

Abstract
Epigenetic DNA modification impacts gene expression, but the underlying molecular mechanisms are only partly understood. Adding a methyl group to a cytosine base locally modifies the structural features of DNA in multiple ways, which may change the interaction with DNA-binding transcription factors (TFs) and trigger a cascade of downstream molecular events. Cells can be probed using various functional genomics assays, but it is difficult to disentangle the confounded effects of DNA modification on TF binding, chromatin accessibility, intranuclear variation in local TF concentration, and rate of transcription. Here we discuss how high-throughput in vitro profiling of protein-DNA interactions has enabled comprehensive characterization and quantification of the methylation sensitivity of TFs. Despite the limited structural data for DNA containing methylated cytosine, automated analysis of structural information in the Protein Data Bank (PDB) shows how 5-methylcytosine (5mC) can be recognized in various ways by amino-acid side chains. We discuss how a context-dependent effect of methylation on DNA groove geometry can affect DNA binding by homeodomain proteins, and how principled modeling of ChIP-seq data can overcome the confounding that makes the interpretation of in vivo data challenging. The emerging picture is that epigenetic modifications affect TF binding in a highly context-specific manner, with a direction and effect size that depends critically on their position within the TF binding site and the amino-acid sequence of the TF. With this improved mechanistic knowledge, we have come closer to understanding how cells use DNA modification to acquire, retain, and change their identity.
Keywords: Epigenetic modification of DNA, quantifying the effect of cytosine methylation on transcription factor binding, high-throughput in vitro assays, structural mechanisms including the effect of methylation on DNA shape, confounding effects in the analysis of in vivo binding data (ChIP-seq)
Graphical Abstract
Introduction
Chemical modification of DNA bases, the most prevalent of which is DNA methylation, is an ancient mark found in all three kingdoms of life. In prokaryotes, methylation of adenines is used as part of the restriction-modification system to protect against foreign viral DNA [1]. In plants and mammals, the dominant methylation mark is 5-methylcytosine (5mC) (Figure 1A), which occurs in both CpG and non-CpG contexts, but intermediate products from enzymatic oxidation of 5mCs, including 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-foC), and 5-carboxylcytosine (5-caC), have also been detected [2].
Figure 1: Structural basis of how TFs recognize methylated DNA.

(A) The structure of 5-methylcytosine. (B) Schematic overview of the processing pipeline to extract structural information about the different readout mechanisms TFs use to recognize methylated DNA. SNAP is used to search PDB entries for 5mC-containing DNA-protein complexes and extract all amino acids interactions formed with respective methylated cytosines. Structural representations are generated using DSSR [79] and rendered in PyMol (https://pymol.org). (C,D) Two common 5mC-specific readout mechanisms. (C) Superimposed view of four distinct examples of a 5mC-Arg contact, extracted from PDB entries 4M9E [80], 4R2E [81], 6MG4 [82], and 3C2I [83]. In all four structures the guanidinium group of the arginine is “stacked” above the methyl-group (top), and thus is perfectly positioned to form hydrogen bonds with the guanine base of the CpG dinucleotide step (bottom). (D) Superimposed view of four distinct examples of a hydrophobic functional group location within VdW distance from the C5-methyl group, extracted from PDB entries 5EF6 [45], 5T01 [84], 5EGO ([45]; contains two 5mC groups), and 6MG3 [47]). The hydrophobic groups are scattered around the methyl group, within the plane spanned by the cytosine base (top). Each contact is between 3.5–4.1 Å away from the C5-methyl group (bottom).
Studies of the methylation system in plants and mammals (for a detailed review see [3] and [4]) have revealed that epigenetic patterns are inherited through generations [5], with the rate of spontaneous epimutations at methylated (5mC) versus unmethylated CpGs estimated to be roughly five orders of magnitude higher than that of spontaneous nucleotide mutations (10−4 versus 10−9 per base pair per generation) [6]. This rate is high enough to allow uncoupling of genetic and epigenetic variation, yet low enough to allow for selection across generations [3]. Given that methylation patterns vary both temporally and spatially, it is tempting to speculate that their purpose is to record information about extrinsic stimuli (environmental or tissue-specific) and to communicate it to subsequent generations. Such a transient role for CpG methylation is consistent with a lack of evidence that CpG islands are under purifying selection [7]. However, it remains difficult to establish a causal link between DNA methylation and gene regulatory network function.
The lack of consensus on the global function of such a prominent epigenetic mark as 5mC is surprising, given that the mechanisms of establishing and erasing DNA methylation marks in mammals are fairly well documented [8–10], and that several high-throughput methods to profile genome-wide methylation patterns have been developed [11–15]. Depending on the context, studies can come to seemingly contradictory conclusions: On the one hand, a lack of coupling between DNA methylation and gene expression has been observed [16], arguing for a subordinate role for methylation in gene regulation. In support of this view, the removal of DNA methyltransferase 1 (Dnmt1) [8] or even all known methyltransferases [17] in murine stem cells does not seem to lead to activation of repressed genes, nor does it have a wide-spread effect on genome accessibility [17]. On the other hand, studies have linked methylation with gene repression, and demethylation with the derepression of repetitive DNA elements [18]. In addition, a complete knockout of Dnmt1 in mice is embryonic lethal [8], and results in a loss of differentiation capacity in embryonic stem (ES) cells [4]. In fact, although some eukaryotes such as Drosophila appear to have completely lost DNA methylation [19, 20], there is no known somatic cell type in vertebrates that is viable without the methylation machinery.
In a possible reconciliation of this discrepancy, serum-cultured ESCs were found to have much higher methylation levels than the inner cell mass (ICM) they are derived from [21, 22]. Thus, methylation may be dispensable for ES cell maintenance [19], yet required for ES cell differentiation where epigenetic plasticity induces cell state transitioning. Consistently, aberrant DNA methylation patterns play a role in the loss of allele-specific expression of imprinted genes [23], neuronal transcript stability [24], and the onset of cancer [25, 26]. Although we currently still lack concrete evidence, it is plausible that a major function of mammalian DNA methylation is transitioning between, rather than maintaining cellular states. This line of thinking raises the question how methylation marks are read out by the gene regulatory machinery. Since DNA methylation alters the biochemical properties of DNA, it likely impacts DNA recognition by DNA-binding proteins, including transcription factors (TFs). The addition of the bulky methyl group allows cytosines to mimic a thymine base to some extent (Figure 1A) and thus attract or repel TFs depending on the steric and hydrophobic environment of their cytosine interaction surface. However, given the complex relationships between the various layers of the regulatory network – binding affinity, genomic TF occupancy, local and global chromatin organization, gene expression, etcetera – it has remained challenging to dissect the causal mechanisms that govern methylation-dependent cell-state transitioning in vivo. Consequently, there has recently been a focus on new in vitro-based methods, which rely on high-throughput binding assays complemented by computational modeling to accurately quantify the impact of DNA methylation on TF binding.
This review will primarily cover in vitro-based methods that quantify how TFs respond to cytosine methylation, and to what extent the results of these assays have improved the interpretability of in-vivo studies. Specifically, we describe how DNA methylation impacts DNA geometry, and how this alters the biophysical properties of the interaction surface seen by TFs. We describe what is currently known about the mechanistic repertoire TFs utilize to recognize epigenetic marks and which high-throughput methods are currently available to measure the impact of methylated DNA on TF binding. By relating position-dependent methylation effects to structural studies, we further spell out key steps required to mechanistically interpret the observed changes in binding free energy derived from these methods. We conclude by detailing both the progress made and the challenges encountered in translating in vitro-derived quantitative insight to in vivo findings, and by laying out what remains to be done to unequivocally establish or rule out a role for TFs as readers and mediators of DNA methylation.
DNA methylation alters DNA structure
Addition of a hydrophobic methyl group to the carbon at position 5 of the cytosine on the major-groove side affects the structure of the DNA double helix. It is generally believed that 5mC widens the major groove on average and, in turn, narrows the minor groove [27]. The magnitude of these effects depends on the sequence environment; for some sequence contexts, the effect of DNA methylation on groove width can be very small [28]. However, structural information from X-ray crystallography or NMR spectroscopy is sparse for methylated DNA. The Protein Data Bank (PDB;[29]) contains about 50 times more unmethylated protein-DNA complexes than ones containing 5mC [28].
Given this sparse coverage in the PDB, studies of the effect of DNA methylation on DNA structure rely in large part on computational predictions and molecular simulations. Force field parameters for atomic-resolution molecular dynamics [30] and Monte Carlo simulations [31] have been derived for 5mC using quantum chemical calculations [32] and employed in simulation studies to probe the structural impact of this modification [27]. Since DNA is a very flexible molecule, the addition of a methyl group changes the conformational dynamics of the double helix. The average value of structural features has been used to describe the methylation effect on specific structural features of DNA in the methyl-DNAshape method [28]. Among the four DNA shape parameters considered, roll and propeller twist are the more strongly affected by DNA methylation [28]. These two parameters are closely related to base pairing and stacking, and larger changes in them indicate steric and electronic structure effects of the methyl group. The decreased helical twist of methylated CpG dinucleotides observed in molecular dynamics simulations has been associated with increased helix stiffness of methylated DNA [30]. The extent of these effects on DNA structure also depends on the base sequence context: for example, methyl groups adjacent to rigid A-tracts generally have a larger effect [28]. A summary of DNA shape profiles for both methylated and unmethylated TF binding sites as derived from high-throughput binding assays can be found in the TFBSshape database [33, 34].
Mechanisms that TFs use to recognize methylation marks, as identified by x-ray crystallography
DNA modifications not only affect the structure of the DNA double-helix, but also add chemical moieties to DNA bases that would otherwise not be present. Both structure- and functional group-mediated effects may modulate DNA recognition by a DNA-binding protein such as a TF. Although its limited number of entries makes it challenging to predict how structural modulations impact TF binding, the PDB provides sufficient information on readout mechanisms that involve direct contacts between protein amino acids and modified DNA bases. To provide an overview of and systematically classify known readout mechanisms (Figure 1B), we first extracted all protein-DNA complexes in the PDB (4,819 entries, based on the September 26, 2019 release), excluding any RNA- or DNA-RNA hybrid-containing structures as well as those classified as enzyme complexes, of low (>3.0 Å) resolution, or solved by NMR. We next analyzed the spatial relationship between DNA bases and protein sidechains in each structure using SNAP (structure of nucleic acids and proteins), which is an established component of the 3DNA suite of programs [35], and was used as a building block in resources such as DNAproDB [36][37].
Our automated SNAP analysis of the PDB allowed us to systematically identify all 63 protein-DNA structures containing at least one 5mC base contacted by one or more amino acid sidechains. For these, we created an interactive online table (http://snap-5mc.x3dna.org) that provides detailed annotation in terms of the specific 5mC readout mechanism(s) observed. An example for PDB entry 4M9E is shown in Figure 1B: In addition to an overview of the TF-DNA complex, detailed views of the 5mC-containing base pairs are included, capturing all relevant interactions with amino-acids. PDB files with coordinate systems centered on the 5mC in a consistent orientation can be downloaded directly for each cluster of 5mC-amino acids interactions. Interactive views rendered using 3Dmol.js [38] are also available. We refer to the tutorial page (http://snap-5mc.x3dna.org/tutorial) for further details.
Comparing the various 5mC-centered structural motifs, two main recognition modes emerge, each of which is described in detail in the respective crystal structures: (i) a 5mC-arginine-guanine triad [39–41], with arginine (Arg) sidechains forming hydrogen bonds with the guanine (G) base 3’ the 5mC in a CpG step, and with the guanidino group positioned above the methyl group in a stereotypical manner (Figure 1C) [42]; and (ii) van der Waals (VdW) interaction between the 5mC methyl group and methyl groups of hydrophobic amino acids or methylene groups of charged amino acids (about 4 Å between atoms; Figure 1D). In many instances, the carbon-5 methyl group is not unique to 5mC, but could also be acquired by replacing the 5mC with a thymine (T) base [41, 43–46]. TFs for which a recognition mode involving a T-Arg-G triad has been postulated should therefore be considered as potentially methyl-sensitive binders. Examples include Zinc finger proteins such as Kaiso and Zpf57 [41], but also methyl-binding proteins (MBDs), including MECP2 [43] and the tumor suppressor protein p53 [44].
More generally, whenever the thymine carbon-5 methyl group is directly contacted in the absence of more specific recognition of a T base or T/A base pair, it is likely that methylation of cytosines will rescue the reduction in complex stability resulting from a change from T to C. Prominent examples that involve such “methyl group only” readout – where the contribution to TF binding specificity at a given position in the DNA ligand is based on contact with the methyl group only – include homeodomain TFs such as HoxB13 [45] and bZIP TFs such as C/EBPβ or AP-1 [47]. In these cases, the binding free energy difference associated with a T to C substitution should be numerically close to that of a 5mC to C substitution. This phenomenon has been dubbed “thymine mimicry” [44].
Only very few structures are available where the 5mC recognition mode involves a different type of interaction. A prominent example is the ZFP57, which in addition to the stereotypic 5mC-Arg-G triad (cf. Figure 1B) displays contacts between the C5 methyl group and the carbon atom of the conserved glutamate carboxy group [48].
Together, these various examples convey that many TFs across several families have the potential to specifically recognize methylated DNA, and that TFs containing a TpG dinucleotide or even a single T in their optimal DNA ligand sequence might well be sensitive to DNA methylation.
High-throughput methods to detect and quantify the impact of DNA methylation on TF binding
Structural studies have been useful in identifying 5mCpG readout mechanisms, as outlined above. However, they rarely provide a direct and quantitative comparison across DNA ligands with identical sequence, yet differential cytosine carbon-5 methyl group status. Traditional assays that measure TF binding free energies (e.g., Isothermal titration calorimetry (ITC), circular dichroism (CD) or EMSA [43, 49]) can provide such information but are limited in throughput, rendering it challenging to make large-scale functional predictions on the direction and effect size of DNA modification on TF binding. To address this, a number of different high-throughput methods that quantify how TF binding preferences are modified by DNA methylation have been developed over the past few years [44–46, 50–52]. The challenge compared to standard TF binding assays is that two distinct randomized libraries – one with and one without modified bases – need to be contrasted directly in order to infer methylation-specific effects.
To add to the challenge, DNA modifications in vivo rarely occur uniformly across the genome. This makes it difficult to construct libraries that are modified neither too sparsely (e.g. only CpG dinucleotides are modified) nor too densely (every base of the same kind is modified). Three distinct approaches have been described: The methyltransferase M.SssI, which modifies both cytosines in the context of a CpG dinucleotide, has been used on randomly synthesized, unmethylated, and double-stranded DNA libraries [39, 44, 45, 50, 51]. To probe methylation that does not occur in a CpG context, or oxidized derivates of methylated cytosine, typically only one DNA strand can harbor the epigenetic mark, introduced by replacing the cytosine base with modified deoxy-nucleotides (i.e. deoxy-5hmC) during the synthesis process [46]. Finally, to specifically modify both strands in a targeted manner, libraries of reduced complexity can be created using a special library synthesis protocol [51].
In the case of enzymatic 5mCpG methylation, four distinct experimental platforms have been used to assay TF binding: (i) solution-based, highly multiplexed assays such as methyl-HT-SELEX [45]; (ii) EMSA-based methods such as EpiSELEX [44] or methyl-Spec-seq [51]; (iii) protein binding microarrays (methyl-PBM; [46, 50]), and (iv) genomic fragmentation followed by immunoprecipitation of DNA bound TFs (ampDAP-seq; [52, 53]). The last of these is distinct in that bisulfite sequencing is required in order to determine methylation status. The first three approaches rely on DNA ligands synthesized in vitro with a known methylation status, which is tracked either with barcodes in the flanking region (EpiSELEX and Spec-Seq) or by performing two separate assays (methyl-HT-SELEX and methyl-PBM). In all methods, the TF of interest is incubated with a modified, unmodified, or mixed DNA library, and the TF-bound fraction is separated, amplified, and sequenced. The last step is to infer DNA binding specificity based on the enrichment observed from the initial to the bound library (in the case of methyl-HT-SELEX or EpiSELEX) or the contrast between the bound and unbound library (in the case of methyl-Spec-seq). An overview of the different platforms, outlining advantages and disadvantages, is given in Figure 2 and Table 1.
Figure 2: Overview of methods that directly quantify TF binding preferences to methylated DNA in vitro.

Four main assay types are shown, each highlighting important features: Left: EpiSELEX-seq and methyl-Spec-seq both infer quantitative 5mC effect sizes from mixed (barcoded) pools of methylated and unmethylated DNA ligands sequenced after one round of TF binding enrichment. Right: Methyl-HT-SELEX and methyl-PBM estimate 5mC effect sizes by comparing TF binding enrichment scores between unmethylated or methylated DNA ligands. For a detailed description see Table 1.
Table 1.
Detailed description of in vitro methods that can be used to directly quantify the methylation sensitivity of TF binding.
| Assay | Methodology | Advantages | Challenges | Ref |
|---|---|---|---|---|
| Methyl-HT-SELEX | • bead-based (automated) • random DNA ligand pool • 5mC probes assayed separately • ∼104 DNA reads per TF |
• hundreds of parallel assays • universal probe design • allows for multiple rounds |
• lower read count per TF due to high degree of multiplexing • risk of biased quantification due to separate ligand pools |
[45] |
| EpiSELEX-seq | • EMSA-based (manual) • random DNA ligand pool • 5mC probes barcoded and assayed in pooled sample • input DNA and bound DNA sequenced separately • ∼106 DNA reads per TF |
• accurate quantification of relative enrichment • full sequence spectrum covered |
• single TF (complex) assayed • limited to single round of enrichment |
[44] |
| Methyl-Spec-seq | • EMSA-based (manual) • random DNA ligand pool • 5mC probes barcoded and assayed in pooled sample • bound and unbound DNA sequenced separately • ∼106 DNA reads per TF |
• EMSA-based (manual) • dedicated probe design |
• single TF (complex) assayed • limited to single round of enrichment |
[51] |
| Methyl-PBM | • DNA microarrays • fluorescently labeled TF • ∼105 dsDNA probes |
• no DNA sequencing needed | • DNA microarray needed | [39, 46, 50] |
| (amp)DAP-seq | • fragments of genomic DNA • recombinant TF proteins |
• natural epigenetic context | • quantification challenging • methylome data required |
[52, 53] |
| Classic low-throughput methods | isothermal calorimetry (ITC) or electromobility shift assay (EMSA) or DNase footprinting |
accurate quantification (Kd) and flexible sequence design |
low throughput |
Methylation is lost during PCR amplification, and therefore SELEX can be repeated over multiple rounds only if unmodified and modified libraries are assayed separately. SELEX variants in which unmethylated and methylated DNA ligands are mixed prior to selection for TF binding necessarily rely on a single round of affinity-based selection. The benefit, however, of mixing unmethylated and methylated DNA ligands is an increased accuracy of effect size quantification that stems from the internal control provided by comparing DNA ligands that lack CpGs and therefore cannot be methylated across both libraries. Accuracy is especially important when trying to detect position- or strand-specific methylation effects or quantify low-affinity binding. Although a protocol has not been reported so far, microfluidics-based approaches that have been shown to produce high-quality TF-binding data – BET-seq [54] and SMILE-seq [55]) – should also be suitable for assaying the methylation sensitivity of TF-DNA binding. Finally, the few studies that have considered DNA modifications other than CpG methylation have either focused on hemi-methylation (to avoid overly spares modification patterns) [46] or ligands with modifications targeted to specific positions [47, 49, 56].
SELEX methods reveal highly context-dependent methylation effects on TF binding
The development of high-throughput SELEX methods has made it possible to classify numerous TFs or TF complexes in terms of the degree (effect on binding free energy) and direction (reduced or increased) with which DNA modifications impact their binding. An important insight from these studies is that TFs do not respond uniformly to epigenetic marks when occurring in different sequence contexts. In other words, knowing whether a TF is sensitive to methylation or not is not sufficient to predict the direction of response to genomic methylation. Rather, it is essential to have information on the exact position of a methylation mark with respect to both position and DNA strand along the TF-DNA interface.
The most extensively studied case are heterodimeric complexes formed between two types of bZIP TFs: cAMP response element-binding protein (CREB type; e.g., ATF4) and CCAAT/enhancer-binding protein (C/EBP type; e.g., C/EBPβ). Several independent studies employing either PBM- or SELEX-based assays have shown that these complexes show either increased or decreased binding affinity to methylated DNA, depending on whether the CpG dinucleotide is located in the center of the two half sites or in the CREB flank [44, 46, 50].
More recently, it has been shown that by boosting the affinity of weak binding sites, CpG methylation can even affect the binding of TFs that lack CpG dinucleotides in their core consensus sequence [44, 45]. The most prominent example are homeodomain TFs, which typically prefer AT-rich sequences [57, 58]. Another is the tumor-suppressor protein p53, whose canonical dimeric half-site (RRRCATGYYY) does not harbor a CpG dinucleotide, yet methylation sensitivity was demonstrated for p53, across three classes of CpG-containing low-affinity sites [44]. As with bZIPs, the response direction and effect size were both strongly position-dependent.
To tackle the question whether the protein’s orientation relative to the double-stranded DNA is important, methyl-Spec-seq was applied with both fully and hemi-methylated dsDNA to study the binding preference of the zinc finger protein ZPF57 [51]. Indeed, a clear strand-specificity was detected, which is perhaps little surprising when considering that TF binding breaks the symmetry between the two methyl groups embedded in a fully methylated CpG base pair step, the most likely scenario for a monomerically binding TF such as ZFP57 being that it only engages directly with one of the methyl groups.
Distinct structural mechanisms can underlie methylation-dependent TF readout
The electrostatic environment along the TF-DNA interface varies with both base identity on the DNA side and amino-acid (aa) identity on the TF side. As a result, any effect a methyl-group might have on TF binding will be highly context-specific. Multiple distinct readout mechanisms can coexist within a single TF-DNA complex. In some cases, the effect of methylation can be interpreted by analyzing the aa identity along the binding interface. When individual structures are not available (typically only a few structures per TF family exist), hypotheses can be generated by comparing cytosine methylation effects between paralogs and considering differences in aa identity at residue positions close to the cytosine base in the 3D structure.
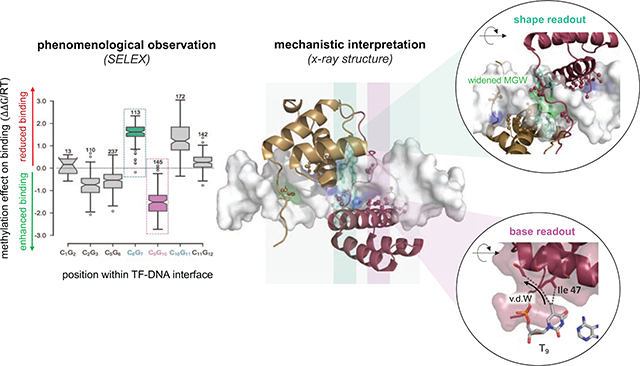
For example, complexes formed between human TF Pre-B-cell leukemia TF 1 (PBX1) and each of three distinct Hox factors (A1, A5, A9) exhibited either a stabilizing or a destabilizing effect associated with methylation of a specific CpG base pair step (Figure 3A), depending on the exact position within the binding site [44]. Moreover, the degree of this methylation sensitivity is paralog-specific [44]. Aligning and comparing protein sequences among Hox paralogs and orthologs revealed a conserved sequence polymorphism two residues upstream of the hydrophobic isoleucine-47 (Ile47). The latter could therefore be regarded as a plausible mechanistic determinant given that “VdW readout” is among the two major readout mechanisms described above. Indeed, a direct comparison of the structures for Pbx1-HoxA9 and Pbx1-HoxB1 (a paralog closely related to HoxA1 and within the same class) showed that this amino acid polymorphism directs the positioning of Ile47, which in turn influences how the 5mCpG modification is read out at base pair position 9 (Figure 3B).
Figure 3: Connecting the impact of 5mC on TF binding to distinct readout mechanisms.

(A) An example 5mC effect size quantification is shown for the TF complex formed between the human Pbx1 and HoxA1 proteins (left). Both enhanced (pink) and reduced (green) binding are observed depending on the positioning of the methylated CpG within the protein-DNA interface. (B) Enhanced binding stems from a stabilizing VdW contact between the 5mC base at DNA position 9 and Ile-47 within helix-3 of the Hox homeodomain (“base readout”). (C) Reduced binding can be attributed to a widening of the DNA minor groove at the consensus AY base step that directly neighbors the methylated C6G7. The MGW readout of the N-terminal arm of the Hox homeodomain is weakened as a result (“shape readout”).
Many observations of enhanced binding upon CpG methylation that were made using high-throughput methods can be explained by one of the two major mechanisms described in the section above. TFs that rely on VdW-mediated readout include homeodomain proteins [44, 45] and bZIP TFs [47]. The T/5C-Arg-G triad is found among ZFPs, proteins with a canonical methyl-binding domain, and specialized TFs such as p53 [40, 44]. In general, however, identifying the readout mechanism underlying a specific methylation effect observed in a high-throughput binding assay is not straightforward. This is particularly true when methylation leads to decreased binding and cannot be explained by direct steric hindrance. For instance, it is not entirely clear why CpG methylation of the core dinucleotide within homodimeric CREB (ATF4) or heterodimeric CREB/CEBP (ATF4-CEBPβ) complexes decreases binding [44, 46, 50], when it boosts binding of homodimeric CEBP (CEBPα or CEBPβ) complexes [45].
A potential explanation is that DNA modifications indirectly affect TF binding affinity by modulating the geometry of neighboring base pairs. This idea was first proposed when analyzing differences in DNase I cleavage rates for unmethylated and methylated hexamers [31]: DNA methylation was predicted to narrow the DNA minor grove width (MGW), which in turn increased enzymatic cleavage efficiency. More recently, a concrete example demonstrated that methyl-induced changes in DNA shape can also be important for TF binding: Analyzing how DNA methylation altered the shape of DNA sequences in the context of the Pbx-Hox binding site (NTGAYNNAYNNN) revealed that the methyl group addition to the CpG dinucleotide downstream of the AY dinucleotides (NTGAYCGAYNNN or NTGAYNNAYCGN) leads to a subtle but significant widening of the DNA minor groove at this exact position [28]. Since Hox proteins read out MGW at positions A4Y5 [59], the methyl-induced widening is detrimental to Pbx-Hox binding. Thus, the methylation effect on binding for sequences of type AYCG is due to readout of DNA shape and electrostatic potential, rather than direct major grove contacts (Figure 3C).
Most published assays and structural analyses of readout mechanisms have been limited to 5mC marks. However, other forms of DNA modification are equally likely to evoke a wide variety of effect sizes. For instance, the addition of a hydroxy-group not only impacts the electrostatic potential experienced by proteins directly engaging with the DNA major groove, but it is also likely to impact DNA shape. Very few TFs have been assayed in terms of their binding preferences to DNA ligands containing modifications other than 5mC [46, 47, 49]. An example of a TF that shows binding modulation to 5-hydroxy-methylation (5hmC) is the Epstein-Barr Virus bZIP Protein Zta [46]. Both enhanced and reduced binding can be observed, depending on the position of the 5hmC mark within the binding interface. Moreover, effect size varies extensively among bZIP paralogs.
Quantifying the impact of DNA methylation on TF binding in vivo
High-throughput studies and structural considerations point to an important role for intra-binding-site position and strand in determining a TF’s response to DNA modification. The TF-DNA binding energetics that govern molecular interaction in an in vitro setting are expected to also be a major determinant of their in vivo behavior. However, it has remained challenging to demonstrate this explicitly. One ostensibly sensible and straightforward approach would be to look for differences in TF occupancy, as measured by ChIP-seq, between methylated and unmethylated sites; alternatively, one might ask whether occupied methylated regions display motif enrichment or depletion relative to unmethylated regions (Figure 4A).
Figure 4: The challenge of quantifying the effect of cytosine methylation on TF binding in vivo.

(A) Conventional approaches typically consider whole-genome bisulfite sequencing (WGBS) and TF occupancy data (ChIP-seq) in conjunction to infer TF binding preferences for methylated binding sites in vivo. Two approaches are shown that either compare the ChIP-seq signal at unmethylated and methylated CpG containing ChIP-seq peaks (top-left), or the total overlap between WGBS sites and TF peaks split by CpG methylation status (top-right). (B) Confounding features that influence TF occupancy (IP signal) at any given site are typically not considered in conventional analysis of in vivo data. These include (i) variation in local TF concentration (likely the result of transient, phase-separated TF hub assemblies), and (ii) the affinity of each individual binding site. (C) To isolate the effect of individual methylation marks from among these confounding site-specific factors, conventional models need to be updated. Local TF concentration cannot be estimated directly, but features such as DNA accessibility or regional methylation level can be used to control for its variation.
Although easy to conceptualize, such approaches fail to capture the true nature of the challenge: DNA methylation seldom evokes a severe response; rather it modulates binding affinity in a relatively subtle way. Furthermore, methylation can have opposite effects at different positions within the same binding site, while at the same time the methylation status of nearby cytosines is often highly correlated, which can cause the net effect of methylation on TF binding to be weaker than the effect for individual cytosines. For example, a ChIP-seq profile for the bZIP heterodimer ATF4/CEBPβ might on average show a decreased IP signal at methylated sites. However, this signal is likely driven by the negative methylation effect that high-affinity TGAC|GCAA sites display, which will mask any increase in TF binding at the subset of lower-affinity CGAC|ACAA sites that contribute less to the overall IP signal.
Let us assume that we assign a separate indicator for each cytosine position within a TF binding site and compare IP signals only across position-matched methylated and unmethylated sites. Regulatory regions tend to be unmethylated [60], so we are likely to observe far more ChIP-seq peaks overlapping unmethylated binding sites than methylated ones. Indeed, the zinc finger protein ZBTB33, which prefers methylated binding sites in vitro, nevertheless was observed to preferentially occupy unmethylated sites in vivo [61]. One might erroneously conclude that methylated sites are not relevant for TF binding in vivo based on the observation that methylation marks are depleted among the genomic regions that are bound by TFs.
One way to resolve this paradox is to consider that genomic methylation patterns are read out by TFs in a highly dynamic context. As mentioned in the Introduction, methyl-dependent binding may occur transiently and indeed, several TFs have been found to bind methylated genomic regions, leading to regional demethylation [62–65]. Not every TF is made equal, and only a subset – which are often referred to as “pioneering TFs” – can cause changes in DNA methylation and accessibility [66]. Existing studies, however, do not provide sufficient information to assess whether a TF relies on prior methylation to be recruited to its binding sites, or whether it tends to target unmethylated sites within heavily methylated regions. Indeed, it is difficult to assign both TF binding and methylation status of single CpGs at the same time, especially when trying to capture transient states that only occur within a small number of cells at any given time.
Nevertheless, recent studies that focused on TFs previously demonstrated to preferentially bind methylated DNA in vitro, indicate that, at least for some TFs, methylation may also positively drive genomic binding in vivo: Performing both 5mC and 5hmC profiling and following the epigenetic response to stimulation with reprogramming factors over time, it was found that enhanced in vivo binding by specific TFs, including KLF4 and CEBP, is associated with genomic regions that undergo demethylation upon stimulation. More specifically, KLF4 was suggested to recruit the demethylase Tet2 to methylated enhancers, leading to the accumulation of hydroxymethylation, and a subsequent increase in DNA accessibility [67]. A related study analyzed a KLF4 mutation that specifically abolishes its methylation readout, and showed that ChIP-seq peaks bound specifically by the wild-type protein were enriched for CpG dinucleotides [68]; the same study found that this methylation-dependent readout affects specific gene regulatory programs, and that the difference between wild-type and mutant KLF4 was largely recapitulated when DNA methylation was blocked by 5-aza-2′-deoxycytidine treatment, thus clearly demonstrating that KLF4 relies on CpG methylation to exert some of its functions. In yet another in vivo study, the reverse was observed, namely that DNA methylation can actively block the binding of the transcription factor NRF1 [17], again underscoring that DNA methylation can have a biologically meaningful impact on TF-dependent gene regulation in vivo.
More generally, a connection between CpG methylation and transitioning between cellular states has been made previously, for instance by demonstrating that embryonic stem cells stripped of all de novo and maintenance DNA methyltransferases (DNMT1, 3a, and 3b) lose their differentiation potential, yet maintain their “ground state” (as measured by DNA accessibility changes) [17]. However, the observation that enzymatic oxidation precedes demethylation and changes in DNA accessibility suggests a direct role for TFs in reading epigenetic landscapes and inducing transitional states. Figure 5 illustrates this idea in the form of a “seesaw” model that explains cell fate conversion in the context of Waddington’s well-known epigenetic landscape.
Figure 5: Transient TF-mediated 5mC readout to transition between cellular states.

Our proposed “seesaw” model connects methylation-dependent TF readout and cell fate conversion within Waddington’s epigenetic landscape: Binding of methylation-stabilized pioneer TFs to methylated regions confers structural rearrangements such as demethylation or recruitment of remodeling factors to persistently open up previously inactive regions. Such transient recognition of methylated regions by TFs would explain both the requirement of methylation for cell differentiation and the apparent lack of TF binding to methylated regions from bulk data. Note that the transient state is short-lived and thus only applies to small fraction of cells at any given time, which makes it hard to detect.
Measuring multiple parallel genome-wide profiles, and doing so in a way that captures relatively fast dynamics, is exceedingly challenging. The few studies that unequivocally established a connection between TF binding and CpG methylation in vivo [17, 68] were each a technical tour de force. It is therefore highly desirable to find alternative ways to gain insight into transient TF binding behavior from static data (i.e., ChIP-seq and WGBS data). To this end, we first need to question the implicit assumptions we make when comparing differences in TF occupancy between methylated and unmethylated sites: (i) that IP signal is independent of the underlying sequence affinity and the position of CpG methylation, (ii) that local TF concentrations is uniform across sites, and (iii) that peak-calling is not biased towards sites with high accessibility. All three assumptions in fact are false: (i) TFs display a wide range of binding affinities, (ii) they form liquid-like phase-separated assemblies [69] and are thus non-uniformly distributed within the nucleus [70] (Figure 4B), and (iii) they can bind regions that are predominantly inaccessible [71], making them less likely to be captured in ChIP-seq or related assays due to the library size-selection step in most protocols.
To properly account for these confounding effects, and to validate the effects observed in vitro in a cellular context without the need for additional experiments, we must analyze in vivo genomics data such as ChIP-seq data in a more sophisticated manner. Assigning an “affinity” score is relatively straightforward, thanks to numerous efforts to map the binding specificities of many TFs in vitro [72, 73]. Dealing with variation in local TF concentration is more challenging, as there is no direct way to measure local TF concentration genome-wide. A solution is to perform a “motif-centric” analysis of ChIP-seq data that avoids peak calling altogether [44]: Instead of relying on large differences in effect size, statistical power is achieved by small, yet consistent differences across a large number of binding sites, each of which has a different set of attributes (including affinity, accessibility, methylation status of cytosines within the binding site, and regional methylation level). This allows estimation of effect sizes associated with “proxy” features – such as DNA accessibility, CpG density (CpG islands), or regional methylation levels – that correlate with, but do not necessarily determine, local TF concentration (Figure 4C). The latter is particularly important, as intra-binding-site methylation status will inevitably correlate with regional methylation levels.
Intuitively, to estimate the direct effect of methylation on TF binding, genomic motif matches that do not harbor any CpG base pair steps and therefore by definition cannot respond to changes in methylation status, can be used to control for regional effects. The confounding factors thus having been accounted for, in vivo binding preferences for methylated DNA can be accurately inferred. In practice, such analyses are best performed using generalized linear models (GLM) based on discrete distributions such as the binomial distribution, which provides a natural way to jointly analyze pairs of ChIP-seq profiles that differ from each other in a subtle way (e.g., TF induced vs. not induced; or wild-type vs. mutant).
One example where this approach has been successful is the tumor suppressor p53 [44]: Because high-affinity p53 binding sites do not harbor any CpG steps, it was straightforward to estimate the regional (negative) effect of methylation on accessibility and thus expected ChIP-seq read coverage. At the same time, by only trying to explain the distribution of read counts between (nutlin-)induced and uninduced cells across countless pairs of matching genomic loci, the analysis of [44] completely avoided the hard problem of explaining differences in ChIP-seq coverage between different loci in the same sample. As expected, positive coefficients were obtained for methylation-blind affinity and CpG density predictors, and a negative coefficient for regional methylation. Accounting for these confounding effects revealed that in vivo p53 occupancy indeed recapitulates the effects observed in in vitro studies [44]. Although the majority of sites that harbored a methylated CpG did not meet the threshold of being called a “peak”, position-specific 5mCpG model coefficients were statistically significant. It is possible that only a small fraction of cells contributed to the signal, with the majority of cells having progressed to a different, post-p53-induction state. Supporting this argument, p53 has been shown to have pioneer activity, allowing it to access and activate otherwise closed genomic regions [74].
Final Remarks
Although the field has made important progress in analyzing the effects of cytosine methylation on transcription factor binding in vitro and, to a lesser extent, in vivo, there is still much room for improvement. In vitro, better analysis methods are needed to be able to accurately quantify binding free energy differences, which tend to be different for each position within the binding site. Microfluidics-based platforms such as SMILE-seq [55] are particularly promising in this regard. It would also be valuable to have a larger number of high-resolution co-crystal structures available for methylated and unmethylated versions of the same DNA ligand, both bound and unbound by transcription factors. In the context of the living cell, it is even more difficult to tease apart the many direct and indirect effects of methylation on gene expression. Obtaining single-cell resolution data on the methylation status of DNA sequences on a genome-wide scale [75] may be particularly useful, especially paired with other modalities of single-cell data such as gene expression, protein binding [76], histone modification status [77], or chromatin accessibility [78]. In vivo imaging experiments in which methylated and unmethylated binding sites can be visualized in intact nuclei so that their localization into TF hubs and interactions with the relevant TFs can be monitored, would be a valuable complement to the biochemical assays that were the main focus of the current review. Finally, other forms of DNA modification than 5mCpG may also have significant impact on TF binding and function in vivo. The tools developed for analyzing the impact of 5mCpG should therefore be expanded to analyze these additional types of modification.
Research Highlights.
The causal mechanisms that determine how epigenetic DNA modifications such as 5-methylcytosine (5mC) impact gene expression in the context of the living cell remain obscure
High-throughput in vitro (SELEX) assays based on massively parallel DNA sequencing have recently made it feasible to characterize the effect on DNA methylation on transcription factor (TF) binding
The impact of cytosine methylation (5mC) is different for each TF, and highly dependent on the position of the modified base within the TF-DNA interface
A rich variety of structural mechanisms, including effects mediated by changes in DNA minor groove width, has been found to underly the methylation sensitivity of TF-DNA interaction
A generalized linear modeling approach that avoids peak calling shows the best prospects for dealing with confounding effects when translating in vitro insight to an in vivo context (e.g., when comparing SELEX and ChIP-seq data for the same TF)
Acknowledgements
This work was supported by NIH grants R01HG003008 to H.J.B., R01GM096889 to X.J.L., R01GM106056 and R35GM130376 to R.R., and R35GM118336 to R.S.M.
Footnotes
Declarations of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE--a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015;43:D298–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18:517–34. [DOI] [PubMed] [Google Scholar]
- [3].Huang SC, Ecker JR. Piecing together cis-regulatory networks: insights from epigenomics studies in plants. Wiley Interdiscip Rev Syst Biol Med. 2018;10:e1411. [DOI] [PubMed] [Google Scholar]
- [4].Ambrosi C, Manzo M, Baubec T. Dynamics and Context-Dependent Roles of DNA Methylation. J Mol Biol. 2017;429:1459–75. [DOI] [PubMed] [Google Scholar]
- [5].Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].van der Graaf A, Wardenaar R, Neumann DA, Taudt A, Shaw RG, Jansen RC, et al. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc Natl Acad Sci U S A. 2015;112:6676–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cohen NM, Kenigsberg E, Tanay A. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell. 2011;145:773–86. [DOI] [PubMed] [Google Scholar]
- [8].Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. [DOI] [PubMed] [Google Scholar]
- [9].Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Edwards JR, O’Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010;20:972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Consortium BLUEPRINT. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat Biotechnol. 2016;34:726–37. [DOI] [PubMed] [Google Scholar]
- [16].Bestor TH, Edwards JR, Boulard M. Notes on the role of dynamic DNA methylation in mammalian development. Proc Natl Acad Sci U S A. 2015;112:6796–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schubeler D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528:575–9. [DOI] [PubMed] [Google Scholar]
- [18].Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. [DOI] [PubMed] [Google Scholar]
- [19].Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S, Sakaue M, Matsuoka C, et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006;11:805–14. [DOI] [PubMed] [Google Scholar]
- [20].Sakaue M, Ohta H, Kumaki Y, Oda M, Sakaide Y, Matsuoka C, et al. DNA methylation is dispensable for the growth and survival of the extraembryonic lineages. Curr Biol. 2010;20:1452–7. [DOI] [PubMed] [Google Scholar]
- [21].Habibi E, Brinkman AB, Arand J, Kroeze LI, Kerstens HH, Matarese F, et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013;13:360–9. [DOI] [PubMed] [Google Scholar]
- [22].Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, Grabole N, et al. Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol. 2013;20:311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–9. [DOI] [PubMed] [Google Scholar]
- [24].Lagger S, Connelly JC, Schweikert G, Webb S, Selfridge J, Ramsahoye BH, et al. MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet. 2017;13:e1006793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Klutstein M, Nejman D, Greenfield R, Cedar H. DNA Methylation in Cancer and Aging. Cancer Res. 2016;76:3446–50. [DOI] [PubMed] [Google Scholar]
- [26].Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. 2018;15:459–66. [DOI] [PubMed] [Google Scholar]
- [27].Dantas Machado AC, Zhou T, Rao S, Goel P, Rastogi C, Lazarovici A, et al. Evolving insights on how cytosine methylation affects protein-DNA binding. Brief Funct Genomics. 2015;14:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rao S, Chiu TP, Kribelbauer JF, Mann RS, Bussemaker HJ, Rohs R. Systematic prediction of DNA shape changes due to CpG methylation explains epigenetic effects on protein-DNA binding. Epigenetics Chromatin. 2018;11:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Berman HM, Bhat TN, Bourne PE, Feng Z, Gilliland G, Weissig H, et al. The Protein Data Bank and the challenge of structural genomics. Nat Struct Biol. 2000;7 Suppl:957–9. [DOI] [PubMed] [Google Scholar]
- [30].Perez A, Castellazzi CL, Battistini F, Collinet K, Flores O, Deniz O, et al. Impact of methylation on the physical properties of DNA. Biophys J. 2012;102:2140–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lazarovici A, Zhou T, Shafer A, Dantas Machado AC, Riley TR, Sandstrom R, et al. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc Natl Acad Sci U S A. 2013;110:6376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Aduri R, Psciuk BT, Saro P, Taniga H, Schlegel HB, SantaLucia J. AMBER Force Field Parameters for the Naturally Occurring Modified Nucleosides in RNA. J Chem Theory Comput. 2007;3:1464–75. [DOI] [PubMed] [Google Scholar]
- [33].Yang L, Zhou T, Dror I, Mathelier A, Wasserman WW, Gordan R, et al. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2014;42:D148–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chiu TP, Xin B, Markarian N, Wang Y, Rohs R. TFBSshape: an expanded motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Research. 2019; doi.org/ 10.1093/nar/gkz970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lu XJ, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc. 2008;3:1213–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sagendorf JM, Berman HM, Rohs R. DNAproDB: an interactive tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 2017;45:W89–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sagendorf JM, Markarian N, Berman HM, Rohs R. DNAproDB: an expanded database and web-based tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 2019; doi.org/ 10.1093/nar/gkz889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rego N, Koes D. 3Dmol.js: molecular visualization with WebGL. Bioinformatics. 2015;31:1322–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hu S, Wan J, Su Y, Song Q, Zeng Y, Nguyen HN, et al. DNA methylation presents distinct binding sites for human transcription factors. Elife. 2013;2:e00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hashimoto H, Zhang X, Vertino PM, Cheng X. The Mechanisms of Generation, Recognition, and Erasure of DNA 5-Methylcytosine and Thymine Oxidations. J Biol Chem. 2015;290:20723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu Y, Zhang X, Blumenthal RM, Cheng X. A common mode of recognition for methylated CpG. Trends Biochem Sci. 2013;38:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Armstrong CT, Mason PE, Anderson JL, Dempsey CE. Arginine side chain interactions and the role of arginine as a gating charge carrier in voltage sensitive ion channels. Sci Rep. 2016;6:21759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu K, Xu C, Lei M, Yang A, Loppnau P, Hughes TR, et al. Structural basis for the ability of MBD domains to bind methyl-CG and TG sites in DNA. J Biol Chem. 2018;293:7344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kribelbauer JF, Laptenko O, Chen S, Martini GD, Freed-Pastor WA, Prives C, et al. Quantitative Analysis of the DNA Methylation Sensitivity of Transcription Factor Complexes. Cell Rep. 2017;19:2383–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tillo D, Ray S, Syed KS, Gaylor MR, He X, Wang J, et al. The Epstein-Barr Virus B-ZIP Protein Zta Recognizes Specific DNA Sequences Containing 5-Methylcytosine and 5-Hydroxymethylcytosine. Biochemistry. 2017;56:6200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yang J, Horton JR, Wang D, Ren R, Li J, Sun D, et al. Structural basis for effects of CpA modifications on C/EBPbeta binding of DNA. Nucleic Acids Res. 2019;47:1774–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu Y, Toh H, Sasaki H, Zhang X, Cheng X. An atomic model of Zfp57 recognition of CpG methylation within a specific DNA sequence. Genes Dev. 2012;26:2374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sayeed SK, Zhao J, Sathyanarayana BK, Golla JP, Vinson C. C/EBPbeta (CEBPB) protein binding to the C/EBP|CRE DNA 8-mer TTGC|GTCA is inhibited by 5hmC and enhanced by 5mC, 5fC, and 5caC in the CG dinucleotide. Biochim Biophys Acta. 2015;1849:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mann IK, Chatterjee R, Zhao J, He X, Weirauch MT, Hughes TR, et al. CG methylated microarrays identify a novel methylated sequence bound by the CEBPB|ATF4 heterodimer that is active in vivo. Genome Res. 2013;23:988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zuo Z, Roy B, Chang YK, Granas D, Stormo GD. Measuring quantitative effects of methylation on transcription factor-DNA binding affinity. Sci Adv. 2017;3:eaao1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bartlett A, O’Malley RC, Huang SC, Galli M, Nery JR, Gallavotti A, et al. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat Protoc. 2017;12:1659–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].O’Malley RC, Huang SC, Song L, Lewsey MG, Bartlett A, Nery JR, et al. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell. 2016;166:1598. [DOI] [PubMed] [Google Scholar]
- [54].Le DD, Shimko TC, Aditham AK, Keys AM, Longwell SA, Orenstein Y, et al. Comprehensive, high-resolution binding energy landscapes reveal context dependencies of transcription factor binding. Proc Natl Acad Sci U S A. 2018;115:E3702–E11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Isakova A, Groux R, Imbeault M, Rainer P, Alpern D, Dainese R, et al. SMiLE-seq identifies binding motifs of single and dimeric transcription factors. Nat Methods. 2017;14:316–22. [DOI] [PubMed] [Google Scholar]
- [56].Golla JP, Zhao J, Mann IK, Sayeed SK, Mandal A, Rose RB, et al. Carboxylation of cytosine (5caC) in the CG dinucleotide in the E-box motif (CGCAG|GTG) increases binding of the Tcf3|Ascl1 helix-loop-helix heterodimer 10-fold. Biochem Biophys Res Commun. 2014;449:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Noyes MB, Meng X, Wakabayashi A, Sinha S, Brodsky MH, Wolfe SA. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res. 2008;36:2547–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Merabet S, Mann RS. To Be Specific or Not: The Critical Relationship Between Hox And TALE Proteins. Trends Genet. 2016;32:334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Abe N, Dror I, Yang L, Slattery M, Zhou T, Bussemaker HJ, et al. Deconvolving the recognition of DNA shape from sequence. Cell. 2015;161:307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schubeler D Function and information content of DNA methylation. Nature. 2015;517:321–6. [DOI] [PubMed] [Google Scholar]
- [61].Blattler A, Yao L, Wang Y, Ye Z, Jin VX, Farnham PJ. ZBTB33 binds unmethylated regions of the genome associated with actively expressed genes. Epigenetics Chromatin. 2013;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].de la Rica L, Rodriguez-Ubreva J, Garcia M, Islam AB, Urquiza JM, Hernando H, et al. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013;14:R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, et al. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun. 2013;4:2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Suzuki T, Shimizu Y, Furuhata E, Maeda S, Kishima M, Nishimura H, et al. RUNX1 regulates site specificity of DNA demethylation by recruitment of DNA demethylation machineries in hematopoietic cells. Blood Adv. 2017;1:1699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Fujiki K, Shinoda A, Kano F, Sato R, Shirahige K, Murata M. PPARgamma-induced PARylation promotes local DNA demethylation by production of 5-hydroxymethylcytosine. Nat Commun. 2013;4:2262. [DOI] [PubMed] [Google Scholar]
- [66].Suzuki T, Maeda S, Furuhata E, Shimizu Y, Nishimura H, Kishima M, et al. A screening system to identify transcription factors that induce binding site-directed DNA demethylation. Epigenetics Chromatin. 2017;10:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sardina JL, Collombet S, Tian TV, Gomez A, Di Stefano B, Berenguer C, et al. Transcription Factors Drive Tet2-Mediated Enhancer Demethylation to Reprogram Cell Fate. Cell Stem Cell. 2018;23:905–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wan J, Su Y, Song Q, Tung B, Oyinlade O, Liu S, et al. Methylated cis-regulatory elements mediate KLF4-dependent gene transactivation and cell migration. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Alberti S, Gladfelter A, Mittag T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell. 2019;176:419–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Liu Z, Tjian R. Visualizing transcription factor dynamics in living cells. J Cell Biol. 2018;217:1181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mayran A, Drouin J. Pioneer transcription factors shape the epigenetic landscape. J Biol Chem. 2018;293:13795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell. 2018;172:650–65. [DOI] [PubMed] [Google Scholar]
- [73].Rastogi C, Rube HT, Kribelbauer JF, Crocker J, Loker RE, Martini GD, et al. Accurate and sensitive quantification of protein-DNA binding affinity. Proc Natl Acad Sci U S A. 2018;115:E3692–E701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sammons MA, Zhu J, Drake AM, Berger SL. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 2015;25:179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Karemaker ID, Vermeulen M. Single-Cell DNA Methylation Profiling: Technologies and Biological Applications. Trends Biotechnol. 2018;36:952–65. [DOI] [PubMed] [Google Scholar]
- [76].Krebs AR, Imanci D, Hoerner L, Gaidatzis D, Burger L, Schubeler D. Genome-wide Single-Molecule Footprinting Reveals High RNA Polymerase II Turnover at Paused Promoters. Mol Cell. 2017;67:411–22 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ku WL, Nakamura K, Gao W, Cui K, Hu G, Tang Q, et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat Methods. 2019;16:323–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lu XJ, Bussemaker HJ, Olson WK. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015;43:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liu Y, Olanrewaju YO, Zheng Y, Hashimoto H, Blumenthal RM, Zhang X, et al. Structural basis for Klf4 recognition of methylated DNA. Nucleic Acids Res. 2014;42:4859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hashimoto H, Olanrewaju YO, Zheng Y, Wilson GG, Zhang X, Cheng X. Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. 2014;28:2304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Morgan GJ, Yan NL, Mortenson DE, Rennella E, Blundon JM, Gwin RM, et al. Stabilization of amyloidogenic immunoglobulin light chains by small molecules. Proc Natl Acad Sci U S A. 2019;116:8360–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29:525–31. [DOI] [PubMed] [Google Scholar]
- [84].Hong S, Wang D, Horton JR, Zhang X, Speck SH, Blumenthal RM, et al. Methyl-dependent and spatial-specific DNA recognition by the orthologous transcription factors human AP-1 and Epstein-Barr virus Zta. Nucleic Acids Res. 2017;45:2503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]