

Abstract
In the fetal mouse testis, PIWI Interacting RNAs (piRNAs) guide PIWI proteins to silence transposons, but after birth, most post-pubertal pachytene piRNAs map to the genome uniquely and are thought to regulate genes required for male fertility. In human males, the developmental classes, precise genomic origins, and transcriptional regulation of post-natal piRNAs remain undefined. Here, we demarcate the genes and transcripts that produce post-natal piRNAs in human juvenile and adult testes. As in mouse, human A-MYB drives transcription of both pachytene piRNA precursor transcripts and the mRNAs encoding piRNA biogenesis factors. Although human piRNA genes are syntenic to those in other placental mammals, their sequences are poorly conserved. In fact, pachytene piRNA loci are rapidly diverging even among modern humans. Our findings suggest that during mammalian evolution, pachytene piRNA genes are under few selective constraints. We speculate that pachytene piRNA diversity may provide a hitherto unrecognized driver of reproductive isolation.
In animal germ cells, PIWI-interacting RNAs (piRNAs) guide PIWI proteins to silence transposons, maintain genome integrity, and promote fertility. Unlike microRNAs or small interfering RNAs, piRNAs are processed from long single-stranded precursor transcripts and have 2′-O-methyl-modified 3′ termini1–5. Transposon and viral silencing is likely the ancestral role of piRNAs6–9, and, in the mouse fetal testis, piRNAs silence active transposons10,11. Unlike other animals, placental mammals also produce abundant piRNAs from transposon-depleted, unique non-coding sequences transcribed by RNA polymerase II (RNAP II)1,2,12–17. In mice, mutations in the proteins required to make these “pachytene” piRNAs lead to male sterility, but loss of piRNAs from individual loci rarely has a consequence15,18,19.
Immediately after birth, mouse germ cells produce 26–27 nt long, MILI bound, pre-pachytene piRNAs derived from the coding sequences or 3′ untranslated regions (3′ UTRs) of mRNAs1,10–12,14,20. At the onset of the pachytene stage of meiosis, the transcription factor A-MYB induces pachytene piRNA precursor transcription, and pachytene piRNAs largely replace pre-pachytene piRNAs, coming to account for ~95% of all piRNAs in the adult mouse testis14. A-MYB drives the coordinated transcription of ~100 pachytene piRNA genes, as well as genes encoding proteins required for pachytene piRNA production, cell cycle progression and meiosis14,21. Genes activated by A-MYB include A-Myb itself and Miwi, which binds 30 nt pachytene piRNAs2,14,20. A-MYB also maintains MILI expression in pachytene spermatocytes; ~84% of pachytene piRNAs are bound to MIWI and ~16% to MILI14,20.
Human piRNA-producing loci have been roughly mapped using piRNA sequences1,2,22,23, but the classes, precise genomic locations, or transcription factors regulating human piRNA loci remain unknown. Here, we use 5′ methyl-guanosine cap sequencing (Cap-seq), poly(A) site-sequencing (PAS-seq), and chromatin immunoprecipitation (ChIP) of histone H3 trimethylated on lysine 4 (H3K4me3) to define the juvenile and adult piRNA-producing genes and transcripts in human testis. As in mouse, human pre-pachytene piRNAs mainly derive from mRNA coding sequences or 3′ UTRs, whereas pachytene piRNAs derive from long non-coding RNAs, often divergently transcribed from a central promoter. Interestingly, ChIP data suggest that A-MYB coordinates the transcription of ~55% of human pachytene piRNA-producing genes and macaque piRNA-producing genes. A-MYB also binds the promoters of ~74% of genes encoding piRNA biogenesis proteins in macaques but only ~30% in humans. One-third of human pachytene piRNA-producing genes can be found at the syntenic location in the genomes of other Eutherian mammals; half are restricted to primates; ~18% are human-specific. While pachytene piRNA gene promoters are well conserved, piRNA sequences themselves have diverged rapidly among placental mammals. Moreover, the sequences of pachytene piRNAs vary within the human population more than any other genomic feature. We speculate that pachytene piRNA diversity may provide a hitherto unrecognized driver of Eutherian reproductive isolation.
Results
Three classes of piRNA-producing loci in post-natal human testis
To begin to classify the piRNAs produced during human spermatogenesis, we used strand-specific RNA sequencing to assemble the RNA transcripts and small (25–31 nt) RNA sequencing to define the piRNA repertoire of the post-natal human testis (Supplementary Table 1). De novo assembly identified 306,503 mRNA and long non-coding RNA (lncRNA) transcripts mapping to 125,526 discrete genomic loci (Extended Data 1a). Mapping the piRNA sequences from one juvenile and seven adult testes to the testis transcriptome identified 9,891 piRNA-producing transcripts corresponding to 724 genomic loci. Comparing the juvenile and adult abundance of piRNAs from each of these loci revealed three distinct classes of temporal expression (Extended Data 1b): (1) 618 loci (9,065 transcripts) whose piRNAs changed <2-fold between juvenile and adult samples, consistent with little or no change in piRNA concentration14; (2) 10 loci (147 transcripts) whose piRNA abundance increased ≥2-fold but less than 4-fold in adult testis; and (3) 96 loci (679 transcripts) whose piRNA abundance in adult was ≥4-fold higher than in juvenile. These three classes of human piRNA-producing loci are analogous to the three well-defined piRNA types in the mouse post-natal testis: pre-pachytene, hybrid, and pachytene14.
To polish our annotations, we manually curated the annotations for the 100 most abundant pre-pachytene loci and all 10 hybrid and 96 pachytene loci. We removed 17 pre-pachytene and seven pachytene loci that overlapped with small nucleolar RNAs or had unusually long transcripts likely generated by transcript assembly errors. The resulting 182 loci—83 pre-pachytene loci, 10 hybrid, and 89 pachytene—together account for ~92% of piRNAs in the adult human testis (Fig. 1a). Supporting our classification of piRNAs that increase <2-fold from juvenile to adult, re-analysis of published RNA-seq data for purified germ cell subtypes24 showed that the steady-state abundance of precursor transcripts for these 83 pre-pachytene piRNA-producing loci remained constant from quiescent spermatogonia to late pachytene spermatocytes. In contrast, transcript abundance for the 89 pachytene piRNA loci increased ~4-fold from quiescent spermatogonia (median = 5.9 rpkm) to pachytene spermatocytes (median = 22.7 rpkm).
Figure 1. Three classes of human post-natal piRNA genes.

(a) Heatmap representation of A-MYB occupancy on the promoters of piRNA-producing genes and of the abundance of piRNA precursor transcripts and mature piRNAs from seven adult and one juvenile testis (mean of two technical replicates; Correlation for technical replicates: piRNA sequencing, Spearman’s ρ = 0.99, Pearson’s ρ = 0.99; RNA sequencing, Spearman’s ρ = 0.91, Pearson’s ρ = 0.96). Rpkm: reads per million unique mapped reads per thousand nucleotides; rpm: reads per million. (b) Heatmap representation of the abundance of piRNA precursor transcripts and mature piRNAs from a second set of human testis samples. (c) The distance from the nearest A-MYB peak to the transcription start sites (TSS) for each gene class: piRNA-producing, piRNA-biogenesis protein encoding, other protein-coding, or lncRNA-producing. Whiskers show 95% confidence intervals. (d) A-MYB ChIP-seq signal on the promoter of a divergently transcribed human pachytene piRNA gene and of the A-MYB and HIWI genes. Only uniquely mapping reads were analyzed.
To further test our set of 182 annotated piRNA-producing genes, we obtained and analyzed an additional two juvenile and seven adult human testes. Analysis of the abundance of piRNAs in these samples using our annotated 182 loci recapitulated the differential expression of pre-pachytene, hybrid, and pachytene piRNA genes between juvenile and adult (Fig. 1b and Extended Data 1c). As in mice, all human pachytene piRNA genes reside on autosomal chromosomes, likely because most genes on the sex chromosomes are silenced during meiosis25 (Extended Data 1d and Supplementary Table 2).
Of the 83 pre-pachytene piRNA loci, 75 correspond to protein-coding genes (Extended Data 2a). Pre-pachytene piRNAs dominate the juvenile testis piRNA pool, comprising 93% of all piRNAs (median = 630 rpm), but just 9.5% of piRNAs in adult testis (median = 333 rpm). Conversely, pachytene piRNAs dominate adult piRNA production, accounting for ~90% of all piRNAs in adult human testes. The median abundance of piRNAs mapping to the 89 pachytene piRNA genes was 82-fold greater in adult (931 rpm) than in juvenile testis (11 rpm). Most pachytene piRNA genes (75 of 89) reside in genomic regions that do not encode proteins; 35 of 89 loci are divergently transcribed from a bidirectional central promoter (Extended Data 2b). None of the 83 annotated pre-pachytene piRNA genes show evidence of bidirectional transcription. We conclude that, like mice, humans produce piRNAs from discrete loci that are transcribed, spliced, and processed into pachytene piRNAs when primary spermatocytes enter the pachytene stage of meiosis.
Human piRNA-producing loci are transcribed by RNAP II
We performed chromatin immunoprecipitation sequencing (ChIP-seq) of histone H3 trimethylated at lysine 4 (H3K4me3), Cap-seq, and PAS-seq to curate the location of transcription start sites (TSS) and transcript 3′ ends for the 182 piRNA-producing loci (Extended Data 3a). As in mice, canonical RNAP II transcription generates human piRNA precursors: in human post-natal testes, pre-pachytene, hybrid, and pachytene piRNA precursor transcripts contained 5′ caps and 3′ poly(A) tails (Extended Data 3a and 3b). Moreover, human piRNAs derive from spliced transcripts (Extended Data 3c): median piRNA density within the exons of pre-pachytene piRNA genes (26 rpkm) was 65-fold higher than in introns (0.4 rpkm), and median piRNA density within the exons of the pachytene piRNA genes (260 rpkm) was 144-fold greater than that of the introns (1.8 rpkm).
The 182 piRNA-producing loci defined here account for >2 million distinct piRNA species in adult and ~0.7 million in juvenile testis. Previous annotations of human piRNA-producing loci relied on piRNA density2,22,23. Our annotations, based on piRNA abundance and transcript and gene structure, represent fewer genomic base pairs (2,601,201 bp), but account for ~92% of all uniquely mapped piRNAs with at least two reads in adult testis (Extended Data 4a).
Human pachytene piRNA exons are depleted of transposons
One ancestral function of piRNAs is to silence germline transposons1,2,6–8,10–12,26–28. As expected, the transposon content of human pre-pachytene piRNA precursors (median = 7%), which are typically mRNAs, resembled that of other mRNAs (Extended Data 4b). Hybrid (median = 20%) and pachytene (median = 28%) piRNA precursors contained more transposon sequence than mRNAs, but less than lncRNAs (median = 29%). Notably, the introns of pachytene piRNA precursors, which are removed before piRNAs are generated, have a transposon content (median = 41%) similar to the introns of mRNAs (median = 43%) and lncRNAs (median = 44%). We observed that LTR retrotransposons (14.2%) were more prevalent within pachytene piRNA genes than other classes of transposable elements (Extended Data 4c). However, LINE elements were particularly depleted from the pre-pachytene piRNA genes, suggesting selection against insertion of LINE elements into coding sequences (Extended Data 4c). The depletion of transposon sequences from the exons of pachytene piRNA genes suggests that these piRNAs have functions beyond transposon silencing.
Further supporting this idea, transposons in the exons of human pachytene piRNA genes have greater divergence rates (average divergence rate = 0.193) than transposons in pre-pachytene piRNA genes (0.176) or transposons across the genome as a whole (average divergence rate = 0.186). Because older transposons experience greater sequence drift than young, active transposons, we conclude that the transposons in the exons of pachytene piRNA-producing loci are, on average, older than those in pre-pachytene piRNAs or the genome as a whole. Older transposons are less likely to be active, consistent with the idea that the primary function of pachytene piRNAs is to regulate gene expression rather than to silence transposons.
A-MYB regulates human pachytene piRNA production
In mice, the transcription factor A-MYB (MYBL1) drives the expression of most pachytene piRNA loci and many mRNAs that encode piRNA biogenesis factors14,21. To test whether A-MYB regulates pachytene piRNA production in humans, we performed A-MYB ChIP-seq from adult human testis (Fig. 1a). A-MYB peaks (FDR <0.05) were located within 500 bp of the transcription start sites of 655 genes (Supplementary Table 3), including 49 pachytene piRNA genes (16 bidirectional, 33 unidirectional; ~55% of all pachytene piRNA genes; Fig. 1a and Extended Data 2b). The median distance from pachytene piRNA transcription start sites to the nearest A-MYB peak was 0 bp (Fig. 1c). We used ChIP-seq of H3K4me3 to map the promoters of the 182 pre-pachytene, hybrid, and pachytene piRNA-producing loci (Extended Data 3a). All 49 pachytene piRNA gene promoters bound by A-MYB were enriched for the canonical A-MYB-binding motif, CAGTT, beneath the ChIP-seq peak (E = 1 × 10−44; Fig. 1d). Human A-MYB also bound the promoters of HIWI, HENMT1, HILI2, DDX39A, and FKBP6, all genes encoding proteins required for piRNA production, as well the A-MYB promoter itself (Fig. 1d and Supplementary Table 3). Unlike mice, where A-MYB binds the promoters of 99% of pachytene piRNA genes and 95% of known piRNA biogenesis protein genes14, the promoters of ~45% of human pachytene piRNA-producing genes and 70% of genes encoding proteins known to participate in piRNA biogenesis were not bound by A-MYB. Accordingly, the promoters of ~45% of human pachytene piRNA genes lack an identifiable A-MYB-binding motif, compared to just 11% in mice. (Similar numbers of piRNA biogenesis genes lack an A-MYB-binding motif in both humans and mice.) Thus, additional transcription factors may be required to coordinate transcription of human pachytene piRNA precursors and biogenesis factors.
To test the idea that additional transcription factors may act with A-MYB to drive pachytene piRNA production, we sequenced piRNAs from purified spermatogonia and primary spermatocytes from mice homozygous for the A-Mybrepro9 mutation21, in which a point mutation within exon 6 leads to alternative splicing that truncates the A-Myb mRNA (Extended Data 5a). For comparison, we sequenced the piRNAs in primary spermatocytes of Miwi−/− mutant mice, which lack the 30 nt pachytene piRNAs normally bound to MIWI but retain the 26–27 nt pachytene piRNAs associated with MILI1,10–12. Overall pachytene piRNA abundance in A-Mybrepro9 primary spermatocytes was reduced ~25-fold (median = 112 rpm) compared to wild-type primary spermatocytes (median = 2801 rpm). Nonetheless, pachytene piRNA abundance was 2.1-fold greater in A-Mybrepro9 mutant primary spermatocytes than in mutant spermatogonia (median = 53 rpm), suggesting that even without A-MYB, some pachytene piRNA loci are transcriptionally activated as spermatogonia differentiate into primary spermatocytes (Extended Data 5b). The abundance of MIWI-bound, 30-nt piRNAs increased just 1.8-fold in A-Mybrepro9 primary spermatocytes (median = 16 rpm vs. 9 rpm for spermatogonia), whereas the abundance of MILI-bound, 26–27-nt piRNAs increased 2.6-fold in mutant primary spermatocytes (median = 45 rpm vs. 17 rpm). This likely reflects the 50-fold reduced transcription of Miwi in A-Mybrepro9 (Ref. 14), making less MIWI available for piRNA loading. We conclude that in both mice and humans, the regulation of pachytene piRNA transcription involves additional factors beyond A-MYB.
Pachytene piRNA precursor expression defines three groups of adult testis samples
Unsupervised hierarchical clustering of the transcriptomes (Fig. 2a) of all 14 adult samples revealed three groups with distinct patterns of pachytene piRNA abundance (Fig. 2b) or modal piRNA length (Extended Data 6a). Group 1 testes had the most pachytene piRNAs (median = 1924 rpm) compared to group 2 (median = 971 rpm) or group 3 (median = 395 rpm). Median piRNA abundance in the juvenile samples was 14 rpm. While 30 nt piRNAs predominated in group 1 (median of average piRNA length = 28.2 nt), group 2 testes contained fewer 30 nt and more 26 nt piRNAs (median of average piRNA length = 27.5). Group 3 testes contain few 30 nt piRNAs (median of average piRNA length = 27.2), even though they are from post-pubertal men (Extended Data 6a). Group 3 had ~2.1-fold more 26–27 nt pre-pachytene piRNAs and ~4.5-fold fewer 30 nt pachytene piRNAs than group 1, while group 2 had twice as many 26–27 nt pre-pachytene piRNAs and half as many pachytene piRNAs as group 1 (Fig. 3a). For all three groups of adult testes, piRNA precursor transcript abundance mirrored piRNA levels (Fig. 2c), suggesting that group 2 and group 3 testes make fewer pachytene piRNAs at least partly because they transcribe fewer pachytene piRNA precursors.
Figure 2. Three groups of adult samples.
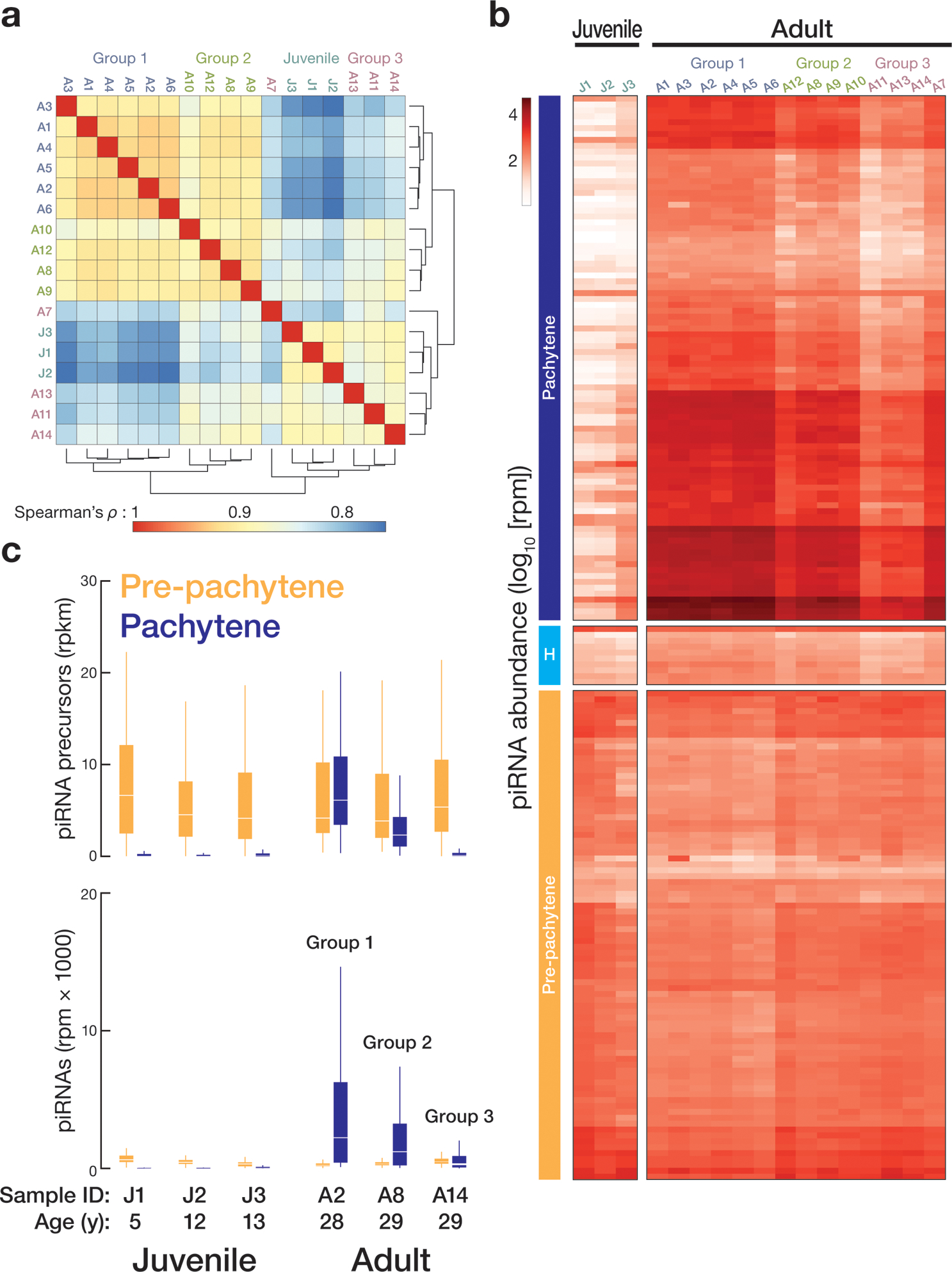
(a) Unsupervised hierarchical clustering of samples based on the abundance of protein-coding and non-coding transcripts. Three groups of adult samples were defined according to their transcriptome. (b) Heatmap representation of the abundance of piRNAs from human testis samples analyzed in this study. (c) Top: changes in the abundance of pre-pachytene and pachytene precursor transcripts between juvenile and adult samples. Bottom: box plots display the change in pre-pachytene and pachytene piRNA abundance for three juvenile samples. YO, year old. Whiskers show 95% confidence intervals.
Figure 3. Dysregulated expression of A-MYB and HIWI in group 3 adult testes.
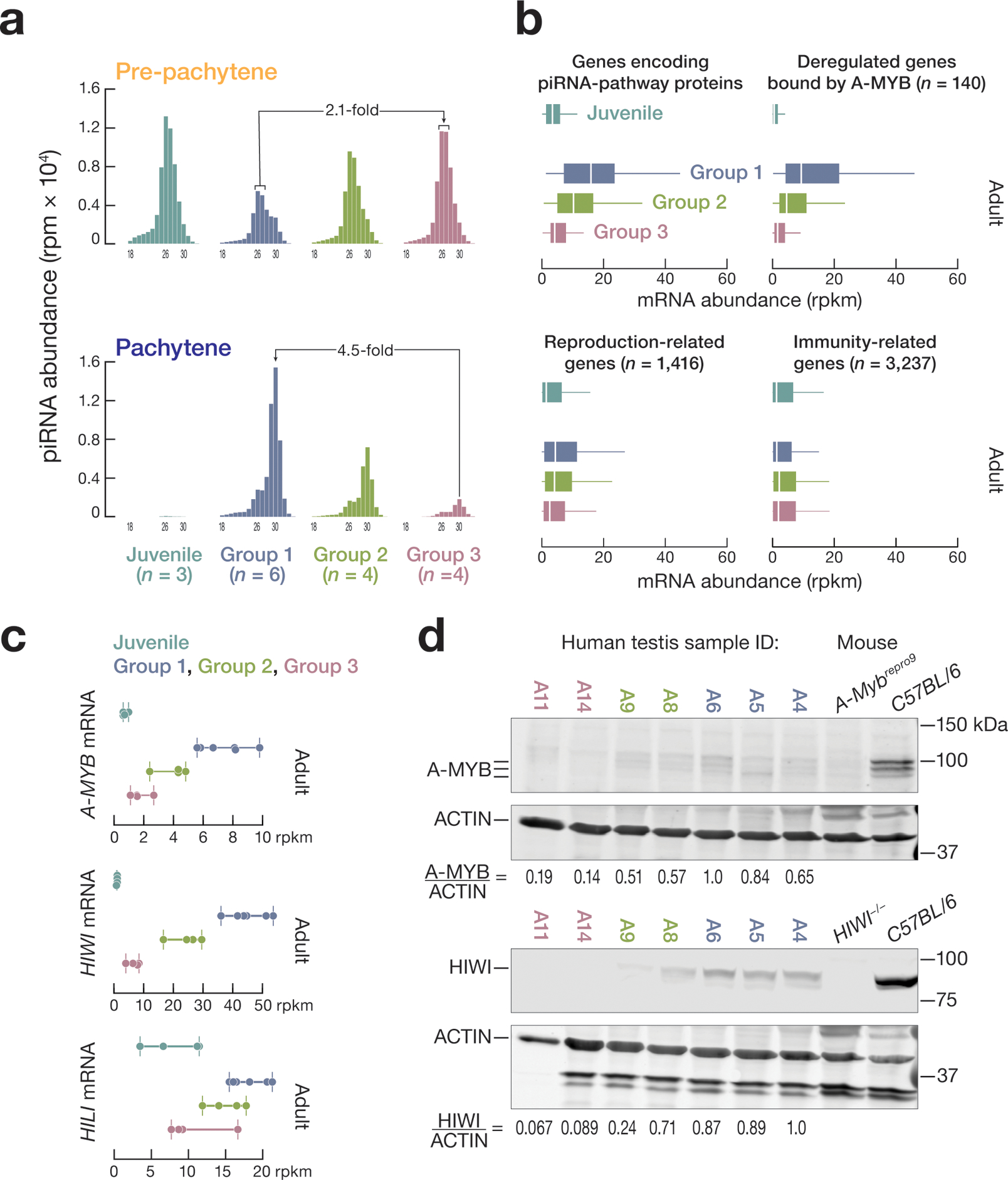
(a) Change in abundance of HILI-bound 26–27 nt piRNAs and HIWI-bound 30 nt piRNAs mapping to pre-pachytene and pachytene piRNA-producing genes among different groups of adult and juvenile samples. (b) Box plots showing the mRNA abundance among the different groups of adult and juvenile samples for genes encoding piRNA biogenesis factors, 140 additional A-MYB-regulated genes, as well as reproduction-related genes and immune response-related genes. (c, d) A-MYB and HIWI mRNA (c) and protein abundance (d).
Impaired A-MYB expression in Group 3 testis
Our data suggest that A-MYB drives transcription of pachytene piRNA genes, HIWI, and other piRNA biogenesis genes in human testis (Fig. 1a, 1c, and 1d). In addition to reduced piRNA precursor transcript abundance, group 2 and 3 testes showed reduced expression of 140 genes regulated by A-MYB, including three genes encoding piRNA biogenesis proteins (Fig. 3b, and Supplementary Table 3), suggesting that A-MYB function might be compromised in these testes. Indeed, compared to group 1, A-MYB mRNA levels were reduced ~1.7-fold in group 2 and ~4.8-fold in group 3. Consistent with the reduced levels of 30 nt pachytene piRNAs, HIWI mRNA levels were similarly reduced compared to group 1: ~1.7-fold lower in group 2 and ~6.2-fold lower in group 3 (Fig. 3c). HILI mRNA abundance in group 3 was half that of group 1. In mice, A-MYB increases transcription of Mili14. Both A-MYB and HIWI protein levels were reduced in group 2 and barely detectable in group 3 testes (Fig. 3d, and Extended Data 6b and 6c).
In addition to reduced steady-state transcript abundance for genes producing pachytene piRNAs and genes encoding piRNA pathway proteins, group 3 testes had a ~2.3-fold lower median mRNA abundance for 1,416 reproduction-related genes (median abundance = 2.5 rpkm) compared to group 1 (median abundance = 4.4 rpkm; Fig. 3b). In contrast, the median mRNA abundance for 3,237 immunity-related genes was comparable among all three groups (Fig. 3b). Group 3 testes appear to be azoospermic. The cause of this azoospermia is not known; RNA sequencing did not detect non-synonymous mutations in the A-MYB or HIWI coding sequences in the samples. Because information about the health of the testes donors is unavailable, we cannot exclude that the individuals were exposed to mutagens or chemotherapy toxic to the male gonad29.
Pachytene piRNA expression accompanies the progression of human spermatogenesis
The three groups of testes afforded an opportunity to examine the developmental timing of pachytene piRNA expression in human spermatogenesis (Extended Data 7a). All germ cell types were present in group 1 testes, whereas group 2 testis had disorganized seminiferous tubules and contained fewer primary spermatocytes as well as fewer round and elongating spermatids. Group 2 testis were arrested early in spermatogenesis; the most abundant group 3 germ cells were spermatogonia adjacent to the basal lamina.
The transcript levels of 5,988 genes decreased and 2,172 increased in group 3 testes compared to group 1 (>2-fold change and FDR <0.001; Extended Data 7b). Among the RNAs with decreased abundance in group 3, 44% (2,605) corresponded to protein-coding genes, and these were enriched for genes involved in male reproduction (597), including 81 genes related to meiosis and 111 genes related to cilium organization, assembly, and movement (Extended Data 7c). Among mRNAs with significantly increased abundance in group 3 testes, 22 genes were related to transposition (n = 8) or viral defense (n = 14), suggesting that group 3 comprises unhealthy testis samples. Therefore, we reanalyzed the change in expression of the 182 annotated piRNA-producing genes between juvenile and adult samples using only Groups 1 and 2. Analysis of the abundance of piRNAs using our annotated 182 genomic loci recapitulated the differential expression of pre-pachytene, hybrid, and pachytene piRNA genes between juvenile and healthy adult samples (Extended Data 8a and 8b). We conclude that as in mice, pachytene piRNA expression accompanies the progression of spermatogenesis through meiosis in human testis.
In mice, A-Myb and Miwi mRNAs and pachytene piRNAs appear at the onset of pachytene spermatocyte development14,21,30. Similarly, human group 1 testes showed strong nuclear localization of A-MYB in pachytene spermatocytes and cytoplasmic localization of HIWI in pachytene and round spermatids (Extended Data 7a), suggesting that the feedforward regulation of piRNA production orchestrated by A-MYB is evolutionarily conserved. A-MYB and HIWI proteins were below the limit of detection for immunohistochemistry in group 2 and group 3 testes.
Feedforward regulation of piRNA production by A-MYB is conserved in macaque
A-MYB is conserved across vertebrates31,32. A-MYB ChIP-seq of Macaca mulatta (rhesus macaque) testis identified 18,579 genomic regions with significant A-MYB peaks (FDR <0.05). These include 7,209 protein-coding genes with A-MYB peaks within 500 bp upstream of their annotated transcription start sites (Supplementary Table 3). As in humans and mice, the promoters of macaque A-MYB, Macaca mulatta PIWIL1 and other piRNA biogenesis genes were bound by A-MYB (Fig. 4a and Supplementary Table 3). For the 12 M. mulatta genes both known to function in piRNA production and bearing an A-MYB peak, the median distance between the transcription start sites and the nearest A-MYB peak was 0 bp (Fig. 4b).
Figure 4. Feedforward regulation of piRNA production by A-MYB is conserved in macaque.
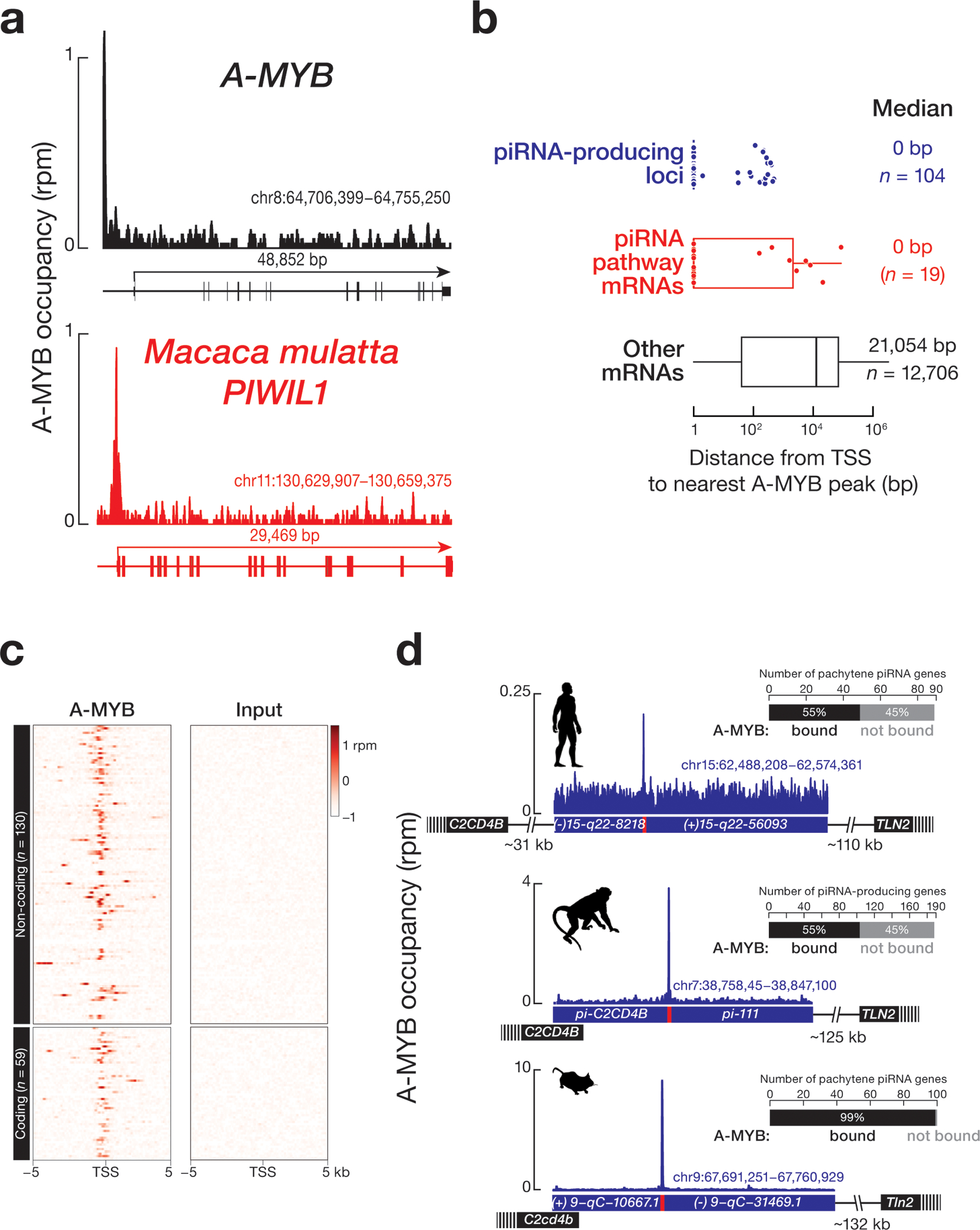
(a) A-MYB ChIP-seq signal at the transcription start sites of macaque A-Myb and PIWIL1. (b) The distance from the transcription start site of each piRNA-producing, piRNA-biogenesis protein encoding, or other protein-coding genes. (c) Heatmap showing A-MYB occupancy around the transcription start sites of macaque piRNA-producing loci. (d) A-MYB ChIP-seq peak on the promoter of a divergently transcribed, piRNA-producing locus from macaque, compared with major syntenic pachytene piRNA genes from human and mouse. Bar graphs show the number of A-MYB-bound pachytene piRNA genes in humans, macaque, and mouse.
We used piRNA density to define 189 piRNA-producing loci in M. mulatta (Supplementary Table 4): A-MYB was bound to ~55% (33 protein-coding and 71 non-coding) of piRNA-producing loci (Fig. 4c and Supplementary Table 3). Among those piRNA-producing loci bound by A-MYB, the median distance between the transcription start site and the nearest A-MYB peak was 0 bp (Fig. 4b). Fig. 4d shows an example of A-MYB occupancy on the promoter of a bidirectionally transcribed, non-coding, M. mulatta piRNA cluster that is syntenic to piRNA clusters in both humans and mice.
Human pachytene piRNA genes evolve rapidly despite conservation of their promoters
To investigate how human piRNA genes have diverged from piRNA-producing loci in other mammals, we used piRNA density to define the piRNA-producing loci in marmoset (Callithrix jacchus) and rat (Rattus norvegicus), both placental mammals; platypus (Ornithorhynchus anatinus), a monotreme; and gray short-tailed opossum (Monodelphis domestica), a marsupial. Our analyses identified 172 piRNA-producing loci in marmoset, 124 in rat, 95 in platypus, and 207 in opossum (Supplementary Table 4). Many of these piRNA-producing genes appear to be non-coding: 69% (130) in macaque, 77% (133) in marmoset, 92% (114) in rat, 95% (89) in platypus, and 94% (194) in opossum.
Post-natal piRNA sequences are highly diverse1,2,14, suggesting that they evolve rapidly. We compared the conservation among 46 placental mammals of the 5′ UTRs, coding sequences, 3′ UTRs, and introns of protein-coding genes; the exons and introns of lncRNA genes; the exons and introns of piRNA-producing genes; and randomly selected, non-transcribed sequences that do not produce piRNAs (Fig. 5a and Extended Data 9a). As expected, protein-coding sequences not making piRNAs were the most conserved transcribed feature (median phastCons score = 0.74) among 46 Eutheria (Fig. 5a and Extended Data 9b). Given that most human and mouse pre-pachytene piRNAs derive from protein-coding genes, the coding sequence of pre-pachytene genes (median phastCons score = 0.81) was conserved at a similar rate as the coding sequence of other protein-coding genes, suggesting that mRNAs are exposed to similar selective pressures regardless of whether they produce piRNAs (Fig. 5a and Extended Data 9b).
Figure 5. Comparative genomic analysis of human piRNA genes.
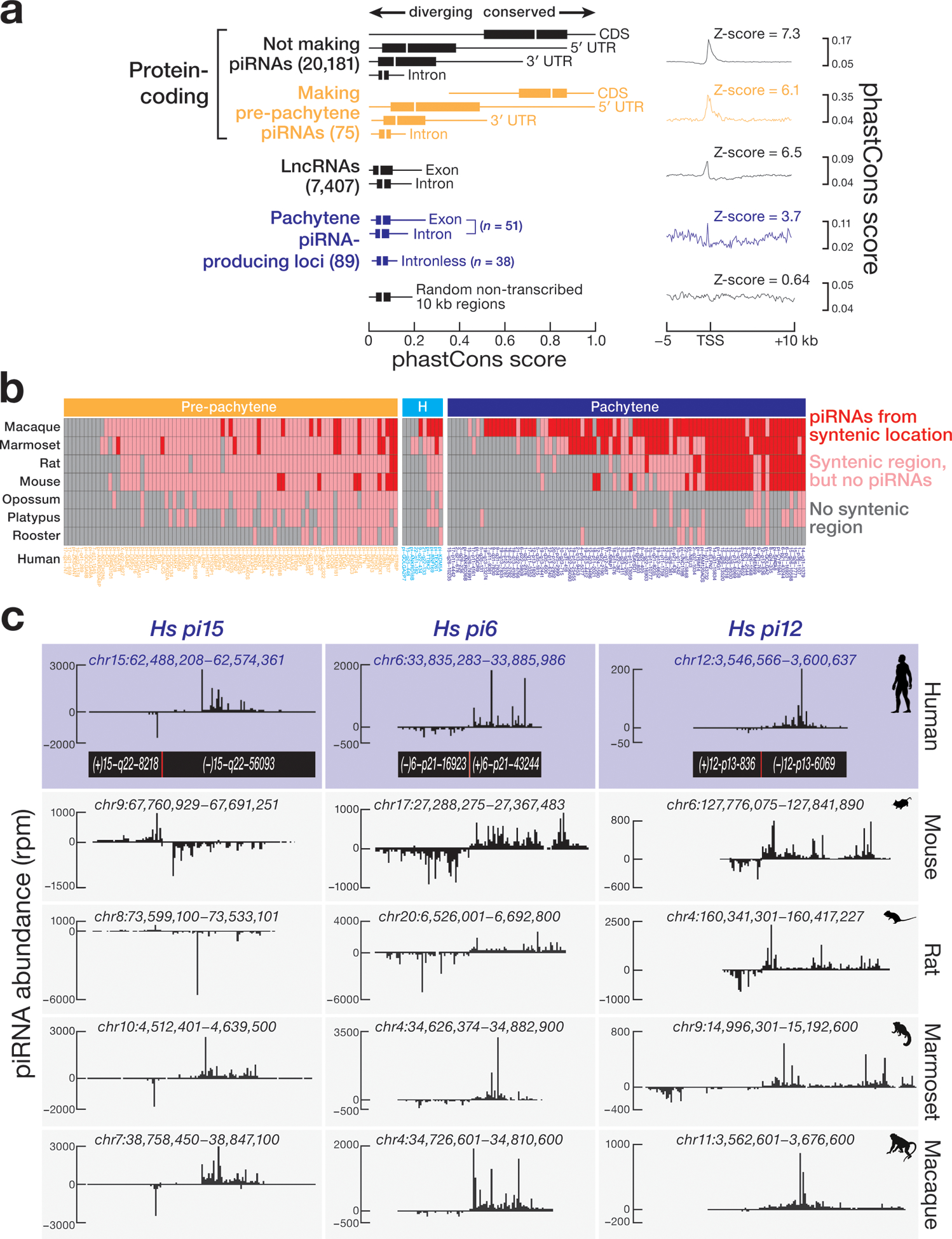
(a) DNA sequence conservation across 46 Eutherian mammals for human piRNA genes compared with other genomic features. Randomly selected 22,604, 10-kb non-transcribed genomic regions, which do not produce piRNAs, was used as a background control. (b) Synteny analysis reveals conservation of location of human piRNA genes across placental mammals, non-placental mammals, and chicken. Mouse and chicken piRNA-producing regions were defined previously14. (c) The syntenic genomic coordinates of three human bi-directional pachytene piRNA genes for mouse, rat, marmoset, and macaque.
The promoters of human pachytene piRNA genes are far more conserved than their transcribed sequences (Fig. 5a; right). In fact, pachytene piRNA precursor transcripts appear to be among the least well conserved transcribed genomic feature we examined, including the exons of lncRNAs and randomly selected non-transcribed sequences (Fig. 5a and Extended Data 9b). Across placental mammals, the lack of DNA sequence conservation of nucleotides making pachytene piRNAs reflects the immense diversity of pachytene piRNA sequences.
Most pre-pachytene piRNA genes started producing piRNAs after the divergence of marsupials and placental mammals
The sequences of the 83 human pre-pachytene piRNA loci are often found at syntenic sites in other placental mammals: of the 74 that reside in regions of synteny in at least one other mammal, 55 are present at the syntenic location in not only placental mammals, but also in opossum, platypus, or rooster. Yet none of these 55 protein-coding genes produces piRNAs in non-placental mammals or birds, suggesting that the ability to produce piRNAs from these mRNAs was acquired after the divergence of placental and non-placental mammals, ~160 million years ago33. In fact, just nine of the 83 human pre-pachytene piRNA-producing genes produce piRNAs at the syntenic location in placental mammals other than primates. Another 10 human pre-pachytene piRNA-producing genes generate piRNAs at the syntenic location only in primates (Fig. 5b and Supplementary Table 5). We currently cannot explain why some mRNAs produce piRNAs in humans but not in other Eutheria. We note that differences in mRNA abundance are unlikely to explain this phenomenon: mRNAs of comparable abundance often produce piRNAs in humans but not other species (Extended Data 10a). Extended Data 10b provides examples of genes producing pre-pachytene piRNAs in humans, indicating the evolutionary depth of synteny and piRNA abundance in other species.
Many human pachytene piRNA genes arose after primates diverged from other placental mammals
Among the 89 human pachytene piRNA-producing genes, 29 produce piRNAs from the syntenic location in both primates and at least one additional placental mammal (Figs. 5b and 5c, and Supplementary Table 5), suggesting that these 29 human piRNA-producing genes antedate the divergence of primates from other placental mammals, ~90 million years ago. Another 44 loci produce piRNAs from the syntenic location only in other primates. The remaining 16 pachytene piRNA loci produce piRNAs only in humans. The absence of these loci in macaque and marmoset implies that the loci are no older than ~30 million years. Notably, more deeply conserved human pachytene piRNA loci produce more piRNAs than younger loci: the abundance of pachytene piRNAs from human loci conserved in at least one other placental mammal (median = 5,188 rpm) was twice the abundance of piRNAs from loci conserved only in primates (median = 2,184 rpm) and 11-fold higher than loci unique to humans (median = 460 rpm) (Extended Data 10c).
Pachytene piRNA genes are highly variable among humans
Among 2,504 human genomes sequenced by the 1000 Genomes Project34, the mean derived-allele frequency—a measure of sequence divergence among individuals within a species—for human pachytene piRNA precursor exons was higher than that of any other transcribed feature we examined and was comparable to the derived-allele frequency of randomly selected, non-transcribed sequences (Fig. 6a). To further test the results of the derived-allele frequency analysis, we measured the nucleotide diversity (π value) at each nucleotide position of human piRNA genes and other transcribed features. Consistently, the π value of human pachytene piRNA precursor exons was higher than that of any other transcribed feature (Fig. 6a). Counterintuitively, loci with synteny among placental mammals are diverging faster than loci with synteny only among primates or loci unique to humans (Fig. 6a), a phenomenon typically associated with old transposons and pseudogenes, not genes under positive selection to retain function. In fact, loci with synteny are diverging faster than even randomly selected, non-transcribed sequences (Fig. 6a). Moreover, the exons of pachytene piRNA genes—the sources of piRNAs—were more polymorphic than the introns of pachytene piRNA genes, which are removed before piRNA precursors are processed into piRNAs (Figs. 6a and 6b). The slower divergence of pachytene piRNA introns likely reflects conservation of splicing signal sequences (Fig. 6b), suggesting that intron removal is required for piRNA processing or function.
Figure 6. Sequence variation for different genomic features within the human population.
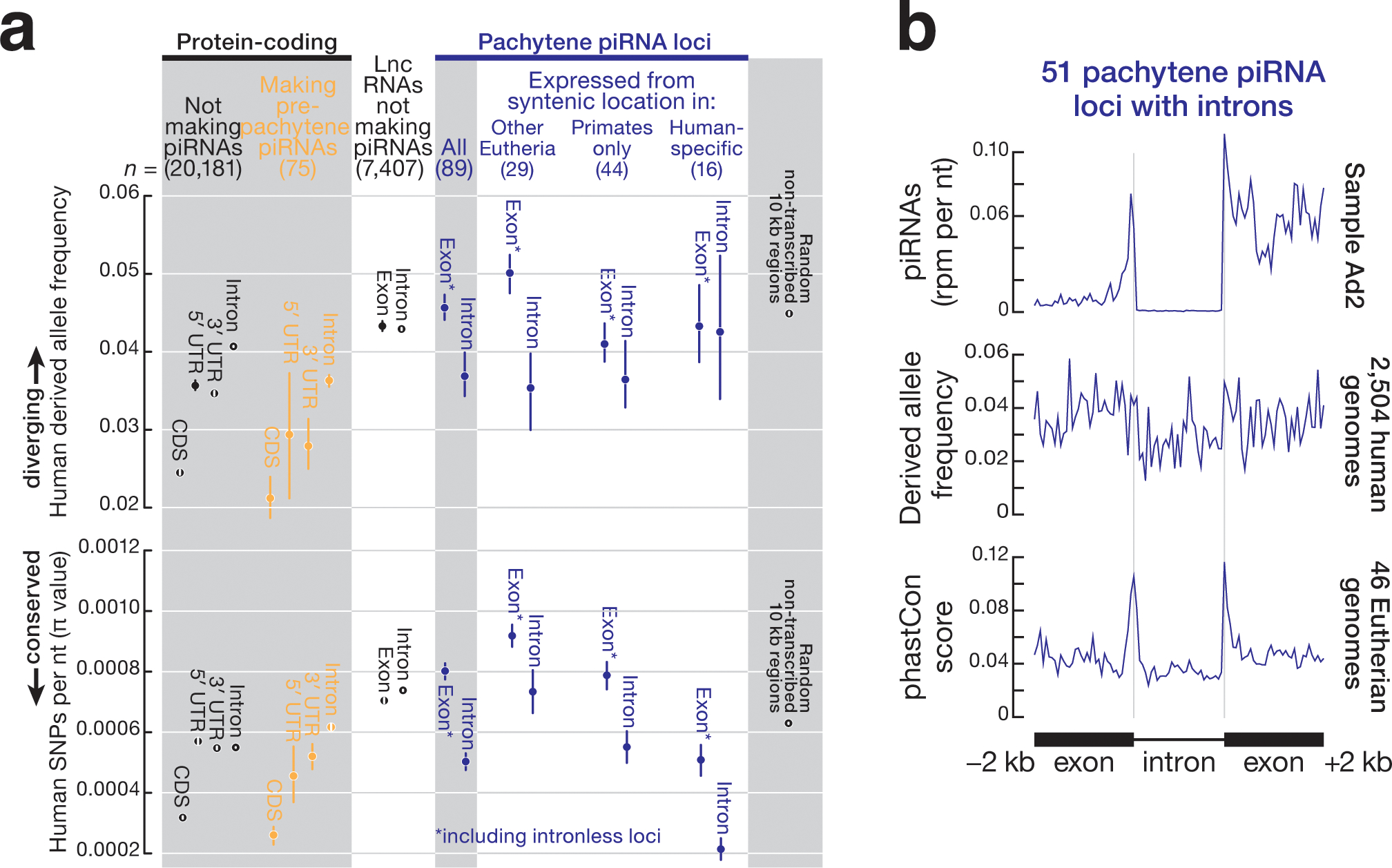
(a) Mean derived-allele frequency of autosomal single-nucleotide polymorphisms (SNPs) and mean nucleotide diversity (π value) for 2,504 human genomes representing 26 different human populations34. Whiskers show 95% confidence generated by random sampling 100 times. Derived-allele frequency from 22,604, 10 kb-long randomly selected non-transcribed genomic regions served as a control. (b) Metagene plots representing the piRNA abundance, derived-allele frequency, and conservation score for intron-containing pachytene piRNA genes (n = 51).
Discussion
Here, we report the structure of the piRNA-producing genes expressed in the post-natal human testis. We use these data to examine the evolutionary conservation of the transcriptional mechanisms that regulate piRNA production and of the sequences of the piRNAs themselves.
A-MYB regulation is conserved across mammals
In mouse testis, pachytene piRNA genes are transcribed at the onset of the pachynema when the A-Myb gene is activated14,21. Three lines of evidence suggest A-MYB coordinates the expression and processing of pachytene piRNAs across Eutherian mammals, including humans. First, A-MYB protein is readily detectable in human pachytene spermatocytes but not spermatogonia. Second, A-MYB is bound to the promoters of many human pachytene piRNA genes, genes encoding piRNA biogenesis proteins, and the A-MYB promoter itself. Third, A-MYB binds to the promoters of macaque piRNA-producing genes, genes encoding piRNA biogenesis proteins, and A-MYB. Given that A-MYB regulation of piRNA-producing genes can be detected in rooster testes14, control of piRNA production by this transcription factor appears to have emerged at least ~300 million years ago, in the last common ancestor of birds and mammals33, long before the emergence of pachytene piRNAs in Eutheria.
Nevertheless, A-MYB does not account for all pachytene piRNA production. At least half the promoters of human pachytene piRNA and macaque piRNA-producing genes are not occupied by A-MYB, and some pachytene piRNAs are still made in mouse or human testes with little or no A-MYB. The transcription of pachytene piRNA genes might therefore be sustained by cooperative or compensatory transcription factors that, together with A-MYB, ensure the burst of piRNA production at the pachytene stage of meiosis.
In Drosophilids, germline dual-strand piRNA clusters are transcribed by a non-canonical, heterochromatin-dependent, promoter-independent mechanism that distinguishes the transcription of piRNA precursors from the promoter-dependent, canonical transcription of mRNA6,37–40. In contrast, Eutherian pachytene piRNA genes appear to be canonical euchromatic transcription units regulated by A-MYB. But A-MYB also regulates transcription of many genes that are not sources of piRNAs, including genes required for meiosis and genes that encode piRNA biogenesis factors14,21. How then are pachytene piRNA precursor transcripts distinguished from mRNAs? What marks these genes or their transcripts to produce pachytene piRNAs? If the fate of a piRNA precursor transcript is determined by cis-acting RNA elements, these must be more complex than simply promoter architecture, because several mammalian pachytene piRNA genes are transcribed divergently from a central promoter, yet produce an mRNA or lncRNA from one arm and a piRNA precursor from the other14,41,42.
Rapid evolution of pachytene piRNA-producing genes
The genomic locations and promoter sequences of many pachytene piRNA genes are conserved among placental mammals. Nevertheless, the transcribed sequences of the genes have diverged rapidly, suggesting that they are under little selective pressure. Different pachytene piRNA sequences emerge from syntenic pachytene piRNA genes in different species and presumably regulate different targets. Indeed, a mouse expressing the human pi6 pachytene piRNA gene is male sterile because it inappropriately silences an mRNA essential for spermatogenesis13. We envision that rapid divergence of piRNA sequences may lead to acquisition of distinct set of targets during mammalian evolution.
Pachytene piRNAs are among the most variable sequences in the human population. Our finding that modern human pachytene piRNA loci with deeper evolutionary conservation are diverging more rapidly than younger loci implies that increased sequence variation plays an intrinsic role in piRNA function. We speculate that the sequences recognized by piRNAs in specific mRNAs drift in parallel with the overall drift of the pachytene piRNA sequences among individuals. Testes express a more complex repertoire of transcripts than other tissues43, allowing a large fraction of the human transcriptome to interact with pachytene piRNAs. In theory, such interactions might probe the compatibility of the paternal and maternal genomes. For example, target RNA cleavage directed by piRNAs from the paternal genomic haplotype could decrease the abundance of mRNA sequence variants encoded by the maternal genome. Such piRNA-dependent transcriptional repression may reduce or enhance the fitness of an individual’s sperm. We speculate that pachytene piRNA diversity may therefore be a hitherto unrecognized driver of Eutherian reproductive isolation.
Methods
Human tissue samples
De-identified, frozen human testis and other tissue samples included in the study were obtained from the University of Massachusetts Medical School Tissue and Tumor Bank except for two adult and two juvenile testis tissue samples that were a gift of Kyle Orwig (University of Pittsburg, USA). Additionally, one sample of human testis total RNA was from Ambion (ThermoFisher; AM7972). The maximum delay from excision of human testis specimens to flash freezing was ~2 h. After receiving frozen samples, they were stored at −80°C. Tissues were handled on dry ice. Seventeen adult and three pre-pubertal (juvenile) human testis samples were included in the study (Supplementary Table 1).
Mice
All mice were maintained and used according to the guidelines of the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School. C57BL/6J mice (RRID: IMSR_JAX:000664) were used as wild-type controls. A-Myb mutant mice (B6(C3)-A-Mybrepro9/JEbfJ, IMSR Cat# JAX:027889; Ref. 21) and Miwi (B6;129-Miwitm1Hfl/Mmmh, RRID:MMRRC_029995-MU; Ref. 30) mutant mice were genotyped as previously described20.
To characterized the repro9 mutation21 residing within the exon 6 of A-Myb, we isolated total RNA from wild-type adult and 17 dpp testes and 14.5 dpp heterozygous and homozygous A-Mybrepro9 mutant testes. After oligo(dT) primed cDNA synthesis, the region spanning the repro9 mutation was amplified by PCR. Sanger sequencing of the amplicon revealed that the C-to-A Mybl1repro9 mutation creates a strong splice donor at the 5′ end of exon 6, leading to an out-of-frame deletion in the mRNA (Extended Data 5a).
Isolation of mouse germ cells by FACS
Germ cell sorting was performed as previously described20. In brief, isolated testes were de-capsulated and incubated in 1× Gey′s Balanced Salt Solution (GBSS, Sigma, G9779) containing 0.4 mg/ml collagenase type 4 (Worthington; LS004188) for 15 min at 33°C, rotating at 150 rpm. The seminiferous tubules were washed twice with 1× GBSS and incubated with 1× GBSS containing 0.5 mg/ml Trypsin and 1 μg/ml DNase I for15 min at 33°C, rotating at 150 rpm. After Trypsin and DNase treatment, tubules were homogenized on ice by gentle, repeated pipetting through a Pasteur pipette for 3 min. Thereafter, Trypsin was inactivated with fetal bovine serum (FBS; 7.5% f.c., v/v). The cell suspension was passed through a 70 μm pre-wetted cell strainer, and cells collected by centrifugation at 300 × g at 4°C for 10 min. The pelleted cells were further resuspended in 1× GBSS containing 5% (v/v) FBS, 1 μg/ml DNase I, and 5 μg/ml Hoechst 33342 (Thermo Fisher, 62249) and incubated for 45 min at 33°C, rotating at 150 rpm. Thereafter, Propidium iodide (0.2 μg/ml, f.c.; Thermo Fisher, P3566) was added, and cells were passed through a 40 μm pre-wetted cell strainer. Finally, cells were sorted at UMass Medical School FACS Core as previously described44.
Testis histology and immunohistochemistry
Paraffin embedded human testis tissue fixed with 4% (v/v) formaldehyde were obtained from the UMass Medical School Tissue and Tumor Bank or Kyle Orwig. Embedded tissues were sectioned at 4 μm thickness and stained with hematoxylin and eosin (H&E) by the UMass Medical School Morphology Core Facility.
Immunohistochemical (IHC) staining was performed using standard protocols. Briefly, testis sections were de-paraffinized with xylene, dehydrated with ethanol, and heated with 1 mM citrate buffer (pH 6.0) to retrieve antigen. Endogenous peroxidase activity was inactivated with 3% (w/v) hydrogen peroxide for 10 min at room temperature. Tissues were blocked with 5% (v/v) horse serum using ImmPRESS HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection Kit (Vector labs; MP-7401). Sections were then incubated with rabbit anti-A-MYB (Sigma, HPA008791; 1:400 dilution) or rabbit anti-MIWI (Abcam, ab12337; 1:400 dilution) antibody overnight at 4°C. Secondary HRP anti-rabbit antibody (Vector labs; MP-7401) was applied for 1 h at room temperature, followed by incubation with substrate/chromogen (Fisher Scientific, TA-125-QHDX). Finally, slides were counterstained with hematoxylin, dehydrated and sealed with a coverslip. H&E and IHC images were captured using a Leica DMi8 microscope.
Western blotting
Frozen testis tissue was homogenized in RIPA buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% (v/v) NP-40, 1% (w/v) sodium deoxycholate, and 0.1% (w/v) SDS) containing 1× E-64 protease inhibitor (Sigma; E3132). Lysed tissue was sonicated (Branson Digital Sonifier; 450 Cell Disruptor) to break nuclei, then samples were centrifuged at 20,000 × g for 30 min at 4°C. The supernatant was transferred to a new tube, and the protein concentration measured (Pierce BCA Protein Assay Kit; ThermoFisher; 23225). For each sample, 75 μg total protein was mixed with 1/3 volume loading dye (106 mM Tris-HCl, pH 6.8, 141 mM Tris base, 2% SDS, 10% glycerol, 0.51 mM EDTA, 0.22 mM SERVA Blue G and 0.175 mM Phenol Red) containing 0.2 M DTT and heated at 95°C for 6 min. After denaturing proteins, samples were resolved by electrophoresis through a 4–20% gradient polyacrylamide/SDS gel (Thermo Fisher, XP04205BOX) and electro-transferred to PVDF membrane (Millipore, IPVH00010). The membrane was blocked (Blocking Buffer; Rockland Immunochemicals, MB-070) for 1 h at room temperature and incubated with rabbit anti-A-MYB (Sigma, HPA008791; 1:700 dilution) or rabbit anti-MIWI (Abcam, ab12337; 1:1000 dilution) antibody overnight at 4°C. The membrane was washed three times with 1× PBS-T (0.1% (v/v) Tween-20 in 1× PBS) for 30 min at room temperature and then incubated with secondary donkey anti-rabbit IRDye 680RD antibody (LI-COR, 926–68073; 1:5000 dilution) for 1.5 h at room temperature. Afterwards, the membrane was washed three times with 1× PBS-T for 30 min at room temperature, and signal detected (Odyssey Infrared Imaging System; LI-COR). As a loading control, membrane was incubated with mouse anti-ACTIN (Santa Cruz Biotechnology, sc-47778; 1:3000 dilution) antibody for 2 h at room temperature and then goat anti-mouse IRDye 800RD secondary antibody (LI-COR, 926–32210; 1:5000 dilution) as described.
Small-RNA library construction and analysis
Total RNA was extracted from frozen human testis tissue using mirVana miRNA isolation kit (Thermo Fisher, AM1560). Small RNA library construction was performed as described45. In brief, 18–34 nt was isolated by separating total RNA by denaturing electrophoresis through a 15% polyacrylamide gel. To prevent microRNAs and siRNAs entering to the library, small RNAs were oxidized with sodium periodate (NaIO4) prior to 3′ adaptor ligation. The 3′ adaptor (5′-rApp NNN TGG AAT TCT CGG GTG CCA AGG/ddC/−3′ or 5′-rApp TGG AAT TCT CGG GTG CCA AGG/ddC/−3′; Supplementary Table 1) was ligated to the RNA in the presence of 50% (w/v) PEG-8000 (Rigaku, 1008063). After 3′ ligation, ligated small RNAs (42–60 nt) were purified by denaturing electrophoresis (15% PAGE). Thereafter, 5′ adaptor was ligated, and libraries were sequenced with 79 nt single-end reads (NextSeq500; Illumina).
The 3′ adaptor sequence was removed from the reads. and those reads with PHRED score <5 were mapped to human genome hg19 using piPipes46 with bowtie 2.2.5 (Ref. 47) allowing one mismatch. We quantified the length distribution of sRNA reads by normalizing to genome mapping reads reported as reads per million (rpm). Juvenile sample J1 was sequenced twice (correlation for technical replicates: Pearson’s ρ = 0.99). Supplementary Table 1 reports small RNA sequencing statistics.
Long RNA library construction and analysis
Total RNA was extracted from frozen tissues (mirVana miRNA isolation kit; Thermo Fisher, AM1560). Library construction was as described48 except rRNA was removed using a pool of antisense oligonucleotides. Briefly, 186 different antisense DNA oligonucleotides (AS oligos) complementary to rRNA sequences were designed and pooled (0.05 μM each;49,50). Total RNA (1 μg) was mixed with 1 μl pooled rRNA AS oligos in the presence of 100 mM Tris-HCl (pH 7.4) and 200 mM NaCl at 95°C and cooled to 22°C (−0.1°C/sec) then held at that temperature for 5 min. The DNA:RNA hybrids were incubated with 10 U Thermostable RNase H (Epicentre, H39500) in 50 mM Tris-HCl (pH 7.4), 0.1 M NaCl, and 20 mM MgCl2 at 45°C for 30 min. Next, samples were incubated with 4 U Turbo DNase (Thermo Fisher, AM2238) at 37°C for 20 min, then purified with RNA Clean & Concentrator-5 (Zymo Research, R1015) to isolate RNA >200 nt. RNA libraries were sequenced as 79 nt paired-end reads (NextSeq500; Illumina).
rRNA reads were removed using Bowtie 2.2.5 with default parameters47 and the remaining reads mapped to human genome (hg19) using STAR 2.3 (Ref. 51). Duplicate reads were removed using SAMtools 1.8 (Ref. 52). Finally, mapped reads were assigned to protein-coding genes, lncRNAs, or piRNA genes using HTSeq 0.9.1 (Ref. 53). Transcript abundance was reported as reads per million uniquely mapped reads per thousand nucleotides mapped (RPKM). Unsupervised hierarchical clustering based on the abundance of protein-coding and non-coding transcripts was performed as described54. Supplementary Table 1 reports RNA sequencing statistics.
PAS-seq library construction and analysis
Total RNA was extracted from frozen tissues using mirVana miRNA isolation kit (Thermo Fisher, AM1560). Poly(A) Site Sequencing (PAS-seq) library construction was performed as described55. Briefly, poly(A)+ RNA was extracted from total RNA using Dynabeads mRNA purification beads (Ambion, 61006), then fragmented to 60–80 nt at 70°C for 7 min by chemical hydrolysis. Fragmented RNA was reverse transcribed using barcoded oligo(dT) oligonucleotides containing sites for forward and reverse Illumina sequencing primers separated by a hexa-ethyleneglycol (Sp18) linker. The oligonucleotides contained unique molecular identifiers (UMIs), to enable duplicate removal, preceded by 5′p-GG for efficient cDNA circularization. cDNA was further circularized using Circligase ssDNA ligase (Illumina, CL4111K) at 60°C for 4 h. Following cDNA circularization, fourteen cycles of PCR amplification was performed using KAPA HiFi library amplification kit (Kapa Biosystems, KK2611), and libraries sequenced as 79 nt single-end reads (NextSeq500; Illumina).
Poly(A) tracts and residual adaptor sequences were trimmed from the 3′ ends of reads passing quality checks. Reads were mapped to human genome hg19 using Bowtie 2.2.5 (Ref. 47) with parameters –m –best –p4. Poly(A) sites were determined as described55. We filtered out internal priming events caused by genomic poly(A) tracts using a naïve Bayes classifier approach56. Supplementary Table 1 reports poly(A) site sequencing statistics.
Cap-seq library construction and analysis
Total RNA was extracted from frozen tissues using mirVana miRNA isolation kit (Thermo Fisher, AM1560). Cap-seq libraries were prepared as described with slight modifications57. Briefly, following rRNA removal, poly(A)+ RNA was extracted from total RNA using Dynabeads mRNA purification beads (Ambion, 61006). RNA was heated to 68°C for 5 min, then immediately chilled on ice. To prevent 5′ phosphorylated RNA entering the library, RNA samples were treated with 10U Antarctic Phosphatase (NEB, M0289) at 37°C for 1h. Antarctic Phosphatase was inactivated at 70°C for 5 min, and then RNA extracted with phenol:chloroform:isoamyl alcohol (25:24:1; pH 6.7). The 5′ cap was removed using 20U Tobacco Decapping Enzyme (Enzymax, LLC, 87) in the presence of 1 mM MnCl2 at 37°C for 2 h followed by phenol:chloroform:isoamyl alcohol extraction. After 5′ ligation, RNA was reverse transcribed using a primer containing 5′-NNN NNN NN-3′ followed by a 3′ adaptor sequence, then amplified by PCR for two cycles with left (5′-CTA CAC GTT CAG AGT TCT ACA GTC CGA-3′) and right (5′-GCC TTG GCA CCC GAG AAT TCC A-3′) primers using 2× NEBNext Q5 High-Fidelity DNA Polymerase Master Mix (NEB; M0541). PCR products 200–400 bp long were purified from a 1% agarose gel run in TAE buffer (QIAquick; QIAGEN, 28706). After ten additional PCR cycles, Cap-seq libraries were sequenced as 79 nt single-end reads (NextSeq500; Illumina). Supplementary Table 1 reports Cap-seq statistics.
Chromatin immunoprecipitation and sequencing
Chromatin Immunoprecipitation (ChIP) was performed as described14 except using frozen tissue. Briefly, frozen tissue was cut into small pieces on dry ice and transferred to 1.7 ml tubes containing ice-cold PBS. Samples were cross-linked with 2% (w/v) formaldehyde at room temperature for 30 min using tumbling end-over-end. Thereafter, fixed tissues were crushed in ChIP lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1) using 40 strokes with a “B” pestle in a Dounce homogenizer (Kimble-Chase, Vineland, USA), then sonicated (Ultrasonicator, Covaris, E220) to shear the chromatin to 150–200 bp. Lysate was then diluted 1:10 with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-1001.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl) and immunoprecipitated using 5.5 μg anti-A-MYB (Sigma, HPA008791) or 4.5 μg anti-H3K4me3 (Abcam, ab8580). DNA was extracted from the immunoprecipitate with phenol:chloroform:isoamyl alcohol (25:24:1) (pH 8), and libraries generated as described48, and sequenced as 79 nt paired-end reads (NextSeq500, Illumina).
Raw reads were mapped to the genome using Bowtie 2.2.5 (Ref. 47) using the parameter –very-sensitive. Duplicate reads were removed using SAMtools 1.8 (Ref. 52). We used model-based analysis of ChIP-seq (MACS 1.1.2; Ref. 58) with parameter −q 0.01 to detect A-MYB peaks that were significantly enriched relative to input (1,296 genomic regions; false discovery rate [FDR] < 0.05). Supplementary Table 1 reports ChIP-seq statistics.
Human piRNA precursor transcript annotation
After mapping reads to the genome using STAR, we used StringTie 1.3.3 (Ref. 59) to perform genome-based transcriptome assembly. Gene annotations were from Ensembl 90. One juvenile (two technical replicates) and seven adult testes samples were used individually for transcriptome assembly, and then each assembly was merged into a final transcript assembly using the merge function with parameter −m 100 −F 1 −T 1. We retained those transcripts both present in more than one sample and producing ≥100 rpm piRNAs in more than one sample.
Human piRNA gene nomenclature
As in mouse14, human piRNA-producing genes overlapping ≥50% with annotated protein-coding genes were named pi-(GeneName); non-coding piRNA genes were named according to their genomic location and piRNA abundance: (chromosome)-(chromosome segment)-(piRNA rpm).
piRNA gene annotation in other species
We defined piRNA genes in rhesus, marmoset, rat, opossum and platypus as previously described6 using sRNA-seq data from testis downloaded from GEO (rhesus, GSE40499; marmoset, GSE52927; rat GSE19054; opossum, GSE40499; platypus, GSE40499). Briefly, we scanned the whole genome using 5 kb sliding windows tiled every 1000 nt to capture the regions mapped by 24–32 nt long sRNA at a density of more than 100 rpm. We then merged 5 kb windows within 20 kb of each other. We defined two types of genomic sources of piRNAs: (1) piRNA-producing genomic regions that overlap an annotated protein-coding gene and (2) piRNA-producing sequences unlikely to encode protein, i.e., non-coding RNA-producing genes. Genomic sources of piRNAs overlapping a protein-coding gene were named pi-(GeneName). For piRNA clusters derived from non-coding sequences, clusters were named pi-(suffix), where the suffix indicates the rank order of the genomic coordinates of the piRNA cluster.
Synteny analysis
Synteny analysis was performed using liftOver from UCSC toolkit. Human (hg19) piRNA genes were aligned to rhesus (rheMac8), marmoset (calJac3), mouse (mm10), rat (rn6), opossum (monDom5) and platypus (ornAna1) genomes with the command liftOver-minMatch=0.1. piRNA-producing genes which were successfully identified in another genome were considered to be syntenic. Loci producing piRNAs from a syntenic location in other species were considered to be evolutionarily conserved.
Calculation of derived-allele frequency and nucleotide diversity (π value)
Vcf files of 2,504 human genomes containing the derived allele frequency (DAF) of each single nucleotide polymorphism (SNP) were downloaded from the 1000 Genomes Project Consortium34. In brief, the 1000 Genomes Project Consortium considers those alleles that are different from the ancestral allele—compared to chimpanzee—as derived alleles. We then took those alleles in non-CpG autosomal-chromosome regions and calculated the frequency of derived alleles in each human genome. Mean nucleotide diversity60 (π) is defined as 2 × DAF × (1 – DAF).
Statistics
Statistics were calculated using R console (https://www.rstudio.com); graphs were generated using R console and Igor Pro 6.37. For box plots, 95% confidence intervals were calculated using Wilcoxon rank-sum test.
Extended Data
Extended Data Fig. 1. Strategy for defining human post-natal piRNA genes.
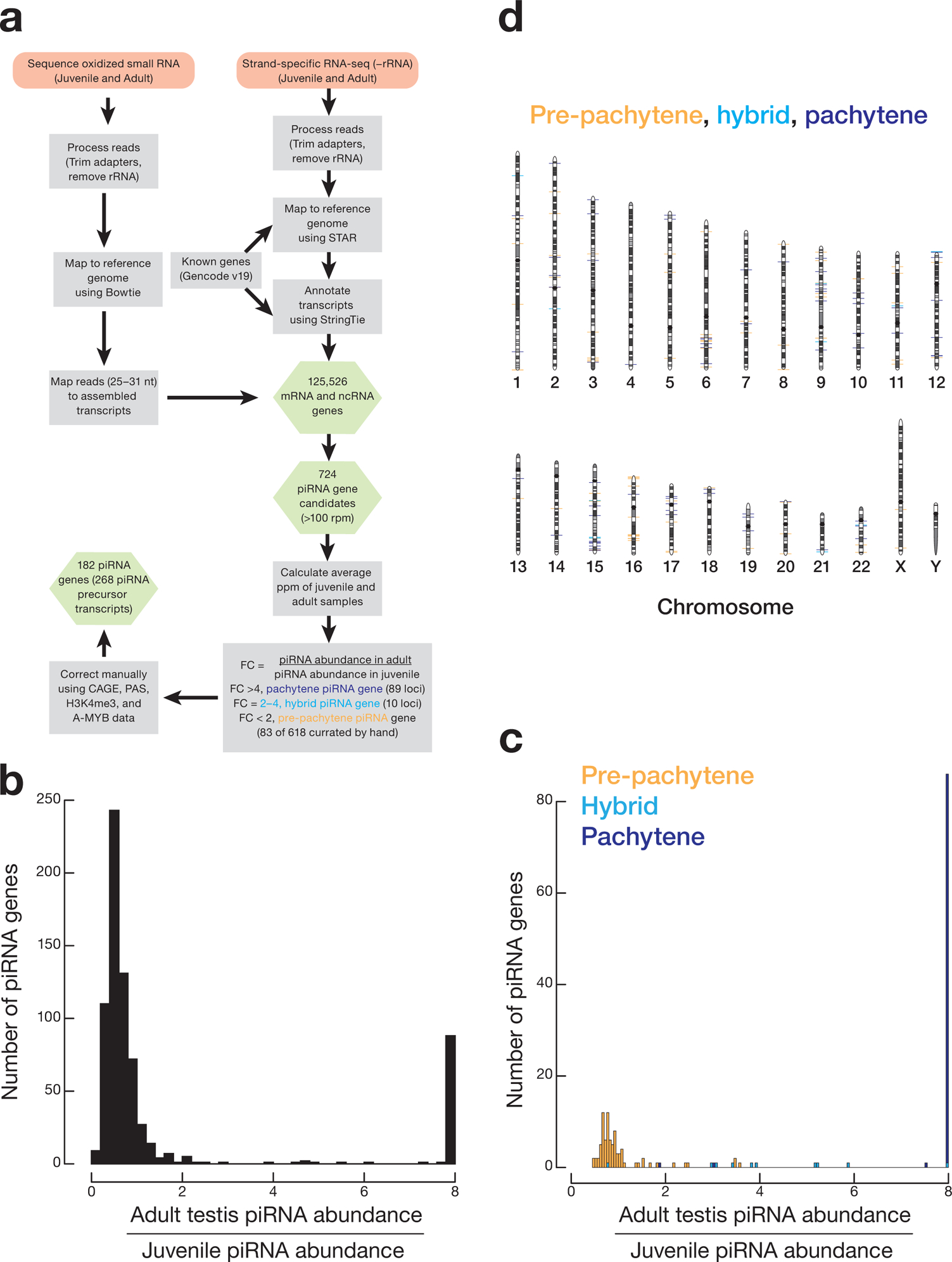
a) Strategy for transcriptome assembly, identification of piRNA precursor transcripts, and annotation of genomic loci. Pink rectangles: input for analyses; gray rectangles: bioinformatic tools. Green hexagons: output from analyses. (b, c) Histograms showing the change in the abundance of piRNAs produced from piRNA genes between juvenile and adult samples. The analysis was conducted twice: once with the set of samples used to define the piRNA genes (b) and then with an independent set of samples to validate the annotations (c). (d) Genomic positions of 182 human piRNA-producing loci on the 22 autosomal chromosomes. Two pre-pachytene piRNA genes were identified on the X chromosome.
Extended Data Fig. 2. Examples of human post-natal piRNA genes.

(a) An exemplary pre-pachytene piRNA gene. (b) uni- and bidirectional pachytene piRNA genes. Human piRNA cluster boundaries annotated in Ref. 10 (dark green) and Ref. 11 (purple).
Extended Data Fig. 3. Analyses of transcription start sites (TSS) and transcript 3′ ends for human piRNA genes.
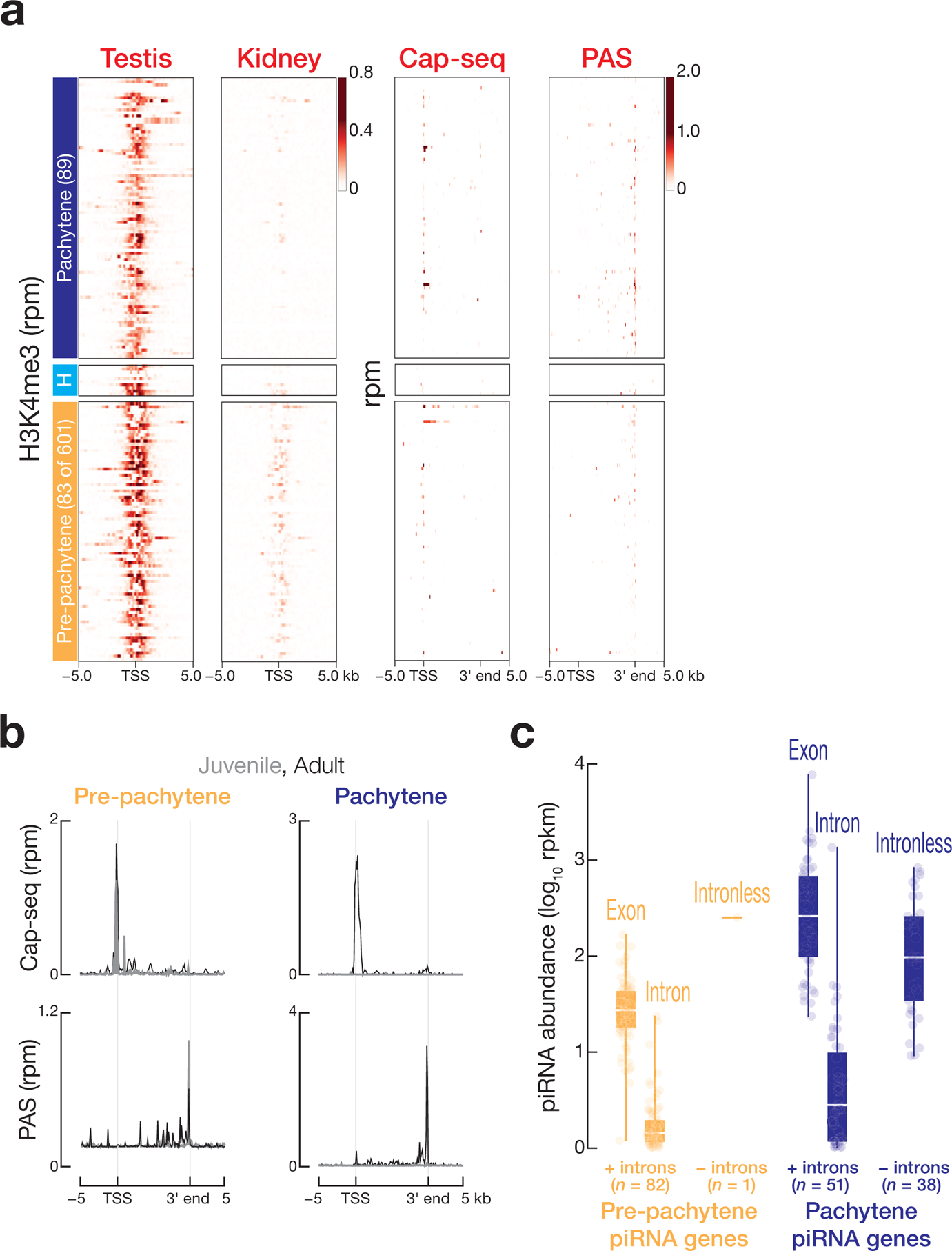
(a) Heatmaps displaying the H3K4me3 ChIP-seq signal around the transcription start sites of piRNA-producing genes from adult testis and the results of Cap-seq and PAS-seq around TSS and transcript 3′ ends for piRNA-producing genes from adult testes. Kidney serves as a negative control. Data are reported as uniquely mapping reads per million reads (rpm). (b) Metagene plot of Cap-seq and PAS-seq signals 5 kb upstream of the TSS and 5 kb downstream of the transcript 3′ ends of pre-pachytene and pachytene piRNA genes for juvenile and adult testis samples. Graphs report trimmed mean (i.e., lowest and highest 5% removed). (c) piRNA density per 1 kb within exons and introns of the pre-pachytene and pachytene piRNA genes defined by our analysis.
Extended Data Fig. 4. Three classes of human post-natal piRNA genes.
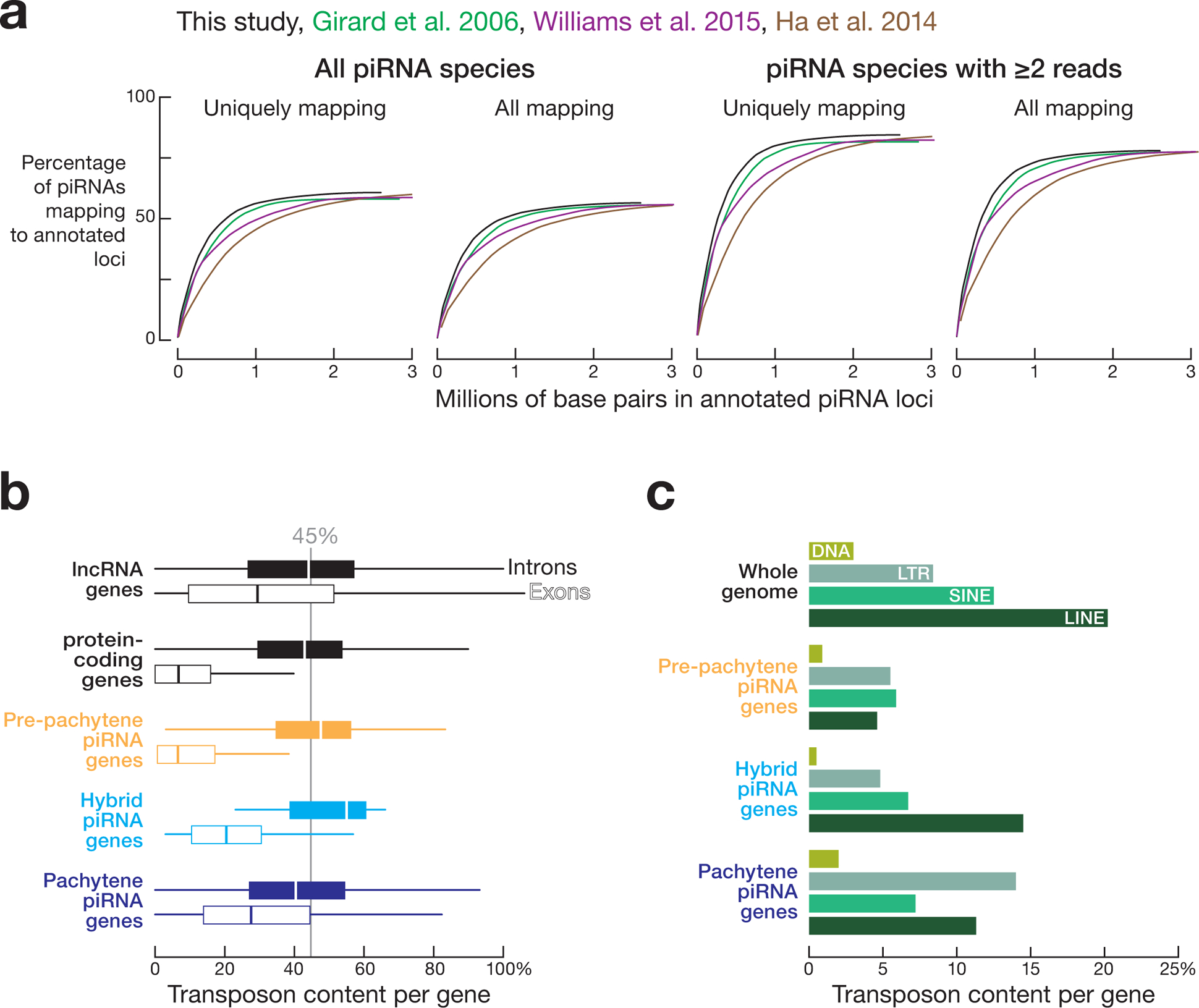
(a) Cumulative distributions of the percentage of 25–31 nt long piRNAs explained by the length of annotated genomic sequence for the piRNA-producing loci defined here or previously10–12. (b) Percentage of transposon sequences present in piRNA-producing, protein-coding, and lncRNA genes. Gray line indicates the transposons content of the entire human genome. (c) Percentage of different classes of transposons within the piRNA-producing genes.
Extended Data Fig. 5. Characterization of the mouse repro9 mutation within exon 6 of A-Myb.
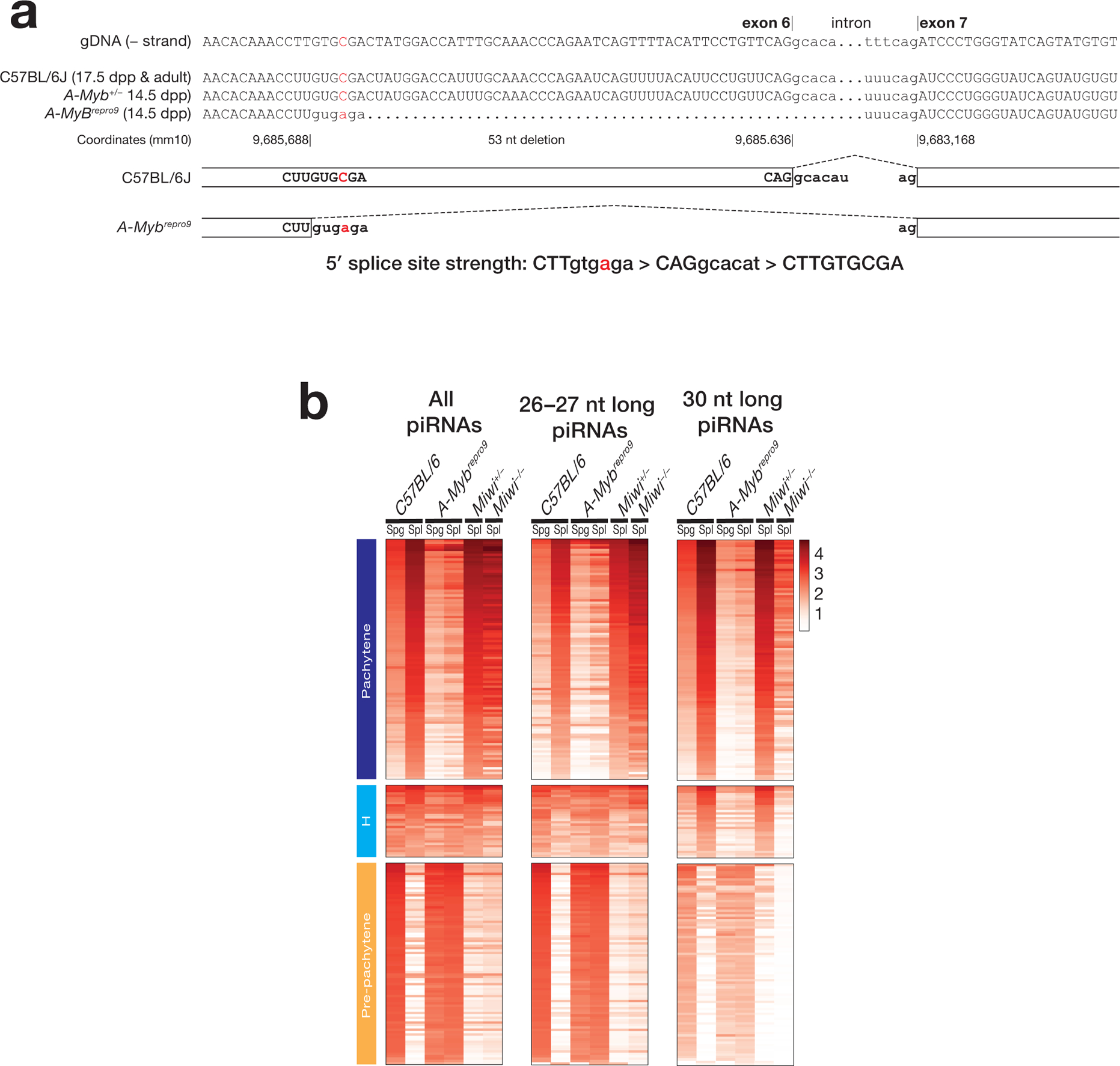
(a) The repro9 mutation creates a stronger 5′ splice site within exon 6 of A-Myb, leading to a truncated mRNA. Splice site strength was determined using MaxEntScan13. (b) Heatmaps of the relative abundance of mouse piRNAs mapping to previously defined piRNA genes14. piRNA abundance was normalized to the total number of mapped reads. Spg: spermatogonia; SpI: primary spermatocytes.
Extended Data Fig. 6. Three groups of adult testes defined by length distribution of total piRNAs and A-MYB and HIWI expression.

(a) The abundance of piRNAs was normalized to the total number of genome-mapping reads. Relative protein abundance of A-MYB (b) and HIWI (c) in adult testis samples. ACTIN serves as a loading control, while mouse A-Mybrepro9 and Miwi−/− mutant testis lysates provide negative controls. Each lane contained 75 μg protein of testis lysate.
Extended Data Fig. 7. Molecular characterization of group 3 testes.
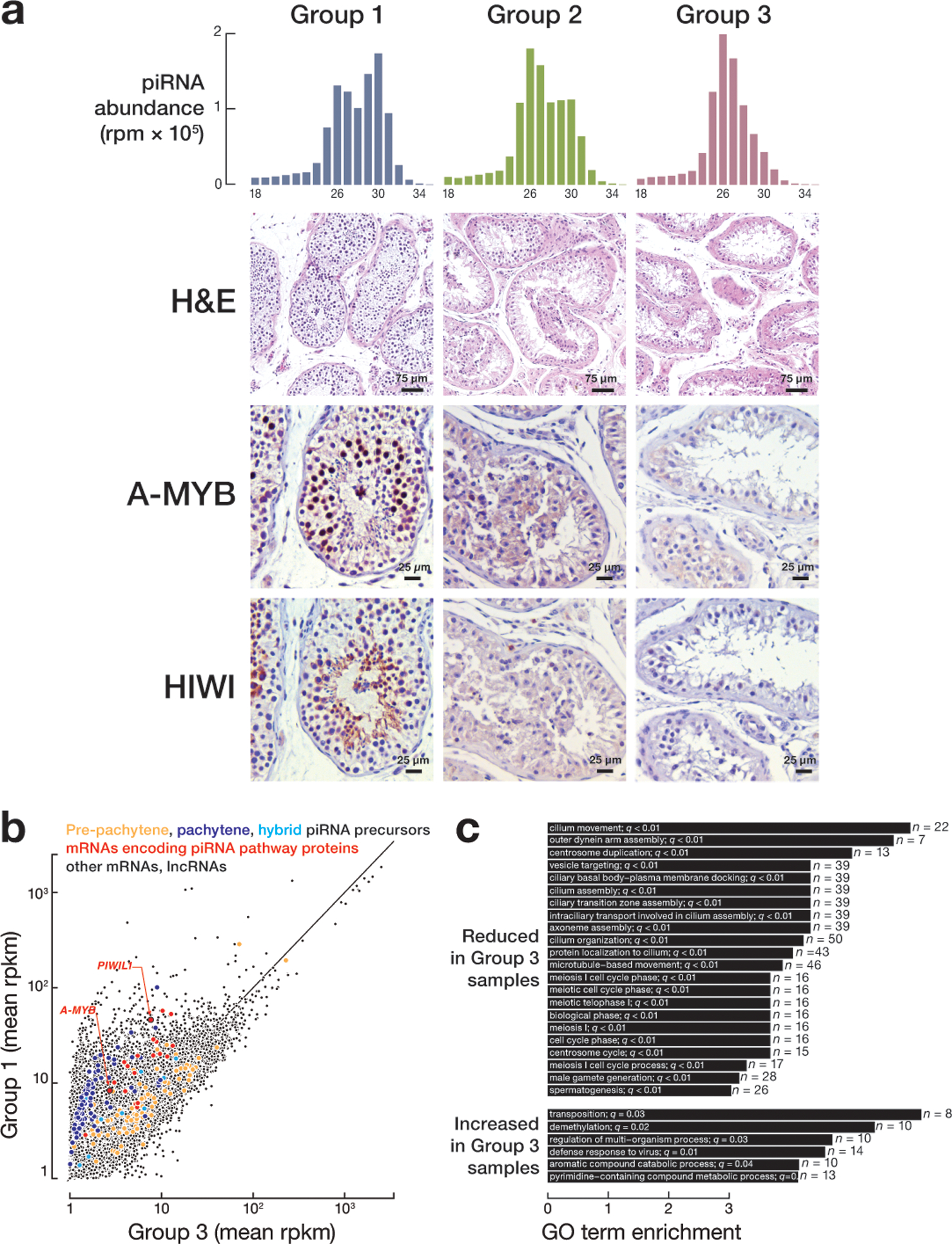
(a) piRNA length distribution, hematoxylin and eosin (H&E) stained testis sections, and immunohistochemical detection of A-MYB and HIWI for representative samples from groups 1, 2, and 3. (b) Scatter plot of steady-state transcript abundance of transcripts in group 1 versus group 3 testes. Each dot represents mean abundance of an mRNA. (c) Gene ontology analysis of mRNAs detected in group 3 samples and whose abundance changed >3-fold (FDR <0.05) compared to group 1.
Extended Data Fig. 8. Three classes of human post-natal piRNA genes expressed by group 1, group 2, and juvenile testis samples.

(a) Histogram shows the change in the abundance of piRNAs produced from piRNA genes between juvenile and healthy adult testis samples (groups 1 and 2). (b) MA plot showing change in mean piRNA abundance comparing healthy adult (groups 1 and 2) to juvenile testis samples for 182 annotated piRNA-producing loci.
Extended Data Fig. 9. Comparative analysis of human piRNA-producing genes.
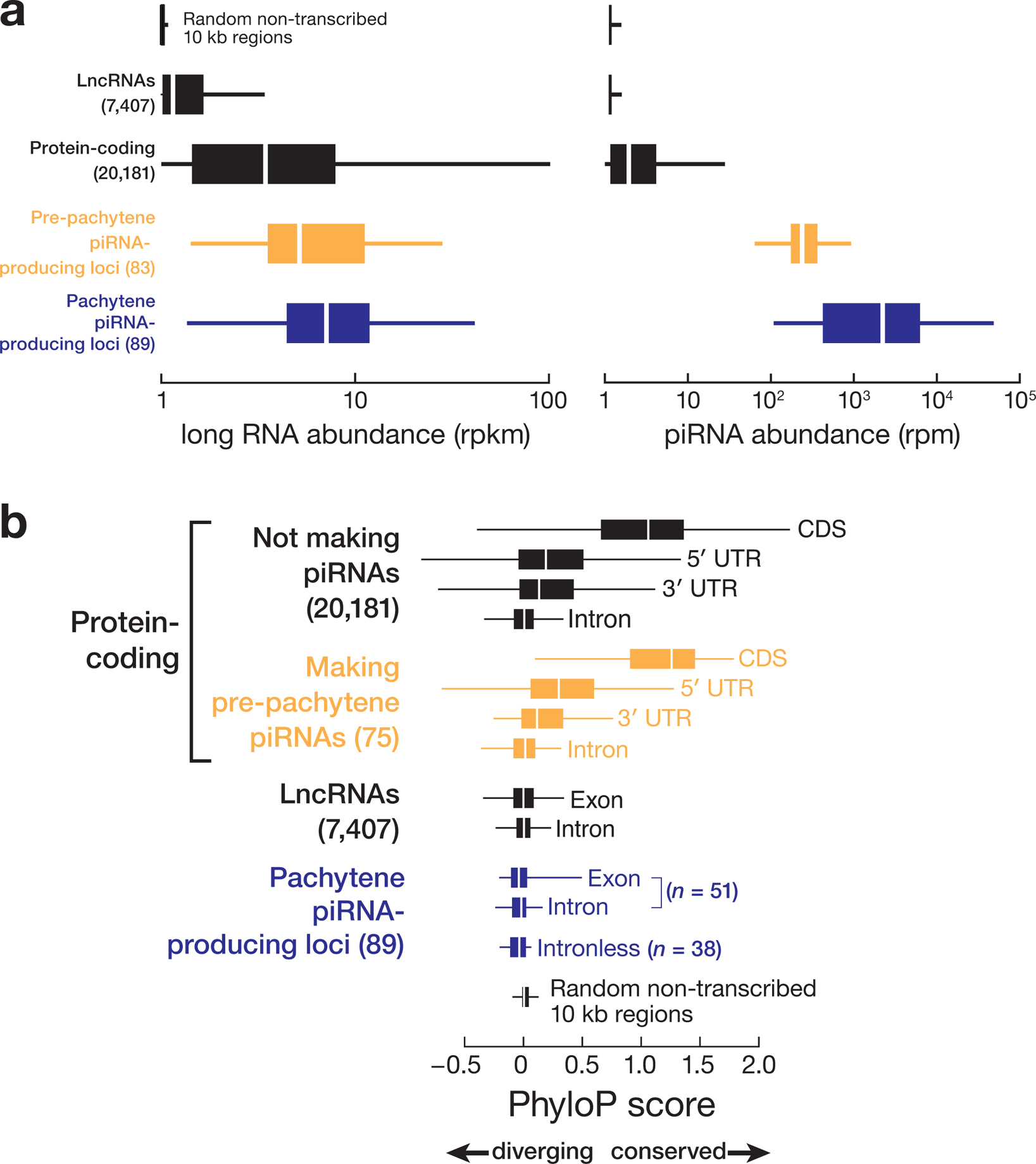
(a) Long RNA and piRNA abundance for different genomic features including piRNA-producing genes; protein-coding genes; lincRNA genes; and 22,604 randomly selected, 10 kb, non-transcribed genomic regions. (b) DNA sequence conservation of different genomic features for 46 eutherian mammals calculated using PhyloP15. The 22,604 randomly selected, 10 kb, non-transcribed genomic regions, which do not produce piRNAs, provide a background control.
Extended Data Fig. 10. mRNA abundance of transcripts from orthologous genes that produce pre-pachytene piRNAs in humans and evolutionary classes of human pachytene piRNA genes.

(a) Transcript abundance in representative primate, rodent, marsupial, and bird species for mRNAs that produce piRNAs in placental mammals and for mRNAs expressed in other species but that make piRNAs only in humans. Expression data for species other than human testis was obtained from EMBL-EMI Expression Atlas. (b) An exemplary pre-pachytene piRNA gene that is syntenic across other species, but piRNA source in placental mammals only, in primates only, in humans only. (c) Abundance of piRNAs from the syntenic locations for human pachytene piRNA loci in other animals.
Supplementary Material
Acknowledgements
We thank Darryl Conte and members of the Zamore laboratory for discussions and comments on the manuscript, and Kyle Orwig for providing testis specimens. The opossum silhouette was drawn by Sarah Werning (https://creativecommons.org/licenses/by/3.0/), and the rat silhouette was drawn by Rebecca Groom (https://creativecommons.org/licenses/by-sa/3.0/). The silhouettes have not been altered in any way.
Funding
This work was supported in part by National Institutes of Health grants R37GM062862 (P.D.Z.) and P01HD078253 (Z.W. and P.D.Z.)
Footnotes
Data availability
Sequencing data are available from the National Center for Biotechnology Information Sequence Read Archive using accession number PRJNA506245 and from the Gene Expression Omnibus using accession number GSE135791.
Competing interests statements
The authors declare no competing interests.
References
- 1.Aravin A et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442, 203–207 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Girard A, Sachidanandam R, Hannon GJ & Carmell MA A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442, 199–202 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Grivna ST, Beyret E, Wang Z & Lin H A novel class of small RNAs in mouse spermatogenic cells. Genes Dev 20, 1709–1714 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lau NC et al. Characterization of the piRNA complex from rat testes. Science 313, 363–367 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Vagin VV et al. A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313, 320–324 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Brennecke J et al. Discrete Small RNA-Generating Loci as Master Regulators of Transposon Activity in Drosophila. Cell 128, 1089–1103 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Fu Y et al. The genome of the Hi5 germ cell line from Trichoplusia ni, an agricultural pest and novel model for small RNA biology. eLife 7, e31628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis SH et al. Pan-arthropod analysis reveals somatic piRNAs as an ancestral defence against transposable elements. Nat Ecol Evol 2, 174–181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ozata DM, Gainetdinov I, Zoch A, O’Carroll D & Zamore PD PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet 20, 89–108 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Aravin AA et al. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31, 785–799 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuramochi-Miyagawa S et al. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 22, 908–917 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K & Hannon GJ Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 316, 744–747 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Goh WS et al. piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Genes Dev 29, 1032–1044 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li XZ et al. An Ancient Transcription Factor Initiates the Burst of piRNA Production during Early Meiosis in Mouse Testes. Mol Cell 50, 67–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu PH, Fu Y, Cecchini K, Ozata DM, Weng Z, Zamore PD. An Evolutionarily Conserved piRNA-producing Locus Required for Male Mouse Fertility. bioRxiv (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vourekas A et al. Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nat Struct Mol Biol 19, 773–781 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang P et al. MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res 25, 193–207 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homolka D et al. PIWI Slicing and RNA Elements in Precursors Instruct Directional Primary piRNA Biogenesis. Cell Rep 12, 418–428 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Xu M et al. Mice deficient for a small cluster of Piwi-interacting RNAs implicate Piwi-interacting RNAs in transposon control. Biol Reprod 79, 51–57 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Gainetdinov I, Colpan C, Arif A, Cecchini K & Zamore PD A Single Mechanism of Biogenesis, Initiated and Directed by PIWI Proteins, Explains piRNA Production in Most Animals. Mol Cell 71, 775–790.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolcun-Filas E et al. A-MYB (MYBL1) transcription factor is a master regulator of male meiosis. Development 138, 3319–3330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ha H et al. A comprehensive analysis of piRNAs from adult human testis and their relationship with genes and mobile elements. BMC Genomics 15, 545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams Z et al. Discovery and Characterization of piRNAs in the Human Fetal Ovary. Cell Rep 13, 854–863 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Jan SZ et al. Unraveling transcriptome dynamics in human spermatogenesis. Development 144, 3659–3673 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turner JM Meiotic sex chromosome inactivation. Development 134, 1823–1831 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Carmell MA et al. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12, 503–514 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Saito K et al. Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev 20, 2214–2222 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aravin AA et al. The small RNA profile during Drosophila melanogaster development. Dev Cell 5, 337–350 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Meistrich ML Effects of chemotherapy and radiotherapy on spermatogenesis in humans. Fertil Steril 100, 1180–1186 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng W & Lin H Miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev Cell 2, 819–830 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Katzen AL, Kornberg TB & Bishop JM Isolation of the proto-oncogene c-myb from D. melanogaster. Cell 41, 449–456 (1985). [DOI] [PubMed] [Google Scholar]
- 32.Lipsick JS et al. Functional evolution of the-myb oncogene family. Blood Cells Mol Dis 27, 456–458 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Kumar S, Stecher G, Suleski M & Hedges SB TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol Biol Evol 34, 1812–1819 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Auton A et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reuter M et al. Miwi catalysis is required for piRNA amplification-independent LINE1 transposon silencing. Nature 480, 264–267 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Wang W et al. Slicing and Binding by Ago3 or Aub Trigger Piwi-Bound piRNA Production by Distinct Mechanisms. Molecular Cell 59, 819–830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andersen PR, Tirian L, Vunjak M & Brennecke J A heterochromatin-dependent transcription machinery drives piRNA expression. Nature 549, 54–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klattenhoff C et al. The Drosophila HP1 homolog Rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell 138, 1137–1149 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohn F, Sienski G, Handler D & Brennecke J The Rhino-Deadlock-Cutoff Complex Licenses Noncanonical Transcription of Dual-Strand piRNA Clusters in Drosophila. Cell 157, 1364–1379 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Zhang Z et al. The HP1 Homolog Rhino Anchors a Nuclear Complex that Suppresses piRNA Precursor Splicing. Cell 157, 1353–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gainetdinov I, Skvortsova Y, Kondratieva S, Funikov S & Azhikina T Two modes of targeting transposable elements by piRNA pathway in human testis. RNA 23, 1614–1625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirano T et al. Small RNA profiling and characterization of piRNA clusters in the adult testes of the common marmoset, a model primate. RNA 20, 1223–1237 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soumillon M et al. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep 3, 2179–2190 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Bastos H et al. Flow cytometric characterization of viable meiotic and postmeiotic cells by Hoechst 33342 in mouse spermatogenesis. Cytometry A 65, 40–49 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Han BW, Wang W, Li C, Weng Z & Zamore PD Noncoding RNA. piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science 348, 817–821 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han BW, Wang W, Zamore PD & Weng Z piPipes: a set of pipelines for piRNA and transposon analysis via small RNA-seq, RNA-seq, degradome- and CAGE-seq, ChIP-seq and genomic DNA sequencing. Bioinformatics 31, 593–595 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Z, Theurkauf WE, Weng Z & Zamore PD Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence 3, 9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adiconis X et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods 10, 623–629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morlan JD, Qu K & Sinicropi DV Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS One 7, e42882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anders S, Pyl PT & Huber W HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Priness I, Maimon O & Ben-Gal I Evaluation of gene-expression clustering via mutual information distance measure. BMC Bioinformatics 8, 111 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ashar-Patel A et al. FLT1 and transcriptome-wide polyadenylation site (PAS) analysis in preeclampsia. Sci Rep 7, 12139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sheppard S, Lawson ND & Zhu LJ Accurate identification of polyadenylation sites from 3′ end deep sequencing using a naive Bayes classifier. Bioinformatics 29, 2564–2571 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Z, Bruno DP, Martens CA, Porcella SF & Moss B Genome-wide analysis of the 5′ and 3′ ends of vaccinia virus early mRNAs delineates regulatory sequences of annotated and anomalous transcripts. J Virol 85, 5897–5909 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pertea M et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keinan A, Mullikin JC, Patterson N & Reich D Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat Genet 39, 1251–1255 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.