

Abstract
Transcriptome compartmentalization by the nuclear membrane provides both stochastic and functional buffering of transcript activity in the cytoplasm, and has recently been implicated in neurodegenerative disease processes. Although many mechanisms regulating transcript compartmentalization are also prevalent in brain development, the extent to which subcellular localization differs as the brain matures has yet to be addressed. To characterize the nuclear and cytoplasmic transcriptomes during brain development, we sequenced both RNA fractions from homogenate prenatal and adult human postmortem cortex using poly(A)+ and Ribo-Zero library preparation methods. We find that while many genes are differentially expressed by fraction and developmental expression changes are similarly detectable in nuclear and cytoplasmic RNA, the compartmented transcriptomes become more distinct as the brain matures, perhaps reflecting increased utilization of nuclear retention as a regulatory strategy in adult brain. We examined potential mechanisms of this developmental divergence including alternative splicing, RNA editing, nuclear pore composition, RNA-binding protein motif enrichment, and RNA secondary structure. Intron retention is associated with greater nuclear abundance in a subset of transcripts, as is enrichment for several splicing factor binding motifs. Finally, we examined disease association with fraction-regulated gene sets and found nuclear-enriched genes were also preferentially enriched in gene sets associated with neurodevelopmental psychiatric disorders. These results suggest that although gene-level expression is globally comparable between fractions, nuclear retention of transcripts may play an underappreciated role in developmental regulation of gene expression in brain, particularly in genes whose dysregulation is related to neuropsychiatric disorders.
Human brain development involves precisely timed gene expression changes across the life span, the largest occurring at the prenatal to postnatal transition (Colantuoni et al. 2011; Jaffe et al. 2015; Li et al. 2018). One factor governing these changes is RNA compartmentalization by the nuclear membrane into nuclear and cytoplasmic fractions. A snapshot of each compartment's composition captures factors of both chance and purpose. For instance, because most splicing occurs cotranscriptionally (Djebali et al. 2012; Tilgner et al. 2012), pre-mRNA and longer genes that take more time to be transcribed/exported are often overrepresented in nucleus compared to cytoplasm (Pandya-Jones et al. 2013). Recent studies have also highlighted the role of the nuclear membrane as a transcriptional noise buffer, filtering stochastic bursts of gene expression from the cytoplasm by retaining selected mature mRNA transcripts in the nucleus (Bahar Halpern et al. 2015). Still, cells can also use nuclear retention to regulate the timing of cytoplasmic activity of a transcript (Mauger et al. 2016) as well as perform quality control by sequestering aberrant transcripts in the nucleus and targeting them for degradation.
Many of these RNA trafficking mechanisms are particularly prevalent in brain cells and some have been found to play a role in brain development. For example, alternative splicing—particularly intron retention (IR)—has been shown to regulate RNA localization as a means to suppress lowly and aberrantly expressed transcripts (Boutz et al. 2015). IR is common in neuronal lineages and serves to down-regulate genes involved in other lineage fates during neuronal differentiation (Yap et al. 2012; Wong et al. 2013; Braunschweig et al. 2014). RNA editing is also developmentally regulated in human brain, with a subset of editing sites associated with neuronal maturation (Hwang et al. 2016). In at least one example, RNA editing was shown to regulate activity-dependent nuclear transcript retention, although global characterization of RNA editing patterns by subcellular fraction shows that RNA editing is not broadly necessary for nuclear retention (Prasanth et al. 2005; Chen 2013). Likewise, nuclear pore composition is also developmentally dynamic, and the RNA-binding proteins (RBPs) that regulate many of the aforementioned processes and shepherd RNA from the nucleus to the cytoplasm are also under developmental control (D'Angelo et al. 2012; Gardiner et al. 2015).
Although nuclear and cytoplasmic transcriptomes have been assessed in vitro, subcellular fractions have not yet been characterized in human cortical tissue and not across age dimensions. Given the increasingly frequent use of nuclear RNA in single cell– and cell population–based studies of human brain as a result of the difficulty of dissociating frozen postmortem brain tissue to a single-cell suspension (Lake et al. 2016), understanding the compositional differences between RNA compartments over human brain development would help inform future studies using nuclear RNA without a comparable cytoplasmic fraction. Given also that disruption of proper nucleocytoplasmic transport of proteins and RNA is increasingly implicated in normal aging as well as neurodegenerative disorders such as Huntington's disease and amyotrophic lateral sclerosis (Mertens et al. 2015; Zhang et al. 2015; Gasset-Rosa et al. 2017), and that mechanisms affecting RNA localization are related to developmental processes particularly in brain cells, characterizing the dynamics of RNA localization between fractions may help clarify what role if any nuclear sequestration of transcripts may play in the etiology of brain disorders with developmental components. To address these questions, we have characterized the nuclear and cytoplasmic transcriptomes in early developing and mature human prefrontal cortex.
Results
We sequenced nuclear and cytoplasmic RNA isolated from three prenatal and three adult human dorsolateral prefrontal cortex (DLPFC) samples, constructing libraries using two strategies for depleting ribosomal RNA, the dominant species in total RNA [poly(A)+ and Ribo-Zero] (Supplemental Fig. S1; Supplemental Table S1). Together, poly(A)+ library preparation (selecting polyadenylated transcripts via a pull-down step) and Ribo-Zero library preparation (using an rRNA depletion step) capture the transcriptomic diversity in these compartments attributable to their respective preferences for mature mRNA and nonpolyadenylated transcripts (e.g., ncRNA or pre-mRNA) (Supplemental Fig. S2A; Cui et al. 2010; Li et al. 2014; Sultan et al. 2014). In total, we profiled 43,610 genes that were expressed across the eight groups of samples (Adult/Cytoplasm, Adult/Nucleus, Prenatal/Cytoplasm, and Prenatal/Nucleus with both library types) (Supplemental Fig. S1).
The quality of fractionation was confirmed by determining that genes known to localize either to nucleus (MALAT1) or cytoplasm (ACTB) (Bahar Halpern et al. 2015) were significantly enriched in the appropriate compartment (P-value adjusted by false discovery rate [FDR] < 0.01) (Supplemental Fig. S2B), although prenatal samples showed less enrichment for these genes than adult samples (t = −10.4, P = 2.4 × 10−4 and t = 12.8, P = 1 × 10−4 for ACTB and MALAT1 in adult, versus t = −4.4, P = 3.4 × 10−3 and t = 2.9, P = 1.7 × 10−2 in prenatal, respectively). A similar RNA amount was collected from the nuclear and cytoplasmic fractions in prenatal and adult samples, also suggesting adequate fractionation across ages (FDR > 0.05) (Supplemental Fig. S2C). Further, comparing expression patterns between fractions to previously available fractionated data from ENCODE in the top 296 genes differentially expressed across compartments in ENCODE showed strong agreement in localization between groups, although as expected, Ribo-Zero libraries showed larger differences between the fractions (Supplemental Fig. S2D).
Developmental gene expression changes in human cortex are similarly detectable in nuclear and cytoplasmic RNA
We first defined the differences between subcellular transcriptomes and replicated many previously described characteristics (Bhatt et al. 2012; Djebali et al. 2012; Solnestam et al. 2012; Tilgner et al. 2012; Zaghlool et al. 2013; Bahar Halpern et al. 2015; Reddy et al. 2017). Genes significantly greater expressed in nucleus were overall longer than genes more abundant in cytoplasm, perhaps because of the longer temporal requirement for transcription and diffusion through the nuclear pore (Supplemental Fig. S2E). The proportion of reads aligning to introns was greater in nucleus than cytoplasm in both poly(A)+ and Ribo-Zero samples (t > 4.7, FDR < 6.0 × 10−3), indicating a greater proportion of immature pre-mRNA or unannotated transcripts (Supplemental Fig. S2F). Although the majority (83.5%) of all fraction-regulated genes were protein-coding (Supplemental Fig. S2G), a larger proportion of genes enriched in the nuclear fraction were noncoding than those enriched in the cytoplasm (OR = 0.25, P = 2.2 × 10−16). Because Ribo-Zero libraries do not require polyadenylation for sequencing, a greater proportion of differentially expressed genes across fractions were noncoding in Ribo-Zero samples (Supplemental Fig. S2H,I).
Expression patterns were overall similar between fractions at the gene level. Principal component analysis showed that age and library type were the largest contributors to transcriptomic diversity, explaining 53% and 35% of the variance, respectively (Fig. 1A). Developmental expression changes (between prenatal and adult) were highly correlated between the fractions (ρ = 0.89, t = 335.8, P < 2.2 × 10−16) (Fig. 1B). A large portion (41%–63%) of genes differentially expressed between developmental stages were detected across all four fraction/library groups (Fig. 1C). Again, fractions in poly(A)+ samples were more similar than Ribo-Zero samples (i.e., 1244 vs. 545 developmental genes shared in Fig. 1C) owing to the expected larger proportion of immature and noncoding transcripts—two RNA species known to have different compartmental abundance—reflected in Ribo-Zero preparations.
Figure 1.

Developmental gene expression changes in human cortex are similarly detectable in nuclear and cytoplasmic RNA. (A) Principal component analysis. PC1 separates the samples by age and PC2 separates the samples by library type so that nuclear and cytoplasmic samples from the same donors cluster together. (B) Log2 fold change of gene expression across age measured in cytoplasmic and nuclear RNA [poly(A)+ library preparation shown only]. Red dots indicate FDR ≤ 0.05. (C) Venn diagram of differentially expressed genes by age [FDR ≤ 0.05; abs(log2 fold change) ≥ 1] measured in both fractions and library types. The total number of genes for each group is listed in parentheses.
Because the differentially expressed genes identified in these homogenate cortical samples likely reflect cell-type-specific dynamics, we next explored which cell types may underlie these differences in compartmentalization. Fraction-regulated genes were expressed in a variety of cell types: 25.9%–29.3% of both cytoplasmic and nuclear-enriched genes were highest expressed in neurons based on prior single-cell RNA-seq (Darmanis et al. 2015), whereas 18.1%–19.7% were highest expressed in astrocytes, 11.1%–12.7% in oligodendrocytes or their precursors, 4.0%–4.5% in microglia, and 4.6%–6.1% in epithelial cells (Supplemental Fig. S3A). At least in the major dichotomy of brain cell types—that between neurons and glia—there was no discernable relationship between expression patterns by fraction and by cell type (Supplemental Fig. S3B). Further cell-specific enrichment analysis (CSEA) of fraction-regulated genes identified enrichment of genes preferentially expressed in adult nuclei in rod cells (Benjamini–Hochberg corrected P-value [BH] = 3.8 × 10−12), cone cells (BH = 2.6 × 10−8), DRD1+ medium spiny neurons (BH = 3.7 × 10−9), and DRD2+ medium spiny neurons (BH = 2.8 × 10−11) (Supplemental Fig. S3C; Benjamini and Hochberg 1995). Nuclear overrepresentation of genes specific to these cells not found in DLPFC may reflect nuclear sequestration of gene expression inappropriate for translation in DLPFC cell types.
Prenatal and adult human cortex show distinct patterns of RNA localization across the nuclear membrane
Prenatal and adult cortex showed substantially different localization patterns across the nuclear membrane. We identified 1894 genes differentially expressed by fraction in adult, but only 40 genes differentially expressed in prenatal cortex in the poly(A)+ samples [abs(log2 fold change) ≥ 1; FDR ≤ 0.05] (Fig. 2A). This difference was also seen in Ribo-Zero samples (Supplemental Fig. S4). Unlike in adult samples in which differentially expressed genes by fraction (FDR ≤ 0.05) were equally likely to be nuclear or cytoplasmic, most fraction-regulated genes in prenatal samples were more abundant in the nuclear compartment (Supplemental Table S2). Despite fewer genes being differentially expressed by fraction in prenatal cortex, subcellular expression patterns were correlated between prenatal and adult (ρ = 0.60, t = 125.9, P < 2.2 × 10−16) (Fig. 2B).
Figure 2.

Prenatal and adult human cortices show distinct patterns of RNA localization across the nuclear membrane. (A) MA plots of prenatal and adult gene expression differences across fraction. (B) Log2 fold change of gene expression across fraction in adult poly(A)+ samples (x-axis) or prenatal poly(A)+ samples (y-axis). (C) Log2 fold change between fraction and age for six developmentally regulated gene sets: Colors indicate developmental expression direction, and columns indicate the fraction in which the developmental expression difference is detected. The y-axis shows the log2 fold change between fractions stratified by the age in which the fraction differences are measured, and the x-axis shows the log2 fold change between ages as measured in cytoplasm. Although these developmentally regulated gene sets are similar across prenatal fractions, down-regulated genes in adult are more sequestered in the nuclear compartment.
The association of fraction expression with changing developmental expression depended on the fraction in which the developmental changes were measured. This can be seen when exploring the differences in fraction expression in six developmentally regulated gene sets (Fig. 2C; Supplemental Table S3). Genes with changing developmental expression detected in both fractions showed no difference in expression between fractions. In contrast, genes with decreasing expression from prenatal to adult when measured in cytoplasm (i.e., developmentally decreasing expression in cytoplasm only) were more likely to be higher expressed in adult nucleus than adult cytoplasm (OR = 38.0, FDR = 1.9 × 10−20), whereas developmentally decreasing genes detected in nucleus were less likely to be expressed in adult nucleus compared with adult cytoplasm (OR = 0.071, FDR = 1.7 × 10−10). In other words, prenatally enriched genes may be relatively sequestered in the nuclear compartment in adult cortex to regulate the translation of these more fetal-relevant genes in adulthood. Likewise, genes with greater expression in adult than prenatal cytoplasm were less expressed in adult nucleus (OR = 0.038, FDR = 4.7 × 10−24), whereas developmentally increasing genes in nucleus were more likely to be greater expressed in adult nucleus than cytoplasm (OR = 19.3, FDR = 5.5 × 10−10). This pattern was not seen in prenatal fractions, because expression differences between fractions were more muted in this age. Taken together, these patterns suggest an inverted relationship between developmental gene expression changes and subcellular compartmentalization, in which genes with decreasing expression across development are progressively more likely to be retained in the adult nucleus.
A subset of introns regulated by fraction and age is associated with decreasing developmental expression and increasing nuclear localization
We sought to provide context for these gene expression patterns by examining several mechanisms associated with RNA transport across the nuclear membrane, beginning with alternative splicing. Although the proportion of reads spanning splice junctions was overall lower in nuclear compared to cytoplasmic samples in both library preparations (as expected), the proportion was not significantly different between the nuclear and cytoplasmic fractions in the poly(A)+ samples, as pre-mRNAs were overall depleted by poly(A)+ selection (t = −1.0, FDR = 0.34) (Fig. 3A). In contrast, in the Ribo-Zero samples the proportion of spliced alignments was much lower in nuclear than cytoplasmic samples, suggesting greater representation of pre-mRNA in these libraries (t = −4.3, FDR = 0.016) and overall relative reduction of mature RNA in the nucleus when depleting rRNA. All following splicing analyses were therefore performed in the poly(A)+ samples.
Figure 3.

Intron retention patterns in prenatal and adult human cortex associate with RNA distribution. (A) Proportion of junction reads per sample by group. Prenatal cortex had a higher proportion of splice junctions than adult, and although Ribo-Zero samples had significantly lower junction proportion in nuclear samples (FDR ≤ 0.05), poly(A)+ samples showed nonsignificantly different proportions between fractions. (B) Percentage of introns passing QC in all groups with an IR percentage above each threshold in at least one sample of each group. (C) The IR ratio of introns differentially retained by fraction when measured in adult and prenatal samples, plotted by gene expression measured as log(reads per kilobase per million mapped [RPKM] + 1). Samples are colored by their age:fraction group. (D) Overlap of genes containing introns that were differentially retained by fraction or age (FDR ≤ 0.05).
After surveying the diversity of splicing events in both RNA fractions and ages (Supplemental Fig. S5A,B), we focused on IR, examining 166,661–173,125 introns per sample that represented 15,345–15,389 unique genes (Fig. 3B). Greater IR was neither associated with the localization of a transcript by fraction overall (Supplemental Fig. S5C), nor correlated with developmental changes to gene expression overall (Supplemental Fig. S5D). However, a small subset of individual introns was differentially retained by fraction and age and showed distinct relationships with gene expression that suggested differing activities by these variables (Supplemental Fig. S5E). For instance, fraction-regulated introns in adult but not prenatal samples were negatively correlated with the expression of their host genes (ρ < −0.44, t < −5.0, FDR < 1.8 × 10−5) (Fig. 3C). One interpretation of this result is that increasing IR down-regulates expression in adult but not prenatal by signaling for nuclear degradation in adult of these genes. Indeed, increasing IR is positively correlated with nuclear enrichment of the intron-containing genes (ρ > 0.45, t > 3.9, FDR < 1.0 × 10−3) (Supplemental Fig. S5F). Differentially retained introns by age showed no relationship between IR and expression levels except in prenatal cytoplasm, where increasing IR was also correlated with decreasing expression (ρ = −0.69, t = −5.1, FDR = 1.2 × 10−4) (Supplemental Fig. S5G).
Genes including differentially retained introns by fraction (FDR ≤ 0.05) were more likely also to include significantly differentially retained introns by age (OR = 88.4, FDR = 6.5 × 10−4). Developmentally regulated introns were also more likely to be in genes that were significantly differentially expressed by fraction and vice versa (OR > 2.8, FDR < 0.032) (Fig 3D). These intron-containing genes—particularly those containing nuclear-enriched introns in adult and prenatal-enriched introns in nuclear RNA—were enriched for ontology terms associated with neuron-specific cellular compartments (FDR ≤ 0.05) (Supplemental Fig. S6). Although most introns are not associated with gene expression patterns by fraction or age, a subset showed decreasing expression of their host genes, genes that were often implicated in both fraction- and age-regulation as IR increased.
Assessment of RNA editing and nuclear pore components by fraction across cortical development
We next profiled RNA editing across fractions and ages, identifying 3064–5840 editing sites per sample and 25,051 unique sites (Supplemental Fig. S7A–E). Of these unique sites, 75.5% were A-to-I edited sites (appears as A:G or T:C in our sequencing data) (Fig. 4A). Of the 18,907 A-to-I edited sites, 1025 were shared by all four groups, representing 729 unique genes (Fig. 4B). To assess the relationships between subcellular localization and RNA editing, we first assessed editing rate changes across fraction and age in the 1025 A-to-I sites shared among the four groups. The distribution of P-values suggested that age but not fraction influenced overall editing rates (Supplemental Fig. S7F).
Figure 4.

RNA editing sites unique to an age/fraction group associate with expression levels. (A) Number of unique editing sites identified in each group stratified by editing context. (B) Venn diagram demonstrating the overlap of edited genes between groups of fraction and age samples. (C) Log2 fold change of expression by age in genes that include an editing site present in all adult but no prenatal samples (Adult Only) or all prenatal but no adult samples (Prenatal Only), as measured in cytoplasm and nucleus. (D) Log2 fold change of expression by fraction in adult samples (y-axis) in genes that include an editing site present in all cytoplasmic but no nuclear samples (right) or all nuclear and no cytoplasmic samples (left) in either adult (red) or prenatal (blue) samples.
We next focused on the sites found consistently in every sample of one of the four preceding poly(A)+ groups and never in a contrasting group, thinking that these sites may be functionally important to that group's compartment and age time point (1235 unique sites) (Supplemental Table S4; summarized in Supplemental Table S5). Genes containing a site in this subset had significantly more edited sites than other genes (t > 3.3, FDR < 6.9 × 10−3) and were higher expressed in the compartment or age containing the edited site. For instance, genes significantly greater expressed in adult samples were enriched for sites consistently and exclusively edited in adult samples (OR = 8.9, FDR = 6.3 × 10−19), whereas genes that were significantly greater expressed in prenatal cortex were enriched for sites consistently and exclusively edited in prenatal samples (OR = 25.9, FDR = 2.1 × 10−26) (Fig. 4C). The sites found in all nuclear but no cytoplasmic samples (i.e., the 159 in adult and 65 in prenatal nuclear RNA from Supplemental Table S5) were more likely to occur in genes significantly higher expressed in nuclear RNA than other editing sites (OR > 2.9, FDR < 2.3 × 10−2) (Fig. 4D; Supplemental Fig. S7G), raising the possibility that these nuclear-unique editing sites help in signaling nuclear sequestration. Although these editing site subsets were found exclusively in one compared to a contrasting group, 86.49%–100% of the introns and 96.55%–100% of the exons including these editing sites were expressed unedited in the contrasting groups, meaning the sequence being edited was usually expressed unedited in other fractions and ages.
We also measured developmental expression patterns of genes associated with nuclear pore complexes (NPCs) and identified 42 NPC genes with decreasing, and nine NPC genes with increasing, expression in either subcellular RNA compartment as the brain matures (FDR ≤ 0.05) (see the final column of Supplemental Table S3). Several of the NPC factors with increasing expression are involved with the NUP107-160 nuclear pore subcomplex, a conserved component of the NPC that is involved in mRNA export and interacts with chromatin to establish new NPCs after mitosis (Supplemental Fig. S8; Walther et al. 2003).
Nuclear-enriched genes contain motifs for distinct sets of RNA-binding proteins and have more stably structured 3′ UTRs
We examined the enrichment of 1157 sequence motifs corresponding to 140 RBPs in genes differentially regulated by fraction in adult and prenatal cortex (FDR ≤ 0.05) (Supplemental Table S6). We found that of the 86–123 unique RBPs with motifs enriched in each of the four gene sets (cytoplasmic in adult, cytoplasmic in prenatal, nuclear in adult, and nuclear in prenatal; FDR ≤ 0.05 for genes’ inclusion in the set, FDR ≤ 0.01 for motif enrichment), the majority was shared across age and fraction (61.8%–88.4% of RBPs per gene set) (Fig. 5A). Many of the RBPs exclusively enriched in one fraction have previously been implicated in neurodevelopment, and their fraction of enrichment corresponded to the RBP's most prevalent location of action. For instance, motifs for ELAVL2 and ELAVL3 were found exclusively in cytoplasmic-enriched genes compared to nuclear genes, and both RBPs act in neuronal dendrites to regulate neuronal development and function (Bryant and Yazdani 2016). Likewise, AGO1 functions in the cytoplasm as part of the RISC complex and was exclusively enriched in cytoplasmic genes. On the other hand, motifs for many nuclear-acting factors including CELF4 and PTBP2—RBPs involved in splicing, mRNA stability, and transport that are implicated in neuronal differentiation, plasticity, and transmission—are exclusively enriched in nuclear-enriched genes (Bryant and Yazdani 2016). In fact, many of the nuclear-enriched RBPs were especially relevant to splicing decisions occurring in neuronal nuclei, such as the neuron-specific splicing factors RBFOX1 and RBFOX2. RBFOX1 is implicated in developmental control of neuronal excitability and synaptic transmission (Bryant and Yazdani 2016). Given the localization and functional implications of the enriched RBPs, it is possible that RBP binding plays a part in the differential expression of these genes across fractions.
Figure 5.

RNA-binding protein motif enrichment, 3′-UTR length, and predicted secondary 3′-UTR structure of fraction-regulated genes. (A) Overlap of RBPs with sequence motifs found (FDR ≤ 0.01) in four groups of genes differentially expressed by fraction measured in adult and prenatal samples (FDR ≤ 0.05). RBPs discussed in the text are listed in purple. (B) Length in kilobases of the highest expressed 3′ UTR for each gene differentially expressed by fraction in both adult and prenatal, only prenatal, or only adult brain. (C) Minimum predicted free energy (MFE) of the secondary structure for the aforementioned 3′ UTRs.
Because 3′-UTR sequence stability is critical to RBP binding (Mayr 2017), we measured the minimum free energy (MFE) of the predicted secondary structure for the highest expressed 3′ UTR for each gene differentially expressed by fraction and found that genes higher expressed in nuclear RNA had significantly lower MFE than cytoplasmic genes, meaning that nuclear-enriched gene 3′ UTRs were putatively more thermodynamically stable than 3′ UTRs of cytoplasmically enriched genes (t < −3.9, FDR < 2.7 × 10−4) (Fig. 5B). This result was not based on differing 3′-UTR length, because 3′ UTRs of nuclear genes from both ages were of comparable size (t = 0.36, FDR = 0.72), whereas adult-specific nuclear-enriched genes had longer 3′ UTRs (t = 7.3, FDR = 2.7 × 10−12) and prenatal-specific nuclear-enriched genes had shorter 3′ UTRs (t = −3.4, FDR = 1.8 × 10−2) (Fig. 5C). Having a more stable 3′-UTR RNA secondary structure may therefore potentially contribute to the localization patterns of these nuclear-enriched genes.
Genes differentially expressed by fraction are overrepresented in gene sets associated with brain disease
We performed Disease Ontology Semantic and Enrichment (DOSE) analysis on the sets of genes differentially expressed by fraction and age, finding that genes with an interaction between fraction and age are enriched for genes associated with Alzheimer's and Parkinson's Diseases, in agreement with previous work implicating nucleocytoplasmic transport in neurodegenerative disease (Supplemental Fig. S9A,B).
We then assessed the relationship between the groups of fraction-associated genes (Supplemental Table S2) with gene sets for neurodevelopmental, neurodegenerative, and psychiatric disorders curated from several sources, including genome-wide association (GWAS), copy number variation (CNV), and single-nucleotide variation (SNV) studies (Supplemental Methods; Supplemental Tables S7, S8). Intellectual disability genes were enriched for cytoplasmic genes in both ages (OR = 16.4, FDR = 0.014). Neurodegenerative genes were enriched for nuclear genes in adults (OR = 2.7, FDR = 8.2 × 10−4), consistent with the importance of nucleocytoplasmic transport to this set of disorders. Nuclear genes in both ages were highly enriched for CNV genes associated with Autism Spectrum Disorder (ASD; OR = 6.7, FDR = 4.9 × 10−4) and schizophrenia (SCZ; OR = 6.2, FDR = 0.02). Bipolar Affective Disorder (BPAD) and other SCZ and ASD gene sets were also associated with genes greater expressed in nuclear RNA in adult cortex but not prenatal cortex (1.7 < OR < 2.5, 0.049 < FDR < 8.2 × 10−4) (Fig 6A).
Figure 6.
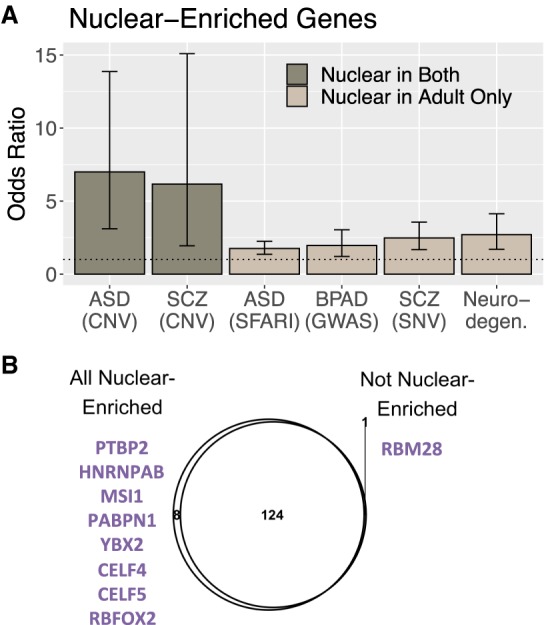
Disease association. (A) Odds ratios for enrichment of genes significantly greater expressed in nuclear RNA in brain disease gene sets (FDR ≤ 0.05). (B) Overlap of RBPs with sequence motifs found (FDR ≤ 0.01) in gene sets enriched in the nuclear compartment (listed in Fig. 6A) compared to the three gene sets that were not. Nonoverlapping RBPs are listed in purple.
We explored the relationship between disease-associated gene sets and mechanisms that regulate RNA localization. Genes in the six nuclear-enriched sets in Figure 6A were significantly longer than those in nonenriched gene sets (t = 16.1, FDR = 1.3 × 10−55) (Supplemental Fig. S9C). We found no difference in 3′-UTR length or predicted MFE or number of A→I editing sites between the nuclear-enriched disease-associated gene sets and nonenriched sets (Supplemental Fig. S10A,B). However, introns within genes in sets enriched in the “Nuclear in both” fraction set had greater IR ratios than in nonenriched sets (t = 10.1, FDR = 4.4 × 10−24) (Supplemental Fig. S10C). In other words, disease gene sets enriched in the nuclear compartment across ages had greater levels of intron retention. This result was not because of the nuclear-enriched sets having longer or more introns, as disease gene sets that were nuclear enriched in both ages had significantly shorter introns (t = −30.0, FDR = 3.8 × 10−196) and similar intron numbers to those in the nonenriched gene sets. Examining RBP motif enrichment in the disease-associated genes showed that eight RBPs had motifs that were uniquely enriched in nuclear disease gene sets versus nonnuclear sets, many of which were also enriched in nuclear genes and serve as splice factors as described above (FDR ≤ 0.01) (Fig. 6B). Splicing (or lack thereof) may therefore be a mediating component to the increased localization of ASD-, SCZ-, and BPAD-associated genes in the nucleus.
To explore these results in an expanded cellular context, we compared profiles of cytoplasmic and nuclear RNA from cell lines sequenced by the ENCODE Consortium (Djebali et al. 2012). H1, a human embryonic stem cell line, showed far fewer differences by fraction than the more differentiated cell types tested (9 of 51,502 genes differentially expressed at FDR ≤ 0.05) (Supplemental Fig. S11). SK-N-SH, a cell line derived from neuroblastoma, was among the most distinct by fraction (13,985 of 51,502 genes differentially expressed at FDR ≤ 0.05). In the ENCODE data, genes with significantly greater nuclear expression when controlling for cell type (FDR ≤ 0.05) were enriched for several disease gene sets, including those for ASD, SCZ, BPAD, syndromal neurodevelopmental disorders, and neurodegenerative disorders (1.5 < OR < 2.8, 0.02 < FDR < 5.8 × 10−10) (Supplemental Table S9). SCZ genes from CNV studies were enriched in genes from both subcellular compartments (OR = 1.9, FDR = 0.02).
Nuclear-enriched genes in individual cell lines, however, only variably held to this pattern (Supplemental Table S9). Overall, many more nuclear-associated disease gene sets were enriched in nuclear genes in each cell line than in the nonassociated sets or in cytoplasmic genes. Of the 61 cell-type-specific fraction gene set/disease gene set comparisons that were significantly enriched, 38 were enriched in nuclear genes in the cell type and nuclear-associated disease gene sets. These results indicate that nuclear enrichment of psychiatric disease genes is prevalent not just in brain but also in many other cellular contexts, suggesting that regulation of nuclear export of these transcripts may be a feature across many cell types.
Discussion
Here, we have characterized a snapshot of RNA compartmentalization in developing and mature human postmortem prefrontal cortex. We find that despite the presence of pre-mRNA, the nuclear RNA compartment alone can be used as a relatively high-fidelity stand-in for the whole transcriptome when focusing on gene-level expression. The use of poly(A)+ library preparation also minimizes the difference between subcellular fractions.
Differences in expression between fractions were much more muted in prenatal compared to adult cortex. We identified greater than 47 times more genes differentially expressed by fraction in adult than prenatal cortex in poly(A)+ samples. Transcription has been shown previously to be more widespread in prenatal brain than at more mature time points, with 4.1% of the prenatal genome transcribed compared to 3.1% of the adult genome (Jaffe et al. 2015). We also show that prenatal samples had a higher proportion of splice junctions, indicating that a greater volume of prenatal transcription is being processed. Given that the cellular composition of prenatal cortex includes a higher proportion of neural progenitor cells and embryonic stem cells and that these immature cells have a more plastic epigenome (Jaffe et al. 2016), it is tempting to speculate that as the brain matures, relative nuclear retention of RNA becomes a more utilized regulatory strategy in cells of the brain. This hypothesis is supported by the H1 embryonic stem cell line showing fewer differentially expressed genes by fraction than the other more differentiated cell lines profiled by ENCODE. It has also been shown that nuclear pore composition changes as cells differentiate and mature (D'Angelo et al. 2012), so it may be that nuclear pores and transport mechanisms are less mature in fetal brain and passage from nucleus to cytoplasm is less restricted.
At the gene level, trends in developmental and subcellular compartment expression patterns suggest nuclear sequestration of developmentally down-regulated RNA. Genes with greater cytoplasmic expression in one age tended to be higher expressed in that age, whereas nuclear-enriched genes tended to be higher expressed in the opposite age, suggesting that these nuclear genes, most of which are protein-coding, are being down-regulated at least in part by RNA not being exported to the cytoplasm for translation. Although this pattern must be tested in individual cell types to be confirmed, it suggests an added layer of regulation to be considered in the design of high-throughput sequencing studies. It is worth noting that nuclear-enriched genes in adult are overrepresented in non-DLPFC cell types, suggesting that genes in this set may also reflect cell-type-specific nuclear sequestration.
IR has been shown recently to be a common splice variant type that increases during development in several cell types including neurons (Yap et al. 2012; Wong et al. 2013; Mauger et al. 2016). Here, we confirm that although the majority of introns are constitutively spliced, IR is an abundant splice variant type—particularly in nuclear RNA—and increasing nuclear IR in a subset of introns correlates with increasing nuclear expression compared to cytoplasm and decreasing expression overall. Like global gene expression, specific splice variants seemed to pass more readily through the nuclear membrane in prenatal cortex than in adult, leading the decreasing relationship to be lacking in prenatal brain. It is unclear why increasing retention in age-associated introns is negatively correlated with expression only in prenatal cytoplasm samples, particularly as 28 of the 31 developmentally retained introns were more retained in prenatal than adult samples in either compartment, counter to the expectation that increased IR could be sequestering these transcripts in the nucleus as the brain matures. Further work in specific cell types or single cells with increased sequencing depth will be required to resolve these relationships. Nevertheless, because differentially retained introns by fraction were also more likely to be differentially retained by age, IR provides a link between developmental and compartmental expression changes in the data.
Identifying motifs of several known splicing factors with neurodevelopmental functions—such as RBFOX1 and RBFOX2—to be exclusively enriched by RNA fraction further strengthens the link between splicing and subcellular localization. We also find that 3′ UTRs of nuclear-enriched genes are predicted to be more thermodynamically stable than cytoplasmic genes, suggesting that there may be a conformational similarity between these RNAs that leads to their increased nuclear expression. However, motif enrichment alone is a weak predictor of in vivo RBP binding, as many RBPs have been shown to bind similar motifs, but show more diversity in their preference for other supplementary factors such as neighboring sequence, RNA secondary structure, and use of bipartite motifs (Dominguez et al. 2018). Although future work will be needed to validate the role of these RBPs in regulating nuclear transcript levels, this preliminary work further implicates splicing to be a critical step in developmentally regulated mRNA transport in human cortex.
Finally, we found that nuclear-enriched genes were also preferentially enriched in gene sets associated with neurodegenerative and neurodevelopmental psychiatric diseases. Previous work has identified the importance of proper nucleocytoplasmic transport in brain diseases, particularly neurodegenerative diseases (Mertens et al. 2015; Zhang et al. 2015). We found that genes associated with neurodevelopmental psychiatric diseases like ASD, SCZ, and BPAD were more likely to have higher expression in the nucleus at both ages tested, but particularly in adult where more nuclear sequestration in general was found. This preference for nuclear localization of developmental neuropsychiatric disorder gene sets extended to other immortalized cell types profiled by ENCODE. Nuclear-associated gene sets also showed increased IR compared to the others and shared enriched RBP motifs associated with splicing. These results suggest that these genes may be undergoing extra processing or regulation in the nucleus that may make them more vulnerable to dysregulation, potentially mediated by alternative splicing and RBP binding and particularly during early adult life. That these sets of genes appear to have easier exit from the nucleus during fetal life is consistent with other data that expression of these disease gene sets tends to be greater during fetal than postnatal life (Birnbaum et al. 2014; Jaffe et al. 2015).
Nonetheless, this study is limited by lack of single-cell or cell-type-specific insight into these patterns. By using bulk human postmortem brain tissue, we trade improved clinical validity and sequencing depth for reduced resolution of nucleocytoplasmic expression patterns. As mentioned previously, prenatal and adult cortices are populated by different cell types in different proportions, each with different proliferation, potency, and connectivity patterns that may influence the import–export decisions across the nuclear membrane. Cortical dissections were, however, matched for cell type composition at the tissue level within an age, lessening the potential for confounding by composition differences. Despite having to average the signal across cells and cell types, the fact that we still see this association between nuclear-expressed genes and psychiatric disease genes suggests that further study of this relationship is warranted.
Methods
Postmortem brain samples
Three prenatal and three adult human postmortem brains were selected from the repository of the Lieber Institute for Brain Development. DLPFC from adult and PFC from prenatal was acquired, dissected, and characterized as described previously (Lipska et al. 2006; Jaffe et al. 2015).
Cytoplasmic and nuclear RNA purification and sequencing
We used the Norgen Biotek Corp. Cytoplasmic and Nuclear RNA Purification Kit (21000, 37400) following the manufacturer's protocol including DNase I treatment, lysing 15 mg of tissue using Lysis Buffer J, and isolating fractions by centrifugation before extracting RNA using spin column chromatography.
RNA-sequencing libraries were prepared using poly(A)-selection (“poly(A)”; Illumina TruSeq Stranded Total RNA Library Prep Kit, RS-122-2201) and rRNA depletion (“Ribo-Zero”; Illumina Ribo-Zero Gold Kit [Human/Mouse/Rat] MRZG126) protocols. Libraries were sequenced on one Illumina HiSeq 2000 lane. Illumina Real Time Analysis (RTA) module performed image analysis and base calling, running the BCL converter (CASAVA v1.8.2), generating FASTQ files containing the sequencing reads. One adult nuclear Ribo-Zero sample failed quality control and was discarded. “Br5339C1_polyA” and “Br5340C1_polyA” FASTQ files were downsampled to 24 million total reads by concatenating FASTQ files across sequencer lanes for each sample, keeping read 1 and read 2 files separate, using the shuf unix command to shuffle the reads then printing the top 24 million shuffled records to new FASTQ files.
Data processing and quality control
Raw sequencing reads were mapped to the hg19/GRCh37 human reference genome with HISAT2 (Kim et al. 2015) v.2.0.4. Feature-level quantification based on GENCODE (release 25, lift 37) annotation was run using featureCounts (subread version 1.5.0-p3) (Liao et al. 2014). The hg19 version of GENCODE v25 annotation was originally created in hg38 and released in March 2016, therefore remapping to hg38 would have little effect on results. Exon–exon junction counts were extracted from the BAM files using RegTools (https://regtools.readthedocs.io/en/latest/) v0.1.0 and “bed_to_juncs” from TopHat2 (Kim et al. 2013) to retain the number of supporting reads. Annotated transcripts were quantified with Salmon (Patro et al. 2017) v.0.7.2. Alignment/processing metrics and the featureCounts results for genes, exons, exon–exon splice junctions, and annotated transcripts were read in and structured into analyzable matrices using R v.3.3.1 (R Core Team 2016). Raw FASTQ files were run through FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). All samples passed quality control for FastQC metrics and alignment, gene assignment, and mitochondrial mapping rates.
ENCODE samples (Djebali et al. 2012), NeuN+ and NeuN− nuclear RNA samples (Price et al. 2019), and single-cell RNA-seq samples (Darmanis et al. 2015) were processed as above.
Gene expression analysis
We did principal component analysis using the plotPCA function from DESeq2 (Love et al. 2014). Read distribution was calculated using read_distribution.py from RSeQC (Wang et al. 2012). Annotation features were assigned as follows: (CDS) exons→UTR exons→introns→intergenic regions.
Gene expression differences were measured using DESeq2. Samples were segregated by library, fraction, and age and compared using several linear models. Gene expression was first modeled by library type in the 11 nuclear samples (“∼ Age + Library”). Adult and prenatal samples from each library separately were assessed by fraction (“∼ Fraction”), and nuclear and cytoplasmic samples from each library were separately assessed by age (“∼ Age”), culminating in eight sets of results. Fractionation quality was checked by comparing expression of a priori localizing genes ACTB and MALAT1 using a paired one-tailed t-test, as well as comparing the t-statistics (paired, two-tailed) in the homogenate cortical samples with those from the fractionated ENCODE samples in a set of the top 296 differentially expressed genes by fraction in the ENCODE samples (see section “Cell-type-specific analyses”).
For subsequent analyses, a gene was considered significantly differentially expressed if FDR ≤ 0.05. We subset these genes according to whether they were in agreement across ages if measuring expression by fraction, or across fractions if measuring changes by age, resulting in eight groups (e.g., by fraction: both nuclear, both cytoplasmic, nuclear in prenatal only, nuclear in adult only, cytoplasmic in prenatal only, cytoplasmic in adult only, nuclear in prenatal but cytoplasmic in adult, and cytoplasmic in prenatal but nuclear in adult). “Interaction” genes were those FDR ≤ 0.05 using the model “∼ Age + Fraction + Age:Fraction” in the 12 poly(A)+ samples.
Gene and Disease Ontology (GO and DO, respectively) enrichments were calculated using the compareCluster function from clusterProfiler (Yu et al. 2012). To assess enrichment in brain disease gene sets, we used updated versions of the sets defined previously (Birnbaum et al. 2014). We calculated enrichment of the nine groups of fraction-associated genes described above using Fisher's exact test with all expressed genes as background.
Nucleoporin genes were those included in the following terms from the AmiGO 2 Gene Ontology Database (http://amigo.geneontology.org/grebe): GO:0005643, GO:0044613, GO:0044611, GO:0044614, GO:0044615, GO:0031080, GO:0070762, GO:1990876.
Cell-type-specific analyses
Each cytoplasmic (including “both cytoplasmic,” “cytoplasmic in prenatal only,” and “cytoplasmic in adult only”) and nuclear (including “both nuclear,” “nuclear in prenatal only,” and “nuclear in adult only”) gene was assigned a cell type based on the type with the maximum reads per million mapped (RPM) as measured in 466 single-cell RNA-seq samples (Darmanis et al. 2015). The proportion of cell types represented was calculated based on the total number of cytoplasmic or nuclear genes assessed.
We also compared neuron/nonneuron and fraction expression by assessing differential expression between NeuN+ and NeuN− nuclear RNA-seq samples from human cortex (Price et al. 2019) using the linear model “∼ Age + Cell type,” and comparing the resulting t-statistic and log2 fold change for each gene to that measured in homogenate cortex between RNA fractions.
Cell-specific expression analysis (CSEA) on the fraction-regulated gene sets was performed by inputting the gene symbols for the nine fraction-regulated gene sets in the CSEA web tool and using a Specificity Index threshold of 0.05 (Dougherty et al. 2010; Xu et al. 2014).
ENCODE expression patterns were first analyzed together using the linear model “∼ Cell type + Fraction + Cell type:Fraction” to identify genes differentially expressed by fraction across cell types, then cell-type-specific patterns were assessed using the model “∼ Fraction” in each of the 11 cell types measured. Enrichment of cytoplasmic and nuclear-enriched genes (defined as FDR ≤ 0.05) from these comparisons in brain disease gene sets was assessed with Fisher's exact test using all expressed genes in this data set as background.
Splicing analysis
The proportion of splice junctions per sample was calculated by dividing the number of reads overlapping a known or predicted splice junction by the total number of reads. To characterize splice variant type use across the poly(A)+ samples, we used SGSeq (Goldstein et al. 2016). We extracted features using getBamInfo(), then used the analyzeFeatures function to predict and quantify splicing events based on GENCODE (release 25, lift 37). We analyzed and summarized that output using analyzeVariants(), setting the minimum denominator to 10. The number of unique splice variants of each type were counted by extracting the types using variantType(). We calculated differential splice variant use by fraction and age using DEXSeq (Anders et al. 2012). We used the variant IDs as the featureID and the event IDs as the groupID in DEXSeqDataSet(). We subset the 12 poly(A)+ samples by fraction and age and compared differential splice variant expression by fraction using the full model “∼ sample + exon + fraction:exon” and the reduced model “∼ sample + exon.” We compared splice variant expression by age using the full model “∼ sample + exon + age:exon” and the reduced model “∼ sample + exon.” We then stratified these results by splice variant type and used Fisher's exact test to calculate the enrichment of each type in each fraction and age.
To assess IR in the poly(A)+ samples, we filtered introns from the IRFinder-IR-nondir.txt output of IRFinder (Middleton et al. 2017) run on the Human-hg19-release75 reference for each sample. We excluded introns with the “NonUniformIntronCover” warning and those that had anything but “clean” listed in the GeneIntronDetails output column (i.e., excluding “anti-near,” “anti-over,” “known-exon + anti-near,” “known-exon,” and “known-exon + anti-near + anti-over”). This step is where most of the introns filtered were lost. Introns were further filtered to exclude introns with fewer than four reads spanning the splice junction or a junction using either the 5′ or 3′ exon–intron boundary, or with fewer than four reads supporting intron inclusion at each exon–intron boundary. To assess the relationship between gene expression and IR, we assigned the maximum IR ratio per sample for each gene from this filtered set of introns and compared IR ratios of genes regulated by fraction and age (FDR ≤ 0.05) using Student's t-test and Fisher's exact test.
To quantify differential retention of individual introns, we subset the samples by fraction and age and filtered the IRFinder-IR-nondir.txt output to create four new lists, first filtering to only include the “clean” introns (from the GeneIntronDetails output column), then filtering constitutively spliced introns by group (i.e., adult, prenatal, nuclear, and cytoplasmic). We then used these new files as input to analysisWithLowReplicates.pl from IRFinder to calculate differential intron retention between fraction in prenatal and adult, and by age in nucleus and cytoplasm, using the Audic and Claverie test. We calculated the false discovery rate using the p.adjust function and setting the n parameter to the total number of clean, nonconstitutively spliced introns in each comparison. The relationship between intron retention by fraction and age and gene expression was further examined by comparing counts of each using Fisher's exact test.
Intron conservation was tested by extracting per base GERP scores for all “clean” introns from the UCSC Table Browser (hg19), calculating the mean score per intron, and comparing the means of groups of introns using Student's t-test. Repetitive elements in introns were analyzed by downloading the RepeatMasker (Smit et al. 1996) track from the UCSC Table Browser (hg19) and finding overlaps using the findOverlaps function from GenomicRanges (Lawrence et al. 2013).
RNA editing analysis
RNA editing sites were called in the 12 poly(A)+ samples as described previously (Hwang et al. 2016). We annotated the editing sites to genomic features using GenomicFeatures (Lawrence et al. 2013) and a transcription database object built on GENCODE (release 25, lift 37). Overlap with repetitive sequences was assessed using RepeatMasker and the findOverlaps function from GenomicRanges. We compared the editing sites identified in this study with previously identified editing sites using findOverlaps(). We examined the effect of fraction, age, and the interaction of the two on editing rate in the 1025 sites present in all samples by filtering the sites to those with a finite and non-NA logit-transformed editing rate in at least five samples and with at least one adult, prenatal, nucleus, and cytoplasm represented and then using the model “∼ Age + Fraction + Age:Fraction.” We compared the pattern of editing in our data set of the 576 developmentally increasing editing sites previously identified (Hwang et al. 2016) using Fisher's exact test.
We defined the sets of fraction- and age-specific editing sites by sites present in all samples of the first group that were not found in the second group. For instance, the “Adult Only” sites were present in all six adult samples but no prenatal samples. We assigned each editing site to the nearest gene using the distanceToNearest function from GenomicRanges and compared the site location by fraction or age with the expression enrichment using the Fisher's exact test. We identified KEGG pathway enrichment using the compareCluster function for the 10 groups of unique editing sites. Annotation enrichment for these unique sites was assessed using the Fisher's exact test. The major 3′-UTR isoform was based on which 3′ UTR had the highest read coverage per gene.
RNA-binding protein motif enrichment analysis
We downloaded position weight matrices for human RNA-binding proteins from the ATtRACT database (https://attract.cnic.es/; v0.99B). Using the makeBackground function from PWMEnrich (https://www.bioconductor.org/packages/release/bioc/html/PWMEnrich.html), we calculated a lognormal background based on 1000 randomly selected cDNAs. For the RBP enrichment by fraction, we created FASTA files for four groups: cDNA for genes significantly higher expressed in nucleus or cytoplasm in adult or prenatal samples (FDR ≤ 0.05). For the disease gene set analysis, we created FASTA files using cDNA for genes in the six nuclear-enriched sets versus the three remaining gene sets. For each of the groups we called the motifEnrichment function to calculate the enrichment for each motif, and then the groupReport function to summarize the results over each gene set. RBPs with a motif that passed a significance threshold of FDR ≤ 0.01 in the group report were considered enriched in the gene set.
3′-UTR secondary structure and length analysis
We selected the highest expressed 3′ UTR for each gene by annotating exons using the threeUTRsByTranscript function from GenomicFeatures v.1.34.1 (Lawrence et al. 2013) and choosing the one with highest mean expression across all samples per gene. We extracted cDNA sequence for these regions and calculated the minimum free energy using RNAfold from ViennaRNA v.2.4.11 (Lorenz et al. 2011). We assessed the difference between MFE in groups defined in the RBP motif methods section using Student's t-test.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Sequence Read Archive (BioProject; https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA595606. Code is available through GitHub (https://github.com/LieberInstitute/BrainRNACompartments), Zenodo (https://doi.org/10.5281/zenodo.3475697), and as Supplemental Code.
Supplementary Material
Acknowledgments
The authors thank Jeffrey D. Rothstein for his insightful comments. This work was funded directly from the Lieber Institute for Brain Development and the Maltz Research Laboratories and from National Institutes of Health grant R21MH105853.
Author contributions: A.J.P.: conceptualization, formal analysis, visualization, writing—original draft preparation, writing—review and editing. T.H.: formal analysis. R.T.: investigation. E.E.B.: data curation, writing—review and editing. A.R.: data curation, writing—review and editing. J.H.S.: supervision, writing—review and editing. T.M.H.: data curation, resources, writing—review and editing. J.E.K.: data curation, resources, writing—review and editing. A.E.J.: conceptualization, supervision, writing—review and editing. D.R.W.: conceptualization, funding acquisition, supervision, writing—review and editing.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.250217.119.
References
- Anders S, Reyes A, Huber W. 2012. Detecting differential usage of exons from RNA-seq data. Genome Res 22: 2008–2017. 10.1101/gr.133744.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar Halpern K, Caspi I, Lemze D, Levy M, Landen S, Elinav E, Ulitsky I, Itzkovitz S. 2015. Nuclear retention of mRNA in mammalian tissues. Cell Rep 13: 2653–2662. 10.1016/j.celrep.2015.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Bhatt DM, Pandya-Jones A, Tong AJ, Barozzi I, Lissner MM, Natoli G, Black DL, Smale ST. 2012. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150: 279–290. 10.1016/j.cell.2012.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum R, Jaffe AE, Hyde TM, Kleinman JE, Weinberger DR. 2014. Prenatal expression patterns of genes associated with neuropsychiatric disorders. Am J Psychiatry 171: 758–767. 10.1176/appi.ajp.2014.13111452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Bhutkar A, Sharp PA. 2015. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev 29: 63–80. 10.1101/gad.247361.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 24: 1774–1786. 10.1101/gr.177790.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Yazdani N. 2016. RNA-binding proteins, neural development and the addictions. Genes Brain Behav 15: 169–186. 10.1111/gbb.12273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. 2013. Characterization and comparison of human nuclear and cytosolic editomes. Proc Natl Acad Sci 110: E2741–E2747. 10.1073/pnas.1218884110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, et al. 2011. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478: 519–523. 10.1038/nature10524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui P, Lin Q, Ding F, Xin C, Gong W, Zhang L, Geng J, Zhang B, Yu X, Yang J, et al. 2010. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics 96: 259–265. 10.1016/j.ygeno.2010.07.010 [DOI] [PubMed] [Google Scholar]
- D'Angelo MA, Gomez-Cavazos JS, Mei A, Lackner DH, Hetzer MW. 2012. A change in nuclear pore complex composition regulates cell differentiation. Dev Cell 22: 446–458. 10.1016/j.devcel.2011.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, Hayden Gephart MG, Barres BA, Quake SR. 2015. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci 112: 7285–7290. 10.1073/pnas.1507125112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. 2018. Sequence, structure, and context preferences of human RNA binding proteins. Mol Cell 70: 854–867.e9. 10.1016/j.molcel.2018.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Schmidt EF, Nakajima M, Heintz N. 2010. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res 38: 4218–4230. 10.1093/nar/gkq130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner AS, Twiss JL, Perrone-Bizzozero NI. 2015. Competing interactions of RNA-binding proteins, microRNAs, and their targets control neuronal development and function. Biomolecules 5: 2903–2918. 10.3390/biom5042903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasset-Rosa F, Chillon-Marinas C, Goginashvili A, Atwal RS, Artates JW, Tabet R, Wheeler VC, Bang AG, Cleveland DW, Lagier-Tourenne C. 2017. Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron 94: 48–57.e4. 10.1016/j.neuron.2017.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LD, Cao Y, Pau G, Lawrence M, Wu TD, Seshagiri S, Gentleman R. 2016. Prediction and quantification of splice events from RNA-seq data. PLoS One 11: e0156132 10.1371/journal.pone.0156132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T, Park CK, Leung AK, Gao Y, Hyde TM, Kleinman JE, Rajpurohit A, Tao R, Shin JH, Weinberger DR. 2016. Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci 19: 1093–1099. 10.1038/nn.4337 [DOI] [PubMed] [Google Scholar]
- Jaffe AE, Shin J, Collado-Torres L, Leek JT, Tao R, Li C, Gao Y, Jia Y, Maher BJ, Hyde TM, et al. 2015. Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat Neurosci 18: 154–161. 10.1038/nn.3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, Kleinman JE. 2016. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci 19: 40–47. 10.1038/nn.4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S, et al. 2016. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352: 1586–1590. 10.1126/science.aaf1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. 2013. Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Tighe SW, Nicolet CM, Grove D, Levy S, Farmerie W, Viale A, Wright C, Schweitzer PA, Gao Y, et al. 2014. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat Biotechnol 32: 915–925. 10.1038/nbt.2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, et al. 2018. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362: eaat7615 10.1126/science.aat7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Lipska BK, Deep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, Kleinman JE. 2006. Critical factors in gene expression in postmortem human brain: focus on studies in schizophrenia. Biol Psychiatry 60: 650–658. 10.1016/j.biopsych.2006.06.019 [DOI] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26 10.1186/1748-7188-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauger O, Lemoine F, Scheiffele P. 2016. Targeted intron retention and excision for rapid gene regulation in response to neuronal activity. Neuron 92: 1266–1278. 10.1016/j.neuron.2016.11.032 [DOI] [PubMed] [Google Scholar]
- Mayr C. 2017. Regulation by 3′-untranslated regions. Annu Rev Genet 51: 171–194. 10.1146/annurev-genet-120116-024704 [DOI] [PubMed] [Google Scholar]
- Mertens J, Paquola ACM, Ku M, Hatch E, Böhnke L, Ladjevardi S, McGrath S, Campbell B, Lee H, Herdy JR, et al. 2015. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17: 705–718. 10.1016/j.stem.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton R, Gao D, Thomas A, Singh B, Au A, Wong JJ, Bomane A, Cosson B, Eyras E, Rasko JE, et al. 2017. IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol 18: 51 10.1186/s13059-017-1184-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, Bhatt DM, Lin CH, Tong AJ, Smale ST, Black DL. 2013. Splicing kinetics and transcript release from the chromatin compartment limit the rate of Lipid A-induced gene expression. RNA 19: 811–827. 10.1261/rna.039081.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, Spector DL. 2005. Regulating gene expression through RNA nuclear retention. Cell 123: 249–263. 10.1016/j.cell.2005.08.033 [DOI] [PubMed] [Google Scholar]
- Price AJ, Collado-Torres L, Ivanov NA, Xia W, Burke EE, Shin JH, Tao R, Ma L, Jia Y, Hyde TM, et al. 2019. Divergent neuronal DNA methylation patterns across human cortical development reveal critical periods and a unique role of CpH methylation. Genome Biol 20: 196 10.1186/s13059-019-1805-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Reddy AS, O'Brien D, Pisat N, Weichselbaum CT, Sakers K, Lisci M, Dalal JS, Dougherty JD. 2017. A comprehensive analysis of cell type–specific nuclear RNA from neurons and glia of the brain. Biol Psychiatry 81: 252–264. 10.1016/j.biopsych.2016.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 1996. RepeatMasker Open-3.0. http://www.repeatmasker.org/.
- Solnestam BW, Stranneheim H, Hällman J, Käller M, Lundberg E, Lundeberg J, Akan P. 2012. Comparison of total and cytoplasmic mRNA reveals global regulation by nuclear retention and miRNAs. BMC Genomics 13: 574 10.1186/1471-2164-13-574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M, Amstislavskiy V, Risch T, Schuette M, Dökel S, Ralser M, Balzereit D, Lehrach H, Yaspo ML. 2014. Influence of RNA extraction methods and library selection schemes on RNA-seq data. BMC Genomics 15: 675 10.1186/1471-2164-15-675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, Curado J, Snyder M, Gingeras TR, Guigó R. 2012. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res 22: 1616–1625. 10.1101/gr.134445.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther TC, Alves A, Pickersgill H, Loïodice I, Hetzer M, Galy V, Hülsmann BB, Köcher T, Wilm M, Allen T, et al. 2003. The conserved Nup107-160 complex is critical for nuclear pore complex assembly. Cell 113: 195–206. 10.1016/S0092-8674(03)00235-6 [DOI] [PubMed] [Google Scholar]
- Wang L, Wang S, Li W. 2012. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28: 2184–2185. 10.1093/bioinformatics/bts356 [DOI] [PubMed] [Google Scholar]
- Wong JJ, Ritchie W, Ebner OA, Selbach M, Wong JW, Huang Y, Gao D, Pinello N, Gonzalez M, Baidya K, et al. 2013. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 154: 583–595. 10.1016/j.cell.2013.06.052 [DOI] [PubMed] [Google Scholar]
- Xu X, Wells AB, O'Brien DR, Nehorai A, Dougherty JD. 2014. Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J Neurosci 34: 1420–1431. 10.1523/JNEUROSCI.4488-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap K, Lim ZQ, Khandelia P, Friedman B, Makeyev EV. 2012. Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev 26: 1209–1223. 10.1101/gad.188037.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaghlool A, Ameur A, Nyberg L, Halvardson J, Grabherr M, Cavelier L, Feuk L. 2013. Efficient cellular fractionation improves RNA sequencing analysis of mature and nascent transcripts from human tissues. BMC Biotechnol 13: 99 10.1186/1472-6750-13-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, et al. 2015. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525: 56–61. 10.1038/nature14973 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.