

Abstract
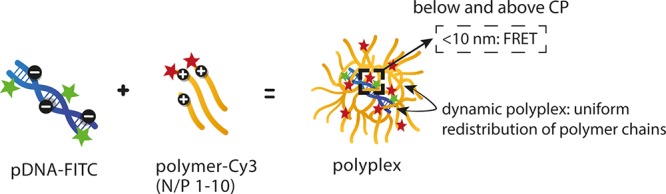
Combining multiple stimuli-responsive functionalities into the polymer design is an attractive approach to improve nucleic acid delivery. However, more in-depth fundamental understanding how the multiple functionalities in the polymer structures are influencing polyplex formation and stability is essential for the rational development of such delivery systems. Therefore, in this study the structure and dynamics of thermosensitive polyplexes were investigated by tracking the behavior of labeled plasmid DNA (pDNA) and polymer with time-resolved fluorescence spectroscopy using fluorescence resonance energy transfer (FRET). The successful synthesis of a heterofunctional poly(ethylene glycol) (PEG) macroinitiator containing both an atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain-transfer (RAFT) initiator is reported. The use of this novel PEG macroinitiator allows for the controlled polymerization of cationic and thermosensitive linear triblock copolymers and labeling of the chain-end with a fluorescent dye by maleimide–thiol chemistry. The polymers consisted of a thermosensitive poly(N-isopropylacrylamide) (PNIPAM, N), hydrophilic PEG (P), and cationic poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA, D) block, further referred to as NPD. Polymer block D chain-ends were labeled with Cy3, while pDNA was labeled with FITC. The thermosensitive NPD polymers were used to prepare pDNA polyplexes, and the effect of the N/P charge ratio, temperature, and composition of the triblock copolymer on the polyplex properties were investigated, taking nonthermosensitive PD polymers as the control. FRET was observed both at 4 and 37 °C, indicating that the introduction of the thermosensitive PNIPAM block did not compromise the polyplex structure even above the polymer’s cloud point. Furthermore, FRET results showed that the NPD- and PD-based polyplexes have a less dense core compared to polyplexes based on cationic homopolymers (such as PEI) as reported before. The polyplexes showed to have a dynamic character meaning that the polymer chains can exchange between the polyplex core and shell. Mobility of the polymers allow their uniform redistribution within the polyplex and this feature has been reported to be favorable in the context of pDNA release and subsequent improved transfection efficiency, compared to nondynamic formulations.
1. Introduction
The use of polymeric carriers is a well-known approach to promote the delivery and uptake of nucleic acids by target cells.1,2 Typically, cationic polymers are used because they are able to efficiently condense negatively charged nucleic acids by electrostatic interactions into polymer–nucleic acid complexes, which are referred to as polyplexes.3 In the last decades, stimuli-responsive polymers have gained interest as a unique class of polymers because their self-assembly behavior can be influenced by biological and/or external physical triggers.4−7 By combining multiple stimuli-responsive functionalities into the polymer design, like pH- and thermosensitivity, the efficiency of nucleic acid delivery could be modulated.8 For this purpose, the synthesis of well-defined linear block copolymers of the AB, ABA, or ABC type, have gained much attention because of their unique structure with multiple different homopolymer blocks. For example, the thermosensitive blocks can be used to anchor polyplexes in a thermosensitive hydrogel to achieve local and sustained delivery of nucleic acids.9,10
In a previous study, the physical properties and in vitro transfection behavior of such cationic and thermosensitive ABC triblock copolymer-based polyplexes were compared with structurally related nonthermosensitive polymers. The size, surface charge, and stability of the formed polyplexes were studied as a function of polymer block composition and molecular weight, charge ratio, and temperature. The results showed a critical balance between the electrostatic and hydrophobic interactions of the multifunctional polymer and plasmid DNA (pDNA) at temperatures above the cloud point (CP) to enable formation of stable nanoparticles. If the molecular weight of the cationic block was above 20 kDa and the N/P charge ratio >1, the electrostatic interactions between the pDNA and the cationic block of the polymer were dominating over the hydrophobic thermosensitive interactions resulting in preservation of the polyplex structure at 37 °C.11 However, more in-depth fundamental understanding how the multiple functionalities in the polymer structures are influencing polyplex formation, structure, and stability is essential for the rational development of such delivery systems. For example, structural studies of polymer–pDNA complexes have shown that poly(ethylenimine) (PEI)-based polyplexes were much more dynamic than poly(l-lysine) (PLL)-based ones, which can help understanding the observed difference in the pDNA transfection efficiency of these polyplexes.12,13 The dynamic feature of the PEI chains in the polyplexes translates into an easier dissociation and thus release of pDNA in the cytosol, which in turn yields higher transfection compared to PLL-polyplexes.14−16 In these studies, the use of time-resolved fluorescence resonance energy transfer (FRET) fluorescence spectroscopy showed to be a powerful tool to visualize the interactions between pDNA and cationic polymer molecules. FRET is a distance-dependent process by which energy is transferred nonradiatively from the donor fluorophore in its excited state to the acceptor fluorophore by intermolecular long-range dipole–dipole interactions.17−19 The requirements for efficient energy transfer are (1) the donor fluorescence spectrum must overlap with the acceptor absorption spectrum; (2) the distance between the donor and acceptor molecules must be less than 10 nm; and (3) the fluorescence quantum yield of the donor as well as the absorption coefficient of the acceptor must be sufficiently high. In the present study, the known fluorophore FRET pair FITC and Cy312,20 was selected to study the interactions between plasmid DNA and polymer by time-resolved fluorescence spectroscopy.
The aim of the present work is two-fold. First, the development of a synthesis method for controlled polymerization of linear thermosensitive and cationic polymers with poly(ethylene glycol) (PEG) as midblock enabling fluorescent labeling of the polymer chain-ends. Second, it aims at investigating the structural dynamics of thermosensitive polyplexes and providing insights for the design of polymers for nucleic acid delivery systems. We report the synthesis of a novel heterofunctional PEG macroinitiator containing both an atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain-transfer (RAFT) initiator. This PEG macroinitiator was used to prepare linear NPD triblock copolymers consisting of a thermosensitive poly(N-isopropylacrylamide) (PNIPAM, N), a hydrophilic PEG (P), and a cationic poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA, D) block, of which the chain-end of the latter block was fluorescently labeled. In the second part of this study, the dynamics of the polyplexes at different temperatures were studied by tracking the behavior of labeled pDNA and polymer using time-resolved fluorescence spectroscopy.
2. Materials and Methods
2.1. Materials
All materials were obtained from Sigma-Aldrich (Zwijndrecht, The Netherlands) and used as received unless noted otherwise. 4-(Dimethylamino)pyridinium-4-toluene-sulfonate (DPTS) was prepared according to a literature procedure.21 Triethylamine (TEA), 2,4,6-trinitrobenzenesulfonic acid (TNBSA) solution (5% w/v), and 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB, Ellman’s reagent) were obtained from Thermo Fisher Scientific (Bleiswijk, The Netherlands). Peptide grade dichloromethane (DCM), N,N-dimethylformamide (DMF), tetrahydrofuran (THF), and diethyl ether were purchased from Biosolve (Valkenswaard, The Netherlands). All solvents were dried by molecular sieves for 24 h before use. To remove the monomethyl ether hydroquinone inhibitor, 2-(dimethylamino)ethyl methacrylate (DMAEMA) was passed through a column of alumina prior to use. Slide-A-lyzer dialysis cassettes (molecular weight cutoff (MWCO) 3.5–10 kDa) were obtained from Thermo Fisher Scientific (Bleiswijk, The Netherlands). The pGL3-control reporter vector, encoding for firefly luciferase, with simian virus 40 (SV40) promoter was purchased from Promega (Leiden, The Netherlands). The plasmid (5256 bp) was amplified with DH5α competent E. coli bacteria cells and purified using NucleoBond PC2000 DNA purification kit (Macherey-Nagel, Bioke, Leiden, The Netherlands). Label IT Nucleic Acid Labeling Kit Fluorescein was obtained from Mirus Bio LLC (WI). Maleimide Cyanine3 was purchased from Lumiprobe GmbH (Hannover, Germany). Linear polyethylenimine (l-PEI, Mw 25 kDa) was obtained from Polysciences (Hirschberg an der Bergstraße, Germany). Human epithelial ovarian carcinoma cells (HeLa) were originally obtained from the American Type Culture Collection (ATCC) (Maryland). The Luciferase assay kit and CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) kit were purchased from Promega (Leiden, The Netherlands).
2.2. Synthesis of Heterofunctional PEG Macroinitiator
The synthesis of a PEG based macroinitiator suitable for subsequent polymerization by ATRP and RAFT was achieved following a two-step synthesis route as described in detail below (Scheme 1).
Scheme 1. Two-Step Synthesis Route Yielding a Heterofunctional PEG Macroinitiator with Both an Atom Transfer Radical Polymerization (ATRP) Initiator and a Chain-Transfer Agent (CTA) for Subsequent Reversible Addition–Fragmentation Chain-Transfer (RAFT) Polymerization.

2.2.1. Characterization of Starting Compound NH2-PEG-OH
Prior to the synthesis, the commercially obtained O-(2-aminoethyl)polyethylene glycol (NH2-PEG-OH) was characterized by 1H NMR by the addition of trichloroacetyl isocyanate (TAIC)22 to determine the number-average molecular weight (Mn) of the polymer (according to manufacturer Mn = 5 kDa). This compound (3 mg) was dissolved in deuterated chloroform (1 mL), and a few drops of TAIC were added. After 15 min incubation at room temperature (RT), the 1H NMR spectrum was recorded. TAIC reacts with the free hydroxyl group of the NH2-PEG-OH, thereby causing a downfield shift to δ = 4.4 ppm of the peak of the methylene group adjacent to the hydroxyl group.23,24 Subsequently, the Mn of the polymer was calculated by comparing the integrals of the peaks at δ = 4.4 ppm and δ = 3.8–3.5 ppm (PEG) assuming the presence of one OH group per PEG molecule, and this value was used for further calculations in the synthesis steps. Furthermore, the number of free primary amines per polymer chain was determined by the TNBSA assay, as described in section 2.5.3.
2.2.2. Synthesis of Br-C(CH3)2-CO-NH-PEG-OH
Functionalization of PEG with α-bromoisobutyryl bromide yields an ATRP macroinitiator, and the procedure was based on a previously described method with some modifications.25 First, 500 mg (1 equiv) of NH2-PEG-OH was dissolved in 7 mL of H2O/THF (1:1) in a two-neck 25 mL glass round-bottom vial and placed on ice. Next, 25 μL (2 equiv) of α-bromoisobutyryl bromide was added to the mixture while monitoring and adjusting the pH > 9.0 by adding TEA (in total, 3 equiv of TEA was added). The mixture was left overnight at RT, and the next day it was concentrated under vacuum and dissolved in water. The product, Br-C(CH3)2-CO-NH-PEG-OH, was further purified by extensive dialysis against water (dialysate was changed 3 times a day) for 2 days at 4 °C (MWCO, 3.5 kDa) and subsequently freeze-dried.
1H NMR (CDCl3): δ (ppm) 3.8–3.5 (478H, (CH2)2O), 1.94 (6H, (CH3)2CBr) (Figure S2).
2.2.3. Synthesis of Br-C(CH3)2-CO-NH-PEG-CTA
Br-C(CH3)2-CO-NH-PEG-OH (453 mg, 1 equiv) was dissolved in 5.0 mL of dry DCM together with 68 mg (2 equiv) of 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (RAFT chain-transfer agent, CTA) and 8 mg (0.3 equiv) of DPTS.26 The mixture was cooled on ice, and 5.0 mL of 0.05 mM (3 equiv) of N,N′-dicyclohexylcarbodiimide (DCC) dissolved in DCM was added dropwise under N2 atmosphere. Next, the reaction mixture was incubated overnight at RT under an N2 atmosphere. Afterward, the mixture was filtered through a 0.2 μm nylon filter to remove the formed dicyclohexyl urea (DCU), and the product was precipitated in cold diethyl ether. For further purification and to remove unreacted RAFT-CTA, the precipitate was dissolved in DMSO and dialyzed against DMSO for 1 day, followed by dialysis against water for 3 days at 4 °C (MWCO, 3.5 kDa). The final PEG macroinitiator was recovered by freeze-drying.
1H NMR (CDCl3): δ (ppm) 3.8–3.5 (478H, O(CH2)2O), 3.32 (2H, S(C=S)SCH2), 2.75 (2H, CH2(CH2)9CH3), 2.65 (2H, (C=O)CH2CH2C(CN)), 2.52–2.37 (2H, (C=O)CH2CH2C(CN)), 1.94 (6H, (CH3)2CBr), 1.88 (3H, CH3C(CN)), 1.41–1.22 (18H, CH2(CH2)9CH3), 0.88 (3H, CH2(CH2)9CH3) (Figure S3).
2.3. Synthesis of NPD Block Copolymers
NPD triblock copolymers (N = PNIPAM, P = PEG, D = PDMAEMA) were synthesized following a two-step procedure. The first step involved atom transfer radical polymerization (ATRP) followed by reversible addition–fragmentation chain-transfer (RAFT) polymerization (Scheme 2).
Scheme 2. Schematic Overview of the Synthesis Route of NPD Triblock Copolymers Using the Heterofunctional PEG Macroinitiator (P Polymer).
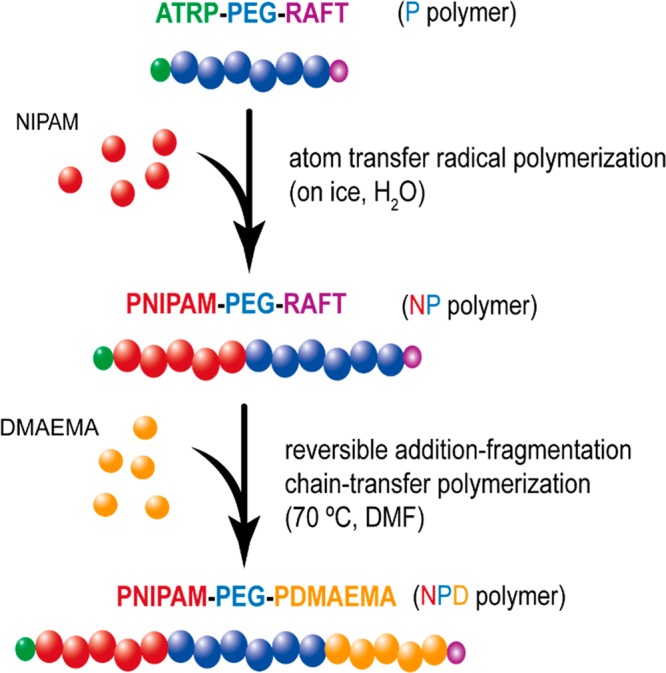
Atom transfer radical polymerization (ATRP) is used in the first step to polymerize N-isopropylacrylamide (NIPAM, N) yielding the intermediate NP polymer. Next, 2-(dimethylamino)ethyl methacrylate (DMAEMA, D) is polymerized by reversible addition–fragmentation chain-transfer (RAFT) polymerization to obtain the final NPD polymer.
2.3.1. Synthesis of PNIPAM-PEG (NP) Polymers by Atom Transfer Radical Polymerization (ATRP)
The PEG macroinitiator (50 mg, 8.7 μmol), NIPAM (146 mg, 1.29 mmol), and CuBr (1.7 mg, 0.01 mmol) were dissolved in water (1.5 mL) in an airtight screw-cap glass vial. The reaction mixture was flushed with nitrogen for 15 min at RT and subsequently another 15 min on ice. The reaction was started by adding 100 μL of 100 mM (0.01 mmol) tris[2-(dimethylamino)ethyl]amine (Me6TREN) solution in water, which changed the color of the mixture immediately from colorless to blue/green. The polymerization reaction was carried out for 4 h on ice. Next, the polymer solution was transferred into a dialysis cassette and dialyzed against water for 48 h at 4 °C (MWCO, 10 kDa), while changing the dialysate three times a day. Finally, the resulting NP polymer was recovered by freeze-drying and analyzed by 1H NMR spectroscopy and GPC (sections 2.5.1 and 2.5.2, respectively).
2.3.2. Synthesis of PNIPAM-PEG-PDMAEMA (NDP) Polymers by Reversible Addition–fragmentation Chain-Transfer (RAFT) Polymerization
The NP-CTA polymer (50 mg, 1 equiv) and DMAEMA (104 mg, 280 equiv) were dissolved in 1.0 mL of dry DMF in an airtight Schlenk flask. Next, 18 μL of 5 mg/mL (90 μg, 0.2 equiv) AIBN stock solution in dry DMF was added. At least three freeze–pump–thaw cycles were applied to degas the solution, after which the reaction mixture was placed in an oil bath at 70 °C and stirred for 16 h under a N2 atmosphere. Next, the polymer solution was transferred into a dialysis cassette (MWCO, 10 kDa) and dialyzed against water for 48 h at 4 °C, while changing the dialysate three times a day. The final NPD polymer was recovered by freeze-drying and analyzed using 1H NMR spectroscopy and GPC. Nonthermosensitive polymers, lacking the PNIPAM block, were synthesized as a control. For this, the commercially available PEG-CTA macroinitiator (poly(ethylene glycol) methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate)) (101 mg, 1 equiv) together with DMAEMA (873 mg, 297 equiv) were dissolved in 8.0 mL of dry DMF. Next, 122 μL of 5 mg/mL (610 μg, 0.2 equiv) AIBN stock solution was added, and the reaction mixture was subjected to the same steps as described above. The synthesized polymers were analyzed using 1H NMR spectroscopy and GPC, and the cloud point was determined for thermosensitive polymers (sections 2.5.1, 2.5.2, and 2.5.4, respectively).
2.4. Cy3-Labeling of NPD and PD Block Copolymers
The trithiocarbonate end group (CTA) of the NPD-CTA and PD-CTA polymers was removed via aminolysis following a previously described method with some modifications.27,28 The polymer (100 mg), which was dissolved in 2.0 mL of dry THF was treated with excess n-butylamine (30 equiv) for 24 h at RT. A few drops of n-tributylphosphine were added to the mixture to minimize disulfide formation. Subsequently, the polymer was recovered by 2-fold precipitation in cold hexane and dried overnight under vacuum at RT. In the next step, 50 mg of thiol terminated polymer (NPD-SH and PD-SH) was dissolved in 2 mL of dry DMF in a screw-capped glass vial and allowed to dissolve overnight at RT under N2 atmosphere. Next, an excess of maleimide-Cy3 dye (2 equiv) in dry DMF was added to the solution and allowed to react for 48 h at 37 °C. The reaction mixture was transferred into a dialysis bag (MWCO, 10 kDa) and dialyzed against DMSO for 3 days, followed by dialysis against water for 1 day at 4 °C. The final Cy3-labeled polymers were recovered by freeze-drying.
2.5. Polymer Characterization
2.5.1. 1H NMR Spectroscopy
The PEG macroinitiator and synthesized polymers were characterized with 1H NMR spectroscopy using an Agilent 400 MR-NMR spectrometer (Agilent Technologies, Santa Clara, CA). Chemical shifts are referred to the residual solvent peak (δ = 7.26 ppm for CDCl3 and δ = 4.80 ppm for D2O). Data analysis was performed using MestReNova Software version 10.0.1-14719.
2.5.2. Gel Permeation Chromatography (GPC)
The PEG macroinitiator and synthesized polymers were characterized by GPC using a Waters Alliance System (Waters Corporation, Milford, MA) equipped with a refractive index (RI) and UV/vis detector. As an eluent, DMF containing 10 mM LiCl was used and 50 μL samples (3 mg/mL) were injected into a PLgel 5 μm MIXED-D column (Polymer Laboratories). The column temperature was set to 65 °C and the flow rate to 1.0 mL/min. Calibration was performed using PEG standards of narrow and defined molecular weights. Data analysis was performed using Empower 3 Software 2010.
2.5.3. Quantification of Primary Amines (TNBSA Assay)
Free primary amine groups of the starting compound (NH2-PEG-OH), intermediate product (Br–-C(CH3)2-CO-NH-PEG-OH) and final PEG macroinitiator (Br-C(CH3)2-CO-NH-PEG-CTA) were quantified by a TNBSA assay following an established procedure.29 Samples were dissolved in sodium bicarbonate buffer (0.1 M, pH 8.5) with a concentration ranging from 1 to 4 mg/mL. Glycine standards were prepared for calibration at concentrations ranging from 0 to 0.15 mM in the same buffer. Next, 250 μL of a freshly prepared 0.01% TNBSA solution in buffer was added to 500 μL of a sample or standard solution, and the solutions were incubated for 2 h at 37 °C. Afterward, 250 μL of 10% SDS and 125 μL of 1 M HCl were added to each solution, and the absorbance at 335 nm was measured in triplicate using a BMG SPECTROstar Nano wellplate reader (BMG Labtech, de Meern, The Netherlands). Data analysis was performed using MARS Data analysis software version 2.22 (BMG Labtech).
2.5.4. Determination of Cloud Point (CP) of Thermosensitive Polymers
The CP of thermosensitive polymers was determined by light scattering using a Jasco FP-8300 spectrophotometer (JASCO, Easton, MD). The polymers were dissolved overnight at a concentration of 1 mg/mL in 20 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) buffer, pH 7.4. Next, 1 mL of the polymer solution was transferred into a clean glass cuvette and the scattering intensity was measured at 640 nm while increasing the temperature from 10 to 50 °C with a heating rate of 1 °C/min. The cloud point was taken as the onset point of increasing scattering intensity.30
2.5.5. UV/Visible Spectroscopy
After the aminolysis step, the polymers were dissolved in THF (1 mg/mL) and UV/vis spectra were recorded using a 10 mm path-length quartz cuvette in a Shimadzu UV 2450 spectrophotometer from 250 to 400 nm with 0.5 nm resolution. After the Cy3-labeling step, polymers were dissolved in DMF (3 mg/mL) and UV/vis spectra were recorded from 400 to 700 nm with 0.5 nm resolution.
2.5.6. Ellman’s Assay
Ellman’s assay was performed to detect free thiol groups at the polymer chain-ends after aminolysis following an established procedure.31 Cysteine hydrochloride monohydrate standards were prepared at concentrations ranging from 0 to 1 mM in a 0.1 M sodium phosphate buffer (pH 8.0) supplemented with 1 mM ethylenediaminetetraacetic acid (EDTA). The polymers were dissolved in the same reaction buffer at a concentration of 30 mg/mL. Next, 50 μL of freshly prepared Ellman’s reagent (4 mg/mL), 2.5 mL of reaction buffer, and 250 μL of each test sample or standard were mixed and incubated at RT for 15 min. The absorbance of the samples at 412 nm was measured using a Shimadzu UV 2450 spectrophotometer.
2.6. FITC-Labeling of pDNA
pDNA was labeled with FITC using the Label IT Nucleic Acid Labeling Kit Fluorescein following the manufacturer’s protocol with labeling ratio dye/DNA of 0.1:1. The labeling density of the final product (pDNA-FITC) was calculated from the absorption spectrum using the extinction coefficient 68 000 cm–1 M–1 of FITC32 attached to pDNA at 495 nm and pH 8. The labeling density was 0.49 mol % of bases for pDNA-FITC, meaning that pDNA contains 1 fluorophore per 220 base pairs.
2.7. Polyplex Preparation
For the constant temperature measurements (Scheme 3A), polyplexes were prepared stepwise. First, polymer stock solutions (23 μg/mL for NPD(-Cy3) and 16 μg/mL for PD(-Cy3) and pDNA stock solution (150 μg/mL) were prepared in HBS buffer (20 mM HEPES, 150 mM NaCl, pH 7.4) and cooled at 0 °C on ice or warmed at 37 °C in a water bath. Subsequently, 333 μL polymer solution was added to 167 μL of pDNA solution at an N/P ratio of 1, and the mixture was vortexed for 10 s. The polyplexes, with a final pDNA concentration of 20 μg/mL, were allowed to form at either 0 or 37 °C for 30 min before further use. After dynamic light scattering or fluorescence measurements at either 4 or 37 °C, the next N/P ratio was reached by adding the corresponding amount of concentrated polymer solution (15.5 mg/mL for NPD(-Cy3) and 11.0 mg/mL for PD(-Cy3)) while keeping the sample at the same temperature (4 or 37 °C). The N/P ratios of the analyzed polyplex dispersion ranged from 1 to 10 (mol/mol). For the temperature cycle measurements (Scheme 3B), polyplexes of a N/P 10 were directly formed at either 0 or 37 °C for 30 min before further use.
Scheme 3. Schematic Overview of Experimental Design for Time-Resolved Fluorescence Spectroscopic Analysis of Polyplexes Based on pDNA and NP Diblock and NPD Triblock Copolymers.

(A) Fixed temperature measurements: polyplexes were prepared with pDNA-FITC and unlabeled polymer up to N/P 2, followed by addition of either more unlabeled polymer (a, unlabeled) or Cy3-labeled polymer (b, mixed) up to N/P 10. Alternatively, polyplexes were prepared with pDNA-FITC and Cy3-labeled polymer for all N/P ratios (c, fully labeled). Preparation of the polyplexes and measurements were performed at either 4 or 37 °C. (B) Temperature cycle measurements: polyplexes were prepared with pDNA-FITC and unlabeled polymer (N/P 10) at either 4 or 37 °C. Subsequently, polyplexes were subjected to a temperature cycle, including cooling and heating series as described in detail in section 3.3.3.
2.8. Fluorescence Measurements of Polyplexes
The formulations investigated using fluorescence experiments are listed in Table 1 and Scheme 3. Steady-state excitation and fluorescence spectra were recorded using a FLS1000 photoluminescence spectrometer (Edinburgh Instrument, Livingston, U.K.). The excitation wavelength was 483 nm, the emission wavelength was 520 or 580 nm, and the spectra were automatically corrected using a correction function provided by the manufacturer.
Table 1. Polyplex Composition: Unlabeled pDNA (pDNA) or FITC-Labeled pDNA (pDNA-FITC) Complexed with Unlabeled Polymers (NPD and PD) and/or Cy3-Labeled Polymers (NPD-Cy3 and PD-Cy3)a.
| NPD
polymer |
PD polymer |
||||
|---|---|---|---|---|---|
| constant temperature (°C)b | pDNA | N/P 1–2 | N/P 5–10 | N/P 1–2 | N/P 5–10 |
| 4 and 37 (a) | pDNA-FITC | NPD | NPD | PD | PD |
| 4 and 37 (b) | pDNA-FITC | NPD | NPD-Cy3 | PD | PD-Cy3 |
| 4 and 37 (c) | pDNA-FITC | NPD-Cy3 | NPD-Cy3 | PD-Cy3 | PD-Cy3 |
| NPD polymer | PD polymer | ||||
|---|---|---|---|---|---|
| temperature cycle (°C) | pDNA | N/P 10 | N/P 10 | ||
|
pDNA-FITC | NPD | PD | ||
|
pDNA-FITC | NPD | PD |
Time-resolved fluorescence was measured using a time-correlated single photon counting (TCSPC) system (Pico-Quant GmBH, Chaussee, Germany) consisting of a PicoHarp 300 controller and a PDL 800-B driver. The samples were excited with the pulsed diode laser head LDH-P-C-485 at 483 nm at a time resolution of 130 ps. The signals were detected with a microchannel plate photomultiplier tube (Hamamatsu R2809U). The influence of the scattered excitation light was reduced with a cutoff filter (transmission > 490 nm) in front of the monitoring monochromator. Fluorescence decays were collected at 520 nm (donor). The instrumental response function (IRF) was measured separately, and the decays were deconvoluted and fitted by applying the iterative least-squares method to the sum of 1–2 exponents (eq 1) either globally or by single curve fitting depending on the system.
| 1 |
In this equation, τi is the global lifetime and ai is the local amplitude (pre-exponential factor). The mean amplitude weighted lifetime ⟨τ⟩ from two-exponential fittings for each N/P ratio was calculated using eq 2. Since the Cy3 dye does not fluoresce at 520 nm, it was assumed that both calculated lifetimes were assigned to FITC.
| 2 |
2.9. In Vitro Transfection and Cytotoxicity Studies
In vitro studies were performed according to the recommendations as previously described.33 HeLa cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing glucose (4.5 g/L glucose) and supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidified atmosphere containing 5% CO2. HeLa cells were seeded at a density of 7000 cells/well in 96-well plates 24 h before transfections. The following day, cells were washed once with 100 μL of PBS and incubated with various formulations in complete medium for 6 h at 37 °C. pDNA-loaded polyplexes (0.50 pDNA/well) were prepared with NPD and PD polymers at different N/P ratios at 4 °C. In addition, NPD and PD polyplexes (N/P 10) were prepared at 4 or 37 °C and subsequently subjected to a temperature cycle (4 → 37 → 4 °C and 37 → 4 → 37 °C, respectively) before addition to HeLa cells. As a positive control for nucleic acid delivery, l-PEI (25 kDa) was used at an optimal N/P ratio of 6,33 and naked pDNA was included as a negative control. Each condition was measured 8 times. After 6 h, all transfection mixtures were replaced by 200 μL of fresh medium and the plates were incubated for another 24 h at 37 °C. The next day, cells were lysed with 100 μL of lysis buffer (25 mM tris(hydroxymethyl)aminomethane (Tris), 2 mM dithiothreitol (DTT), 2 mM 1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid (DCTA), 1% Triton X-100, 10% glycerol) on a shaking board at RT for 15 min. Afterward, 50 μL of lysate was transferred into a white luminescence plate. Luciferase Assay Reagent was injected in each well (50 μL) using a FLUOstar OPTIMA microplate reader (BMG Labtech, Ortenberg, Germany) equipped with an injection pump, and after 2 s, luminescence was measured for 10 s according to the supplier’s recommendation. An MTS assay was performed in parallel on a separate plate to assess the cytotoxicity of all tested formulations. The transfection protocol was similar as described above, except that instead of adding lysis buffer 100 μL of fresh medium was added to each well followed by 20 μL of MTS Assay Reagent. Plates were incubated for 1–2 h at 37 °C, and subsequently the absorbance at 490 and 690 nm was measured using the iMark Microplate Absorbance Reader (Bio-Rad Laboratories Inc., Hercules, CA), and cell viability was calculated relative to untreated cells.
3. Results and Discussion
3.1. Heterofunctional PEG Macroinitiator Synthesis and Characterization
The heterofunctional PEG macroinitiator enables a new synthesis strategy for the preparation of linear cationic and thermosensitive triblock copolymers having a PEG midblock by two-step controlled radical polymerization (ARTP for the NIPAM block and RAFT for the DMAEMA block). The commercially obtained starting compound NH2-PEG-OH was analyzed by 1H NMR (Figure S1), and after addition of TAIC, a number-average molecular weight (Mn) of 5.2 kDa was determined, which is in agreement with the Mn obtained by gel permeation chromatography (GPC) of 5.5 kDa (Table 2). In addition, results from the TNBSA assay show that within the experimental error the polymer chains indeed have one free amine terminus. The PEG macroinitiator was synthesized following a two-step synthesis route, starting with functionalization of the amine groups of NH2-PEG-OH with an ATRP initiator. The second step consisted of a DCC-mediated coupling of RAFT-CTA to the free OH group (Scheme 1). In the first step, NH2-PEG-OH was functionalized with α-bromoisobutyryl bromide under Schotten-Baumann conditions to preferentially react the amine groups, leaving the hydroxyl groups free for the second step of the synthesis route with RAFT-CTA (Scheme 1, step 1). After the first step, the 1H NMR spectrum showed a sharp peak at δ = 1.94 ppm, which corresponds to the protons of the methyl groups of bromoisobutyryl (Figure S2). The integral of this peak was compared with that of the peak at δ = 3.8–3.5 ppm (PEG), and the results showed that the amines were quantitatively derivatized with the bromoisobutyryl-group (Table 2). Furthermore, no free primary amines could be detected using the TNBSA assay, confirming that the amine groups had reacted quantitatively with α-bromoisobutyryl bromide. Importantly, addition of TAIC resulted in a shift of the CH2 protons of PEG adjacent to the OH end to 4.4 ppm corresponding to the initial peak intensity of the starting NH2-PEG-OH, indicating that indeed only the amine groups had reacted leaving the terminal OH groups for subsequent derivatization (Figure S2). In the next step, the RAFT-CTA was coupled to the PEG macroinitiator by a DCC-mediated esterification between the carboxylic acid groups of RAFT-CTA and the hydroxyl groups of PEG (Scheme 1, step 2). 1H NMR analysis showed the appearance of characteristic peaks corresponding to the CH2 and CH3 groups of the RAFT-CTA, indicating successful coupling to the PEG macroinitiator (Figure S3). Comparing the integrals of those peaks (δ = 1.88 (3H methyl group CTA), 1.4–1.2 (18H, methylene groups CTA), 0.88 (3H, methyl group CTA)) to the integral of the peak at δ = 3.8–3.5 ppm (PEG) showed a quantitative CTA-functionalization. After addition of TAIC, no peak shift to 4.4 ppm was observed in the 1H NMR spectrum, confirming that indeed the hydroxyl groups of PEG had reacted quantitatively with the RAFT-CTA. In addition, the characteristic peak of the methyl groups of bromoisobutyryl was still present (Figure S3) and no free amines were detected with the TNBSA assay (Table 2), demonstrating that the bromoisobutyryl-functionality was still intact in the final PEG macroinitiator. Furthermore, no significant shift in the molecular weight distributions was observed in the GPC chromatograms of all polymer products (Figure 1A). GPC analysis also showed that the trithiocarbonate group present on the CTA, which has a characteristic absorbance at 310 nm, overlaps with the RI-signal of the final PEG macroinitiator. This clearly demonstrates the presence of the CTA group on the final polymer and the absence of free RAFT-CTA (Figure 1B).
Table 2. Characteristics of the Starting Compound (NH2-PEG-OH), Intermediate Product (Br-C(CH3)2-CO-NH-PEG-OH), and Final PEG Macroinitiator (Br-C(CH3)2-CO-NH-PEG-CTA).
| product | yield (%) | Mn(kDa)a | PDIa | free amines (%)b | % Br functionalizationc | % CTA functionalizationc |
|---|---|---|---|---|---|---|
| NH2-PEG-OH | n.a.d | 5.5 | 1.1 | 99 | n.a.d | n.a.d |
| Br-C(CH3)2-CO-NH-PEG–OH | 92 | 5.3 | 1.1 | 0 | 96 | n.a.d |
| Br-C(CH3)2-CO-NH-PEG-CTA | 71 | 5.4 | 1.1 | 0 | 96 | 99 |
Determined by GPC.
Determined by TNBSA assay.
Determined by 1H NMR.
n.a. = not applicable
Figure 1.

(A) GPC chromatograms (RI-detection) of the starting compound NH2-PEG-OH (solid line), intermediate product Br-C(CH3)2-CO-NH-PEG-OH (dashed line) and final PEG macroinitiator Br-C(CH3)2-CO-NH-PEG-CTA (dotted line). (B) GPC analysis with dual RI (black) and UV–vis (at 310 nm, yellow) detection of final PEG macroinitiator (solid line) and CTA compound (dashed line).
3.2. Synthesis and Characterization of NPD Triblock Copolymers
The heterofunctional PEG macroinitiator was used to synthesize triblock copolymers, following a two-step synthesis route (Scheme 2). In the present study, NPD polymers consisting of a PEG midblock flanked by PNIPAM and PDMAEMA blocks to introduce thermosensitive and cationic properties to the polymer structure, respectively, were synthesized (Figure 2A). The PNIPAM block was obtained by polymerization of NIPAM monomers by ATRP, and the resulting NP (N = PNIPAM, P = PEG) polymer was obtained with a monomer conversion of 94% based on 1H NMR analysis (Table 3) and a yield of 74%. GPC analysis further confirmed the successful synthesis of NP polymers, as the peak of the macroinitiator (15 min) was almost completely shifted to lower retention times (12–14 min), which corresponds to a higher Mn of the formed polymer (Table 3). In the next step, DMAEMA was polymerized by RAFT polymerization to yield the final NPD (N = PNIPAM, P = PEG, D = PDMAEMA) triblock copolymer. The monomer conversion was 76% as determined with 1H NMR (Table 3, Figure S4), and the polymer was obtained in a yield of 71%. The thermosensitive properties of the NPD polymer were investigated by light scattering, and the cloud point temperature of the polymer was 35 °C (Table 3). To make sure that the desired polymer block composition is obtained and that ATRP can be carried out in the presence of the RAFT initiator, without initiating or disrupting the RAFT initiator, control reactions were performed (Table S1). Therefore, ATRP of NIPAM was carried out in a mixture containing PEG with the ATRP initiator (Br-PEG-Br, 6 kDa) and PEG with a RAFT initiator (CTA-PEG-CTA, 10 kDa). The GPC chromatogram clearly showed a shift of the ATRP PEG macroinitiator (15 min) to lower retention times while the peak of the RAFT PEG macroinitiator (14 min) was still visible (Figure S5). In addition, the peak at 14 min was shifted to lower retentions times after RAFT polymerization, indicating that the RAFT PEG macroinitiator was still able to initiate the polymerization of DMAEMA (Figure S5). PD (P = PEG, D = PDMAEMA) diblock copolymers lacking the thermosensitive PNIPAM block were synthesized, which served as control polymers in further experiments (Figure 2B). For this, a commercially available PEG-CTA macroinitiator was used and a similar monomer conversion of DMAEMA and polymer yield were obtained, as for the NPD polymers (77% and 70%, respectively) (Table 3, Figure S6). Both polymers, NPD and PD, were obtained with acceptable molecular weight distributions (PDI of 1.6–1.8) as determined by GPC, which is lower than the PDIs reported for similar triblock copolymers synthesized by a combination of ATRP and free radical polymerization.11
Figure 2.
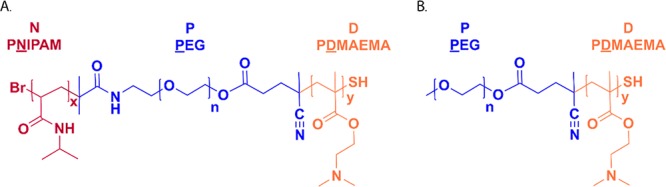
(A) Chemical structure of NPD triblock polymer consisting of a PEG midblock (P), flanked by blocks of PNIPAM (N) and PDMAEMA (D). (B) Chemical structure of PD diblock polymer consisting of a PEG block (P) and a PDMAEMA (D) block.
Table 3. Characteristics of NPD Triblock and PD Diblock Copolymers Synthesized by Radical Polymerizationa.
| name | Mn N block (kDa)b | Mn P block (kDa)b | Mn D block (kDa)b | total Mn (kDa)b | total Mn (kDa)c | PDIc | cloud point (°C)d |
|---|---|---|---|---|---|---|---|
| NP-CTA | 16 | 5 | n.a.e | 21 | 39 | 1.4 | n.a.e |
| NPD-CTA | 16 | 5 | 34 | 55 | 52 | 1.8 | 35 |
| NPD-SH | 16 | 5 | 34 | 55 | 51 | 1.7 | 35 |
| NPD-Cy3 | 16 | 5 | 34 | 55 | 52 | 1.8 | 35 |
| PD-CTA | n.a.e | 5 | 36 | 41 | 33 | 1.8 | n.a.e |
| PD-SH | n.a.e | 5 | 36 | 41 | 34 | 1.6 | n.a.e |
| PD-Cy3 | n.a.e | 5 | 36 | 41 | 34 | 1.7 | n.a.e |
The polymer names are abbreviated according to the block composition (N = PNIPAM, P = PEG, D = PDMAEMA).
Determined by 1H NMR.
Determined by GPC.
Determined by light scattering at 640 nm.
n.a. = not applicable.
The advantage of using RAFT polymerization encompasses the presence of the trithiocarbonate functional group on the final polymer after the polymerization process, which can subsequently be used for end-group modification.34,35 First, this group was converted into a thiol functional group by aminolysis and this reaction was followed via UV/vis spectroscopy since the trithiocarbonate group absorbs at 310 nm, whereas the thiol group does not absorb at this wavelength. After 24 h, a clear decrease of the absorbance peak at 310 nm was observed, indicating nearly complete removal of the trithiocarbonate group (Figure 3A). In addition, no shift in the molecular weight distribution was observed in the GPC chromatograms, confirming that disulfide bridge formation between two thiol terminated polymers did not occur (Table 3). Further functionalization of the polymer chain-end was achieved by coupling of a reactive maleimide-Cyanine3 (Cy3) dye to the thiol functionality. GPC analysis showed that both signals from RI and UV–vis detection at 550 nm overlapped, demonstrating covalent attachment of the Cy3 dye to the polymer (Figure 3B). Quantification of the Cy3 label was achieved via UV–vis spectroscopy, and the results showed that 6% of the polymer chains were labeled with the Cy3 fluorophore (Table S2). The presence of free thiols on the final polymer could result in unwanted disulfide formation during polyplex preparation. As determined by the Ellman’s assay, 11% of the polymer chains carry a thiol group after aminolysis (Table S2). It is reported that at high polymerization conversions, the RAFT end groups can be lost due to radical side reactions, which might explain the lower percentage of thiol groups.28,35 Taking into account that 6% of the chain ends were labeled with Cy3, 5% of the polymer chains contain a free thiol group. Additionally, no change in the molecular weight distribution as determined by GPC was observed for the polymers before and after aminolysis as well as after Cy3 labeling (Table 3), indicating no significant disulfide bond formation. Similar results for trithiocarbonate conversion to a thiol group and subsequent Cy3-labeling were obtained for the PD polymer, which was 5% labeled with the Cy3 dye (Table S2, Figure S7).
Figure 3.

(A) UV–vis spectra of NPD polymer before (solid line) and after (dotted line) aminolysis with n-butylamine for 24 h at RT. (B) GPC analysis with dual RI (black) and UV/vis (at 550 nm, pink) detection of Cy3-labeled NPD polymer (solid line) and free maleimide-Cy3 dye (dashed line).
A similar strategy was reported before, where the synthesis of a PEG macroinitiator containing both an azoinitiator (for classical free radical polymerization) and an ATRP initiator was demonstrated.25 In the present study, RAFT polymerization is used instead of classical free radical polymerization. RAFT polymerization has proven to be an excellent controlled free radical polymerization technique and might provide a better alternative, since the classical free radical polymerization mechanism may result in undesired chain termination reactions, broadening the molecular weight distribution of the synthesized polymer.36 More importantly, retention of both types of initiators on the final polymer chains provides the opportunity to functionalize both ends of the polymer chain.37,38 For example, the living ATRP chain-end can be substituted by an azide which in turn can be used to attach functional groups via click chemistry.39 Additionally, the RAFT chain-transfer agent can conveniently be converted into a free thiol, which can be exploited in further coupling reactions, used here for coupling of the Cy3 fluorophore.27,40 These features of this novel PEG macroinitiator can be exploited for the tailored design and preparation of polymers for biomedical applications, as targeting moieties, drug molecules, or fluorescent labels can be attached to the polymer chains.27,35,40
3.3. Polyplex Formation and Fluorescence Measurements
The synthesized polymers (characteristics shown in Table 3) were used to prepare pDNA–polyplexes, and the effect of the N/P charge ratio, temperature, and polymer block composition on the polyplex properties were investigated. Time-resolved fluorescence measurements in combination with the FRET technique allow detection of core–shell structures of the polyplexes and give valuable information about polymer behavior at higher N/P ratios. Knowledge about the polyplex dynamics is important for its application in gene delivery as it has been shown that dynamic polyplexes can achieve higher transfection efficiency compared to less dynamic systems.12,41,42 First, the results of the fluorescence measurements are presented (section 3.3.1) followed by interpretation of the FRET studies and a more in depth discussion about core–shell structures of polyplexes (section 3.3.2).
3.3.1. FRET Studies of Polyplexes with pDNA-FITC and Unlabeled and/or Labeled Polymers at Fixed Temperatures of 4 or 37 °C (Scheme 3A)
First, the behavior of the labeled species in solution and in polyplexes in the absence of the other FRET partner should be understood. To this end, the fluorescence properties of pDNA-FITC in its soluble form were compared with those of pDNA-FITC complexed with unlabeled polymer (further referred to as unlabeled polyplexes) both at 4 and 37 °C (Table 4). A clear shift to higher wavelengths was observed in the spectra of pDNA-FITC in the presence of the polymers (Figure S8). Furthermore, already at N/P 1, the FITC fluorescence efficiency has decreased considerably (Figure S9). The FITC fluorescence lifetimes were calculated from the decay curves monitored at 520 nm (Figure S10, Table 4). In the absence of NPD and PD polymers, the pDNA-FITC fluorescence decay curve is one-exponential, implying one lifetime. However, in the presence of the polymers two-exponential global fitting was used to calculate the lifetimes. The mean amplitude weighted fluorescence lifetime (⟨τ⟩) of pDNA-FITC in the presence of unlabeled NPD or PD polymers remained constant for all tested N/P ratios. The lifetimes at 4 °C were slightly higher compared to those at 37 °C, independent of the N/P ratio and type of polymer. To further visualize changes in the fluorescence lifetime of pDNA-FITC in the absence and presence of polymers, the τ0/⟨τ⟩ values are plotted as a function of the N/P ratio (Figure 4). In this parameter, τ0 is the fluorescence lifetime of pDNA-FITC in the absence of the polymers and ⟨τ⟩ is the mean amplitude weighted lifetime in the presence of the polymers. For both types of polymers (NPD and PD) and regardless of the temperature (4 and 37 °C), a clear change in the τ0/⟨τ⟩ value was observed upon the addition of polymer at N/P 1, whereas the τ0/⟨τ⟩ values remained constant upon the addition of extra polymer (N/P > 1) (Figure 4, blue triangles).
Table 4. Excitation and Fluorescence Spectrum Maxima (λmax,exand λmax,fl), Fluorescence Lifetimes (τ1, τ2), the Proportion of the Longer Living Component (α1), and the Mean Amplitude Weighted Fluorescence Lifetime (⟨τ⟩) at 4 and 37 °C for pDNA-FITC in the Absence and Presence of Unlabeled Polymers (NPD and PD).
| sample | temperature (°C) | N/P ratio | λmax,ex (nm) | λmax,fl (nm) | τ1 (ns) | α1 (%) | τ2 (ns) | ⟨τ⟩ (ns) |
|---|---|---|---|---|---|---|---|---|
| pDNA-FITC | 4 | n.a.a | 493 | 518 | 4.01 | 100 | n.a.a | 4.01 |
| 37 | n.a.a | 495 | 521 | 3.75 | 100 | n.a.a | 3.75 | |
| pDNA-FITC + NPD | 4 | 1 | 500 | 523 | 4.21 ± 0.07 | 71 | 2.44 ± 0.17 | 3.70 |
| 5 | 68 | 3.65 | ||||||
| 10 | 69 | 3.66 | ||||||
| 37 | 1 | 501 | 525 | 3.39 ± 0.04 | 72 | 1.32 ± 0.11 | 2.81 | |
| 5 | 71 | 2.78 | ||||||
| 10 | 66 | 2.68 | ||||||
| pDNA-FITC + PD | 4 | 1 | 500 | 523 | 4.23 ± 0.07 | 70 | 2.47 ± 0.17 | 3.71 |
| 5 | 69 | 3.69 | ||||||
| 10 | 69 | 3.69 | ||||||
| 37 | 1 | 501 | 525 | 3.65 ± 0.06 | 58 | 1.78 ± 0.10 | 2.86 | |
| 5 | 58 | 2.86 | ||||||
| 10 | 55 | 2.81 |
n.a. = not applicable.
Figure 4.

Fluorescence lifetime ratio of pDNA-FITC as a function of N/P ratio in the presence of unlabeled and/or labeled NPD polymer (A,B) and PD polymer (C,D) at 4 and 37 °C. The fluorescence lifetime of pDNA-FITC (τ0) was used to calculate the fluorescence lifetime ratio (τ0/⟨τ⟩).
The polyplexes, based on both labeled polymer and labeled pDNA (further referred to as fully labeled polyplexes), prepared at different N/P ratios were measured both at 4 and 37 °C similarly to the above-described polyplexes formed with unlabeled polymers. The presence of pDNA-FITC is seen as the fluorescence band at 520 nm and the absorption peak at 500 nm in the excitation spectra. The presence of Cy3-labeled polymers is observed as the fluorescence band at 562 nm and absorption peak at 545 nm (Figure S11). Due to the low labeling density of the polymers and the large overlap of the FITC and Cy3 fluorescence spectra, Cy3 fluorescence could not be monitored without disturbances from FITC fluorescence even at the highest N/P ratio. Therefore, we focused on the changes observed in FITC fluorescence. The fluorescence intensity ratio at 520 nm (i.e., intensity in the absence of the polymers over intensity in the presence of the polymers) is plotted as a function of N/P ratio (Figure S12). No significant difference is observed between the curves in the presence and absence of Cy3, because the major part of the pDNA-FITC fluorescence intensity (91%) was already quenched by the polymer at N/P 1 (Figure S9). The additional effect of Cy3 on the pDNA-FITC intensity was too small to be detected, and therefore further analysis is based on the time-resolved fluorescence data. Since the Cy3 dye does not fluoresce at 520 nm (Figure S13), the decay curves monitored at 520 nm were used to calculate changes in FITC fluorescence lifetime due to the presence of Cy3 (Figure S14). Similar to the unlabeled polyplexes, the decay curves of the fully labeled polyplexes were two-exponential. However, the proportion of the longer-living component decreased with increasing N/P ratio (Table S3). This is also reflected in higher τ0/⟨τ⟩ values for labeled polyplexes (Figure 4, light blue dots), compared to unlabeled polyplexes. Moreover, the τ0/⟨τ⟩ values continuously increased with increasing N/P ratio. Again, no clear differences in pDNA-FITC lifetime between polyplexes consisting of NPD-Cy3 or PD-Cy3 polymers were observed.
To study the dynamics, polyplexes containing both unlabeled and labeled polymers were prepared (further referred to as mixed polyplexes). For this, unlabeled polymer was used to prepare the polyplexes up to N/P 2 and subsequently the N/P was increased by adding Cy3-labeled polymer. As expected, the decay curves of the mixed polyplexes were two-exponential resulting in two fluorescence lifetime populations of FITC (Table S3). At low N/P ratios (<2), the τ0/⟨τ⟩ values of the mixed polyplexes (Figure 4, red squares) were similar to those of the unlabeled polyplexes. At higher N/P ratios, where labeled polymer was added to the polyplex formulations, higher τ0/⟨τ⟩ values were observed for the mixed polyplexes, in a similar trend as for the fully labeled polyplexes.
3.3.2. Interpretation of the FRET Results
Fluorescence resonance energy transfer (FRET) is a distance-dependent excited-state interaction between a donor and acceptor fluorophore.17−19 It can been used as a tool for measuring distance between molecules and monitoring conformational changes in macromolecules. In the present study, the fluorophore pair FITC and Cy3 was selected to obtain insight into the interactions between pDNA and polymer by time-resolved fluorescence spectroscopy. Besides the occurrence of energy transfer, a variety of molecular interactions can lead to a reduction in fluorescence intensity, which is referred to as quenching.43 In polyplexes, fluorescence quenching is also observed due to changes in the microenvironment of the FITC dye upon complexation of pDNA with the polycation. In addition, the fluorescence lifetime of FITC, and of every fluorophore in general, is strongly dependent on the microenvironment and, hence, on the binding between cationic polymers and pDNA in the case of polyplexes.43,44 The negatively charged phosphate groups (P) of pDNA interact with the protonated amine groups (N) of the polymer via electrostatic interactions and the molar ratio between these two is defined as the N/P ratio. At low N/P ratios, the complexation results in conformational changes and the initial condensation of pDNA,45 which is further referred to as the polyplex core. Not necessarily all the phosphate groups of pDNA interact with the polymers, which is also illustrated by a negative ζ-potential at N/P 1 for both the NPD- and PD-based polyplexes and indicates incomplete pDNA shielding (Figure S15). In addition, the negative ζ-potential at N/P 1 can also be explained by the fact that not all DMAEMA units are protonated at pH 7.4.46 At higher N/P ratios, the extra polymer chains most likely saturate pDNA via electrostatic interactions completing the polyplex structure, which is referred to as the polyplex shell. This core–shell structure of polyplexes can be interpreted from the time-resolved fluorescence measurements. Formation of the polyplex core leads to changes in the microenvironment of the FITC dye and subsequently fluorescence quenching. Once this process is completed, the addition of extra polymer does not further affect the FITC fluorescence because of a less strong electrostatic interaction and larger distance between the polycation and pDNA-FITC.12,16,43 The excess of polycations generally results in formation of polyplexes with a positive surface charge. Indeed, the ζ-potentials measured at N/P 10 and at 4 °C were 7.7 ± 0.9 mV and 16.7 ± 1.5 mV for NPD and PD polyplexes, respectively (Figure S15). Noteworthy, the NPD polyplexes showed a lower ζ-potential than the PD-based polyplexes, which is likely due to the additional shielding of the PNIPAM blocks both below and above the CP, as also recently reported.11 For both types of polyplexes, no significant differences in ζ-potential were observed when measured at 4 or 37 °C.
As discussed above, it is important to study the effect of the polymer itself on pDNA-FITC before looking into additional quenching due to FRET. For the unlabeled polyplexes, the addition of polymer resulted in the appearance of two lifetime populations (Table 4). This most likely indicates that in some microenvironments FITC is surrounded by groups which increase or do not affect the fluorescence lifetime and other microenvironments that decrease FITC fluorescence lifetime, due to, e.g., differences in viscosity, charge density, and chemical groups in the immediate vicinity of FITC.43 At the same time, the fluorescence intensity of pDNA-FITC was strongly quenched upon addition of the cationic polymers (Figure S9). Taken together, these results clearly indicate the interaction between pDNA and the polymers are consistent with previous findings for PEI- and PLL-based polyplexes.12,16 The FITC fluorescence quenching can also be observed as an increase in the τ0/⟨τ⟩ value, since the mean amplitude weighted fluorescence lifetime of pDNA-FITC (⟨τ⟩) decreased upon complexation with the polymers compared to soluble pDNA-FITC (τ0) (Figure 4, blue triangles). Because quenching is a competitive process that depopulates the excited state of the fluorophore, this leads to a decrease in the lifetime.43 Taking it one step further, the core–shell structure of the unlabeled polyplexes can be observed as effective quenching of pDNA-FITC fluorescence at N/P ≤ 1 when the nanoparticle core is formed. At higher N/P ratios, the addition of extra polymer did not further affect FITC fluorescence suggesting that the extra polymer forms a more loose positively charged shell around the core structure.16 The presence of some free polymer chains in solution at the higher N/P ratios is not excluded but cannot be confirmed by the method used.
For the fully labeled polyplexes, again two lifetime populations were found, however higher τ0/⟨τ⟩ values were observed compared to unlabeled polyplexes (Figure 4, light blue dots). The difference in τ0/⟨τ⟩ value at the same N/P ratio can be ascribed to additional quenching of FITC due to the presence of Cy3 in the fully labeled polyplexes. This indicates that Cy3 in the fully labeled polyplex formulation leads to stronger quenching of the FITC fluorescence via energy transfer, i.e., FRET, compared to the quenching of the polymer only in the unlabeled polyplexes and confirms that the selected FRET pair is suitable for this study. Interestingly, the τ0/⟨τ⟩ values for fully labeled polyplexes increased with increasing N/P ratio, which was not observed for fully labeled PEI- and PLL-based polyplexes.12,16 These results suggest that even after the polyplex core formation, Cy3-labeled polymers are able to get close enough (∼10 nm) to pDNA-FITC for FRET to take place. Noteworthy, at low N/P ratios (N/P ≤ 2), the difference between the τ0/⟨τ⟩ values for unlabeled polyplexes and fully labeled polyplexes (Figure 4, dark blue triangles and light blue dots) is smaller at 37 °C compared to at 4 °C. In other words, the energy transfer from FITC to Cy3 is more pronounced at 4 °C than at 37 °C as the increase in τ0/⟨τ⟩ for Cy3-labeled polymers is much stronger. Nevertheless, the difference in τ0/⟨τ⟩ between labeled and unlabeled polymer becomes also visible at higher N/P ratios at 37 °C. No clear differences in energy transfer efficiency between NPD-Cy3 and PD-Cy3 polymers were observed, confirming that the presence of the thermosensitive PNIPAM block does not hinder the formation of stable polyplexes within the measured temperature range.
The slopes of the τ0/⟨τ⟩ plots, the K values, were used as a measure of relative quenching efficiency and allowed for a more in-depth analysis of the fluorescence pDNA-FITC quenching in the different polyplex formulations and at different temperatures (4 and 37 °C). Two linear phases can be distinguished in the τ0/⟨τ⟩ plots (Figure 4): the first phase at N/P ≤ 1 and the second at N/P > 1. The K value is the slope of the τ0/⟨τ⟩ plot and the obtained values were normalized against the KN/P≤1 value of pDNA-FITC with unlabeled polymer at 4 °C (Table 5). For the unlabeled polyplexes (pDNA-FITC in the absence of Cy3 acceptor), only the K value for the first part of the τ0/⟨τ⟩ plot (N/P ≤ 1) could be determined and indicates that the polyplex core formation is completed at N/P 1, since the addition of extra unlabeled polymer at higher N/P ratios did not further affect FITC fluorescence. Interestingly, the KN/P≤1 values for the unlabeled polyplexes at 37 °C were 4 times higher compared to those at 4 °C, which indicates that the fluorescence quenching at N/P ≤ 1 is more pronounced at 37 °C (Table 5). Such a difference cannot be explained by only a decrease in viscosity or increased nonradiative relaxation of the excited state at higher temperatures. Although cationic polymer and pDNA complex with each other by electrostatic forces, their structures reorganize upon complex formation leading to further reduced hydration of the initial electrostatic complexes. This assembly phenomenon has been reported to be typically entropy-driven and an endothermic process,47,48 and therefore a temperature increase could favor the formation of polyplex core.49 In addition, the flexibility of pDNA increases with increasing temperature allowing it to adjust better during the polyplex formation.50 Furthermore, polyplexes formed at 4 °C had a hydrodynamic size of around 150 nm, while those formed at 37 °C were about 50 nm bigger in size (Figure S16A,B). Remarkably, these DLS results are contradictory with the differences observed in the KN/P≤1 values, between 4 and 37 °C. Taken all together, these results indicate that the polyplex core formed at 4 or 37 °C differ from each other. This phenomenon was observed for both NPD and PD polymers, making it unlikely that the observed differences in polyplex size and K values at 4 and 37 °C are due to the presence of thermosensitive PNIPAM blocks.
Table 5. Normalized Slopes, K, of the Two Phases Observed in τ0/⟨τ⟩ Plots (Figure 4).
| core | shell | ||
|---|---|---|---|
| sample | temperature (°C) | KN/P≤1a | KN/P>1a |
| pDNA-FITC + NPD (unlabeled) | 4 | 1.0 | n.a.b |
| 37 | 4.0 | n.a.b | |
| pDNA-FITC + NPD-Cy3 (fully labeled) | 4 | 2.2 | 0.10 |
| 37 | 4.4 | 0.19 | |
| pDNA-FITC + NPD(-Cy3) (mixed) | 4 | 1.3 | 0.13 |
| 37 | 3.7 | 0.28 | |
| pDNA-FITC + PD (unlabeled) | 4 | 1.0 | n.a.b |
| 37 | 3.9 | n.a.b | |
| pDNA-FITC + PD-Cy3 (fully labeled) | 4 | 2.1 | 0.14 |
| 37 | 4.5 | 0.19 | |
| pDNA-FITC + PD(-Cy3) (mixed) | 4 | 1.3 | 0.18 |
| 37 | 4.1 | 0.29 |
Normalized against KN/P≤1 value (core) of pDNA-FITC + unlabeled polymer at 4 °C.
n.a. = not applicable.
At both temperatures (4 and 37 °C), the KN/P≤1 values are smaller in the absence of Cy3 (unlabeled polyplexes) than in the presence of Cy3 (fully labeled polyplexes), indicating more pDNA-FITC quenching in the latter formulation and further confirming the occurrence of FRET (Table 5). For the fully labeled polyplexes, the KN/P>1 values are over 20 times smaller (both at 4 and 37 °C) than the values of KN/P≤1 (Table 5). Since the K value indicates the sensitivity of the fluorophore to a quencher, these results further support the core–shell structure of the polyplexes. The difference in K values can be interpreted as two main pDNA-FITC populations with different accessibility to the Cy3-labeled polymers, which also depends on the N/P ratio.43 The higher K value at N/P ≤ 1 corresponds to a closer spacing between the FITC and Cy3 molecules, in other words, between pDNA and polymer chains in the polyplex core. The lower K values at N/P > 1 correspond to less quenching and thus less energy transfer of Cy3 to FITC because of a larger distance between pDNA-FITC and Cy3-polymers (polyplex shell). Similar as for the unlabeled polyplexes, the KN/P≤1 values of fully labeled polyplexes are 2 times higher at 37 °C compared to 4 °C during the core formation (Table 5). This indicates a comparable temperature sensitivity of the core formation step for polyplexes formed with both labeled and unlabeled polymers. In addition, the formation of the shell of the fully labeled polyplexes was also temperature dependent at all tested N/P ratios. To illustrate, the KN/P>1 values at 37 °C are 1.5–2 times higher compared with the KN/P>1 values at 4 °C (Table 5). The finding that the FITC lifetime is also temperature dependent during the shell formation may indicate that the accessibility of the unquenched FITC population to Cy3-labeled polymers in the polyplex shell increases with increasing temperature.
Based on the above discussion and since only the cationic PDMAEMA block is capable of binding pDNA, it is suggested that the structure of the polyplex core would resemble that of a hairy ball (Figure 5A) allowing the shell forming polymers to intercalate between the (N)P-blocks of the core forming polymers. In other words, some of the Cy3-moieties of the shell forming polymer can locate close enough to pDNA-FITC for FRET to take place, even at high N/P ratios. This kind of structure differs from the polyplex structures described for pDNA complexed with PEI and PLL homopolymers. Likely, cationic homopolymers form a more dense core with pDNA as compared with block copolymers, in which the shell polymers are more screened from the pDNA-containing core (Figure 5B).12 This difference in morphology between homo- and block copolymer-based polyplexes can be further supported by earlier reported results on the ratio of radius of gyration (Rg) and hydrodynamic radius (Rh) of the polyplexes measured by static light scattering.11,51,52 The ratio between Rg and Rh provides information about the shape and compactness of particles. For example, this parameter has a value of ∼0.774 for hard solid spheres, ∼1.5 for random coils, and ∼2 for elongated structures.53 For PEI/pDNA polyplexes, Rg/Rh values smaller than 1 are reported, indicating that the polyplexes have a dense hydrophobic core and a more hydrophilic solvent-draining shell formed by the excess of PEI.51,52 Recently, it was shown that Rg/Rh values of NPD- and PD-based polyplexes are greater than 1, indicating more loose, hairy structures.11
Figure 5.
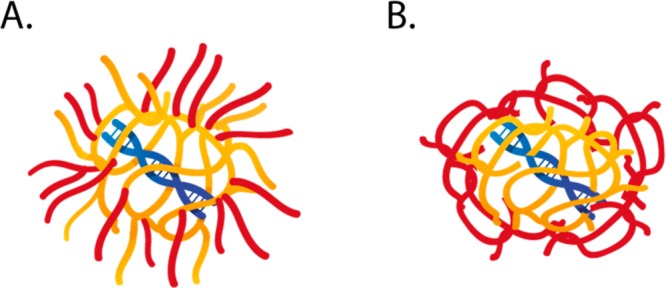
Suggested polyplex structure for studied NPD and PD polymers (A) compared to the structure of the polyplex formed by PEI and PLL (B) as revealed earlier.12 The pDNA is depicted in blue, core polymers in yellow, and shell polymers in red.
For the mixed polyplexes (Figure 4, red squares), the occurrence of energy transfer from FITC to Cy3 was again observed as higher τ0/⟨τ⟩ values, only at the higher N/P ratios where labeled polymer was added to the polyplex formulation. The KN/P≤1 values of the mixed polyplexes are, as expected, similar to those for unlabeled polyplexes. Upon addition of Cy3-labeled polymers at higher N/P ratios, the KN/P>1 values obtained are slightly higher than those for the fully labeled polyplexes. The results from the mixed polyplexes indicate that the Cy3-labeled polymers can interact directly with pDNA-FITC regardless of the presence of unlabeled polymers in the polyplex core. In other words, the exchange between polymers in the core and shell is efficient, and independent of the N/P ratio, the Cy3-labeled polymers are uniformly distributed in the polyplex particles. Therefore, these results demonstrate the dynamic character of the polyplex structure for both NPD and PD polymers. The observed dynamic structure is most likely an intrinsic feature of these polyplexes, which is beneficial for further application as gene delivery vectors. It is known that polyplexes and lipoplexes with secondary and tertiary amines show easier DNA release from the nanoparticle carrier compared to those containing only primary or quaternary amines.14,15,52 Mobility of the polymer molecules between the core and the shell has also been suggested to make the polyplexes more sensitive to dissociation and pDNA release at the cellular level.12 As reported before, NPD-based polyplexes have a better cytocompatibility than PD-based polyplexes with similar efficiencies of delivering its cargo into HeLa cells in the presence of serum.11 Although the synthesis route to obtain NPD polymers was slightly different in that study, the block composition of the polymers used in this study is similar except for the presence of fluorescently labeled chain ends, and therefore no significant differences in transfection efficiency and cytotoxicity are expected, which was also confirmed (Figure 6). PEI-based polyplexes showed around 10 times higher transfection compared to NPD or PD formulations; however, a decrease in cell viability of ∼25% was observed as well (Figure 6). PEGylated polyplexes are taken up to a lower extent than polyplexes based on cationic homopolymers (such as PEI).54 Nevertheless, non-PEGylated polyplexes are not suitable for in vivo applications due to their compromised circulation kinetics and local administration is known to result in low transfection because of their restricted mobility.54,55
Figure 6.

In vitro evaluation of polyplexes on HeLa cells. Cells were transfected with pDNA (0.50 μg/well) formulated in NPD and PD polyplexes at different N/P ratios in serum supplemented culture medium for 6 h. A formulation with l-PEI, 25 kDa (N/P 6) was added as a control. Transfection efficiency was determined by a luciferase reporter assay (A), and cell viability was determined by an MTS assay (B).
3.3.3. Properties of the Polyplexes in Temperature Cycle Measurements (Scheme 3B)
The effect of temperature change on the behavior of the polyplexes (N/P 10) was studied. The polyplexes were first prepared at 4 or 37 °C, after which the temperature was changed in the series of 4 → 22 → 37 → 22 → 4 °C or 37 → 22 → 4 → 22 → 37 °C. Since fluorescence lifetimes are highly dependent on the microenvironment of the fluorophore, time-resolved fluorescent measurements can be used here as a tool for monitoring structural changes in the polyplexes. The mean amplitude weighted lifetimes, ⟨τ⟩, are plotted as a function of temperature for all the NPD- and PD-based polyplexes (Figure 7, Table S4). To note, the ⟨τ⟩ values obtained for polyplexes prepared directly at N/P 10 were slightly smaller than for the polyplexes formed stepwise through a gradual increase of the N/P ratio during the fixed temperature measurements (Figure S17).
Figure 7.

Mean amplitude weighted fluorescence lifetime (⟨τ⟩) of pDNA-FITC as a function of temperature in the presence of unlabeled NPD polymer (A,B, blue) and PD polymer (C,D, blue) at N/P 10. The arrows indicate the direction of the temperature change and points marked with a star (bright blue) indicate changes in the polyplex structure. The mean amplitude weighted fluorescence lifetimes obtained before the temperature change are shown in red dots.
When NPD-based polyplexes were heated from 4 to 37 °C, the ⟨τ⟩ values were nearly the same as those for polyplexes prepared directly and measured at 37 °C (Figure 7A). However, cooling down the polyplexes back to 4 °C resulted in a significantly smaller ⟨τ⟩ value compared with the initial value for the polyplex prepared at 4 °C before the temperature changes (2.77 and 3.38 ns, respectively). Moreover, two-exponential fitting could not be used for the data obtained after cooling down, indicating that irreversible changes in the polyplex structure took place (Table S4). A similar thermosensitive behavior of the NPD polyplexes was observed for the temperature cycle 37 → 22 → 4 → 22 → 37 °C (Figure 7B). Upon cooling the polyplexes from 37 to 4 °C, the ⟨τ⟩ values are in good agreement with the ⟨τ⟩ values of polyplexes prepared directly and measured at 4 °C. However, increasing the temperature again to 37 °C resulted in shorter ⟨τ⟩ values. Although this difference in ⟨τ⟩ values before and after the temperature changes was not as pronounced as seen in Figure 7A, a two-exponential fitting could not be used for the obtained decay curves (Table S4). This indicates that irreversible changes in the polyplex structure have occurred during the temperature cycle. Interestingly, these irreversible changes were not reflected in the size of the polyplexes at different temperatures, as determined by DLS (Figure S16C,D), which was also reported before.11 This illustrates that time-resolved fluorescence measurements can give additional information about the polyplex structure, which cannot be obtained by particle size or ζ-potential measurements. For the nonthermosensitive PD-based polyplexes, no significant differences in ⟨τ⟩ values were observed during both types of temperature cycles measurements (4 → 22 → 37 → 22 → 4 °C and 37 → 22 → 4 → 22 → 37 °C) (Figure 7C,D). The changes in the ⟨τ⟩ values are much smaller for PD-based polyplexes than for those of NPD-based ones and lie within the limits of fitting errors (Table S4). The transfection efficiency and cytotoxicity of polyplexes before and after the temperature changes were evaluated in the presence of serum using HeLa cells. No significant differences in transfection efficiency were observed for NPD and PD polyplexes prepared at 4 or 37 °C (Figure 8A). Moreover, NPD polyplexes that underwent temperature changes before addition to HeLa cells, exhibited similar transfection efficiencies as NPD polyplexes before the temperature cycle. The preparation temperature (4 or 37 °C) as well as the temperature changes did not affect the cytocompatibility for both NPD and PD polyplexes (Figure 8B). These results indicate that although changes in the NPD polyplex structure were observed after temperature changes, they do not translate in a different in vitro bioactivity of the polyplexes. In other words, multiple functionalities can be introduced into the polymer design without compromising the polyplex function.
Figure 8.

In vitro evaluation of polyplexes before and after the temperature cycle on HeLa cells. Cells were treated with pDNA (0.50 μg/well) formulated in NPD and PD polyplexes (N/P 10) in serum supplemented culture medium for 6 h. The polyplexes were prepared at either 4 or 37 °C and subsequently subjected to a temperature cycle (4 → 37 → 4 °C and 37 → 4 → 37 °C, respectively). A formulation with l-PEI, 25 kDa (N/P 6) and naked pDNA were added as positive and negative controls, respectively. Transfection efficiency was determined by a luciferase reporter assay (A), and cell viability was determined by an MTS assay (B).
4. Conclusion
This study shows the successful synthesis of a heterofunctional PEG macroinitiator containing both an ATRP and a RAFT initiator. The use of this novel PEG macroinitiator allows the synthesis of linear triblock copolymers, having a PEG midblock. The combination of two types of controlled polymerization techniques enables the combination of a wide range of monomers into the polymer design. Moreover, the opportunity of using the living chain-ends to further functionalize the final polymer is an attractive tool in the design of polymers for biomedical and pharmaceutical applications. Cationic block copolymers based on PDMAEMA, PEG, and PNIPAM formed complexes with pDNA both at 4 and 37 °C, meaning that the introduction of the thermosensitive PNIPAM block did not compromise the polyplex structure. In addition, thermosensitive NPD polyplexes have a better cytocompatibility than nonthermosensitive PD polyplexes with similar efficiencies of delivering its cargo into HeLa cells, even after multiple temperature changes. Furthermore, results from the time-resolved fluorescence measurements showed that the studied polyplexes have a less dense core compared to polyplexes based on cationic homopolymers such as PEI. Both at 4 and 37 °C, the polyplexes showed to have a dynamic character, meaning that the polymer chains can exchange between the polyplex core and shell. Such a mobility of the polymers allows their uniform redistribution within the polyplex, and this feature is favorable in the context of pDNA release and subsequent improved transfection efficiency, compared to nondynamic formulations. Overall, we demonstrated that different properties can be introduced in the polymer design without compromising the polyplex structure and function. This approach can potentially be used for different applications, such as using the thermosensitive properties to entrap the polyplexes in a thermosensitive hydrogel facilitating local and sustained release of nucleic acids.
Acknowledgments
The Netherlands Organization for Scientific Research (NWO/VIDI Grant 13457 and NWO/Aspasia Grant 015.009.038) is acknowledged for funding as well as the Academy of Finland (Project No. 311362).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.9b00896.
Experimental procedures and characterization of the synthesized macroinitiator and polymers (Tables S1 and S2, Figures S1–S7); excitation and fluorescence spectra and fluorescence decay curves for pDNA(-FITC) in the presence of (Cy3-labeled) polymers (Figures S8–S11, S13, and S14); fluorescence intensity ratio for pDNA-FITC in the presence of (Cy3-labeled) polymers (Figure S12); fitting results of fluorescence decay curves (Tables S3 and S4); experimental procedures and characterization of polyplexes by dynamic light scattering (DLS) and laser Doppler electrophoresis (LDE) (Figures S15 and S16); and fluorescence lifetimes for polyplexes prepared stepwise or directly at N/P 10 (Figure S17) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chakraborty C.; Sharma A. R.; Sharma G.; Doss C. G. P.; Lee S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther.--Nucleic Acids 2017, 8, 132–143. 10.1016/j.omtn.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachelt U.; Wagner E. Nucleic Acid Therapeutics Using Polyplexes: A Journey of 50 Years (and Beyond). Chem. Rev. 2015, 115 (19), 11043–78. 10.1021/cr5006793. [DOI] [PubMed] [Google Scholar]
- Wagner E. Polymers for nucleic acid transfer-an overview. Adv. Genet. 2014, 88, 231–61. 10.1016/B978-0-12-800148-6.00008-0. [DOI] [PubMed] [Google Scholar]
- Stuart M. A. C.; Huck W. T. S.; Genzer J.; Muller M.; Ober C.; Stamm M.; Sukhorukov G. B.; Szleifer I.; Tsukruk V. V.; Urban M.; Winnik F.; Zauscher S.; Luzinov I.; Minko S. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101. 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- Sun H.; Kabb C. P.; Sims M. B.; Sumerlin B. S. Architecture-transformable polymers: Reshaping the future of stimuli-responsive polymers. Prog. Polym. Sci. 2019, 89, 61–75. 10.1016/j.progpolymsci.2018.09.006. [DOI] [Google Scholar]
- Bordat A.; Boissenot T.; Nicolas J.; Tsapis N. Thermoresponsive polymer nanocarriers for biomedical applications. Adv. Drug Delivery Rev. 2019, 138, 167–192. 10.1016/j.addr.2018.10.005. [DOI] [PubMed] [Google Scholar]
- Fu X.; Hosta-Rigau L.; Chandrawati R.; Cui J. Multi-Stimuli-Responsive Polymer Particles, Films, and Hydrogels for Drug Delivery. Chem. 2018, 4 (9), 2084–2107. 10.1016/j.chempr.2018.07.002. [DOI] [Google Scholar]
- Alexander C. Temperature- and pH-responsive smart polymers for gene delivery. Expert Opin. Drug Delivery 2006, 3 (5), 573–81. 10.1517/17425247.3.5.573. [DOI] [PubMed] [Google Scholar]
- Wang L. L.; Burdick J. A. Engineered Hydrogels for Local and Sustained Delivery of RNA-Interference Therapies. Adv. Healthcare Mater. 2017, 6 (1), 1601041. 10.1002/adhm.201601041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliervoet L. A. L.; Engbersen J. F. J.; Schiffelers R. M.; Hennink W. E.; Vermonden T. Polymers and hydrogels for local nucleic acid delivery. J. Mater. Chem. B 2018, 6 (36), 5651–5670. 10.1039/C8TB01795F. [DOI] [PubMed] [Google Scholar]
- Fliervoet L. A. L.; van Nostrum C. F.; Hennink W. E.; Vermonden T. Balancing hydrophobic and electrostatic interactions in thermosensitive polyplexes for nucleic acid delivery. Multifunctional Materials 2019, 2 (2), 024002 10.1088/2399-7532/ab12ee. [DOI] [Google Scholar]
- Lisitsyna E. S.; Ketola T. M.; Morin-Picardat E.; Liang H. M.; Hanzlikova M.; Urtti A.; Yliperttula M.; Vuorimaa-Laukkanen E. Time-Resolved Fluorescence Spectroscopy Reveals Fine Structure and Dynamics of Poly(L-lysine) and Polyethylenimine Based DNA Polyplexes. J. Phys. Chem. B 2017, 121 (48), 10782–10792. 10.1021/acs.jpcb.7b08394. [DOI] [PubMed] [Google Scholar]
- Vinogradov S. V.; Bronich T. K.; Kabanov A. V. Self-Assembly of Polyamine–Poly(ethylene glycol) Copolymers with Phosphorothioate Oligonucleotides. Bioconjugate Chem. 1998, 9 (6), 805–812. 10.1021/bc980048q. [DOI] [PubMed] [Google Scholar]
- Arigita C.; Zuidam N. J.; Crommelin D. J. A.; Hennink W. E. Association and Dissociation Characteristics of Polymer/DNA Complexes Used for Gene Delivery. Pharm. Res. 1999, 16 (10), 1534–1541. 10.1023/A:1015096302720. [DOI] [PubMed] [Google Scholar]
- Ruponen M.; Ronkko S.; Honkakoski P.; Pelkonen J.; Tammi M.; Urtti A. Extracellular glycosaminoglycans modify cellular trafficking of lipoplexes and polyplexes. J. Biol. Chem. 2001, 276 (36), 33875–80. 10.1074/jbc.M011553200. [DOI] [PubMed] [Google Scholar]
- Vuorimaa-Laukkanen E.; Lisitsyna E. S.; Ketola T.-M.; Morin-Pickardat E.; Liang H.; Hanzlíková M.; Yliperttula M. Difference in the core-shell dynamics of polyethyleneimine and poly(l-lysine) DNA polyplexes. Eur. J. Pharm. Sci. 2017, 103, 122–127. 10.1016/j.ejps.2017.03.025. [DOI] [PubMed] [Google Scholar]
- Breunig M.; Lungwitz U.; Liebl R.; Goepferich A. Fluorescence resonance energy transfer: Evaluation of the intracellular stability of polyplexes. Eur. J. Pharm. Biopharm. 2006, 63 (2), 156–165. 10.1016/j.ejpb.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Clegg R. M. Fluorescence resonance energy transfer. Curr. Opin. Biotechnol. 1995, 6 (1), 103–10. 10.1016/0958-1669(95)80016-6. [DOI] [PubMed] [Google Scholar]
- Forster T. Energiewanderung und Fluoreszenz. Naturwissenschaften 1946, 33 (6), 166–175. 10.1007/BF00585226. [DOI] [Google Scholar]
- Guo C.; Dower S. K.; Holowka D.; Baird B. Fluorescence resonance energy transfer reveals interleukin (IL)-1-dependent aggregation of IL-1 type I receptors that correlates with receptor activation. J. Biol. Chem. 1995, 270 (46), 27562–8. 10.1074/jbc.270.46.27562. [DOI] [PubMed] [Google Scholar]
- Moore J. S.; Stupp S. I. Room temperature polyesterification. Macromolecules 1990, 23 (1), 65–70. 10.1021/ma00203a013. [DOI] [Google Scholar]
- De Vos R.; Goethals E. J. End group analysis of commercial poly(ethylene glycol) monomethyl ether’s. Polym. Bull. 1986, 15 (6), 547–549. 10.1007/BF00281766. [DOI] [Google Scholar]
- Loccufier J.; Van Bos M.; Schacht E. Convenient method for the analysis of primary and secondary hydroxyl end groups in polyethers. Polym. Bull. 1991, 27 (2), 201–204. 10.1007/BF00296031. [DOI] [Google Scholar]
- Talelli M.; Rijcken C. J.; Oliveira S.; van der Meel R.; van Bergen en Henegouwen P. M.; Lammers T.; van Nostrum C. F.; Storm G.; Hennink W. E. Nanobody-shell functionalized thermosensitive core-crosslinked polymeric micelles for active drug targeting. J. Controlled Release 2011, 151 (2), 183–92. 10.1016/j.jconrel.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Fliervoet L. A. L.; Najafi M.; Hembury M.; Vermonden T. Heterofunctional Poly(ethylene glycol) (PEG) Macroinitiator Enabling Controlled Synthesis of ABC Triblock Copolymers. Macromolecules 2017, 50 (21), 8390–8397. 10.1021/acs.macromol.7b01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri M.; Bresseleers J.; Varela-Moreira A.; Sandre O.; Meeuwissen S. A.; Schiffelers R. M.; Metselaar J. M.; van Nostrum C. F.; van Hest J. C. M.; Hennink W. E. Effect of Formulation and Processing Parameters on the Size of mPEG- b-p(HPMA-Bz) Polymeric Micelles. Langmuir 2018, 34 (50), 15495–15506. 10.1021/acs.langmuir.8b03576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hembury M.; Beztsinna N.; Asadi H.; van den Dikkenberg J. B.; Meeldijk J. D.; Hennink W. E.; Vermonden T. Luminescent Gold Nanocluster-Decorated Polymeric Hybrid Particles with Assembly-Induced Emission. Biomacromolecules 2018, 19 (7), 2841–2848. 10.1021/acs.biomac.8b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallyn S.; Zhang Z.; Driessen F.; Pietrasik J.; De Geest B. G.; Hoogenboom R.; Du Prez F. E. Straightforward RAFT procedure for the synthesis of heterotelechelic poly(acrylamide)s. Macromol. Rapid Commun. 2014, 35 (4), 405–11. 10.1002/marc.201300690. [DOI] [PubMed] [Google Scholar]
- Habeeb A. F. Determination of free amino groups in proteins by trinitrobenzenesulfonic acid. Anal. Biochem. 1966, 14 (3), 328–36. 10.1016/0003-2697(66)90275-2. [DOI] [PubMed] [Google Scholar]
- Neradovic D.; Hinrichs W. L. J.; Kettenes-van den Bosch J. J.; Hennink W. E. Poly(N-isopropylacrylamide) with hydrolyzable lactic acid ester side groups: a new type of thermosensitive polymer. Macromol. Rapid Commun. 1999, 20 (11), 577–581. . [DOI] [Google Scholar]
- Ellman G. L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82 (1), 70–7. 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Haque F.; Geng J.; Montemagno C.; Guo P. Incorporation of a viral DNA-packaging motor channel in lipid bilayers for real-time, single-molecule sensing of chemicals and double-stranded DNA. Nat. Protoc. 2013, 8 (2), 373–392. 10.1038/nprot.2013.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gaal E. V.; van Eijk R.; Oosting R. S.; Kok R. J.; Hennink W. E.; Crommelin D. J.; Mastrobattista E. How to screen non-viral gene delivery systems in vitro?. J. Controlled Release 2011, 154 (3), 218–32. 10.1016/j.jconrel.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Narain R. Progress of RAFT based polymers in gene delivery. Prog. Polym. Sci. 2013, 38 (5), 767–790. 10.1016/j.progpolymsci.2012.09.008. [DOI] [Google Scholar]
- Willcock H.; O’Reilly R. K. End group removal and modification of RAFT polymers. Polym. Chem. 2010, 1 (2), 149–157. 10.1039/B9PY00340A. [DOI] [Google Scholar]
- Moad G.; Chong Y. K.; Postma A.; Rizzardo E.; Thang S. H. Advances in RAFT polymerization: the synthesis of polymers with defined end-groups. Polymer 2005, 46 (19), 8458–8468. 10.1016/j.polymer.2004.12.061. [DOI] [Google Scholar]
- Matyjaszewski K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30 (23), 1706441. 10.1002/adma.201706441. [DOI] [PubMed] [Google Scholar]
- Averick S.; Mehl R. A.; Das S. R.; Matyjaszewski K. Well-defined biohybrids using reversible-deactivation radical polymerization procedures. J. Controlled Release 2015, 205, 45–57. 10.1016/j.jconrel.2014.11.030. [DOI] [PubMed] [Google Scholar]
- de Graaf A. J.; Mastrobattista E.; van Nostrum C. F.; Rijkers D. T. S.; Hennink W. E.; Vermonden T. ATRP, subsequent azide substitution and ‘click’ chemistry: three reactions using one catalyst in one pot. Chem. Commun. 2011, 47, 6972–6974. 10.1039/c1cc12224j. [DOI] [PubMed] [Google Scholar]
- Scales C. W.; Convertine A. J.; McCormick C. L. Fluorescent Labeling of RAFT-Generated Poly(N-isopropylacrylamide) via a Facile Maleimide–Thiol Coupling Reaction. Biomacromolecules 2006, 7 (5), 1389–1392. 10.1021/bm060192b. [DOI] [PubMed] [Google Scholar]
- Pigeon L.; Goncalves C.; Pichon C.; Midoux P. Evidence for plasmid DNA exchange after polyplex mixing. Soft Matter 2016, 12 (33), 7012–9. 10.1039/C6SM00575F. [DOI] [PubMed] [Google Scholar]
- Sun C.; Tang T.; Uludağ H. Molecular Dynamics Simulations of PEI Mediated DNA Aggregation. Biomacromolecules 2011, 12 (10), 3698–3707. 10.1021/bm2009476. [DOI] [PubMed] [Google Scholar]
- Lakowicz J.Principles of Fluorescence Spectroscopy, 3rd ed.; Springer, 2006. [Google Scholar]
- Cubeddu R.; Comelli D.; D’Andrea C.; Taroni P.; Valentini G. Time-resolved fluorescence imaging in biology and medicine. J. Phys. D: Appl. Phys. 2002, 35 (9), R61–R76. 10.1088/0022-3727/35/9/201. [DOI] [Google Scholar]
- Dunlap D. D.; Maggi A.; Soria M. R.; Monaco L. Nanoscopic structure of DNA condensed for gene delivery. Nucleic Acids Res. 1997, 25 (15), 3095–101. 10.1093/nar/25.15.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering P.; Zuidam N. J.; van Steenbergen M. J.; van der Houwen O. A. G. J.; Underberg W. J. M.; Hennink W. E. A Mechanistic Study of the Hydrolytic Stability of Poly(2-(dimethylamino)ethyl methacrylate). Macromolecules 1998, 31 (23), 8063–8068. 10.1021/ma980689g. [DOI] [Google Scholar]
- Ikonen M.; Murtomaki L.; Kontturi K. Controlled complexation of plasmid DNA with cationic polymers: effect of surfactant on the complexation and stability of the complexes. Colloids Surf., B 2008, 66 (1), 77–83. 10.1016/j.colsurfb.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Zheng M.; Pavan G. M.; Neeb M.; Schaper A. K.; Danani A.; Klebe G.; Merkel O. M.; Kissel T. Targeting the blind spot of polycationic nanocarrier-based siRNA delivery. ACS Nano 2012, 6 (11), 9447–54. 10.1021/nn301966r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungsardthong U.; Ehtezazi T.; Bailey L.; Armes S. P.; Garnett M. C.; Stolnik S. Effect of Polymer Ionization on the Interaction with DNA in Nonviral Gene Delivery Systems. Biomacromolecules 2003, 4 (3), 683–690. 10.1021/bm025736y. [DOI] [PubMed] [Google Scholar]
- Driessen R. P. C.; Sitters G.; Laurens N.; Moolenaar G. F.; Wuite G. J. L.; Goosen N.; Dame R. T. Effect of temperature on the intrinsic flexibility of DNA and its interaction with architectural proteins. Biochemistry 2014, 53 (41), 6430–6438. 10.1021/bi500344j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z. J.; Wu C. How Does DNA Complex with Polyethylenimine with Different Chain Lengths and Topologies in Their Aqueous Solution Mixtures?. Macromolecules 2012, 45 (10), 4346–4353. 10.1021/ma2027963. [DOI] [Google Scholar]
- Hanzlikova M.; Ruponen M.; Galli E.; Raasmaja A.; Aseyev V.; Tenhu H.; Urtti A.; Yliperttula M. Mechanisms of polyethylenimine-mediated DNA delivery: free carrier helps to overcome the barrier of cell-surface glycosaminoglycans. Journal of Gene Medicine 2011, 13 (7–8), 402–9. 10.1002/jgm.1587. [DOI] [PubMed] [Google Scholar]
- Chu B. Structure and Dynamics of Polymers by Synchrotron Saxs. In Solvents and Self-Organization of Polymers; Webber S. E., Munk P., Tuzar Z., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 1996; pp 409–431. [Google Scholar]
- van den Berg J. H.; Oosterhuis K.; Hennink W. E.; Storm G.; van der Aa L. J.; Engbersen J. F. J.; Haanen J. B. A. G.; Beijnen J. H.; Schumacher T. N.; Nuijen B. Shielding the cationic charge of nanoparticle-formulated dermal DNA vaccines is essential for antigen expression and immunogenicity. J. Controlled Release 2010, 141 (2), 234–40. 10.1016/j.jconrel.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Verbaan F. J.; Oussoren C.; Snel C. J.; Crommelin D. J. A.; Hennink W. E.; Storm G. Steric stabilization of poly(2-(dimethylamino)ethyl methacrylate)-based polyplexes mediates prolonged circulation and tumor targeting in mice. J. Gene Med. 2004, 6 (1), 64–75. 10.1002/jgm.475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.