puromycin. Significance was determined using a Wilcoxon–Mann–Whitney rank sum test compared to neg control sample of the respective cell line, or comparing Zic2 and Ehmt1 to Ehmt1/Zic2 depletion. *P < 0.05. Average and SD of five experiments performed in duplicates.
In the absence of Ehmt1, H3K9me2 was limited to DAPI‐rich speckles (Fig EV4M), which is reminiscent of the depletion of euchromatic H3K9me2 and its enrichment at pericentric heterochromatin in Ehmt2 mutants (Tachibana et al, 2002). Since exposure to the Ehmt inhibitors A‐366 and UNC0642 induced dose‐dependent cell lethality (data not shown), we decided to test Ehmt1's enzymatic activity by expressing specific loss‐of‐function alleles in Ehmt1 KO cells: An Ehmt1 protein with mutations in the ankyrin domain (Ehmt1ank), responsible for binding to methylated H3K9 in vitro (Collins et al, 2008), reverted nuclear H3K9me2 distribution (Fig EV4M) and resistance to Rex1 downregulation (Figs 5C and EV4N) to a similar extent as the wild‐type protein did. In contrast, substitutions in the SET domain (Ehmt1NH‐LE) that perturb Ehmt1 methyltransferase in vitro (Tachibana et al, 2008) rescued only partially and a small deletion in the SET domain (Ehmt1ΔNHHC) that additionally ablates binding to Ehmt2 completely abolished rescue. Therefore, both catalytic activity of Ehmt1 and formation of a larger methyltransferase protein complex are implicated in ESC transition.
Chemical inhibition and knockdown of Ehmt enzymes in somatic cells enhances reprogramming (Shi et al, 2008; Sridharan et al, 2013; Rodriguez‐Madoz et al, 2017). Consistently, depletion of Ehmt1 but also of Zic2 in 796.4 and O4GIPGY118F EpiSCs increased Epi‐iPSC formation in the presence of Gcsf (Fig 5D and Appendix Fig S1B). The effect was modest, but enhanced by the combined knockdown of both. Taken together, these results suggest that Ehmt1 and Zic2, similar to Zfp281, drive exit from naïve pluripotency and restrain reprogramming of EpiSCs. Notably, phenotypes induced by co‐depletion of Zic2 and Ehmt1 were weaker than elimination of Zfp281, suggesting the existence of additional Zfp281 effectors that may include other histone‐modifying complexes (Huang et al, 2017; Zhou et al, 2017; Ishiuchi et al, 2019).
Overlapping transcriptional functions of Zfp281 and Ehmt1/Zic2
To test whether the biochemical and functional interaction with Zfp281 is reflected in similar transcriptional outputs, we profiled mRNA expression in Ehmt1, Zic2, and Ehmt1/Zic2 KO cells in 2i and after 32 h of differentiation (Table EV2). Principal component (PC) analysis, including Zfp281 KO, EpiLC (Buecker et al, 2014), and EpiSC (Bao et al, 2018) datasets, of changes relative to WT ESCs identified PC1 to discriminate developmental timing and to separate differentiated cells from ESCs (Fig 6A). Mutant and wild‐type ESCs projected similarly onto PC1 and expressed pluripotency marker genes at similar levels (Fig EV5A), confirming their naïve identity. PC2, in contrast, segregated WT from Zfp281 and, in particular, Ehmt1 KO genotypes. We, indeed, observed 1,274 deregulated genes in Ehmt1 2i cells that were unchanged in Zfp281 2i cells (Fig EV5B). These were enriched for homeostatic and cell adhesion GO terms (Fig EV5B) and likely contribute to the cellular and polarization phenotypes in Ehmt1 KO cells.
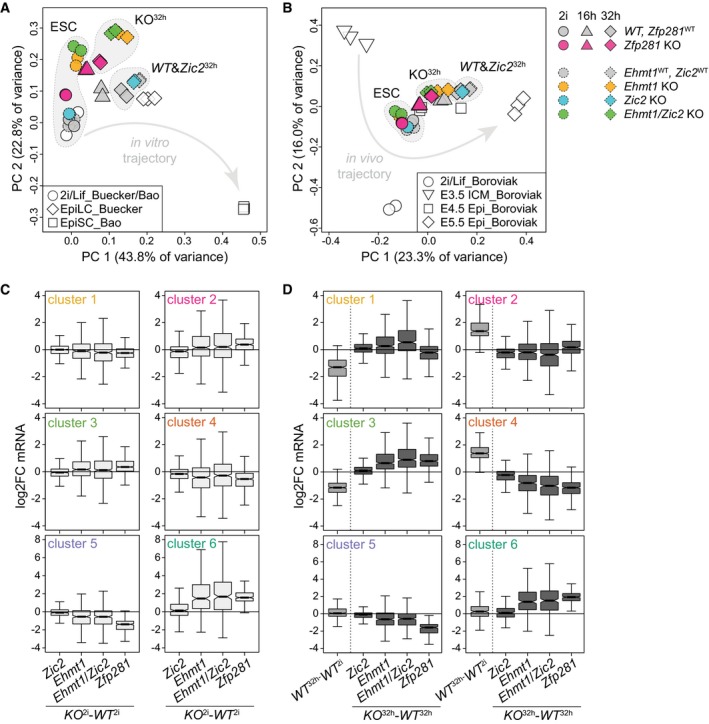
Figure 6. Related transcriptional defects in Ehmt1/Zic2 and Zfp281 KO cells.
-
A, BPC analysis of indicated samples normalized to WT ESCs (A) and all samples (B) within each dataset. Arrows indicate developmental trajectories. Full and dashed outlines indicate independent RNA‐seq experiments.
-
C, DQuantification of cell state‐specific mRNA log2FC of gene clusters 1–6 between indicated genotypes in 2i (C) and 32 h after 2i withdrawal (D). Boxes and number of datapoints as in Fig 3C.
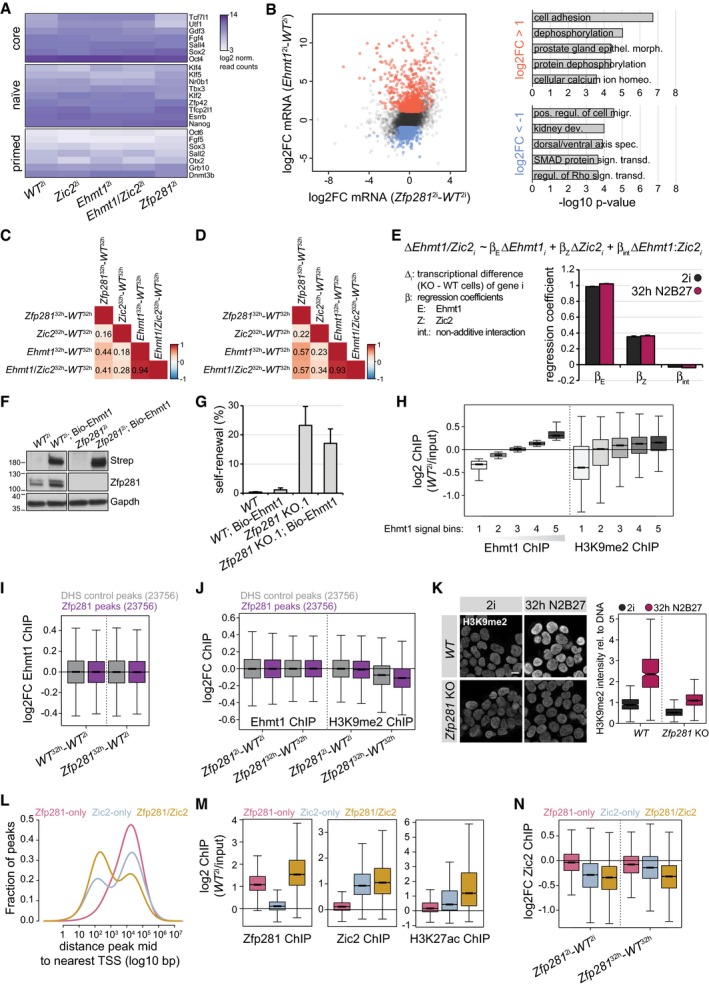
Figure EV5. Ehmt1 and Zic2 transcriptomics and genomics.
-
ALog2‐normalized read counts of selected core, naïve, and primed pluripotency markers in ESCs of indicated genotypes.
-
BScatter plot of mRNA log2FC in Zfp281 2i and Ehmt1 2i cells (left). Top 5 GO terms enriched in genes upregulated (red) and downregulated (blue) specifically in Ehmt1 2i cells (right).
-
C, DPairwise Pearson correlation coefficients of mRNA changes between indicated differentiated cells considering all detected transcripts (C) or gene cluster 1–6 transcripts (D).
-
EEstimated regression coefficients and standard errors for the contribution of Ehmt1 (ΔEhmt1), Zic2 (ΔZic2), and their interaction (ΔEhmt1:Zic2) to cell state‐specific gene expression changes in Ehmt1/Zic2 compound KO cells. All expressed genes (13,096) are taken into account.
-
FWestern blot confirming Ehmt1 biotinylation [probed with Streptavidin (Strep)] in ESCs of indicated genotypes expressing the BirA ligase.
-
GESC self‐renewal of indicated genotypes after 3 days of 2i withdrawal. Average and SD of three experiments performed in duplicates.
-
HLog2 Ehmt1 and H3K9me2 ChIP enrichment in ESCs over matched inputs at five classes of 10‐kb genome‐wide windows binned by increasing Ehmt1 chromatin association. Boxes as in Fig 3C for 42,887 datapoints each.
-
I, JEhmt1 (I, J) and H3K9me2 (J) ChIP log2FC between indicated cell states and genotypes at Zfp281 peaks (purple) or matching and non‐overlapping DHS control peaks (gray) extended to 10‐kb windows. Boxes and number of datapoints as in Fig 7A.
-
KRepresentative immunofluorescence staining of H3K9me2 (left) and quantification relative to DNA (right) in indicated genotypes and conditions. Scale bar is 10 μm. Boxes as in Fig 3C for 453 (WT 2), 465 (WT 32 h), 574 (Zfp281 2), and 792 (Zfp281 32 h) datapoints.
-
LDensity plot showing distance of Zfp281‐only (pink), Zic2‐only (blue), and Zfp281/Zic2 co‐bound peaks (yellow) to nearest TSS.
-
MZfp281 (left), Zic2 (middle), and H3K27ac (right) log2 ChIP enrichment over matched inputs in ESCs at Zfp281‐only (pink), Zic2‐only (blue), and Zfp281/Zic2 co‐bound (yellow) peaks. Boxes and number of datapoints as in Fig 7B.
-
NCell state‐specific Zic2 ChIP log2FC between indicated genotypes and cell states at Zfp281‐only (pink), Zic2‐only (blue), and Zfp281/Zic2 co‐bound (yellow) peaks. Boxes and number of datapoints as in Fig 7B.
Progression of Zfp281 16 h/32 h , Ehmt1 32 h, and Ehmt1/Zic2 32 h cells along PC1 was impaired when compared to matching WT controls (Fig 6A), which we also observed in PC analysis using blastocyst development datasets (Boroviak et al, 2015; Fig 6B). In fact, alterations in Ehmt1 and Zfp281 KO cells correlated during differentiation (Fig EV5C), suggesting similar defects in developmental transcription. This correlation was not strong (R = 0.44), but increased (R = 0.57) when only considering cluster 1–6 gene expression (Fig EV5D). Although we also noted slight deregulation of clusters 1 and 2, transcriptional defects in clusters 3–6 were similar in Ehmt1 32 h and Zfp281 32 h cells (Fig 6C and D). Ehmt2‐dependent H3K9 di‐methylation is associated with gene silencing (Zylicz et al, 2015), consistent with the majority of genes showing increased RNA levels in Ehmt1 2i cells (Fig EV5B). Nevertheless, 38% of target genes were downregulated. Changes in the absence of Ehmt1 are therefore likely consequential to both direct and indirect effects and may also include the contribution of non‐histone Ehmt1 substrates (Sim et al, 2017) to transcription.
Based on mRNA levels, Zic2 32 h cells were not separated from matching control cells (Fig 6A–D). This was surprising, since Zic2 KO cells appeared similarly impaired in exiting self‐renewal as Ehmt1 KO cells (Fig 5A). However, loss of Zic2 in Ehmt1 32 h cells enhanced the deregulation of clusters 1–4 during differentiation (Fig 6D) and induced a shift along PC1 (Fig 6A and B). Linear regression revealed that perturbations in Ehmt1/Zic2 KO cells were predominantly the sum of alterations in single mutants rather than synergistic (Fig EV5E), implying subtle, but functionally relevant, changes in Zic2 KO cells. Ehmt1 and Zic2 therefore regulate transcription independently of each other, aligning with their additive loss‐of‐function phenotypes in differentiation and reprogramming (Fig 5A and D). Taken together, this analysis demonstrates connected functions of Zfp281 and Ehmt1/Zic2 in gene expression during cell state transition.
Ehmt1 and Zic2 act downstream of Zfp281 on chromatin
To identify direct targets and to explore how those relate to the physical interaction with Zfp281 in differentiated cells, we performed Zic2 and Ehmt1 ChIP‐seq, and profiled H3K9me2 as a proxy for Ehmt1 activity. Due to absence of ChIP‐seq compatible Ehmt1 antibodies, we inserted an N‐terminal Flag‐Avi tag at both Ehmt1 alleles in ESCs expressing the BirA biotin ligase (Fig EV5F). This did not perturb exit from self‐renewal (Fig EV5G), indicating expression of a functional Ehmt1 fusion protein (Bio‐Ehmt1). ChIP‐seq using Streptavidin beads identified broad Ehmt1‐occupied chromatin domains that, consistent with an enzyme–substrate relationship, scaled with H3K9me2 genome‐wide (Fig EV5H). To determine overlap with Zfp281, we quantified Ehmt1 and H3K9me2 enrichment at Zfp281‐bound and matching unbound control windows (see Materials and Methods for details). Ehmt1 localization at both sets of regions was unchanged during differentiation or in Zfp281 KO cells (Fig EV5I and J), indicating that Zfp281 is not required for Ehmt1 localization on chromatin. In contrast to Ehmt1, H3K9me2 increased in WT 32 h cells and EpiLCs (Kurimoto et al, 2015), with a more pronounced increase at Zfp281‐occupied loci than control windows (Fig 7A). To test whether these dynamics require Zfp281, we performed immunofluorescence staining, which revealed that mutant cells failed to gain H3K9me2 by 32 h after 2i withdrawal (Fig EV5K). H3K9me2 ChIP‐seq in Zfp281 mutants confirmed that H3K9me2 levels were unaffected in Zfp281 2i cells but did not increase in Zfp281 32 h cells (Figs 7A and EV5J). Impaired gain of H3K9me2 was observed at both Zfp281‐bound and Zfp281‐unbound sites, suggesting that Zfp281 is a differentiation‐specific pervasive activator of Ehmt1 during ESC transition. Zic2 ChIP‐seq identified 28,495 peaks, of which approximately 30% overlapped with Zfp281 (Fig 7B). These were closer to promoters and enriched for H3K27ac compared to Zic2‐only and Zfp281‐only peaks (Fig EV5L and M), suggesting co‐localization of Zfp281 and Zic2 at CREs. In WT 32 h cells, Zic2 increased predominantly at co‐bound peaks (Fig 7B). Although we noted a general reduction of Zic2 on chromatin in Zfp281 mutants, Zic2 localization was particularly perturbed at co‐bound sites in Zfp281 32 h cells (Figs 7B and EV5N). Our findings therefore suggest that Zfp281 engages Ehmt1 and Zic2 during ESC differentiation through chromatin co‐occupancy‐dependent and chromatin co‐occupancy‐independent mechanisms.
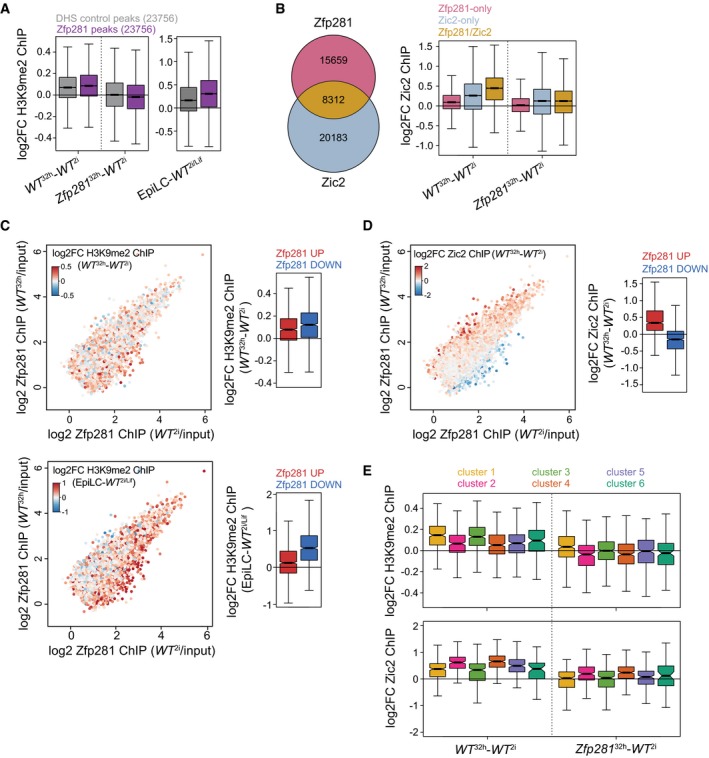
Figure 7. Zfp281 engages with Ehmt1 and Zic2 at developmental CREs.
-
AH3K9me2 ChIP log2FC between indicated cell states and genotypes at 10‐kb windows surrounding Zfp281‐bound (purple) or matching DNase‐hypersensitive site (DHS) control peaks (gray). Boxes as in Fig 3C for 23,756 datapoints each.
-
BOverlap of Zfp281 and Zic2 ChIP peaks (left) and Zic2 ChIP log2FC between specified cell states and genotypes at indicated peak subsets (right). Boxes as in Fig 3C for 15,659 (Zfp281‐only), 20,183 (Zic2‐only), and 8,312 (Zfp281/Zic2) datapoints.
-
C, DSame as in Fig 3F. Coloring is according to H3K9me2 ChIP log2FC between WT 32 h and WT 2i cells (C, top left) and between EpiLCs and WT 2i/Lif cells (C, bottom left) at Zfp281 peaks extended to 10‐kb windows, and according to Zic2 ChIP log2FC between WT 32 h and WT 2i cells (D, left). Quantification of corresponding ChIP changes at top 1,000 Zfp281 peaks with increased (red) or decreased (blue) Zfp281 binding during ESC differentiation (right). Boxes and number of datapoints as in Fig 3F.
-
EH3K9me2 (top) and Zic2 (bottom) ChIP log2FC between indicated cell states and genotypes at all Zfp281 peaks extended to 10‐kb windows (top) or Zfp281/Zic2 co‐bound peaks (bottom) associated with nearest TSSs of cluster 1–6 genes. Boxes and number of datapoints as in Fig 3C.
At Zfp281‐bound peaks, the gain of H3K9me2 and Zic2 occurred mostly at mutually exclusive sets of genomic loci with reduced and increased Zfp281 binding during differentiation, respectively (Fig 7C and D), indicating that these are sites of direct physical interaction that control transcription of nearby genes. We therefore stratified H3K9me2 and Zic2 binding dynamics at Zfp281 peaks by gene cluster association. H3K9me2 increased predominantly at peaks linked to repressed clusters 1 and 3, and Zic2 at peaks belonging to induced clusters 2 and 4 (Fig 7E). Surprisingly, the gain in WT 32 h cells and reduction in Zfp281 32 h cells were indistinguishable between clusters 1 and 3 (for H3K9me2) and clusters 2 and 4 (for Zic2). Hence, Zfp281 catalyzes H3K9me2 and Zic2 deposition at transition‐associated CREs genome‐wide and without any qualitative or quantitative specificity for its transcriptional targets in clusters 3 and 4. Why cluster 1 and 2 gene expression is insensitive to perturbation of H3K9me2 and Zic2 dynamics in Zfp281 KO cells (Figs 3C and 7E) remains to be determined. Additional chromatin regulators may be involved, since transcription of clusters 1 and 2 is also less sensitive to Ehmt1 depletion than of clusters 3 and 4 (Fig 6D). We therefore propose that Zfp281 drives and stabilizes transition‐specific transcription, at least in part, through activation of Ehmt1 at cluster 3 CREs and recruitment or stabilization of Zic2 at cluster 4 CREs.
Discussion
Cellular plasticity in response to injury in vivo or TF overexpression in vitro is frequently accompanied by the reversal of cellular specialization (Merrell & Stanger, 2016). Although single‐cell profiling has shown that this process is not a strict inversion of natural development (Treutlein et al, 2016; Gerber et al, 2018), differentiation and de‐differentiation trajectories may mechanistically intersect. We aimed to uncover such plasticity regulators in pluripotent cells and performed loss‐of‐function screening for genes that both drive exit from ESC self‐renewal and shield EpiSCs from reprogramming into the pluripotent ground state. Within the experimental limitations of this approach, we identified only one gene, the TF Zfp281. Such exclusivity suggests a prominent role in establishing and maintaining the unidirectionality of pluripotent cell state progression in vitro. The former is consistent with perturbed epiblast maturation in Zfp281 mutant embryos (Huang et al, 2017), but whether Zfp281 protects cellular identities against de‐differentiation in vivo remains to be determined. We, however, note that Zfp281 restrains iPSC formation from fibroblasts at a late pre‐iPSC stage (Fidalgo et al, 2012), supporting the notion that resetting of EpiSCs into naïve pluripotency recapitulates a late phase of somatic cell reprogramming (Dunn et al, 2019). Other factors that, similar to Zfp281, drive differentiation and inhibit de‐differentiation of cell states not represented in our ESC‐EpiSC conversion system are likely to exist.
We showed that Zfp281 is important for robust ESC differentiation. This is reminiscent of lineage‐specifying TFs that are specifically expressed in the lineages they instruct (Graf & Enver, 2009). In adult mice, Zfp281 is indeed transcribed strongest in heart tissue and its overexpression in fibroblasts enhances cardiac reprogramming (Zhou et al, 2017). During ESC differentiation, however, Zfp281 neither changes expression nor occupies distinct genomic sites, indicating a facilitating, rather than specifying, function. Using biochemical, genetic, and genomic experiments, we provide evidence that Zfp281 directs sequential gene expression through permissive and instructive mechanisms involving physical interaction with Ehmt1 and Zic2. Cluster 5 and 6 genes are differentially expressed in EpiSCs, but transcriptionally primed by Zfp281 throughout differentiation, including in the naïve pluripotent starting cell state. Cluster 5 contains modulators of cell adhesion that may contribute to polarization of post‐implantation epiblast cells. However, although Zfp281 null embryos degenerate during gastrulation, they do form an epithelial egg cylinder (Huang et al, 2017), suggesting operation of compensatory mechanisms in vivo. Cluster 3 and 4 genes are, in contrast, regulated by Zfp281 predominantly during transition, enriched for developmental functions, and therefore likely responsible for extinguishing ESC identity. Strikingly, Zfp281 quantitatively decreases at CREs associated with repressed cluster 3 genes and, vice versa, increases at CREs associated with induced cluster 4 genes. Concomitant gain of H3K9me2 and Zic2 at cluster 3 and 4 CREs, respectively, suggests that subtle Zfp281 chromatin binding differences are converted into CRE activity changes by reciprocal activation of Ehmt1 and Zic2. Consistently, embryonic arrest in Ehmt1 and Ehmt2 mutant mouse embryos (Tachibana et al, 2002, 2005) has been attributed to reduced H3K9me2 deposition at and impaired silencing of developmental enhancers (Zylicz et al, 2015), while Zic2 triggers neural plate gene expression in EpiSCs through enhancer activation (Iwafuchi‐Doi et al, 2012). The molecular mechanisms inducing differential binding of Zfp281 to Ehmt1 and Zic2 remain to be elucidated. Because of similar overexpression phenotypes in ESCs, we surmise that protein ratios are rate‐determining. Zic2 protein levels, despite unchanged mRNA (Fig 4B), indeed increase during differentiation, while Ehmt1 persists (Fig EV4G). However, Ehmt1 and Ehmt2 interact with additional zinc finger TFs (Shinkai & Tachibana, 2011) that may compete with Zfp281 for binding to Ehmt1 specifically in ground state ESCs. Taken together, we suggest that Zfp281 promotes stable cell state transition by permissively marking cluster 5 and 6 genes for forthcoming developmental regulation, and instructing cluster 3 and 4 gene expression for elimination of naïve pluripotency, the latter through differential engagement with Ehmt1 or Zic2 at CREs.
Zfp281 drives differentiation to and inhibits reprogramming of EpiSCs, and therefore qualifies as a bidirectional cell state regulator with antipodal functions during developmental progression and reversion. Whether this is because Zfp281‐Ehmt1/Zic2 control the same cell state transition and act through the same target genes in mutual directions remains to be determined. We showed that Zfp281 KO cells exposed to differentiation‐promoting conditions are less clonogenic than ESCs and give rise to an equilibrium of Rex1high and Rex1low cell states that features defective coupling of Rex1 downregulation with exit from self‐renewal. It is therefore conceivable that Zfp281 acts after an initial commitment step to induce and stabilize irreversible silencing of naïve identity. During reprogramming, Zfp281 may inversely antagonize induction of naïve pluripotency prior to formation of a Rex1high state, which has recently been shown to mark transition intermediates with full ESC self‐renewal activity and destined to give rise to Epi‐iPSCs (Stuart et al, 2019). Acting at a late stage of reprogramming also reconciles our finding of Zfp281 impairing Epi‐iPSC formation by STAT3 activation and Esrrb overexpression (Figs 1G and EV1E) with the notion that both regimes proceed along distinct transcriptional trajectories before converging on a Rex1high cell state (Stuart et al, 2019). However, Zfp281 depletion in EpiSCs also causes cell cycle arrest and apoptosis (Fidalgo et al, 2016), but not upregulation of naïve pluripotent TFs such as Nanog (Fig EV2K), and is not alone sufficient for reprogramming to Epi‐iPSCs in the presence of 2i (Fig 1F). Zfp281 activates transcription of Nodal signaling components, such as the cluster 5 genes Lefty1 and Lefty2, in ESCs, EpiSCs, and the post‐implantation epiblast (Huang et al, 2017). Nodal signaling is required for EpiSC maintenance (Vallier et al, 2009), but dispensable for exit from ESC self‐renewal (Mulas et al, 2017) and somatic cell reprogramming (Ruetz et al, 2017). We therefore speculate that Zfp281 controls the EpiSC state and reprogramming through different effector genes, the former through regulation of cluster 5/6 genes, e.g., Lefty1, Lefty2 or cell polarity regulators, and the latter through stabilization of cell state transition‐specific cluster 3/4 genes.
In metastable serum/Lif ESCs, Zfp281 is reported to promote DNA methylation and differentiation by recruiting Tet1 and silencing Tet2 (Fidalgo et al, 2016). Upon exit from the naïve ESC state, Zfp281 binds to Dnmt3a, Dnmt3b, and Dnmt3l (Fig 4A), suggesting that Zfp281 controls 5‐methylcytosine turnover through regulating DNA methylating and demethylating enzymes. Furthermore, Dnmt1, Dnmt3a, and Dnmt3b interact with the Ehmt1‐Ehmt2 heterodimer (Estève et al, 2006; Epsztejn‐Litman et al, 2008), H3K9me2 and 5‐methylcytosine marks overlap genome‐wide (von Meyenn et al, 2016), and DNA is hypomethylated upon depletion or catalytic inhibition of Ehmt2 in ESCs (Zhang et al, 2016b). We, however, showed that Zfp281 drives exit from naïve pluripotency independent of Dnmt3a, Dnmt3b, Tet1, and Tet2. Although the specific contributions of 5‐methylcytosine, H3K9me2, and other Ehmt substrates (Sim et al, 2017) to pluripotent cell plasticity remain to be determined, our findings suggest that resolution of naïve pluripotency in vitro is masked or mechanistically distinct in heterogeneous Serum/Lif ESC cultures. Similarly, Zic2 has previously been reported to act as a repressor in metastable serum/Lif ESCs (Luo et al, 2015), but we detect only minor transcriptional defects in naïve Zic2 KO ESCs.
Using the paradigm of pluripotent cell state conversion in vitro, we here identify, in Zfp281, a regulator of cellular plasticity that modulates CRE activity and transcription without reliance on cell type‐restricted expression or chromatin occupancy. We propose that the persistence of Zfp281 at developmental CREs throughout cell state transition provides a molecular platform for ordered remodeling of the cis‐regulatory architecture and further consolidation by lineage‐specifying TFs. These findings demonstrate that differential gene expression is not a necessary criterion of cellular plasticity regulators, and we suggest that this feature may not be limited to pluripotent cells.
Materials and Methods
Cell culture
Embryonic stem cells (male and female RGd2 cells containing a Rex1:GFPd2‐IRES‐blasticidin (Wray et al, 2011), male O4GIP ESCs containing a GFP‐IRES‐puromycin transgene under control of an Oct4 regulatory element (Betschinger et al, 2013), and male E14 cells) were cultured on plastic coated with gelatin or laminin (Sigma) in N2B27 medium [DMEM/F12 (Life Technologies), Neurobasal (Gibco) supplemented with N2 (homemade) and B‐27 serum‐free supplement (Gibco), 2 mM l‐glutamine (Gibco), and 0.1 mM 2‐mercaptoethanol (Sigma)] with 2i [3 μM CHIR99021 and 1 μM PD0325901 (Steward Lab, Dresden)] and, where indicated, with 1 μg/ml doxycycline (Sigma). EpiSCs [O4GIP and OEC2 (Guo et al, 2009) and 796.4 (Yang et al, 2010)] were cultured on plastic coated with fibronectin (Millipore) in N2B27 with bFGF (12 ng/ml) and activin A (20 ng/ml) (FA) (Smith Lab, Cambridge).
For monolayer differentiation, ESCs were seeded on gelatin‐coated plates at 1.5 × 104 cells/cm2 in 2i, and the following day, 2i was withdrawn. Cells were incubated in N2B27 for 32 h or 72 h, or continuously passaged on laminin‐coated plates, as indicated. For EpiLC differentiation for 48 h (Hayashi et al, 2011), medium was adjusted to FA and 1% knockout serum replacement (Thermo Fisher). For embryoid body (EB) differentiation, ESCs were seeded at 2.5 × 104 cells/cm2 on ultra‐low attachment plates (Corning) in serum media [GMEM (Sigma) supplemented with 10% fetal bovine serum (Sigma), 1 mM sodium pyruvate (Gibco), 2 mM l‐glutamine (Gibco), 0.1 mM non‐essential amino acids (Gibco), and 0.1 mM 2‐mercaptoethanol (Sigma)] for 72 h. Exit from pluripotency was quantified by measuring GFP fluorescence in RGd2 cells on a LSRII SORP Analyzer (Becton Dickinson) and analyzed using FlowJo (FlowJo, LLC), and by performing self‐renewal and commitment assays as described before (Betschinger et al, 2013). Briefly, differentiated RGd2 and E14 cells at indicated time points were plated at clonal density on laminin‐coated plates in 2i medium, and resulting colonies were stained for alkaline phosphatase activity (Sigma) according to the manufacturer's instruction and counted. RGd2 cells were additionally selected with 10 μg/ml blasticidin (Thermo Fisher). O4GIP cells were differentiated for 72 h and treated with 2i medium containing 1 μg/ml puromycin (Gibco), and uncommitted cells were quantified after 3 days by adding 1:10 diluted Alamar Blue (Invitrogen) in 2i medium, following by readout on a SpectraMax Gemini EM (Molecular Devices) microplate reader. For cell cycle analysis, cells were fixed in cold 70% ethanol for 30 min at 4°C, washed twice with PBS and 0.1% BSA (Sigma), treated with 5 μg RNaseA (Thermo Fisher) for 15 min at room temperature (RT), and stained with 10 μg propidium iodide (Sigma). Cells were analyzed on a LSRII SORP Analyzer, and cell cycle distributions were determined using FlowJo.
siRNA transfections were performed as described (Betschinger et al, 2013) using 16.7 nM siRNA (detailed in Table EV4) and transfection mixes in OptiMEM (Invitrogen) containing Lipofectamine 2000 or RNAiMAX (Thermo Fisher) for ESCs or EpiSCs, respectively.
For EpiSC reprogramming, cells were plated at 1.5 × 104 cells/cm2 on fibronectin‐coated plates in N2B27 with FA. The next day, medium was changed to 2i and, as indicated, supplemented with 30 ng/ml granulocyte colony‐stimulating factor (Gcsf) (PeproTech), 10 ng/ml Lif (Smith lab, Cambridge), or 1 μg/ml doxycycline. After 4 days (d), medium was changed to 2i with 1 μg/ml puromycin, and Epi‐iPSC colonies were stained for alkaline phosphatase activity and counted. For experiments shown in Figs 1E and EV1A, cells were subjected to self‐renewal assays in 2i after 2 and 4 days of Gcsf supplementation. For experiments shown in Fig EV1D, individual Epi‐iPSC colonies were picked and expanded in 2i with puromycin for further experiments.
Spheroid formation of ESCs was performed as described before (Shahbazi et al, 2017). Briefly, ESCs grown in 2i medium were washed in N2B27, and 7.5 × 103 cells were resuspended in 25 μl ice‐cold growth factor reduced Matrigel (Corning, 356231), plated dropwise on uncoated 96‐well glass plates (Greiner Bio‐One), and aggregated in N2B27 with or without 2i for 3 or 4 days.
EpiSC screen
O4GIPGY118F EpiSCs were reverse‐transfected in fibronectin‐coated 384‐well plates using mixtures of 50 ng esiRNA and 0.075 μl Lipofectamine 2000 in 10 μl OptiMEM. EpiSCs were plated at a density of 5,000 cells/well in 80 μl N2B27 with FA. Each plate included three negative (Luciferase esiRNA) and two positive (Stat3 esiRNA) control wells. The next day, medium was changed to 2i containing 30 ng/ml Gcsf and 4 days later to 2i containing 1 μg/ml puromycin. After 3–4 days, medium was changed to 2i containing 1/10 vol Alamar Blue (Invitrogen) and cell survival quantified on a SpectraMax M2 (Molecular Devices).
Genome editing
CRISPR/Cas9 genome editing was performed by transient co‐transfection of hCas9 and U6‐gRNA plasmids (Mali et al, 2013) (Addgene plasmids 41,815 and 41,824) and a dsRed expression plasmid into E14 or female RGd2 ESCs. Two days later, single dsRed‐positive cells were sorted into gelatin‐coated 96‐well plates containing serum media supplemented with 10 ng/ml Lif and 2i. Clones were genotyped by sequencing amplified target loci and by confirming protein absence in Western blots. For generation of knockout (KO) cell lines, two independent KO clones (specified in Appendix Fig S2) and, in the case of Zfp281, Ehmt1, and Zic2 targeting, one untargeted wild‐type sibling clone were kept for further analysis.
N‐terminal Flag‐Avi tagging of Ehmt1 was performed in WT or Zfp281 KO ESCs constitutively expressing the BirA biotin ligase (see below). The recombination template was generated by cloning homology arms (548 bp upstream and 618 bp downstream of the Ehmt1 transcription start site) into pDONR221 using Gateway technology (Thermo Fisher) and inserting the Flag‐Avi sequence by Seamless Cloning (Thermo Fisher). Targeting was performed as above with hCas9, U6‐gRNA, and dsRed expression plasmids, but included co‐transfection of the recombination template. Targeting was confirmed by sequencing the target locus and by the expression of biotinylated Ehmt1 in Western blots.
gRNA sequences, genotyping primers, and the Ehmt1 recombination template are specified in Table EV4. gRNA sequences targeting Tet1 and Tet2 (Wang et al, 2013) and Dnmt3a and Dnmt3b (Domcke et al, 2015) have been described.
Immunostaining
Cells seeded on laminin‐coated 96‐well glass plates were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 10 min and spheroids grown in Matrigel for 20 min at RT. Samples were blocked for 1 h in blocking solution (PBS, 0.1% Triton X‐100 (Sigma), 3% donkey serum (Sigma), and 1% BSA) and incubated overnight with primary antibodies [Ehmt1 (Abcam, ab41969, 1:300) and H3K9me2 (Abcam, ab1220, 1:300)] at 4°C. After three washes in washing solution (PBS, 0.1% Triton X‐100), secondary antibodies were added, DNA was stained with Hoechst33342 (Life Technologies) and, where indicated, incubated with Alexa Fluor 488 Phalloidin (Life Technologies, A12379, 1:40) for 20 min at RT. Images were acquired using a LSM 710 scanning head confocal microscope (Zeiss) at 20× magnification and handled using Fiji and Adobe Photoshop (Adobe). Imaging of spheroids used for quantification in Fig EV4L was performed with a Yokogawa CV7000S high‐throughput confocal microscope at 20× magnification. Images were acquired in confocal mode as z‐stack multiplane images over z distance of 50 μm with a 5 μm step size, and maximum intensity projections were stored, representatives of which are shown in Fig 5B.
Molecular biology
Coding sequences for Ehmt1, Esrrb, Klf4, and Zfp281 were amplified from ESC complementary DNA (cDNA) and for BirA biotin ligase from a plasmid (gift of Matyas Flemr, Friedrich Miescher Institute, Basel). For Zic2, the coding sequence was synthesized as a double‐stranded gBlock (IDT). Polynucleotides were recombined into pDONR221 using Gateway technology. Ehmt1 point mutations [Ehmt1ΔNHHC: NHHC1198‐1201del (Tachibana et al, 2008), Ehmt1NH‐LE: NH1198‐1199LE (Tachibana et al, 2008), and Ehmt1ank: W872A, W877A, and E880A (Collins et al, 2008)] were introduced by polymerase chain reaction (PCR). Expression destination vectors were pPB‐CAG‐DEST‐pgk‐hph (Betschinger et al, 2013) and pPB‐TRE‐DEST‐rTA‐pgk‐hph (Villegas et al, 2019), and GY118F expression vector as described (Yang et al, 2010). Stable integration into ESCs or EpiSCs after co‐transfection with pBASE (Betschinger et al, 2013) was selected in the presence of 150 μg/ml hygromycin B (Thermo Fisher).
For relative mRNA quantification, total RNA was isolated from indicated samples using RNeasy Mini Kit (Qiagen) and cDNA prepared using SuperScript III reverse transcriptase (Invitrogen). Quantitative PCR was performed using TaqMan Fast Universal PCR Master Mix (Applied Biosystems) with gene‐specific primers, using either the universal probe library (UPL, Roche) or TaqMan system (Applied Biosystems), and a GAPDH probe (Applied Biosystems) for normalization. Oligonucleotide sequences and probes are listed in Table EV4.
Protein methods
Cell lysates for Western blotting were generated in RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, and 0.1% SDS). Primary antibodies were anti‐GAPDH (Sigma, G8795, 1:5,000), anti‐Ehmt1 (Abcam, ab41969, 1:500), anti‐Tet1 (Millipore, 09‐872, 1:1,000), anti‐Tet2 (Abcam, ab124297, 1:300), anti‐Zic2 (Abcam, ab150404, 1:500), anti‐Zfp281 (Bethyl Laboratories, A303‐118A, 1:500), and anti‐Streptavidin coupled to HRP (Sigma, EV2438, 1:1,000).
Nuclear immunoprecipitations (IPs) for Zfp281 were performed in three biological replicates using WT 2i, Zfp281 2i, WT 40 h, and Zfp281 40 h cells. Cells were washed with cold PBS, resuspended in five packed cell volume (pcv) of buffer A (10 mM HEPES pH 7.9, 1.5 mM MgCl2, and 10 mM KCl), incubated for 10 min on ice, and broke open using a Dounce homogenizer. Nuclei were pelleted at 3,300 g for 15 min at 4°C, resuspended in 3 pcv of buffer B (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, and 20% glycerol) supplemented with 420 mM NaCl, Complete Mini protease, PhosSTOP phosphatase inhibitors (Roche), and 250 U/ml Benzonase (Sigma), and incubated for 30 min at 4°C on a rotating wheel. Insoluble material was pelleted at 25,000 g for 30 min at 4°C, and the supernatant diluted with buffer B to a final concentration of 150 mM NaCl and including 0.02% NP40. 1% of the supernatant was kept as input sample and the remainder incubated with 10 μl Dynabeads Protein G (Invitrogen) and 1 μg Zfp281 antibody (Bethyl Laboratories, A303‐118A) for 1 h at 4°C on a rotating wheel. Beads were collected on a magnetic rack for 2–3 min to remove the supernatant, and washed four times in 1 ml buffer B containing 150 mM NaCl and 0.02% NP40 for 10 min each at 4 °C on a rotating wheel. For mass spectrometry, proteins were digested on the beads as described before (Villegas et al, 2019).
Whole‐cell lysate IPs using 1 μg Zfp281 antibody (Bethyl Laboratories, A303‐118A) were performed as described before (Villegas et al, 2019) using Dynabeads.
Chromatin immunoprecipitation
For ChIP of endogenous proteins or histone modifications, 8 × 106 cells per IP were fixed for 10 min with 1.1% formaldehyde in fixing solution (0.1 M NaCl, 1 mM EDTA, 0.5 mM EGTA, and 50 mM HEPES pH 7.5) at RT on a rotating wheel, and neutralized with glycine to a final concentration of 0.125 M for 5 min at RT. Cells were washed three times with ice‐cold PBS by spinning at 1,600 g for 5 min at 4°C, incubated for 10 min at 4°C on a rotating wheel with 1 ml lysis buffer 1 (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP‐40, and 0.25% Triton X‐100), pelleted, and incubated for a further 10 min at 4°C in 1 ml lysis buffer 2 (10 mM Tris pH 8.0, 200 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA). Nuclei were pelleted, resuspended in 140 μl shearing buffer (50 mM Tris pH 8.0, 10 mM EDTA, and 1% SDS), and sonicated in Diagenode 15‐ml Falcon tubes for 25 cycles (30 s ON, 30 s OFF) in ice‐cold water using a Bioruptor Plus (Diagenode). 10% of sonicated DNA was kept as input sample. Lysates were further pelleted at 14,000 g for 10 min at 4°C, and the supernatant diluted 1:10 with ChIP dilution buffer (50 mM Tris pH 8.0, 167 mM NaCl, 1.1% Triton X‐100, and 0.11% Na‐deoxycholate). Lysates were precleared over 10 μl Dynabeads for 2 h and incubated overnight at 4°C on a rotating wheel with the following antibodies: 2 μg H3K27ac (Active Motif, 39135), 2 μg H3K9me2 (Abcam, ab1220), 2 μg Zic2 (Abcam, ab150404), or 2 μg Zfp281 (Bethyl Laboratories, A303‐118A). The next day, 10 μl Dynabeads were added and incubated with lysates for 1 h at 4°C on a rotating wheel. Beads were washed with 1 ml of the following buffers for 5 min each at 4°C: twice with wash buffer 1 (50 mM Tris pH 8.0, 0.1% SDS, 0.1% Na‐deoxycholate, 1% Triton X‐100, 150 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA), once with wash buffer 2 (50 mM Tris pH 8.0, 0.1% SDS, 0.1% Na‐deoxycholate, 1% Triton X‐100, 500 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA), once with wash buffer 3 (50 mM Tris pH 8.0, 250 mM LiCl, 0.5% Na‐deoxycholate, 0.5% NP40, 1 mM EDTA, and 0.5 mM EGTA), and twice with wash buffer 4 (50 mM Tris pH 8.0, 10 mM EDTA, and 5 mM EGTA). Finally, beads were eluted twice with 100 μl elution buffer (0.1 M NaHCO3, and 1% SDS) for 15 min at RT in a shaker at maximum speed, and combined supernatants de‐crosslinked overnight by supplementation to 200 mM NaCl and continuous shaking at maximum speed at 65°C. The same procedure was followed for input samples by adjusting the total volume of elution buffer to 200 μl and 200 mM NaCl. The next day, DNA was purified using MinElute PCR Purification Kit (Qiagen).
Bio‐ChIP for Flag‐Avi‐tagged Ehmt1 was performed as described before (Ostapcuk et al, 2018) with minor modifications. Briefly, 8 × 106 cells per IP were fixed for 8 min with 1% formaldehyde in PBS at RT on a rotating wheel, and neutralized with adjusting glycine to a final concentration of 0.125 M and incubation for 1 min at RT and for 5 min on ice. Cells were washed three times with ice‐cold PBS and pelleted at 1,000 g for 5 min at 4°C. Cells were lysed in lysis buffers 1 and 2 as described above. Nuclei were washed once in 5 ml NUC buffer (15 mM HEPES pH 7.5, 60 mM KCl, 15 mM NaCl, and 0.32 mM sucrose) and resuspended in 1 ml NUC buffer supplemented with Complete Mini protease inhibitors, 3.3 μl 1 M CaCl2, and 2–3 μl Micrococcal Nuclease (Cell Signaling, 10011S). Enzymatic activity was induced for 15 min at 37°C and shaking at 1,000 rpm, and stopped by addition of 50 μl of STOP solution (250 mM EDTA and 500 mM EGTA) and 110 μl of 10× ChIP buffer (167 mM Tris pH 8.0, 1.67 M NaCl, 12 mM EDTA, 10% Triton X‐100, and 0.1% SDS) with a further incubation for 5 min on ice. Nuclei were gently disrupted by sonication in Diagenode 15‐ml Falcon tubes for eight cycles (5 s ON, 5 s OFF) in ice‐cold water using a Bioruptor Plus. Lysates were centrifuged at 14,000 g for 5 min at 4°C, 5% of the supernatant was kept as input sample, and the remaining supernatant precleared for 2 h over 10 μl Dynabeads at 4°C on a rotating wheel. Chromatin was incubated for 1 h with M‐280 Streptavidin‐coupled Dynabeads (Invitrogen) at 4°C on a rotating wheel, and washed with 1 ml of the following buffers for 5 min each at 4°C: twice with TE buffer (10 mM Tris pH 8.0, and 1 mM EDTA) supplemented with 2% SDS, once with high‐salt buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% Triton X‐100, and 0.1% Na‐deoxycholate), once with wash buffer 3 (see above), and twice with TE buffer. Beads were eluted in 60 μl elution buffer (see above) supplemented with 2 μl RNaseA (10 mg/ml stock) and incubated for 30 min at 37°C while mixing. After supplementation to 10 mM EDTA, 10 mM Tris pH 8.0, and 2 μl proteinase K (10 mg/ml, Promega), the bead suspension was further incubated for 3 h at 55°C and overnight at 65°C while shaking. The same procedure was followed for input samples, including RNaseA and proteinase K digestion. DNA was purified using AMPure XP beads (Beckman Coulter).
Sequencing libraries
RNA from ESCs grown in 2i and 16 and 32 h after 2i withdrawal was isolated using RNeasy kit (Qiagen). For Zfp281 KO and corresponding WT cells, total RNA was subjected to ribosomal RNA depletion using Ribozero removal kit (Illumina) followed by library construction using ScriptSeq V2 library preparation kit (Illumina). For Ehmt1, Zic2, Ehmt1/Zic2 KO, and corresponding WT cells, RNA‐seq libraries were prepared using TruSeq mRNA Library Preparation Kit (Illumina). ChIP‐seq libraries were prepared using NEBNext Ultra kit (New England BioLabs) following the manufacturer's recommendations. Sequencing was performed on an Illumina HiSeq2500 machine (50‐bp single‐end reads).
Screen analysis
For the EpiSC reprogramming screen, Z scores were calculated for each plate, excluding the two outer most columns and rows (Table EV1). Screen replicates are presented in Fig EV1B. Average Z scores > 2 were considered as screen hits, identifying 146 genes (Fig 1B). We quantified their role in exit from the ESC state by extracting primary data for these 146 genes from two previous ESC differentiation studies (Betschinger et al, 2013; Li et al, 2018), and computing Z scores on this subset (Table EV1). Sixty‐seven and 129 of the 146 genes were mapped on results from Betschinger et al (2013) and Li et al (2018), respectively, resulting in mapping of 130 genes in total. Average Z scores from both ESC differentiation screens are presented in Fig 1C.
Quantification of immunostaining
Quantification of H3K9me2 was performed in CellProfiler3 (Broad Institute). Nuclei were identified using Hoechst33342 staining and average H3K9me2 and DNA fluorescence intensity per nucleus determined: WT 2i (453 cells), Zfp281 2i (574 cells), WT 32 h (465 cells), Zfp281 32 h (792 cells).
For quantification of d3 and d4 spheroids from two biological replicates, images were stitched to generate a single image per channel and per well, and used for object segmentation with MATLAB (MathWorks). Segmented object outlines were exported and used for feature extraction with software package CellProfiler3. Extracted features (84 features) describing object area, shape, and intensity were normalized within corresponding assay plates using Z score transformation and unified into a cross‐comparable dataset. Normalized features were used to extract 10 principal components (PC), which were further used for unsupervised clustering with the software package PhenoGraph (Levine et al, 2015). Unsupervised clustering returned 13 clusters, and objects belonging to the two sparsest clusters were discarded as outliers based on extreme variance in measured features in the respective classes. To describe radial distribution of F‐actin signal intensity, segmented objects were divided into 20 concentric regions of same width. Distribution of F‐actin staining intensity was described as mean fraction of intensity per region (MeanFrac) whereby the total intensity in the given region was normalized to the fraction of object area corresponding to the region. Mean fractions of intensity per region were not Z scored as these values were normalized per object ad initio. Visualization in Fig EV4L shows the mean of F‐actin signal in a given concentric ring of all d4 spheroids per genotype and medium condition: WT 2i (559 spheroids), Ehmt1 2i (349 spheroids), Zic2 2i (292 spheroids), Ehmt1/Zic2 2i (164 spheroids), Zfp281 2i (324 spheroids), WT 96 h (437 spheroids), Ehmt1 96 h (228 spheroids), Zic2 96 h (202 spheroids), Ehmt1/Zic2 96 h (233 spheroids), Zfp281 96 h (547 spheroids). Heatmap was generated using aheatmap function from the Bioconductor package NMF (Gaujoux & Seoighe, 2010) (package version 0.21.0).
Protein identification and quantification
Relative quantification of mass‐spec data from three biological replicates (Table EV3) was performed with MaxQuant (version 1.5.3.8) using Andromeda as search engine and label‐free quantification as described (Villegas et al, 2019). Briefly, the mouse subset of the UniProt version 2015_01 combined with the contaminant DB from MaxQuant was searched, and the protein and peptide FDR were set to 0.01. For Fig 4A and B, proteins were considered, which passed an interaction threshold of an at least twofold enrichment in IPs from WT 2i or WT 40 h lysates compared to matched Zfp281 KO lysates with a significant P‐value < 0.1, and were quantifiable with at least two unique razor peptides.
RNA‐seq and GRO‐seq analysis
RNA‐seq reads from wild‐type and mutant cells in 2i, and 16 and 32 h after 2i withdrawal were aligned to the mouse GRCm38/mm10 genome using qAlign from the Bioconductor package QuasR (Gaidatzis et al, 2015) (package version 1.22.0) with default parameters except for splicedAlignment=TRUE. Published RNA‐seq from ESCs cultured in 2i/Lif and EpiLC (Buecker et al, 2014) was 36‐bp reads, and therefore, no spliced alignment could be performed. RNA‐seq from ESCs cultured in 2i/Lif and EpiSCs (Factor et al, 2014; Bao et al, 2018), and global run‐on sequencing (GRO‐seq) data from 2i/Lif‐cultured ESCs (Dorighi et al, 2017) were 100‐bp and 50‐bp paired‐end reads, respectively, and therefore, paired=“fr” was used. For in vivo embryo data (Boroviak et al, 2015), preexisting alignments to mouse GRCm38/mm10 genome were downloaded from ArrayExpress (E‐MTAB‐2958) and used. Alignments were quantified for known UCSC genes obtained from the TxDb.Mmusculus.UCSC.mm10.knownGene package (package version 3.4.4) using qCount from the Bioconductor package QuasR with default parameters (Table EV2).
Only transcripts with at least three counts per million in at least two biological samples from this study were considered as expressed genes (total: 13,096 genes). For identification of significantly deregulated genes, edgeR (Robinson & Oshlack, 2010; package version 3.24.0) was used and detected genes were fitted to two generalized linear models:
-
(a)
˜ time + genotype + time:genotype: This model uses WT 2i, WT 16 h, WT 32 h, Zfp281 2i, Zfp281 16 h, and Zfp281 32 h expression datasets. Genes with a significant time coefficient are genes that change either between WT 2i and WT 16 h or WT 2i and WT 32 h cells, genes with a significant genotype coefficient are differentially expressed between Zfp281 2i and WT 2i cells, and genes with a significant interaction term time:genotype are deregulated in Zfp281 KO cells specifically during 16‐h or 32‐h differentiation.
-
(b)
˜ genotype: This model identifies genes that differ between Ehmt1 2i and WT 2i cells.
Raw P‐values were corrected for multiple testing by calculating false discovery rates (FDR). Significant genes were identified as genes with an absolute log2 fold change > 1.0 and an FDR of < 0.01.
For visualization of RNA‐seq data, except principal component analysis (PCA) in Fig 6A and B and heatmap of selected markers in Fig EV5A, log2 fold‐change values were used that were obtained from edgeR by fitting the indicated datasets to the following models:
-
(c)
˜ time (EpiLCs or EpiSCs): EpiLCs (Buecker et al, 2014) or EpiSCs (Factor et al, 2014) compared to WT 2i/Lif (Buecker et al, 2014) and EpiSC compared to WT 2i/Lif (Bao et al, 2018; used for Fig 3A and B).
-
(d)
˜ genotime: Zfp281 16 h or Zfp281 32 h compared to WT 2i (used for Figs 3A and B, and EV3A), where genotime is the combination of genotype and time.
-
(e)
˜ genotype (cell state‐specific): KO cells in 2i compared to WT 2i, or KO cells 16 h or 32 h after 2i withdrawal compared to WT 16 h or WT 32 h, respectively (used for Figs 3C and 6C and D, and EV5B–E).
For heatmap visualization in Fig 3A, only significantly deregulated genes in at least one condition were considered (model (a): time, genotype, or time:genotype; total: 2,495 genes) and the following log2 contrast was used for clustering: WT 16 h‐WT 2i, WT 32 h‐WT 2i, Zfp281 2i‐WT 2i, Zfp281 16 h‐WT 2i, Zfp281 32 h‐ WT 2i, Zfp281 16 h‐WT 16 h, and Zfp281 32 h‐WT 32 h. For heatmap visualization in Fig EV5A, RNA‐seq read counts were normalized (divided by the total number of aligned reads (library size), multiplied with minimal library size, and added with a pseudocount of 8) and log2‐transformed, and the mean of biological replicates was plotted. For visualization in Fig EV5B, significantly deregulated genes in Ehmt1 2i (model (b): genotype) but not Zfp281 2i (model (a): genotype) are highlighted. In Fig 3B, dots represent the median, and shades, the lower and upper quartile of indicated samples. Boxplots were generated using the boxplot function in R with default parameters except outline=FALSE. Correlation plots (Fig EV5C and D) were generated using corrplot function from the Bioconductor package corrplot (https://github.com/taiyun/corrplot) (package version 0.84). Heatmaps (Figs 3A and EV3A, and EV5A) were generated using aheatmap function from the Bioconductor package NMF. For PCA represented in Fig 6A, normalized (see above) and log2‐transformed read counts were centered by subtracting the average of WT ESCs within each of the following four RNA‐seq datasets: (i) Zfp281 KO and corresponding WT samples from this study; (ii) Ehmt1, Zic2, Ehmt1/Zic2 KO, and corresponding WT samples from this study; (iii) WT 2i/Lif cells and EpiLCs (Buecker et al, 2014); and (iv) WT 2i/Lif cells and EpiSCs (Bao et al, 2018). For PCA represented in Fig 6B, normalized (see above) and log2‐transformed read counts were centered by subtracting the average over all samples within each of the following three RNA‐seq datasets: (i) Zfp281 KO and corresponding WT samples from this study; (ii) Ehmt1, Zic2, Ehmt1/Zic2 KO, and corresponding WT samples from this study; and (iii) WT 2i/Lif cells and embryo samples (Boroviak et al, 2015). Centered read counts for each PCA are provided in Table EV2, and the detailed R code is provided in Dataset EV1. PCA was performed using the prcomp function in R. Analyses of enriched gene sets (Figs EV1C, EV3B and EV5B) were performed using DAVID (Huang et al, 2009) (version 6.8) for GO terms of biological processes.
The linear model to estimate synergistic transcriptional effects in Ehmt1 and Zic2 KO cells (Fig EV5E) takes into account all (13,096) expressed genes, and was fitted using lm function in R:
∆Ehmt1/Zic2 i ˜ βE∆Ehmt1 i + βZ∆Zic2 i + βint∆Ehmt1:Zic2 i
∆: transcriptional difference (KO—WT) of gene i
β: regression coefficient:
E: Ehmt1
Z: Zic2
int: non‐additive interaction
ChIP‐seq and DHS‐seq analysis
ChIP‐seq data from this study, published datasets (Buecker et al, 2014; Huang et al, 2017; Ishiuchi et al, 2019), and DNase I hypersensitive site sequencing (DHS‐seq) (Encode; accession number: ENCSR000CMW) reads were aligned to mouse GRCm38/mm10 genome using qAlign from the Bioconductor package QuasR with default parameters. Published H3K4me3, H3K27me3, and H3K9me2 ChIP‐seq data (Kurimoto et al, 2015) were aligned using Bowtie (Langmead et al, 2009) (version 4.4.7) with parameter ‐C in colorspace. Alignments were sorted and indexed using SAMtools (package version 1.2), and all ChIP‐seq data were quantified with qCount from the Bioconductor package QuasR. Read counts were normalized (divided by the total number of aligned reads (library size), multiplied with minimal library size, and added with a pseudocount of 8) and log2‐transformed. For DHS‐seq, reads per million (RPM) were calculated by dividing the total number of aligned reads, multiplying with one million, adding a pseudocount of 8, and log2 transforming the data.
We observed a non‐linear relationship in Zfp281 ChIP‐seq data when comparing read counts in Zfp281 peaks between ChIP‐seq replicates in WT 2i, and therefore performed loess regression using normalizeBetweenArrays function of the Bioconductor package limma (Ritchie et al, 2015) (package version 3.38.2) with method = cyclingloess.
In Ehmt1 ChIP‐seq data, we detected a variable dependency of read counts in genomic tiles on the tile's GC composition (GC bias), which was most pronounced in input samples. In order to reduce this bias, we used a loess‐based normalization method: First, reads were counted in each sample in 10‐kb windows (either genome‐tiling windows or windows centered on Zfp281 peaks and corresponding control windows; see below). The counts were then scaled [divided by the total number of aligned reads (library size)], multiplied with minimal library size, added with a pseudocount of 8, and log2‐transformed. A loess curve was fit to the log2‐transformed counts as a function of the fraction of G+C bases in the window using the R function loess with span = 0.3. This fit robustly captures the global signal dependency on the underlying GC composition. GC‐corrected log2 read counts were then obtained by subtracting the values predicted by the loess fit from the observed log2 read counts (residuals of the fit).
Zfp281 peaks were called on Zfp281 ChIP‐seq reads in WT 2i and WT 32 h cells using Macs2 (Zhang et al, 2008; version 2.1.1.20160309) with default parameters. Peaks that were at least twofold enriched (IP over respective inputs) in at least one of the four Zfp281 ChIP samples were considered (total: 23,756 peaks; Table EV2). For comparison of Zfp281 ChIP samples from this study to ChIP in EpiSCs (Huang et al, 2017) and TSCs (Ishiuchi et al, 2019), peaks were called on Zfp281 ChIP‐seq reads in WT 2i, WT 32 h, EpiSCs, and TSCs. Peaks that were at least twofold enriched (IP over respective inputs) in at least one of the four Zfp281 ChIP samples from this study, in the one Zfp281 ChIP sample in EpiSCs, or in one of the two Zfp281 ChIP samples in TSCs were considered (total: 27,435 peaks) and used for plotting (Fig EV3E and F). Zic2 peaks were called on Zic2 ChIP‐seq reads in WT 2i, WT 32 h, Zfp281 2i, and Zfp281 32 h cells. To quantitatively compare Zfp281 and Zic2 binding, both peak sets were combined and overlapping peak regions were merged using the function reduce from Bioconductor package GenomicRanges (Lawrence et al, 2013) (package version 1.34.0). Fused peaks were classified into single‐ or co‐bound as follows: Peaks that were enriched at least twofold (IP over respective inputs) in at least one of the four Zfp281 ChIP samples and in at least one of the eight Zic2 ChIP samples were considered as co‐bound (total: 8,312 peaks), while those if detected only in Zfp281 ChIP or only in Zic2 ChIP samples were considered as Zfp281‐only (total: 15,659 peaks) or Zic2‐only (total: 20,183 peaks), respectively (Table EV2). Reads of H3K27ac ChIP‐seq datasets were counted in Zfp281, Zic2, or Zfp281/Zic2 co‐bound peaks and normalized as described above.
Zfp281 or Zfp281/Zic2 co‐bound peaks were assigned to genes by calculating the distances of peak midpoint to the nearest transcriptional start site (TSS) using a set of non‐redundant TSSs with a single start site randomly selected for each gene. Zfp281 peaks were classified as proximal if the distance to the nearest TSS was less than 2,000 bp (7,697 peaks) and as distal otherwise (16,059 peaks; Fig EV3G). H3K27ac reads in WT 2i and WT 32 h cells, and H3K4me1 (Buecker et al, 2014) and H3K4me3 (Kurimoto et al, 2015) reads in WT 2i/Lif and EpiLCs were counted in Zfp281 peak regions and normalized as described above. Proximal Zfp281 binding sites with at least 1.5‐fold enrichment of H3K27ac over respective inputs in either WT 2i or WT 32 h cells and with at least twofold enrichment of H3K4me3 over respective inputs in either WT 2i/Lif cells or EpiLCs were considered as associated with active promoters (54% of proximal peaks; total: 4,128). Distal Zfp281 binding sites with at least 1.5‐fold enrichment of H3K4me1 over respective inputs in either WT 2i/Lif cells or EpiLCs were considered as putative enhancers (62% of distal peaks; total: 9,990), of which sites additionally enriched at least 1.5‐fold over respective inputs in H3K27ac in either WT 2i or WT 32 h cells were classified as active enhancers (38% of putative enhancers; total: 3,818). For quantification at target sites differentially bound by Zfp281 during differentiation (Figs 3F, and 7C and D), the 1,000 binding sites with strongest increase (Zfp281 UP) and decrease (Zfp281 DOWN) in Zfp281 ChIP signal in WT 32 h compared to WT 2i were considered.
Due to the broad chromatin distribution of Ehmt1 and H3K9me2, ChIP‐seq reads were first quantified in genome‐tiling windows of 10 kb, which were generated using tileGenome function from Bioconductor package GenomicRanges with tilewidth=10000 and cut.last.tile.in.chrom=TRUE. In Fig EV5H, all 10‐kb genome‐tiling windows were separated in five bins with equal number of tiles but increasing Ehmt1 ChIP log2 enrichment over respective input in WT 2i cells. To quantify Ehmt1 and H3K9me2 enrichment at Zfp281 binding sites, Zfp281 peak regions were extended to 10 kb centered on the peak midpoint using the function resize from the Bioconductor package GenomicRanges. As a control set, DHS‐seq peaks (peak annotation downloaded from ENCODE; accession number: ENCSR000CMW) were extended to 10 kb centered on the peak midpoint and only regions non‐overlapping with 10‐kb extended Zfp281 peaks were considered. The final set of control regions was obtained by randomly sampling one DHS 10‐kb peak per Zfp281 10‐kb peak, such that the distributions of DHS‐seq signal (log2 RPM) and GC content between Zfp281 10‐kb extended peaks and the selected control regions matched closely.
Boxplots were generated using the boxplot function in R with default parameters except outline=FALSE. Genomic profiles for heatmaps centered on the Zfp281 peak midpoint (Fig EV3G) were generated with qProfile from the Bioconductor package QuasR, and visualized using ComplexHeatmap (package version 1.20.0) (Gu et al, 2016). Except for Zfp281 ChIP‐seq and GRO‐seq (Dorighi et al, 2017), the averages of two replicates are shown.
Details for quantification and statistical analysis
Details of experiments are specified in figure legends, including the number of biological or technical replicates and the number of datapoints, and are further detailed in the respective Materials and Methods sections. In brief, experimental data quantification is represented as the mean and standard deviation (SD). Significances in Figs 4C, and 5A and D were determined using a Wilcoxon–Mann–Whitney rank sum test using R function wilcox_test. In Fig 2B, the computational analysis shows dots as the median and shades as the lower and upper quartile. In Figs 3C, F, G, 6C, D, and 7A–E, and EV5H–K and M, N, the computational analysis shows the median as middle line, the 95% confidence interval of the median as notches, the interquartile range as box, and 1.5 times the interquartile range as whiskers. In Fig EV5E, the estimated regression coefficients and standard errors are derived from the computational linear model.
Author contributions
DM, MR, and JB performed experiments. DM and MBS performed bioinformatical analysis. DH performed mass spectrometry. IL performed spheroid image analysis. MW, AS, and FB assisted the esiRNA screen. DM and JB wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Acknowledgements
We would like to thank S. Dessus‐Babus, K. Jacobeit, E. Pandini Figueiredo Moreno, T. Roloff, and S. Smallwood (FMI) for processing sequencing samples; L. Gelman, S. Bourke, and M. Zinner (FMI) for imaging assistance; H. Kohler (FMI) for cell sorting; M. Flemr and P. Knuckles (FMI) for advice on genome editing and ChIP; and P.A. Ginno, L. Hoerner, and A.H.F.M. Peters (FMI) for providing reagents. We are grateful to S. Gasser, M. Michalski, F. Mohn, and A.H.F.M. Peters (FMI), and S. Stricker (Helmholtz Centre) for comments on the manuscript. This work was supported by the EU Seventh Framework Programme Integrated Project SyBoSS (to A.S. and F.B.) and the Novartis Research Foundation (to J.B.). A.S is a Medical Research Council Professor.
The EMBO Journal (2020) 39: e102591
Data availability
Next‐generation sequencing data reported in this study have been deposited at the Gene Expression Omnibus with accession number GSE131017; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131017.
References
- Acampora D, Di Giovannantonio LG, Simeone A (2013) Otx2 is an intrinsic determinant of the embryonic stem cell state and is required for transition to a stable epiblast stem cell condition. Development 140: 43–55 [DOI] [PubMed] [Google Scholar]
- Acloque H, Adams MS, Fishwick K, Bronner‐Fraser M, Nieto MA (2009) Epithelial‐mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 119: 1438–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlani B, Liu Y, Chen T, Ee L‐S, Lopez P, Heguy A, Apostolou E, Kim SY, Stadtfeld M (2018) Nascent induced pluripotent stem cells efficiently generate entirely iPSC‐derived mice while expressing differentiation‐associated genes. Cell Rep 22: 876–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Tang WW, Wu B, Kim S, Li J, Li L, Kobayashi T, Lee C, Chen Y, Wei M et al (2018) Derivation of hypermethylated pluripotent embryonic stem cells with high potency. Cell Res 28: 22–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J, Nichols J, Dietmann S, Corrin PD, Paddison PJ, Smith A (2013) Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 153: 335–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Loos R, Bertone P, Smith A, Nichols J (2014) The ability of inner‐cell‐mass cells to self‐renew as embryonic stem cells is acquired following epiblast specification. Nat Cell Biol 16: 516–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Loos R, Lombard P, Okahara J, Behr R, Sasaki E, Nichols J, Smith A, Bertone P (2015) Lineage‐specific profiling delineates the emergence and progression of naive pluripotency in mammalian embryogenesis. Dev Cell 35: 366–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brons IGM, Smithers LE, Trotter MWB, Rugg‐Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund‐Richter L, Pedersen RA et al (2007) Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448: 191–195 [DOI] [PubMed] [Google Scholar]
- Buckley SM, Aranda‐Orgilles B, Strikoudis A, Apostolou E, Loizou E, Moran‐Crusio K, Farnsworth CL, Koller AA, Dasgupta R, Silva JC et al (2012) Regulation of pluripotency and cellular reprogramming by the ubiquitin‐proteasome system. Cell Stem Cell 11: 783–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, Wysocka J (2014) Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell 14: 838–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacchiarelli D, Trapnell C, Ziller MJ, Soumillon M, Cesana M, Karnik R, Donaghey J, Smith ZD, Ratanasirintrawoot S, Zhang X et al (2015) Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell 162: 412–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantzoura E, Skylaki S, Menendez S, Kim S‐I, Johnsson A, Linnarsson S, Woltjen K, Chambers I, Kaji K (2015) Reprogramming roadblocks are system dependent. Stem Cell Reports 5: 350–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, Cheng X (2008) The ankyrin repeats of G9a and GLP histone methyltransferases are mono‐ and dimethyllysine binding modules. Nat Struct Mol Biol 15: 245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Paszkowski‐Rogacz M, Nitzsche A, Slabicki MM, Heninger A‐K, de Vries I, Kittler R, Junqueira M, Shevchenko A, Schulz H et al (2009) A genome‐scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell 4: 403–415 [DOI] [PubMed] [Google Scholar]
- Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schübeler D (2015) Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 528: 575–579 [DOI] [PubMed] [Google Scholar]
- Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD, Garcia BA, Adelman K, Wysocka J (2017) Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol Cell 66: 568–576.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn S‐J, Li MA, Carbognin E, Smith A, Martello G (2019) A common molecular logic determines embryonic stem cell self‐renewal and reprogramming. EMBO J 38: e100003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebnet K, Kummer D, Steinbacher T, Singh A, Nakayama M, Matis M (2018) Regulation of cell polarity by cell adhesion receptors. Semin Cell Dev Biol 81: 2–12 [DOI] [PubMed] [Google Scholar]
- Epsztejn‐Litman S, Feldman N, Abu‐Remaileh M, Shufaro Y, Gerson A, Ueda J, Deplus R, Fuks F, Shinkai Y, Cedar H et al (2008) De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol 15: 1176–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estève P‐O, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S (2006) Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 20: 3089–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Factor DC, Corradin O, Zentner GE, Saiakhova A, Song L, Chenoweth JG, McKay RD, Crawford GE, Scacheri PC, Tesar PJ (2014) Epigenomic comparison reveals activation of ‘seed’ enhancers during transition from naive to primed pluripotency. Cell Stem Cell 14: 854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festuccia N, Osorno R, Halbritter F, Karwacki‐Neisius V, Navarro P, Colby D, Wong F, Yates A, Tomlinson SR, Chambers I (2012) Esrrb is a direct Nanog target gene that can substitute for Nanog function in pluripotent cells. Cell Stem Cell 11: 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidalgo M, Faiola F, Pereira C‐F, Ding J, Saunders A, Gingold J, Schaniel C, Lemischka IR, Silva JCR, Wang J (2012) Zfp281 mediates Nanog autorepression through recruitment of the NuRD complex and inhibits somatic cell reprogramming. Proc Natl Acad Sci USA 109: 16202–16207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidalgo M, Huang X, Guallar D, Sanchez‐Priego C, Valdes VJ, Saunders A, Ding J, Wu W‐S, Clavel C, Wang J (2016) Zfp281 coordinates opposing functions of Tet1 and Tet2 in pluripotent states. Cell Stem Cell 19: 355–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D, Lerch A, Hahne F, Stadler MB (2015) QuasR: quantification and annotation of short reads in R. Bioinformatics 31: 1130–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R, Seoighe C (2010) A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11: 367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber T, Murawala P, Knapp D, Masselink W, Schuez M, Hermann S, Gac‐Santel M, Nowoshilow S, Kageyama J, Khattak S et al (2018) Single‐cell analysis uncovers convergence of cell identities during axolotl limb regeneration. Science 362: eaaq0681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf T, Enver T (2009) Forcing cells to change lineages. Nature 462: 587–594 [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M (2016) Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32: 2847–2849 [DOI] [PubMed] [Google Scholar]
- Guo G, Yang J, Nichols J, Hall JS, Eyres I, Mansfield W, Smith A (2009) Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development 136: 1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Huang Y, Humphreys P, Wang X, Smith A (2011) A PiggyBac‐based recessive screening method to identify pluripotency regulators. PLoS One 6: e18189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M (2011) Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146: 519–532 [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Balmer S, Yang F, Fidalgo M, Li D, Guallar D, Hadjantonakis A‐K, Wang J (2017) Zfp281 is essential for mouse epiblast maturation through transcriptional and epigenetic control of Nodal signaling. Elife 6: 243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiuchi T, Ohishi H, Sato T, Kamimura S, Yorino M, Abe S, Suzuki A, Wakayama T, Suyama M, Sasaki H (2019) Zfp281 shapes the transcriptome of trophoblast stem cells and is essential for placental development. Cell Rep 27: 1742–1754.e6 [DOI] [PubMed] [Google Scholar]
- Iwafuchi‐Doi M, Matsuda K, Murakami K, Niwa H, Tesar PJ, Aruga J, Matsuo I, Kondoh H (2012) Transcriptional regulatory networks in epiblast cells and during anterior neural plate development as modeled in epiblast stem cells. Development 139: 3926–3937 [DOI] [PubMed] [Google Scholar]
- Jackson SA, Olufs ZPG, Tran KA, Zaidan NZ, Sridharan R (2016) Alternative routes to induced pluripotent stem cells revealed by reprogramming of the neural lineage. Stem Cell Reports 6: 302–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkan T, Olova N, Roode M, Mulas C, Lee HJ, Nett I, Marks H, Walker R, Stunnenberg HG, Lilley KS et al (2017) Tracking the embryonic stem cell transition from ground state pluripotency. Development 144: 1221–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Kaufman‐Francis K, Studdert JB, Steiner KA, Power MD, Loebel DAF, Jones V, Hor A, de Alencastro G, Logan GJ et al (2014) The transcriptional and functional properties of mouse epiblast stem cells resemble the anterior primitive streak. Cell Stem Cell 14: 107–120 [DOI] [PubMed] [Google Scholar]
- Kurimoto K, Yabuta Y, Hayashi K, Ohta H, Kiyonari H, Mitani T, Moritoki Y, Kohri K, Kimura H, Yamamoto T et al (2015) Quantitative dynamics of chromatin remodeling during germ cell specification from mouse embryonic stem cells. Cell Stem Cell 16: 517–532 [DOI] [PubMed] [Google Scholar]
- Ladewig J, Koch P, Brüstle O (2013) Leveling Waddington: the emergence of direct programming and the loss of cell fate hierarchies. Nat Rev Mol Cell Biol 14: 225–236 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ (2013) Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M, Dietmann S, Paramor M, Niwa H, Smith A (2014) Genetic exploration of the exit from self‐renewal using haploid embryonic stem cells. Cell Stem Cell 14: 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir E‐AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER et al (2015) Data‐driven phenotypic dissection of AML reveals progenitor‐like cells that correlate with prognosis. Cell 162: 184–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q et al (2010) A mesenchymal‐to‐epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7: 51–63 [DOI] [PubMed] [Google Scholar]
- Li M, Yu JSL, Tilgner K, Ong SH, Koike‐Yusa H, Yusa K (2018) Genome‐wide CRISPR‐KO screen uncovers mTORC1‐mediated Gsk3 regulation in naive pluripotency maintenance and dissolution. Cell Rep 24: 489–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Gao X, Lin C, Smith ER, Marshall SA, Swanson SK, Florens L, Washburn MP, Shilatifard A (2015) Zic2 is an enhancer‐binding factor required for embryonic stem cell specification. Mol Cell 57: 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA‐guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello G, Sugimoto T, Diamanti E, Joshi A, Hannah R, Ohtsuka S, Göttgens B, Niwa H, Smith A (2012) Esrrb is a pivotal target of the Gsk3/Tcf3 axis regulating embryonic stem cell self‐renewal. Cell Stem Cell 11: 491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell AJ, Stanger BZ (2016) Adult cell plasticity in vivo: de‐differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 17: 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Meyenn F, Iurlaro M, Habibi E, Liu NQ, Salehzadeh‐Yazdi A, Santos F, Petrini E, Milagre I, Yu M, Xie Z et al (2016) Impairment of DNA methylation maintenance is the main cause of global demethylation in naive embryonic stem cells. Mol Cell 62: 848–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulas C, Kalkan T, Smith A (2017) NODAL secures pluripotency upon embryonic stem cell progression from the ground state. Stem Cell Reports 9: 77–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nefzger CM, Rossello FJ, Chen J, Liu X, Knaupp AS, Firas J, Paynter JM, Pflueger J, Buckberry S, Lim SM et al (2017) Cell type of origin dictates the route to pluripotency. Cell Rep 21: 2649–2660 [DOI] [PubMed] [Google Scholar]
- Niwa H, Ogawa K, Shimosato D, Adachi K (2009) A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature 460: 118–122 [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257 [DOI] [PubMed] [Google Scholar]
- Okita Y, Matsumoto A, Yumimoto K, Isoshita R, Nakayama KI (2012) Increased efficiency in the generation of induced pluripotent stem cells by Fbxw7 ablation. Genes Cells 17: 768–777 [DOI] [PubMed] [Google Scholar]
- Osorno R, Tsakiridis A, Wong F, Cambray N, Economou C, Wilkie R, Blin G, Scotting PJ, Chambers I, Wilson V (2012) The developmental dismantling of pluripotency is reversed by ectopic Oct4 expression. Development 139: 2288–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostapcuk V, Mohn F, Carl SH, Basters A, Hess D, Iesmantavicius V, Lampersberger L, Flemr M, Pandey A, Thomä NH et al (2018) Activity‐dependent neuroprotective protein recruits HP1 and CHD4 to control lineage‐specifying genes. Nature 557: 739–743 [DOI] [PubMed] [Google Scholar]
- Pereira L, Yi F, Merrill BJ (2006) Repression of Nanog gene transcription by Tcf3 limits embryonic stem cell self‐renewal. Mol Cell Biol 26: 7479–7491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab S, Klingenstein M, Möller A, Illing A, Tosic J, Breunig M, Kuales G, Linta L, Seufferlein T, Arnold SJ et al (2017) Reprogramming to pluripotency does not require transition through a primitive streak‐like state. Sci Rep 7: 16543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, Mansour AA, Caspi I, Krupalnik V, Zerbib M et al (2013) Deterministic direct reprogramming of somatic cells to pluripotency. Nature 502: 65–70 [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol 11: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Madoz JR, San Jose‐Eneriz E, Rabal O, Zapata‐Linares N, Miranda E, Rodriguez S, Porciuncula A, Vilas‐Zornoza A, Garate L, Segura V et al (2017) Reversible dual inhibitor against G9a and DNMT1 improves human iPSC derivation enhancing MET and facilitating transcription factor engagement to the genome. PLoS One 12: e0190275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruetz T, Pfisterer U, Di Stefano B, Ashmore J, Beniazza M, Tian TV, Kaemena DF, Tosti L, Tan W, Manning JR et al (2017) Constitutively active SMAD2/3 are broad‐scope potentiators of transcription‐factor‐mediated cellular reprogramming. Cell Stem Cell 21: 791–805.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavarchi‐Tehrani P, Golipour A, David L, Sung H‐K, Beyer TA, Datti A, Woltjen K, Nagy A, Wrana JL (2010) Functional genomics reveals a BMP‐driven mesenchymal‐to‐epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7: 64–77 [DOI] [PubMed] [Google Scholar]
- Shahbazi MN, Scialdone A, Skorupska N, Weberling A, Recher G, Zhu M, Jedrusik A, Devito LG, Noli L, Macaulay IC et al (2017) Pluripotent state transitions coordinate morphogenesis in mouse and human embryos. Nature 552: 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Desponts C, Do JT, Hahm HS, Schöler HR, Ding S (2008) Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small‐molecule compounds. Cell Stem Cell 3: 568–574 [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Tachibana M (2011) H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev 25: 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim Y‐J, Kim M‐S, Nayfeh A, Yun Y‐J, Kim S‐J, Park K‐T, Kim C‐H, Kim K‐S (2017) 2i maintains a naive ground state in ESCs through two distinct epigenetic mechanisms. Stem Cell Reports 8: 1312–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A (2017) Formative pluripotency: the executive phase in a developmental continuum. Development 144: 365–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan R, Gonzales‐Cope M, Chronis C, Bonora G, McKee R, Huang C, Patel S, Lopez D, Mishra N, Pellegrini M et al (2013) Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein‐1γ in reprogramming to pluripotency. Nat Cell Biol 15: 872–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart HT, Stirparo GG, Lohoff T, Bates LE, Kinoshita M, Lim CY, Sousa EJ, Maskalenka K, Radzisheuskaya A, Malcolm AA et al (2019) Distinct molecular trajectories converge to induce naive pluripotency. Cell Stem Cell 25: 388–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H et al (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16: 1779–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3‐K9. Genes Dev 19: 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y (2008) G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J 27: 2681–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Sasaki A, Yamamoto M, Nakamura M, Sutou K, Osafune K, Yamanaka S (2014) Induction of pluripotency in human somatic cells via a transient state resembling primitive streak‐like mesendoderm. Nat Commun 5: 3678 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S (2015) A developmental framework for induced pluripotency. Development 142: 3274–3285 [DOI] [PubMed] [Google Scholar]
- Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL, Gardner RL, McKay RDG (2007) New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448: 196–199 [DOI] [PubMed] [Google Scholar]
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SAM, Sim S, Neff NF, Skotheim JM, Wernig M et al (2016) Dissecting direct reprogramming from fibroblast to neuron using single‐cell RNA‐seq. Nature 534: 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiridis A, Huang Y, Blin G, Skylaki S, Wymeersch F, Osorno R, Economou C, Karagianni E, Zhao S, Lowell S et al (2014) Distinct Wnt‐driven primitive streak‐like populations reflect in vivo lineage precursors. Development 141: 1209–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallier L, Mendjan S, Brown S, Chng Z, Teo A, Smithers LE, Trotter MWB, Cho CH‐H, Martinez A, Rugg‐Gunn P et al (2009) Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 136: 1339–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas F, Lehalle D, Mayer D, Rittirsch M, Stadler MB, Zinner M, Olivieri D, Vabres P, Duplomb‐Jego L, De Bont ESJM et al (2019) Lysosomal signaling licenses embryonic stem cell differentiation via inactivation of Tfe3. Cell Stem Cell 24: 257–270 [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One‐step generation of mice carrying mutations in multiple genes by CRISPR/Cas‐mediated genome engineering. Cell 153: 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerman BA, Braat AK, Taub N, Potman M, Vissers JHA, Blom M, Verhoeven E, Stoop H, Gillis A, Velds A et al (2011) A genome‐wide RNAi screen in mouse embryonic stem cells identifies Mp1 as a key mediator of differentiation. J Exp Med 208: 2675–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray J, Kalkan T, Gomez‐Lopez S, Eckardt D, Cook A, Kemler R, Smith A (2011) Inhibition of glycogen synthase kinase‐3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat Cell Biol 13: 838–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane M, Ohtsuka S, Matsuura K, Nakamura A, Niwa H (2018) Overlapping functions of Krüppel‐like factor family members: targeting multiple transcription factors to maintain the naïve pluripotency of mouse embryonic stem cells. Development 145: dev162404 [DOI] [PubMed] [Google Scholar]
- Yang J, van Oosten AL, Theunissen TW, Guo G, Silva JCR, Smith A (2010) Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell Stem Cell 7: 319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Li L, Huang C, Shen C, Tan F, Xia C, Liu P, Rossant J, Jing N (2010) Distinct functions of BMP4 during different stages of mouse ES cell neural commitment. Development 137: 2095–2105 [DOI] [PubMed] [Google Scholar]
- Zhang H, Gayen S, Xiong J, Zhou B, Shanmugam AK, Sun Y, Karatas H, Liu L, Rao RC, Wang S et al (2016a) MLL1 inhibition reprograms epiblast stem cells to naive pluripotency. Cell Stem Cell 18: 481–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Termanis A, Özkan B, Bao XX, Culley J, de Lima Alves F, Rappsilber J, Ramsahoye B, Stancheva I (2016b) G9a/GLP complex maintains imprinted DNA methylation in embryonic stem cells. Cell Rep 15: 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Morales MG, Hashimoto H, Dickson ME, Song K, Ye W, Kim MS, Niederstrasser H, Wang Z, Chen B et al (2017) ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression. Genes Dev 31: 1770–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylicz JJ, Dietmann S, Günesdogan U, Hackett JA, Cougot D, Lee C, Surani MA (2015) Chromatin dynamics and the role of G9a in gene regulation and enhancer silencing during early mouse development. Elife 4: 717 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Data Availability Statement
Next‐generation sequencing data reported in this study have been deposited at the Gene Expression Omnibus with accession number GSE131017; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131017.