

Key Points
Factor IX, but not factor XI, is essential for the postnatal survival of mice under conditions of low TF.
Embryonic factor XI, but not factor IX, prevents enlargement of blood pools in low-TF placentas.
Abstract
The intrinsic tenase complex (FIXa-FVIIIa) of the intrinsic coagulation pathway and, to a lesser extent, thrombin-mediated activation of FXI, are necessary to amplify tissue factor (TF)-FVIIa–initiated thrombin generation. In this study, we determined the contribution of murine FIX and FXI to TF-dependent thrombin generation in vitro. We further investigated TF-dependent FIX activation in mice and the contribution of this pathway to hemostasis. Thrombin generation was decreased in FIX- but not in FXI-deficient mouse plasma. Furthermore, injection of TF increased levels of FIXa-antithrombin complexes in both wild-type and FXI−/− mice. Genetic studies were used to determine the effect of complete deficiencies of either FIX or FXI on the survival of mice expressing low levels of TF. Low-TF;FIX−/y male mice were born at the expected frequency, but none survived to wean. In contrast, low-TF;FXI−/− mice were generated at the expected frequency at wean and had a 6-month survival equivalent to that of low-TF mice. Surprisingly, a deficiency of FXI, but not FIX, exacerbated the size of blood pools in low-TF placentas and led to acute hemorrhage and death of some pregnant dams. Our data indicate that FIX, but not FXI, is essential for survival of low-TF mice after birth. This finding suggests that TF-FVIIa–mediated activation of FIX plays a critical role in murine hemostasis. In contrast, FXI deficiency, but not FIX deficiency, exacerbated blood pooling in low-TF placentas, indicating a tissue-specific requirement for FXI in the murine placenta under conditions of low TF.
Visual Abstract
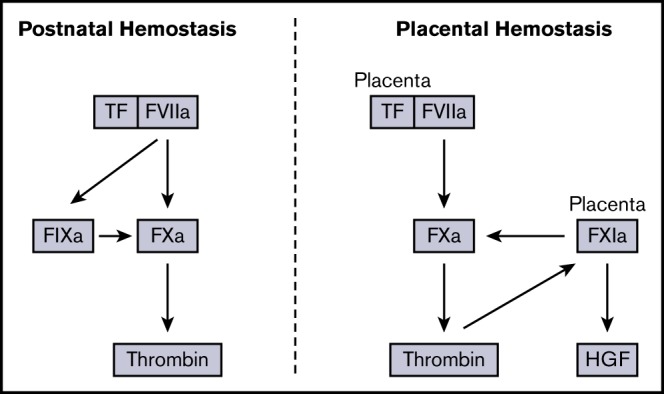
Introduction
The coagulation cascade can be divided into the extrinsic (tissue factor [TF]–factor [F] VIIa complex), intrinsic (FXIIa, FXIa, FIXa, and FVIIIa), and common (FXa, FVa, and thrombin) pathways.1 The extrinsic pathway is the physiologic trigger of blood coagulation and activates both FIX and FX.2-4 TF-FVIIa–mediated activation of FIX was initially described by Francois Josso and colleagues2 and is commonly referred to as the “Josso loop.” The intrinsic pathway also contains a feedback loop in which thrombin activates FXI leading to further generation of FIXa.5,6 These pathways are thought to play an importnant role in thrombin generation, because the TF-FVIIa complex is rapidly inhibited by TF pathway inhibitor (TFPI).7
Humans deficient in FVIII (hemophilia A) or FIX (hemophilia B) have hemostatic defects.8 Similarly, FVIII−/− and FIX−/− mice exhibit increased bleeding after a hemostatic challenge, such as tail transection.9,10 FXI-deficient patients rarely have spontaneous bleeding events, but increased bleeding occurs in some patients on provocation, particularly in tissues with high fibrinolytic activity, such as the mouth, nose, and genitourinary tract.11,12 FXI is thought to stabilize the fibrin clot and protect it from fibrinolysis.13,14 FXI−/− mice have normal bleeding times in the tail vein transection model.15 One study reported a significant increase in bleeding in FXI−/− mice in the saphenous vein injury model,16 but this was not confirmed in an independent study.17 In vitro studies have shown that FXI contributes to thrombin generation in human plasma initiated by low, but not high, concentrations of TF.18-20
In contrast to studies with mice lacking components of the intrinsic pathway, a complete deficiency of components of the extrinsic or common pathways in mice leads to death during embryonic development or shortly after birth.21-28 For instance, 80% to 90% of TF−/− embryos die at embryonic day (E) 9.5-E10.5.21-23 TF−/− mice can be rescued with a transgene that expresses human TF (hTF) regulated by the human TF promoter.29 These “low-TF” mice (murine TF−/− [mTF−/−];hTF+/+) express ∼1% levels of TF. Low-TF mice exhibit spontaneous hemostatic defects in various organs and have significantlyincreased bleeding in the tail vein transection model.1,30-32 Mice expressing ∼1% levels of FVII have phenotypes similar to those of low-TF mice.33,34 It is notable that maternal blood pooling occurs in placentas of low-TF embryos but does not cause any loss of embryos or fatal hemorrhages in the dams.31 The placenta is derived from embryonic tissue, indicating that blood pooling is related to low levels of TF in the embryo.
In the current study, we investigated the contribution of FIX and FXI to thrombin generation in vitro and in vivo under conditions of low TF. Plasma-based thrombin generation assays were used to assess the relative contribution of FIX and FXI to TF-initiated thrombin generation. The ability of the TF-FVIIa complex to activate FIX in vivo was also determined in wild-type and FXI−/− mice. Finally, a genetic approach was employed to determine the effect of a complete deficiency of either FIX or FXI on the survival of mice expressing low levels of TF.
Methods
Mice
FIX−/− and FXI−/− mice were backcrossed 10 generations to the C57BL/6J background.10,15 Low-TF mice (mTF−/−;hTF+/+) were backcrossed 6 generations to the C57BL/6J background.29 Studies complied with National Institutes of Health Guide for the Care and Use of Laboratory Animals and were performed with the approval of the Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
All additional details of experimental procedures are given in the supplemental Methods.
Results
Effect of either FIX or FXI deficiency on TF-initiated thrombin generation in murine plasma
The effect of FIX deficiency on TF-initiated thrombin generation in mouse platelet-poor plasma (PPP) was determined. Endogenous thrombin potential (ETP), time to peak, peak, and lag time were measured. As expected, a significant reduction in peak thrombin generation and ETP was observed in FIX-deficient plasma compared with control plasma when thrombin generation was initiated with a low concentration of TF (Figure 1A; data not shown).
Figure 1.

Effect of deficiency of FIX or FXI on thrombin generation in murine plasma. (A) Thrombin generation in murine FIX-deficient PPP was initiated with low and high concentrations of hTF. Thrombin generation in mFXI-deficient PPP collected in the absence of CTI was initiated with silica (B) or low and high doses of hTF (C). Thrombin generation in mFXI-deficient PPP collected in the presence of CTI was initiated with silica (D) or low and high doses of hTF (E). Thrombin generation in mFXI-deficient PRP collected in the presence of CTI was initiated with silica (F) or low and high doses of hTF (G). Data are presented as individual values with means ± 95% confidence intervals. *P < .05, 2-way analysis of variance with post hoc Bonferroni test (FIX+/+ vs FIX−/−); **P < .001, 1-way analysis of variance with post hoc Bonferroni test (FXI+/+ and FXI+/− vs FXI−/−).
Next, the contribution of FXI to thrombin generation was determined in mouse plasma in which activation of coagulation was initiated by either silica or TF. As expected, silica-initiated thrombin generation was significantly impaired in FXI−/− PPP compared with FXI+/− or FXI+/+ PPP (Figure 1B). In contrast, there was no significant difference in thrombin generation (lag time, time to peak, peak, or ETP) in FXI−/− PPP compared with FXI+/+ or FXI+/− PPP, at a low concentration of TF (Figure 1C; data not shown). To exclude the potential effects of contact pathway activation during blood collection, additional samples were collected in the presence of corn trypsin inhibitor (CTI). However, even in the presence of CTI, despite observing a robust reduction in silica-induced thrombin generation (Figure 1D; data not shown), there was no difference in TF-initiated thrombin generation between FXI+/+, FXI+/−, or FXI−/− PPP (Figure 1E; data not shown).
Platelets provide an important surface for the activation of FXI and may be important in FXI-mediated amplification of thrombin generation.35-37 To account for the potential effects of platelets we assessed thrombin generation in platelet-rich plasma (PRP). When coagulation was initiated with silica, thrombin generation was significantly reduced in PRP from FXI−/− mice compared to FXI+/− or FXI+/+ controls (Figure 1F). In contrast, when coagulation was initiated with TF, there was no difference in thrombin generation in PRP from FXI−/− mice compared to FXI+/− or FXI+/+ controls (Figure 1G; data not shown). Collectively, these data in PPP and PRP show that, although FXI deficiency was detectable in mouse plasma in contact silica-initiated assays, FXI did not contribute to thrombin generation in mouse plasma when initiated by TF.
TF activates FIX in vivo in the absence of FXI
To determine whether TF can activate FIX in vivo, mice were injected with TF and plasma levels of FIXa-antithrombin (AT) complexes measured. The thrombin inhibitor dabigatran was used to prevent death associated with the formation of intravascular thrombi after injection of TF and to block thrombin-mediated activation of FXI. Injection of TF into wild-type mice significantly increased the plasma levels of FIXa-AT complexes compared with controls (Figure 2A). FXI−/− mice were used to eliminate the thrombin-FXIa feedback loop. Similar to the results in wild-type mice, TF injection significantly increased the plasma levels of FIXa-AT complexes in thrombin-inhibited FXI−/− mice (Figure 2B). These results indicate that the TF-FVIIa complex activates FIX in mice.
Figure 2.

TF activates FIX in thrombin-inhibited wild-type and FXI−/−mice. Plasma levels of FIXa-AT complexes were quantified in dabigatran-pretreated wild-type (WT) (A) or FXI−/− (B) mice, administered either vehicle control or TF (n = 6-10 per group). A significant increase in plasma FIXa-AT complexes was observed on administration of TF in both WT and FXI−/− mice. Data are presented as individual values with means ± 95% confidence intervals. *P < .0001; **P < .05; unpaired Student t test.
Effect of FIX deficiency on the survival of low-TF offspring
The FIX gene resides on the X chromosome. Therefore, to study the effect of FIX deficiency on low-TF mice, male mTF−/−;hTF+/+;FIX+/y and female mTF+/−;hTF+/−;FIX+/− were crossed and the allele frequencies of offspring determined at various timepoints. No male low-TF;FIX−/y offspring were observed at postnatal day (P) 21, a significant reduction compared with the expected frequency (Table 1). Next, offspring were analyzed at E14.5 to determine if low-TF;FIX−/y offspring had survived development beyond E9.5 and E10.5 given that embryos lacking various coagulation factors die at this time point. Viable low-TF;FIX−/y offspring were observed at E14.5 (Table 1) and appeared grossly normal by histological analysis (data not shown). Finally, the number of low-TF;FIX−/y at P1 was analyzed. Male low-TF;FIX−/y offspring were observed at P1 (Table 1). However, 2 of the 5 (40%) male P1 low-TF;FIX−/y offspring were dead. One low-TF;FIX−/y P1 pup was pale and had a dark abdomen, which was consistent with abdominal hemorrhage (supplemental Figure 1). One of the 6 (17%) male low-TF;FIX+/y P1 pups was dead with no apparent phenotype. These results indicate that FIX is essential for postnatal survival of low-TF mice.
Table 1.
Distribution of genotypes at weaning from a breeding to generate low-TF;FIX−/− offspring
| Sex | mTF | hTF | FIX | Expected, % | E14.5, n | E14.5 observed, % | E14.5 O/E, % | P1, n | P1 observed, % | P1 O/E, % | P21, n | P21 observed, % | P21 O/E, % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F | +/− | +/(+/−) | +/+ | 12.5 | 7 | 18.4 | 147 | 3 | 6.6 | 52 | 15 | 18.5 | 148 |
| F | +/− | +/(+/−) | +/− | 12.5 | 7 | 18.4 | 147 | 7 | 15.5 | 147 | 13 | 16.0 | 128 |
| M | +/− | +/(+/−) | +/y | 12.5 | 1 | 2.6 | 21 | 10 | 22.2 | 178 | 15 | 18.5 | 148 |
| M | +/− | +/(+/−) | −/y | 12.5 | 4 | 10.5 | 84 | 8 | 17.7 | 142 | 9 | 11.1 | 89 |
| F | −/− | +/(+/−) | +/+ | 12.5 | 5 | 13.2 | 106 | 4 | 8.8 | 70 | 8 | 9.9 | 79 |
| F | −/− | +/(+/−) | +/− | 12.5 | 5 | 13.2 | 106 | 2 | 4.4 | 35 | 12 | 14.8 | 118 |
| M | −/− | +/(+/−) | +/y | 12.5 | 5 | 13.2 | 106 | 6* | 13.3 | 106 | 9 | 11.1 | 89 |
| M | −/− | +/(+/−) | −/y | 12.5 | 4 | 10.5 | 84 | 5† | 11.1 | 89 | 0 | 0 | 0‡ |
| Total | 100.0 | 38 | 100.0 | — | 45 | 100.0 | — | 81 | 100.0 | — | |||
F, female; hTF+/(+/−), hTF+/− or hTF+/+; M, male; O/E, observed/expected.
One of 6 dead.
Two of 5 dead.
P < .05 observed vs expected, 2-tailed Fisher’s exact test.
FXI deficiency does not affect the generation or survival of low-TF offspring
The effect of FXI deficiency on the generation and survival of mice expressing low levels of TF was determined. Male and female F1 intermediates (mTF+/−;hTF+/−;FXI+/−) were crossed, and the allele frequencies of 117 offspring were determined. Six of 117 (5.13%) of the offspring at wean were low TF;FXI−/−, which was not significantly different from the expected frequency of 4.69% (Table 2). Four additional crosses with sires and dams of differing genotypes were conducted to yield larger numbers of low-TF;FXI−/− offspring. Similar to the results observed when crossing F1 intermediates low-TF;FXI−/− mice were generated at a frequency that was not significantly different from the expected frequency in these 4 additional breedings (supplemental Table 1). These observations indicate that, even when overall procoagulant potential is reduced under conditions of low TF, FXI is not necessary for survival in mice.
Table 2.
Distribution of genotypes at weaning from a breeding to generate low-TF;FXI−/− offspring
| mTF | hTF | FXI | Expected, % | Observed, n | Observed, % | O/E, % |
|---|---|---|---|---|---|---|
| +/+ | — | +/+ | 1.56 | 4 | 3.42 | 219 |
| +/+ | — | +/− | 3.13 | 4 | 3.42 | 109 |
| +/+ | — | −/− | 1.56 | 4 | 3.42 | 219 |
| +/+ | +/(+/−) | +/+ | 4.69 | 6 | 5.13 | 109 |
| +/+ | +/(+/−) | +/− | 9.38 | 10 | 8.55 | 91 |
| +/+ | +/(+/−) | −/− | 4.69 | 2 | 1.71 | 36 |
| +/− | — | +/+ | 3.13 | 2 | 1.71 | 55 |
| +/− | — | +/− | 6.25 | 8 | 6.84 | 109 |
| +/− | — | −/− | 3.13 | 3 | 2.56 | 82 |
| +/− | +/(+/−) | +/+ | 9.38 | 16 | 13.68 | 146 |
| +/− | +/(+/−) | +/− | 18.75 | 25 | 21.37 | 114 |
| +/− | +/(+/−) | −/− | 9.38 | 11 | 9.40 | 100 |
| −/− | — | +/+ | 1.56 | 0 | 0.00 | 0* |
| −/− | — | +/− | 3.13 | 0 | 0.00 | 0* |
| −/− | — | −/− | 1.56 | 0 | 0.00 | 0* |
| −/− | +/(+/−) | +/+ | 4.69 | 8 | 6.84 | 146 |
| −/− | +/(+/−) | +/− | 9.38 | 8 | 6.84 | 73 |
| −/− | +/(+/−) | −/− | 4.69 | 6 | 5.13 | 109 |
| Total | 100.00 | 117 | 100.00 | — | ||
Expected embryonic lethality.
Next, the effect of FXI deficiency on the survival of low-TF mice between weaning and 6 months of age was determined. As expected from previous studies,38,39 27% (3 of 11) of low-TF;FXI+/+ mice died during the 6-month survival study, with evidence of pulmonary hemorrhage in all mice. Importantly, survival did not differ significantly between low-TF;FXI−/−, low TF;FXI+/− offspring, and low-TF;FXI+/+ controls (Figure 3).
Figure 3.

Survival of low-TF mice with different levels of FXI. Low-TF;FXI+/+, low-TF;FXI+/−, and low-TF;FXI−/− mice were assessed in a 6-month survival study. As expected, 27% (3 of 11) of low-TF;FXI+/+ mice died by 6 months with evidence of pulmonary hemorrhage. By comparison 10% (2 of 21) of low-TF;FXI+/− and 25% (3 of 12) of low-TF;FXI−/− mice died by 6 months with evidence of pulmonary hemorrhage. No significant difference in survival was observed between groups. P > .05, log-rank test.
Death of pregnant dams bearing low-TF;FXI−/− embryos
While generating low-TF;FXI−/− offspring, the death of pregnant mTF+/−;hTF+/(+/−);FXI+/− (n = 6) and mTF+/−;hTF+/(+/−);FXI−/− (n = 5) dams bearing low-TF;FXI−/− embryos occurred during late gestation (E13.5-15.5) in all 5 breedings (Table 3). In breeding 1, death occurred in 9% (2 of 33) of pregnancies in mTF+/−;hTF+/(+/−);FXI+/− dams. Death of 20% to 40% of pregnant mTF+/−;hTF+/(+/−);FXI+/− dams in breedings 2 and 3 and of 30% to 33% of the pregnant mTF+/−;hTF+/(+/−);FXI−/− dams in breedings 4 and 5 was observed. These results suggest that the death of the pregnant dams was associated with the relative burden of low-TF;FXI−/− embryos and was largely independent of the maternal FXI genotype.
Table 3.
Observed low-TF;FXI−/− offspring at weaning from additional breedings and death of females
| Breeding type | Expected, % | Dam FXI genotype | Death/pregnancy, n/n (%) | Survived | Died | ||
|---|---|---|---|---|---|---|---|
| n/N at P21 (litters) | Observed, % | n/N at E13-15.5 (litters) | Observed, % | ||||
| 1 | 4.69 | +/− | 2/23 (9) | — | — | — | — |
| 2 | 10.26 | +/− | 2/5 (40) | 1/25 (3) | 4.0 | 4/14 (2)* | 29.0 |
| 3 | 10.26 | +/− | 2/10 (20) | 3/40 (6) | 7.5 | 7/19 (2)* | 37.0 |
| 4 | 10.26 | −/− | 2/6 (33) | 2/19 (3) | 10.5 | 2/7 (1) | 29.0 |
| 5 | 20.52 | −/− | 3/10 (30) | 3/22 (7) | 13.6 | 6/17 (3) | 35.3 |
P < .05, difference in observed frequency of mTF−/−;hTF+/(+/−);FXI−/− in surviving mothers vs dead mothers assessed by 2-tailed Fisher’s exact test.
There was a trend toward increased death of the pregnant dams as the burden of low-TF;FXI−/− embryos increased (Table 3). To further investigate whether there was an association between the frequency of low-TF;FXI−/− offspring and maternal death, the burden of low-TF;FXI−/− offspring was determined for pregnancies in which the dam survived vs those in which the dam died. Interestingly, dams that died carried a higher proportion of low-TF;FXI−/− embryos compared to the dams that survived (Table 3).
FXI deficiency is associated with increased size of blood pools in low-TF placentas
Necropsies were conducted on 9 of the 11 dams carrying low-TF;FXI−/− embryos that had either died (n = 8) or had been euthanized due to uterine hemorrhage (n = 1) during late gestation. Free blood was observed in the uterus of 3 of 9 (33%) of the females that died with evidence of ruptured low-TF;FXI−/− placentas, suggesting that some of the pregnant females carrying low-TF;FXI−/− embryos died of acute uterine or placental hemorrhage.
The size of the maternal blood pools in low-TF;FXI−/− placentas was compared to those in low-TF;FXI+/− placentas and low-TF;FXI+/+ placentas removed from dead or euthanized dams (Figure 4A-B). The average size of the maternal blood pools was significantly increased in low-TF;FXI−/− placentas compared with low-TF;FXI+/+ placentas (Figure 4B). In addition, 5 of 17 low-TF;FXI−/− placentas appeared to have ruptured (Figure 4B). Of note, FIX deficiency did not affect the size of the blood pools (Figure 4B). Importantly, no abnormalities were observed in FXI−/− placentas with 50% or 100% levels of mouse TF (Figure 4A).
Figure 4.

Analysis of the contribution of FIX and FXI to placental blood pooling under conditions of low TF. (A) Representative images of placentas from 2 dams from breeding 3 (top row) and 4 (bottom row), each showing a large blood pool in a low-TF;FXI−/− placenta. Genotypes have been abbreviated to FXI+/− (mTF+/−;hTF+/(+/−);FXI+/−), FXI−/− (mTF+/+;hTF+/(+/−);FXI−/−), low TF;FXI+/− (mTF−/−;hTF+/(+/−);FXI+/−), and low TF;FXI−/− (mTF−/−;hTF+/(+/−);FXI−/−). Blood pools are indicated by arrowheads or asterisks in placentas of E14.5 sections stained with hematoxylin and eosin. (B) The percentage of placental area occupied by maternal blood pools in placentas of different genotypes was quantified at E13.5-E15.5 (n = 4-17 per group). Filled circles indicate ruptured placentas. A significant increase in the percentage area occupied by blood pools was observed in low-TF;FXI−/− placentas compared with low-TF;FXI+/+ placentas. *P < .01, Kruskal-Wallis with post hoc Dunn’s tests; low TF;FIX+/y;FXI+/+ (LTF) vs low TF;FXI−/−. Data represented as individual values with median. (C) F9, F11, and F12 gene expression in the placenta at E14.5 was assessed by quantitative polymerase chain reaction and expressed relative to expression of these genes in the liver. Data are the mean ± SD. **P < .0001 unpaired Student t test (liver vs placenta). (D) Representative western blots of TFPI, pro-HGF, and β-actin in tissue lysates from FXI+/+ and FXI−/− placentas. Densitometric analysis of TFPI (E) and pro-HGF (F) in FXI−/− placentas and FXI+/+ controls (n = 8 per group) normalized against β actin expression. Data are presented as individual values with the median. ***P < .01; ****P < .05, Mann-Whitney U test.
Given the observation that FXI deficiency exacerbated placental blood pooling under conditions of low TF, F9, F11, and F12 gene expression was assessed in wild-type mouse placentas. A high level of F11 mRNA, but not of F9 or F12 mRNA, was observed in the placenta (Figure 4C), suggesting that local FXI expression plays a role in the placenta.
The contribution of FIX-independent functions of FXI in the exacerbation of blood pooling in low-TF placentas was investigated. FXIa has been shown to proteolytically inactivate TFPI.40 Conversely, FXIa can proteolytically cleave pro-hepatocyte growth factor (pro-HGF) to active HGF.41 Therefore, levels of TFPI and pro-HGF were analyzed in placentas from FXI−/− and FXI+/+ embryos. If FXIa were a major proteolytic inactivator of TFPI, increased expression of full-length TFPI protein would be expected in FXI−/− placentas. However, TFPI protein expression in FXI−/− placentas was significantly lower than that in FXI+/+ controls (Figure 4D-E). In contrast, levels of pro-HGF protein were significantly higher in FXI−/− placentas than in FXI+/+ controls (Figure 4D, F), suggesting that FXIa regulates activation of pro-HGF in the placenta.
Discussion
The FIXa;FVIIIa complex has been proposed to sustain activation of coagulation and thrombin generation after inhibition of the TF-FVIIa complex by TFPI. Importantly, the TF-FVIIa complex is known to activate both FIX and FX in vitro.1-3,42 Indeed, an in vitro study comparing activation of FIX and FX by the TF-FVIIa complex concluded that FIX is the major substrate.4 In addition, individuals with a point mutation in FIX that reduces interaction with TF-FVIIa but not FXI have a mild form of hemophilia B.43 Furthermore, basal levels of FIXa activation are reduced in FVII-deficient (<7% levels) but not in FXI-deficient (<8% levels) individuals.44 Taken together, these in vitro and in vivo studies indicate that the TF-FVIIa complex is a major activator of FIX.
Consistent with the important contribution of TF-FVIIa–mediated activation of FIX to thrombin generation, a deficiency of FIX markedly blunted TF-initiated thrombin generation in human plasma.45-47 A similar approach has been taken to investigating the contribution of thrombin-mediated FXI activation to thrombin generation. In FXI-deficient human plasma, thrombin generation is impaired at a low, but not a high, initiating concentration of TF.20,48,49 Taken together, these data suggest that, in humans, TF-FVIIa–mediated activation of FIX and, to a lesser extent, thrombin-mediated activation of FXI both contribute to thrombin generation. However, it is unclear whether these amplificatory pathways also contribute to thrombin generation and hemostasis in mice.
In the present study, a deficiency in FIX resulted in a modest, but significant, impairment in thrombin generation in mouse PPP at a low, but not a high, initiating dose of TF. In contrast, no defect in TF-initiated thrombin generation was observed in FXI-deficient mouse PPP at this same low initiating dose of TF. Inhibition of exogenous contact activation by the FXIIa inhibitor CTI at the time of blood collection failed to reveal a contribution of FXI to TF-initiated thrombin generation in PPP. Platelets have been identified as an important physiologic surface for the activation of FXI by thrombin.35-37 TF-initiated thrombin generation, however, was unchanged in FXI-deficient PRP. These data support the involvement of FIX, but not FXI, in TF-initiated thrombin generation in mouse plasma. It is interesting to note that in murine PPP the contribution of FIX to TF-initiated thrombin generation was observed only at 0.05 pM of TF, whereas in human PPP, impaired thrombin generation was observed in FIX-deficient plasma at doses as high as 1 pM. This result suggests that thrombin generation in humans may have a greater dependence on FIX than in mice.
Given the similarity in the components and general mechanism of the hemostatic systems in humans and mice, it is somewhat surprising that FXI does not seem to serve a similar role in the amplification of coagulation in these 2 mammals. Interestingly, it was recently demonstrated that, unlike humans, most of the FXI in mice is localized to the vascular surface through binding to glycosaminoglycans, owing to an additional cluster of basic residues on the Apple 4 domain of mouse FXI.50 This process leads to three- to four-fold lower levels of circulating FXI in mice compared to humans.
To confirm the ability of the TF-FVIIa complex to activate murine FIX in vivo, dabigatran-anticoagulated mice were injected with exogenous TF, and FIXa generation was determined by measurement of plasma levels of FIXa-AT complexes. Injection of TF into wild-type mice resulted in generation of significant levels of FIXa. This finding was also replicated in FXI−/− mice, which eliminated the notion of any contribution of the thrombin-FXI pathway to the observed generation of FIXa. This approach provides the first direct evidence of a functional Josso loop in mice.
Having demonstrated the ability of the TF-FVIIa complex to activate FIX in vivo in mice, we used a complementary genetic approach to investigate the contribution of TF-FVIIa–mediated activation of FIX to murine embryonic development and hemostasis. Mice were crossed to generate offspring in which expression of a low level of TF was combined with a complete deficiency of FIX. Surprisingly, the expected number of male low-TF;FIX−/y embryos was observed at E14.5, indicating that these embryos survived the E9.5 to 10.5 period associated with the death of mTF−/− embryos.21-23 Birth presents a strong hemostatic challenge as pups are squeezed through the birth canal. Mice with a complete absence of different components of the extrinsic (FVII) and common (FV, FX, and prothrombin) pathways exhibit a high rate of death at birth and in the immediate postnatal period.24-27 Some of these pups lacking key coagulation factors appear to die of acute abdominal hemorrhage after birth and appear pale rather than pink, with dark abdomens due to blood pooling.24-27 Consistent with this phenotype, 2 of 5 (40%) of the low-TF;FIX−/y male mice died after birth, and 1 pup was pale with a dark abdomen, consistent with death due to an acute abdominal hemorrhage. In addition, 1 of 6 (17%) of the low-TF male mice also died at birth, which was likely caused by a hemostatic defect.
Low-TF mice backcrossed 7 generations to a C57BL/BJ background demonstrate a spontaneous hemostatic defect that results in the death of 40% to 60% of mice by 6 months of age, primarily as a result of spontaneous hemorrhage in the lung.38,39 In contrast, FIX−/− mice do not exhibit a spontaneous hemostatic defect, but have increased bleeding upon challenge, such as a tail transection, despite the presence of normal levels of TF. The postnatal lethality associated with low TF;FIX−/y mice, therefore, represents a marked exacerbation of the phenotype observed in either low-TF mice or FIX-deficient mice. Importantly, the data suggest that FIX is essential for the survival of low-TF mice during the trauma of birth and in the immediate postnatal period.
To evaluate the contribution of thrombin-mediated activation of FXI, attempts were made to generate low-TF mice lacking FXI. Low-TF;FXI−/− mice were generated at the expected frequency at wean from multiple breedings involving both FXI+/− and FXI−/− dams, in contrast to the attempts to generate low-TF;FIX−/y mice. Moreover, the survival of low-TF;FXI−/− mice was not different from that of low-TF;FXI+/− or low-TF;FXI+/+ mice between wean and 6 months of age. This result differs from the reduced survival of low-TF;PAR4−/− mice during the same period.39 The results from this genetic approach suggest that FIX, but not FXI, contributes to thrombin generation and hemostasis under conditions of reduced TF in mice.
During the generation of low-TF;FXI−/− mice, several pregnant dams died during late gestation. Unexpectedly, in pregnant dams that died, several of the placentas from low-TF;FXI−/− embryos were found to have large maternal blood pools. Previous studies have demonstrated that placentas from low-TF offspring contain small maternal blood pools in the labyrinth layer.31 It is important to note that, over a 20-year period, our group has not observed rupture of placentas from low-TF embryos or death of pregnant mTF+/− dams. No defect was observed in placentas from FXI−/− offspring, in which TF levels were normal, suggesting that a role of FXI in the placenta is revealed only under conditions of low TF.
The death of the pregnant dams appeared to be related to the rupture of low-TF;FXI−/− placentas and subsequent uterine hemorrhage. Both FXI+/− and FXI−/− dams carrying low-TF;FXI−/− embryos died during mid- to late gestation, suggesting that embryonic, but not maternal, FXI limits the size of the placental blood pools in low-TF placentas. A trend toward an increased rate of death of dams was observed as the expected frequency of low-TF;FXI−/− embryos increased in different breedings. In addition, death of the pregnant dams in each of 5 breedings was associated with the percentage of low-TF;FXI−/− embryos carried by the dam. Indeed, pregnant dams that died had a significantly higher percentage of low-TF;FXI−/− embryos than did dams that survived. Interestingly, the murine placenta was found to express high levels of F11 mRNA, but not of F9 or F12 mRNA, which suggests a specific role for FXI in the murine placenta. In contrast, FXI is not expressed in human placenta.51 These contrasting findings provide evidence of another potential species-specific difference in FXI biology between humans and mice. It is possible that FXI expression in the mouse placenta serves as an adaptive response to the reduced levels of circulating FXI.50
We have previously shown that placentas of low-TF embryos contain blood pools.31 Maternal lacunae are separated by a barrier formed of labyrinth trophoblast cells. Ultrastructural analysis of low-TF placentas revealed a reduction in the number of contacts between trophoblasts and a thinning of the barrier spanning the blood space between adjacent trabeculae.31 This mechanism appears to be the one underlying the coalescence of maternal lacunae and formation of blood pools. It was unclear whether the primary defect in low-TF placentas was hemostatic or structural. It is notable that there is also a reduction in contacts between mesoderm and endoderm in the yolk sac of TF−/− embryos at E9.5.21 Interestingly, blood pools in low-TF placentas were reduced by concomitant deletion of TFPI, presumably because of increased local thrombin generation.38
It is possible that FXI contributes to thrombin generation in the placenta. It has recently been shown that human FXI can bypass FIX and restore hemostasis in FIX−/− mice.17 Indeed, 2 studies have shown that FXIa can activate FV, FVIII, and FX.17,52 High levels of FXI in the placenta may drive a similar pathway that enhances local thrombin generation. A deficiency of FXI would further reduce thrombin levels under low-TF conditions and could contribute to the observed increase in maternal blood pools in placentas from low-TF;FXI−/− embryos. In addition, it is possible that levels of TF are lower in the placenta than in other tissues, making this tissue more dependent on thrombin-mediated activation of FXI. Interestingly, FXIa has also been shown to proteolytically inactivate TFPI.40 It is possible that loss of FXI in the placenta leads to increased levels of TFPI that further decrease FXa and thrombin generation in low-TF placentas. However, analysis of placentas from FXI+/+ and FXI−/− embryos revealed that loss of FXI resulted in a paradoxical decrease in TFPI expression that would not explain the observed phenotype.
An alternative explanation for the increase in blood pooling in low-TF placentas lacking FXI is that FXIa regulates factors required for placenta development. For instance, FXIa can also serve as a noncanonical activator of pro-HGF to active HGF.41 HGF has been found to serve as an important regulator of placental development. Deletion of HGF severely impaired placental development between E13.5 and E15.5 resulting in the death of the embryos.53,54 This is the same period during which blood pooling was observed in low-TF and low-TF;FXI−/− placentas. HGF−/− placentas had significantly reduced numbers of labyrinth trophoblast cells with poorly developed maternal lacunae.53 Importantly, significantly higher levels of pro-HGF were observed in FXI−/− placentas, suggesting that FXIa could function as an important activator of HGF during placental development. A reduction in HGF caused by an absence of FXI may explain the observed increase in blood pooling in low-TF placentas.
In summary, in this study, the ability of the TF-FVIIa complex to activate FIX in vivo was confirmed. Furthermore, under conditions of low TF, a complete deficiency in FIX was found to result in postnatal death of mice. This finding is consistent with a critical function for TF-FVIIa–mediated activation of FIX in thrombin generation. In contrast, under conditions of low TF, a complete deficiency in FXI was compatible with murine survival. However, under conditions of low TF, FXI, but not FIX, appeared to be essential for preventing expansion of placental blood pools, supporting a tissue-specific requirement for FXI under conditions of low TF in mice.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Ying Zhang for expert technical assistance and Bentley Midkiff in the Translational Pathology Laboratory for expert technical assistance.
This work was supported by a John C. Parker Professorship (N.M.). S.P.G. is supported by an American Heart Association postdoctoral fellowship (19POST34370026). M.L.P. was supported by a grant from the National Science Foundation (1559922). The UNC Translational Pathology Laboratory is supported in part by grants from the National Institutes of Health, National Cancer Institute (5P30CA016080-42 and U54-CA156733), National Institute of Environmental Health Sciences (3P30 EOS010126-17), the University Cancer Research Fund, and the North Carolina Biotechnology Center (2015-IDG-1007).
Authorship
Contribution: S.P.G., N.M., A.M., A.S.W., A.C.C., M.V., and S.H. designed the experiments; S.P.G., C.M.S., A.C.A., E.B., A.M., M.L.P., A.C.C., and M.V. conducted the experiments and analyzed the data; S.P.G., N.M., A.M., A.S.W., A.C.C., M.V., S.H., J.J.P., H.M.S., S.A., D.G., and R.P. interpreted the data; S.P.G. and N.M. drafted the manuscript; S.P.G., N.M., A.S.W., A.C.C., S.H., H.M.S., S.A., R.P., and D.G. edited the manuscript; and all authors read and approved the manuscript before submission.
Conflict-of-interest disclosure: N.M. and H.M.S. are consultants for Bayer. S.H. is an employee of Bayer. The remaining authors declare no competing financial interests.
Correspondence: Nigel Mackman, Department of Medicine, University of North Carolina at Chapel Hill, 8004B Mary Ellen Jones Building, 116 Manning Dr, Chapel Hill, NC 27599; e-mail: nmackman@med.unc.edu.
References
- 1.Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. 2018;38(4):709-725. [DOI] [PubMed] [Google Scholar]
- 2.Josso F, Prou-Wartelle O. Interaction of tissue factor and factor VII at the earliest phase of coagulation. Thromb Diath Haemorrh Suppl. 1965;17:35-44. [PubMed] [Google Scholar]
- 3.Osterud B, Rapaport SI. Activation of factor IX by the reaction product of tissue factor and factor VII: additional pathway for initiating blood coagulation. Proc Natl Acad Sci USA. 1977;74(12):5260-5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu G, Broze GJ Jr, Krishnaswamy S. Formation of factors IXa and Xa by the extrinsic pathway: differential regulation by tissue factor pathway inhibitor and antithrombin III. J Biol Chem. 2004;279(17):17241-17249. [DOI] [PubMed] [Google Scholar]
- 5.Gailani D, Renné T. The intrinsic pathway of coagulation: a target for treating thromboembolic disease? J Thromb Haemost. 2007;5(6):1106-1112. [DOI] [PubMed] [Google Scholar]
- 6.Gailani D, Broze G Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253(5022):909-912. [DOI] [PubMed] [Google Scholar]
- 7.Wood JP, Ellery PE, Maroney SA, Mast AE. Biology of tissue factor pathway inhibitor. Blood. 2014;123(19):2934-2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361(9371):1801-1809. [DOI] [PubMed] [Google Scholar]
- 9.Bi L, Sarkar R, Naas T, et al. Further characterization of factor VIII-deficient mice created by gene targeting: RNA and protein studies. Blood. 1996;88(9):3446-3450. [PubMed] [Google Scholar]
- 10.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997;90(10):3962-3966. [PubMed] [Google Scholar]
- 11.Bolton-Maggs PH. Factor XI deficiency and its management. Haemophilia. 2000;6:100-109. [DOI] [PubMed] [Google Scholar]
- 12.Salomon O, Steinberg DM, Seligshon U. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia. 2006;12(5):490-493. [DOI] [PubMed] [Google Scholar]
- 13.Zucker M, Seligsohn U, Salomon O, Wolberg AS. Abnormal plasma clot structure and stability distinguish bleeding risk in patients with severe factor XI deficiency. J Thromb Haemost. 2014;12(7):1121-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gidley GN, Holle LA, Burthem J, Bolton-Maggs PHB, Lin FC, Wolberg AS. Abnormal plasma clot formation and fibrinolysis reveal bleeding tendency in patients with partial factor XI deficiency. Blood Adv. 2018;2(10):1076-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gailani D, Lasky NM, Broze GJ Jr. A murine model of factor XI deficiency. Blood Coagul Fibrinolysis. 1997;8(2):134-144. [DOI] [PubMed] [Google Scholar]
- 16.Ay C, Hisada Y, Cooley BC, Mackman N. Factor XI-deficient mice exhibit increased bleeding after injury to the saphenous vein. J Thromb Haemost. 2017;15(9):1829-1833. [DOI] [PubMed] [Google Scholar]
- 17.Mohammed BM, Cheng Q, Matafonov A, Monroe DM, Meijers JCM, Gailani D. Factor XI promotes hemostasis in factor IX-deficient mice. J Thromb Haemost. 2018;16(10):2044-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu S, Travers RJ, Morrissey JH, Diamond SL. FXIa and platelet polyphosphate as therapeutic targets during human blood clotting on collagen/tissue factor surfaces under flow. Blood. 2015;126(12):1494-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leiderman K, Chang WC, Ovanesov M, Fogelson AL. Synergy between tissue factor and exogenous factor XIa in initiating coagulation. Arterioscler Thromb Vasc Biol. 2016;36(12):2334-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kravtsov DV, Matafonov A, Tucker EI, et al. Factor XI contributes to thrombin generation in the absence of factor XII. Blood. 2009;114(2):452-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carmeliet P, Mackman N, Moons L, et al. Role of tissue factor in embryonic blood vessel development. Nature. 1996;383(6595):73-75. [DOI] [PubMed] [Google Scholar]
- 22.Bugge TH, Xiao Q, Kombrinck KW, et al. Fatal embryonic bleeding events in mice lacking tissue factor, the cell-associated initiator of blood coagulation. Proc Natl Acad Sci USA. 1996;93(13):6258-6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toomey JR, Kratzer KE, Lasky NM, Stanton JJ, Broze GJ Jr. Targeted disruption of the murine tissue factor gene results in embryonic lethality. Blood. 1996;88(5):1583-1587. [PubMed] [Google Scholar]
- 24.Rosen ED, Chan JC, Idusogie E, et al. Mice lacking factor VII develop normally but suffer fatal perinatal bleeding. Nature. 1997;390(6657):290-294. [DOI] [PubMed] [Google Scholar]
- 25.Cui J, O’Shea KS, Purkayastha A, Saunders TL, Ginsburg D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature. 1996;384(6604):66-68. [DOI] [PubMed] [Google Scholar]
- 26.Dewerchin M, Liang Z, Moons L, et al. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb Haemost. 2000;83(2):185-190. [PubMed] [Google Scholar]
- 27.Sun WY, Witte DP, Degen JL, et al. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci USA. 1998;95(13):7597-7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue J, Wu Q, Westfield LA, et al. Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proc Natl Acad Sci USA. 1998;95(13):7603-7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parry GC, Erlich JH, Carmeliet P, Luther T, Mackman N. Low levels of tissue factor are compatible with development and hemostasis in mice. J Clin Invest. 1998;101(3):560-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pawlinski R, Fernandes A, Kehrle B, et al. Tissue factor deficiency causes cardiac fibrosis and left ventricular dysfunction. Proc Natl Acad Sci USA. 2002;99(24):15333-15338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erlich J, Parry GC, Fearns C, et al. Tissue factor is required for uterine hemostasis and maintenance of the placental labyrinth during gestation. Proc Natl Acad Sci USA. 1999;96(14):8138-8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pawlinski R, Pedersen B, Erlich J, Mackman N. Role of tissue factor in haemostasis, thrombosis, angiogenesis and inflammation: lessons from low tissue factor mice. Thromb Haemost. 2004;92(3):444-450. [DOI] [PubMed] [Google Scholar]
- 33.Rosen ED, Xu H, Liang Z, Martin JA, Suckow M, Castellino FJ. Generation of genetically-altered mice producing very low levels of coagulation factorVII. Thromb Haemost. 2005;94(3):493-497. [PubMed] [Google Scholar]
- 34.Xu H, Noria F, Sandoval-Cooper MJ, et al. Severe deficiency of coagulation Factor VII results in spontaneous cardiac fibrosis in mice. J Pathol. 2009;217(3):362-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baglia FA, Walsh PN. Prothrombin is a cofactor for the binding of factor XI to the platelet surface and for platelet-mediated factor-XI activation by thrombin. Biochemistry. 2007;46(44):12886-12887. [DOI] [PubMed] [Google Scholar]
- 36.Oliver JA, Monroe DM, Roberts HR, Hoffman M. Thrombin activates factor XI on activated platelets in the absence of factor XII. Arterioscler Thromb Vasc Biol. 1999;19(1):170-177. [DOI] [PubMed] [Google Scholar]
- 37.Walsh PN. Roles of platelets and factor XI in the initiation of blood coagulation by thrombin. Thromb Haemost. 2001;86(1):75-82. [PubMed] [Google Scholar]
- 38.Pedersen B, Holscher T, Sato Y, Pawlinski R, Mackman N. A balance between tissue factor and tissue factor pathway inhibitor is required for embryonic development and hemostasis in adult mice. Blood. 2005;105(7):2777-2782. [DOI] [PubMed] [Google Scholar]
- 39.Bode MF, Mackman N. A combined deficiency of tissue factor and PAR-4 is associated with fatal pulmonary hemorrhage in mice [published correction in Thromb Res. 2017;149:95]. Thromb Res. 2016;146:46-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puy C, Tucker EI, Matafonov A, et al. Activated factor XI increases the procoagulant activity of the extrinsic pathway by inactivating tissue factor pathway inhibitor. Blood. 2015;125(9):1488-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peek M, Moran P, Mendoza N, Wickramasinghe D, Kirchhofer D. Unusual proteolytic activation of pro-hepatocyte growth factor by plasma kallikrein and coagulation factor XIa. J Biol Chem. 2002;277(49):47804-47809. [DOI] [PubMed] [Google Scholar]
- 42.Zur M, Nemerson Y. Kinetics of factor IX activation via the extrinsic pathway. Dependence of Km on tissue factor. J Biol Chem. 1980;255(12):5703-5707. [PubMed] [Google Scholar]
- 43.Taylor SA, Liddell MB, Peake IR, Bloom AL, Lillicrap DP. A mutation adjacent to the beta cleavage site of factor IX (valine 182 to leucine) results in mild haemophilia Bm. Br J Haematol. 1990;75(2):217-221. [DOI] [PubMed] [Google Scholar]
- 44.Bauer KA, Kass BL, ten Cate H, Hawiger JJ, Rosenberg RD. Factor IX is activated in vivo by the tissue factor mechanism. Blood. 1990;76(4):731-736. [PubMed] [Google Scholar]
- 45.Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33(1):4-15. [DOI] [PubMed] [Google Scholar]
- 46.Dargaud Y, Béguin S, Lienhart A, et al. Evaluation of thrombin generating capacity in plasma from patients with haemophilia A and B. Thromb Haemost. 2005;93(3):475-480. [DOI] [PubMed] [Google Scholar]
- 47.Chelle P, Montmartin A, Damien P, et al. Tissue factor pathway inhibitor is the main determinant of thrombin generation in haemophilic patients. Haemophilia. 2019;25(2):343-348. [DOI] [PubMed] [Google Scholar]
- 48.Keularts IM, Zivelin A, Seligsohn U, Hemker HC, Béguin S. The role of factor XI in thrombin generation induced by low concentrations of tissue factor. Thromb Haemost. 2001;85(6):1060-1065. [PubMed] [Google Scholar]
- 49.Pike GN, Cumming AM, Hay CR, Bolton-Maggs PH, Burthem J. Sample conditions determine the ability of thrombin generation parameters to identify bleeding phenotype in FXI deficiency. Blood. 2015;126(3):397-405. [DOI] [PubMed] [Google Scholar]
- 50.Mohammed BM, Cheng Q, Matafonov A, et al. A non-circulating pool of factor XI associated with glycosaminoglycans in mice. J Thromb Haemost. 2019;17(9):1449-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uhlén M, Fagerberg L, Hallström BM, et al. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- 52.Matafonov A, Cheng Q, Geng Y, et al. Evidence for factor IX-independent roles for factor XIa in blood coagulation. J Thromb Haemost. 2013;11(12):2118-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uehara Y, Minowa O, Mori C, et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373(6516):702-705. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt C, Bladt F, Goedecke S, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373(6516):699-702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.