

Abstract
Recent experimental and computational efforts have provided large data sets describing three-dimensional organization of mouse and human genomes and showed the interconnection between the expression profile, epigenetic state, and spatial interactions of loci. These interconnections were utilized to infer the spatial organization of chromatin, including enhancer–promoter contacts, from one-dimensional epigenetic marks. Here, we show that the predictive power of some of these algorithms is overestimated due to peculiar properties of the biological data. We propose an alternative approach, which provides high-quality predictions of chromatin interactions using information on gene expression and CTCF-binding alone. Using multiple metrics, we confirmed that our algorithm could efficiently predict the three-dimensional architecture of both normal and rearranged genomes.
Spatial interactions between promoters and their regulatory sequences are required to maintain a cell type–specific expression pattern (Rao et al. 2014). It is known that enhancers do not necessarily regulate the closest promoters, and enhancer–promoter (EP) interactions often span large genomic distances (Rao et al. 2014; Tang et al. 2015). Although enhancer targets can be directly identified by using high-resolution 3C-methods (Rao et al. 2014), these data are expensive to obtain and currently available only for a small subset of cell types. Besides, experimental identification of enhancer targets does not provide a mechanism explaining target selection.
Several computational tools have been developed to address these challenges. Their task was to predict three-dimensional EP interactions, based on data on one-dimensional genetic and epigenetic marks (Fortin and Hansen 2015; Moore et al. 2015; Chen et al. 2016; Chiariello et al. 2016; Whalen et al. 2016; Zhu et al. 2016; Di Pierro et al. 2017; Al Bkhetan and Plewczynski 2018b; Buckle et al. 2018; Kai et al. 2018; Zeng et al. 2018; Zhang et al. 2018; Ibn-Salem and Andrade-Navarro 2019; Qi and Zhang 2019). All these tools fall into two categories: physical models and statistical approaches. The former rely on knowledge of polymer physics to build a physical model of chromatin and optimize the model parameters to fit experimental (usually Hi-C) data (Chiariello et al. 2016; Di Pierro et al. 2017; Buckle et al. 2018). The optimized model can be used to infer spatial conformation of chromatin, including those regions containing EP interactions. In contrast, statistical methods do not imply any a priori knowledge of polymer physics, aiming to find consistent patterns in epigenetic data which would explain three-dimensional contacts of loci (Moore et al. 2015; Chen et al. 2016; Whalen et al. 2016; Di Pierro et al. 2017; Kai et al. 2018; Zeng et al. 2018; Zhang et al. 2018; Ibn-Salem and Andrade-Navarro 2019). Thus, statistical approaches are able to predict spatial contacts of chromatin even without complete knowledge of the physical mechanisms underlying the three-dimensional organization of the genome.
Here, we aimed to infer the three-dimensional interactions of chromatin, and particularly promoter–enhancer interactions, in normal and rearranged genomes, using available epigenetic data. We benchmarked the existing statistical approach and found that its predictive power is overestimated due to the peculiar properties of the biological data. Thus, we have aimed to develop a new machine-learning algorithm for quantitative prediction of genome architecture based on broadly available epigenetic data sets.
Results
TargetFinder fails to predict EP interactions
Our objective was to develop an algorithm for prediction of enhancer–promoter interactions in normal and rearranged genomes. For this aim, we decided to employ existing TargetFinder algorithm (Whalen et al. 2016), which is of particular interest because of high accuracy, a low false-discovery rate, and reproducibility, demonstrated by an analysis of several human cell types. Since several well-studied examples of chromosomal rearrangements causing changes of chromatin architecture have been investigated using mouse models (Fishman et al. 2018; Spielmann et al. 2018), we aimed to extend the TargetFinder algorithm for prediction of EP interactions in mouse cells.
We annotated promoters and enhancers as interacting and noninteracting using high resolution Hi-C data on mouse embryonic stem (ES) cells (Bonev et al. 2017) and collected a set of 24 genetic and epigenetic predictors. To construct our data sets, we used an original definition of “interacting” promoters and enhancers, proposed in a TargetFinder paper (Whalen et al. 2016), i.e., promoter and enhancer were considered as interacting only if they were located in the anchors of a Hi-C loop. The accuracy of TargetFinder (measured by either precision, recall, or F1-score on a validation data set) was lower than previously reported on human data (Table 1). We found that changing the ratio of interacting to noninteracting EP pairs from 1:20 to 1:1 increases F1-scores; however, obtained values were still below that reported previously (Table 1; Whalen et al. 2016). We additionally ran TargetFinder on mouse cortex and neural progenitor cells (NPC) data (Bonev et al. 2017) using 10 available epigenetic predictors. As in the ES cells data, TargetFinder was not efficient on these data sets (Table 1).
Table 1.
Effect of train/validation split strategy on TargetFinder efficiency

To understand why TargetFinder fails to predict EP interactions, we reprocessed original human data, generating predictors, training, and validation data sets for human GM12878 cells de novo. Running TargetFinder on these reprocessed human data sets resulted in low F1-scores, with only small improvement compared to mouse ES cells data (Table 1).
Comparing our protocol of data processing with the pipeline that was used to generate the original TargetFinder data sets, we noticed the difference in composition of training and validation samples. In the original approach, EP pairs were randomly split to obtain training (∼90% of data) and validation (∼10% of data) data sets. Our pipeline randomly selects two chromosomes and designs all EP pairs on these chromosomes as a validation data set (∼10% of all data) and the rest of EP pairs as a training data set. This difference in design of training and validation data sets is essential, because, when we performed a by-chromosome split of the original TargetFinder data on human GM12878, F1-scores were reduced a lot compared to the random-split and become similar to those obtained for mouse data (Table 1). Moreover, when we used the random-split strategy on mouse data, F1-scores increased substantially (Table 1). Thus, the TargetFinder performance strongly depends on the design of training and validation data sets.
To explain the observed effect of the data splitting strategy on TargetFinder efficiency, we explored the structure of EP data sets. We found that ∼70% of GM12878 promoters interact with multiple enhancers, which are located close to each other. Such EP pairs share a large portion of genomic region between promoter and enhancer (referred to hereafter as “window”). In general, overlaps of EP windows are frequent (>99% of all pairs share a window with at least one other pair) and overlap size is often large. Thus, epigenetic predictors characterizing a window of these pairs are not independent, and EP pairs with a shared window should not be placed in both the training and validation data set (Fig. 1A,B). As this happens when employing the random-split strategy, TargetFinder could match overlapping samples in training and validation sets based on window information and then copy the information about interactions from the pair in the training set to the pair in the validation set. One should note that patterns of spatial contacts of neighboring genomic regions are correlative. Thus, interactions of two EP pairs, one from the training set and another from the validation set, are often similar if both promoters and enhancers are located nearby (i.e., if window overlap is high). To confirm this, for each EP pair we explicitly used interaction of the EP pair with the highest window overlap as a predictor and obtained a high F1-score (∼0.9) for GM12878 cells. Moreover, for NPC and mouse cortex data sets, which contain approximately three times less interacting EP pairs than mouse ES cells and human GM12878 cells (Table 1) and therefore the lowest rate of overlapping windows, we obtained the lowest F1-scores in the random-split design.
Figure 1.
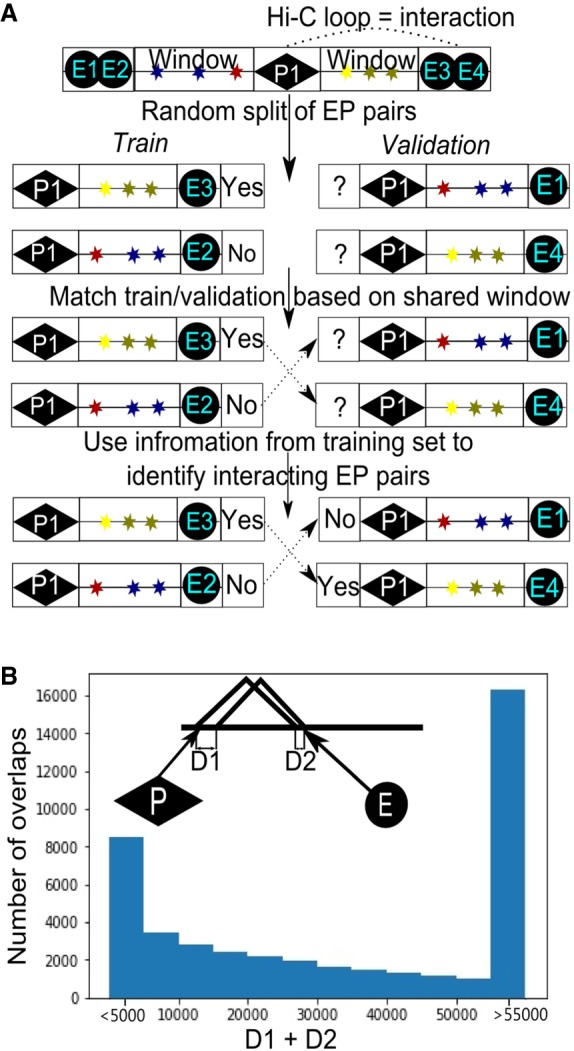
Promoter–enhancer pairs with overlapping windows in training and validation data sets. (A) Schematic illustration showing how information could be shared between training and validation data sets because of overlapping EP windows. (B) Distribution of distances between boundaries of overlapping EP windows. For each EP pair, we found the window of another EP pair, so that the distance between boundaries of their windows (d = D1 + D2) is minimal. Histogram shows distribution of the obtained values of d.
To sum up, the random-split strategy breaks the assumption of independence of samples in the training and validation data sets and thus results in overestimation of the predictive power of machine-learning algorithms. In contrast, when using a by-chromosome splitting strategy, genomic regions never overlap between training and validation data sets, allowing unbiased estimation of algorithms’ efficiency. Considering that TargetFinder efficiency drops when using by-chromosome data splitting, we claim that this algorithm cannot reconstruct relations between 1D-genetic marks and 3D-genome organization. Moreover, while this manuscript was in preparation, Xi and Beer (2018) independently concluded that the local epigenomic state cannot discriminate interacting and noninteracting enhancer–promoter pairs with high accuracy.
We note that other published algorithms also use a random-split design of training and validation data sets (see Discussion). Thus, our results highlighted specific peculiarities of the biological data, which should be considered in the future to prevent overfitting issues and incorrect efficiency estimation of machine-learning approaches focused on predictions of chromatin interactions.
Enhancer–promoter interactions are quantitative rather than qualitative
As we found that TargetFinder cannot efficiently predict EP interactions, we aimed to improve this algorithm. We considered the following enhancements.
First, we decided that epigenetic marks not only between, but also outside of, the promoter and enhancer should be considered. This makes sense in light of recently discovered mechanisms, underlying the spatial organization of chromatin. For example, according to the loop extrusion model (Fudenberg et al. 2017), binding of CTCFs in the converged orientation outside of, but close to, an EP pair will result in increased looping between the promoter and enhancer. Based on the loop extrusion model, we also introduced orientation of CTCF sites as a predictor.
Second, we reconsidered the definition of the enhancer–promoter interaction. TargetFinder and other approaches (Li et al. 2019) define an EP pair as interacting only if the enhancer and promoter occur within anchors of a Hi-C loop. To benchmark this approach, we collected all interacting EP pairs for human monocytes based on SlideBase and GeneHancer databases (see Methods for details). In addition to promoter-capture 3C data, these databases utilize information of co-expression of promoters and regulatory elements, their distance, and other information to define interacting EP pairs. We used human monocytes data because these cells are represented in the SlideBase and GeneHancer database and characterized by high-resolution Hi-C (Phanstiel et al. 2017), which allowed comparison of Hi-C loops and interacting EP pairs. We confirmed that contact frequencies between interacting EP pairs, as well as between loop anchors, are higher than average (Fig. 2A,B). However, the vast majority of interacting EP pairs do not overlap with loops, although they are often located within a reasonable distance from loop anchors (Fig. 2C). Similar results were obtained for human macrophages (Supplemental Fig. S1).
Figure 2.

Hi-C loops do not provide complete information about interacting EP pairs. (A,B) Distribution of row (A) and distance-normalized (B) contact frequencies for interacting EP pairs and loop anchors in human monocytes. (C) Number of interacting EP pairs overlapping loops in monocyte data. Red line represents the number of EP pairs overlapping any Hi-C loop anchors or located within a distance not more than x kb of them, shown as a function of x. Gray line and gray area represent average plus three standard deviations of 100 randomized controls. (D) Chromatin interactions within the region on human Chromosome 8 containing seven experimentally validated MYC enhancers (yellow lines, e1–e7) and Hi-C loops (purple squares). Although both enhancers and loops were identified in the same cell type (K562 cells), they show little overlap.
The set of EP pairs described in SlideBase and GeneHancer is probably not complete, and these databases (partially) rely on the 3C information to infer EP connections. The gold standard for identification of functional EP interactions is direct genetic screening. Such screenings are expensive and time-consuming, thus there is a very limited number of experimentally validated enhancers, which prevents systematic genome-wide analysis of their relations to Hi-C loops. However, individual reports of genetically validated functional EP interactions support our general conclusion. For example, a recent study (Fulco et al. 2016) identified seven distal enhancers of the MYC gene, which form two clusters located 0.16 and 1.9 Mb away from the MYC promoter, respectively. According to Fulco et al. (2016), all of these enhancers affect MYC expression in K562 cells proportionally to the number of contacts between the enhancer and the MYC promoter in this cell type. However, we found that only e6 and e7 enhancers overlap Hi-C loops (Fig. 2D). Moreover, out of five loops containing the MYC promoter, only one contains validated MYC enhancers. Altogether, this means that binary classification of EP interactions guided by the location of Hi-C loop anchors may have poor predictive power. These observations are consistent with the recent model of enhancer–promoter communication (Furlong and Levine 2018), which suggests that loops and domains serve to decrease the effective distance separating enhancers and promoters but are not necessarily formed by EP pairs themselves.
Thus, we concluded that increased interaction frequency, rather than location within loop anchors, should be used to characterize EP interaction. As spatial interactions are quantitative, we aimed to design a quantitative algorithm which predicts frequencies of spatial interactions between genomic loci in general and EP interactions in particular.
Quantitative prediction of EP interactions using machine-learning tool 3DPredictor
We used the following biological information to predict EP and other genomic interactions: ChIP-seq profiles, describing chromatin binding of architectural proteins or histone modifications; RNA-seq profiles, describing gene expression levels; E1 values, classifying chromatin to active (A) and inactive (B) compartments; and, genomic distance, which is an essential factor of three-dimensional contacts. We restricted our algorithm to the prediction of mid-range contacts (≤ 1.5 Mb) since almost all EP interactions occur within this distance. To increase sample size and avoid overfitting, we included contacts of all loci, regardless of the presence of promoters and enhancers, into the training set. We always performed training and validation on different chromosomes and never used chromosome number or genomic coordinates of loci as predictors, to prevent overfitting.
Using recently generated Hi-C and genomic data on mouse hepatocytes and human K562 and GM12878 cells, we compared several forms of predictors parametrization and performance of different machine-learning algorithms (see Supplemental Note; Supplemental Figs. S9–S12; Supplemental Tables S2, S3 for details). To estimate the quality of predictions, we used Pearson's correlation, stratum-adjusted correlation coefficient (SCC) (Yang et al. 2017a), mean squared error (MSE), mean absolute error (MAE), and mean relative error (MRE). As a result, we developed 3DPredictor, a machine-learning tool for computational prediction of chromatin interactions. Analyses of importance of different epigenetic features showed that information about cohesin and CTCF-binding, gene expression, chromatin accessibility, and distance between loci has the greatest contribution to the prediction accuracy (see Supplemental Fig. S2; Supplemental Table S1; Supplemental Note). Moreover, according to the feature importance analysis, epigenetic characteristics of the region between interacting loci are essential for accurate prediction (Supplemental Fig. S2), which supports a previously obtained (Whalen et al. 2016) conclusion that there is significant information relevant to looping interactions outside the interacting loci themselves.
Although various epigenetic information contributed to the prediction of chromatin contacts, it appeared that many predictors are interchangeable. We were able to generate accurate predictions of chromatin interactions in mouse hepatocytes (Pearson's r = 0.92–0.95, SCC = 0.53–0.72, MSE = 0.0017–0.0082, MAE = 0.0010–0.0015, MRE = 0.52–1.74) (Fig. 3A–F), limiting input information to CTCF ChIP-seq data (including orientation of the occupied CTCF sites), RNA-seq data, and distance between loci, and using only one chromosome out of 20 for training. These results can be further improved using multiple chromosomes for training (Fig. 3B–F). Orientation of CTCF sites was among the features with the highest importance, and omitting this information impaired predictions of loops (Supplemental Figs. S2, S3). Thus, we used information about CTCF binding, RNA-seq, and genomic distance for all predictions in this paper.
Figure 3.

3DPredictor efficiently reconstructs spatial interactions based on CTCF occupancy, expression, and genomic distance. (A) Representative region of mouse Chromosome 2 showing predicted and experimentally derived Hi-C interactions in mouse hepatocytes. (B–F) Various metrics of 3DPredictor accuracy for each chromosome of mouse hepatocytes. Circles represent comparison between two replicas; green squares show comparisons between hepatocytes and other cell types. Red triangles display 3DPredictor results obtained using single Chromosome 5 for training; data obtained when validating on the same chromosome are marked with stars. Purple triangles show results of 3DPredictor trained on 10 chromosomes (results for even chromosomes obtained using model trained on odd chromosomes and vice versa).
Chromatin contacts are known to be moderately similar between cell types (Rao et al. 2014; Battulin et al. 2015). To find whether our predictions are cell type–specific, we first compared the chromatin architecture of different cell types using the aforementioned measures. In most cases, results obtained by 3DPredictor differ from real data less than cell types differ from each other (Fig. 3B–F). For example, for 13 chromosomes, 3DPredictor results, judged by the mean average error, resemble experimental hepatocyte's Hi-C data more closely than experimental data derived from other studied cell types. For five more chromosomes (Chromosomes 1, 4, 6, 9, and 15), prediction errors were comparable with the MAE obtained for different cell types, and on Chromosomes 19 and X, predictions were worse than transferring contact counts from other cell types. Similar results were obtained for the MSE and MRE. According to the SCC, 3DPredictor performs approximately at the level of intercellular differences, whereas, according to the Pearson's correlation, predictions were almost always more similar to the hepatocyte's data than other cell types.
We next compared Hi-C data of mouse hepatocytes and NPC and found that some genomic regions show apparently different 3D-organization in these cell types. In most cases, the differences were due to the presence of cell type–specific TADs, borders of which coincide with cell type–specific CTCF sites, as was observed previously (Rao et al. 2014; Bonev et al. 2017). We utilized an insulation-based score to select genomic regions with cell type–specific chromatin architecture (see Methods for details), and ran 3DPredictor for these regions using cell type–specific RNA-seq and CTCF ChIP-seq data. Predicted contact frequencies reflected cell type–specific genome organization (Fig. 4A–C; Supplemental Fig. S4), and correlation of insulation scores of predicted and experimental data was much higher than between cell types (Fig. 4D). We provided an example of an accurate prediction of the cell type–specific TAD boundary in NPC and hepatocytes in Figure 4, B and C, and in Supplemental Figure S4.
Figure 4.

3DPredictor accurately reconstructs cell type–specific chromatin organization. (A) Representative region on Chromosome 3 showing different 3D organization in mouse hepatocytes and NPC. Cell type–specific TAD boundary is marked by arrow. (B,C) Comparison of 3DPredictor results with experimental NPC (B) or hepatocyte (C) data for the same region of Chromosome 3. (D) Insulation scores in 88 NPC cell type–specific regions correlate with insulation scores calculated based on predicted contacts significantly better than with insulation scores based on experimental hepatocyte data (P-value 4 × 10−6).
Finally, we ran 3DPredictor on human GM12878 data. According to all metrics except SCC, predictions fit experimentally derived Hi-C interactions better than data from other cell types, even when using a single chromosome for training (Supplemental Fig. S5). At the same time, however, transferring interaction frequencies from other cell types results in better SCC values compared to the predictions, with only one exception on Chromosome 9, and, in general, SCC values obtained on human data were slightly lower than obtained on mouse data.
When focused on EP contacts, we found that for this specific set of interactions predictions accuracy was the same as for other interactions. The MRE of contact frequencies for interacting (according to SlideBase and GeneHancer databases) EP pairs was slightly lower than for all chromatin interactions predicted in monocytes and the MSE and MAE slightly higher (Fig. S5A; Supplemental Fig. S6). In general, experimentally derived contact frequencies of EP pairs in monocytes were highly correlated with predicted contact frequencies for corresponding loci in these cells (Fig. 5B). We defined cell type–specific EP interactions (see Methods) to examine whether 3DPredictor captures differences in EP interactions between cell types. As for the cell type–specific TADs, the difference between predicted and experimentally measured EP interactions was smaller than between interactions of these enhancers and promoters measured in different cell types (Fig. 5C). These results have also been confirmed using mouse data (Supplemental Fig. S7) and both low-resolution (25 kb) (Fig. 5; Supplemental Fig. S7) and high-resolution (5 kb) (Supplemental Figs. S6, S7) models.
Figure 5.

Accurate prediction of promoter–enhancer interaction frequencies. (A) Prediction accuracy of contact frequencies of EP pairs defined as “interacting” in monocytes according to SlideBase and GeneHancer databases (“EP”), and all other pairs of loci (“all except EP”). (B) Scatterplot displaying predicted (y-axis) and experimentally measured (x-axis) contact frequencies for interacting EP pairs. (C) Distribution of the similarity scores for cell type–specific EP interactions in different cell types (K562 versus monocytes) or experimental and predicted data (K562 experimental versus K562 predicted and monocytes experimental versus monocytes predicted). See Methods for definition of cell type–specific EP interactions and similarity scores. Data in A–C provided for 25-kb resolution.
We next used 3DPredictor trained on mouse hepatocytes data (single chromosome or half of the genome) to predict contact frequencies in mouse NPC (Fig. 6A). Predictions of spatial interactions for the cell type that was not used for training appeared to be as good as when the same cell type was used for training and validation (Fig. 6B–F). From the practical point of view, this indicates that our approach can be used to predict three-dimensional genome organization, including EP contacts, in those cell types where 3C-data are not available. From a fundamental standpoint, these results show that principles of genome architecture are very similar in different cell types.
Figure 6.

3DPredictor accurately reconstructs genome organization of novel cell types. (A) Example of mouse NPC Hi-C contact map derived from experimental data (above diagonal) or obtained using 3DPredictor trained on hepatocyte contacts and provided with epigenetic data relevant for NPC. (B–F) SCC (B), Pearson's correlation (C), MRE (D), MSE (E), or MAE (F) measurements of 3DPredictor accuracy for training and validation on the same (green and purple lines) or different (red line) cell types.
Comparing 3DPredictor with other models
There are several computational tools which could quantitatively predict short- and mid-range chromatin interactions (see Discussion for comprehensive comparison of these tools). For example, MEGABASE + MiChroM (Di Pierro et al. 2017) predicts chromatin interactions at 50-kb resolution using information about epigenetic marks and CTCF loops. Whereas modeling of CTCF-mediated looping interactions requires Hi-C data to infer loop anchors, use of the reduced MiChroM Hamiltonian lacking the term in that energy function that models the CTCF-mediated looping interactions can be used to predict chromatin contacts without any experimental measurements of 3D genome organization (Di Pierro et al. 2017). We benchmarked 3DPredictor against this reduced MEGABASE + MiChroM model and found that 3DPredictor significantly outperforms it, showing much higher SCC (Supplemental Table S5; Supplemental Fig. S13A–C). However, we wish to note that MEGABASE + MiChroM was originally developed to capture long-range interactions mediated by chromatin compartmentalization, and lack of information about CTCF-mediated loops could explain, at least partially, poor performance of short-range interactions prediction.
Qi and Zhang have recently proposed another model based on polymer physics to predict Hi-C interactions using epigenetic data (Qi and Zhang 2019). In contrast to the full MEGABASE + MiChroM model, Qi and Zhang do not use experimental 3C information to define CTCF-mediated loop anchors, requiring only ChIP-seq data and genomic sequence to describe the CTCF binding landscape. When employing the approach proposed by Qi and Zhang to infer chromatin contacts in GM128787 cells, we obtain better results compared to the reduced MEGABASE + MiChroM model (Supplemental Tables S5, S7). However, performance of 3DPredictor on the same data set was even higher, judged by SCC, Pearson's correlation, MSE, and MAE (Supplemental Fig. S15A–C; Supplemental Table S7). It is worth pointing out that the polymer models were developed with 3D structures in mind and are useful for studying compartmentalization and higher-order contacts as well.
A statistical approach showing that CTCF looping and gene expression could explain chromatin contacts in mammalian cells was recently proposed by Rowley et al. (2017). This approach requires a very limited amount of information as an input; however, similarly to the full MEGABASE + MiChroM model, it cannot be used to predict chromatin interactions, because information about CTCF looping should be extracted from experimental Hi-C data. For example, in the region of Сhromosome 4 of GM12878 cells, analyzed by Rowley et al. (2017), their model uses only 63 manually selected CTCF sites, which comprise ∼35.4% of all CTCF-bound sites in this region (Supplemental Fig. S16). Moreover, the Rowley et al. (2017) approach requires Hi-C information to define pairs of interacting CTCF sites. This information cannot be trivially obtained from ChIP-seq data because, in some cases, loops are formed between distal CTCF-bound sites, skipping the nearest CTCF-bound site in convergent orientation (see Supplemental Fig. S16 for representative examples and Kai et al. 2018 for systematic analysis). Nevertheless, we compared 3DPredictor with the Rowley et al. (2017) model and found that the latter gives significantly better results (Supplemental Table S6; Supplemental Fig. S14A–C). Consistent with the fact that Rowley et al. (2017) derived information about CTCF-mediated looping from the experimental data, their model captures experimental loops especially well (Supplemental Fig. S14C). Although 3DPredictor does not require any experimental 3C-information, it also captures approximately half of the looping interactions (Supplemental Fig. S17), and predicted frequencies of contacts between loop anchors were higher than between other genomic regions (Supplemental Fig. S14D).
Predicting effects of chromosomal rearrangements on three-dimensional genome organization
One of the applications of enhancer targets prediction is understanding of EP rewiring after chromosomal rearrangements. There are several well-studied examples of pathological changes in EP contacts caused by deletions, inversions (Lupiáñez et al. 2015), or duplications (Franke et al. 2016). Recently, PRISMR (Bianco et al. 2018) was developed to resolve the chromatin structure of a rearranged genome. Although impressively accurate, PRISMR requires Hi-C data to optimize chromatin model parameters, which limits its usage to cell types with available Hi-C data or genomic regions with three-dimensional structure conserved across cell types. 3DPredictor lacks these limitations, as we have shown that it can predict chromatin packaging of cell type–specific regions and previously unstudied cell types.
We employed recently generated 5C data describing mouse Epha4 rearrangements to find whether 3DPredictor can infer ectopic interactions in the mutated genome. We re-analyzed 5C data generated from wild-type cells, as well as cells carrying homozygous deletion of ∼1.5 Mb encompassing Epha4 gene (Lupiáñez et al. 2015). This deletion (referred to as DelB in Bianco et al. 2018) results in establishment of ectopic contacts between the Pax3 gene and Epha4 enhancers cluster, which is associated with Pax3 misexpression, leading to brachydactyly.
We ran 3DPredictor trained on mouse hepatocytes to infer three-dimensional organization of the rearranged Epha4 locus in hindlimb cells. We did not use any a priori knowledge of the three-dimensional structure of the wild-type Epha4 locus in hindlimb cells, yet 3DPredictor results were very similar to experimental data (Fig. 7A). We used the method described in Bianco et al. (2018) to find ectopic interactions in the rearranged locus. Out of 1561 interactions inferred from the experimental data, 589 were captured by 3DPredictor, including a majority of interactions between the Pax3 gene and Epha4 enhancers (Fig. 7A,B). The overlap between real and predicted ectopic interactions was large and differed significantly from a randomized control (P-value < 5 × 10−6) (Fig. 7C). This shows that our model successfully predicts ectopic interactions in the rearranged genome.
Figure 7.

3DPredictor captures three-dimensional organization of rearranged genomic regions. (A) Contact map of mouse DelB locus, carrying homozygous deletion of ∼1.5 Mb, with experimentally measured contacts in the top and 3DPredictor modeling results in the bottom. White lines correspond to contacts of the deleted locus. Note ectopic interactions between Pax3 and Epha4 TADs (indicated by arrows). These ectopic interactions are even more visible in B, where the same region is plotted and only those interactions which differ between WT and DelB by more than three standard deviations are kept. In A, the color indicates contact counts, whereas in B, the color indicates significance of differences between WT and DelB data. (C) Sizes of observed (red vertical bar) and expected (blue bars) overlaps between experimental and predicted ectopic interactions.
Discussion
Machine-learning approaches are actively employed to capture complex epigenetic signatures underlying chromatin contacts. As we have shown here, biological data may have a specific structure, which should be accounted for when designing computational experiments. For example, pairs of loci with overlapping windows partially share the epigenetic environment and often display similar three-dimensional architecture. This means that these regions cannot represent independent samples in training and validation data sets, and correlations captured by machine-learning approaches do not reflect the causation underlying genome architecture if overlapping regions are present in both training and validation data sets.
We benchmarked TargetFinder because it is often cited as a straightforward tool and employed for prediction of EP interactions (Atlasi and Stunnenberg 2017; Gudmundsson et al. 2017; Moorthy et al. 2017; Stricker et al. 2017; Yang et al. 2017b; Wu et al. 2018; Zhu et al. 2018); however, this is not the sole example of research that does not take into account this peculiarity of the biological data. For instance, recently published EP2vec (Zeng et al. 2018) utilizes the same data set as TargetFinder and constructs training and validation samples in the same way. Another tool aimed to predict CTCF loops, CTCF-MP (Zhang et al. 2018), does not take into consideration nested loops when employing window features. Although both EP2vec and CTCF-MP can generate predictions without window information, performance of such a setup is lower: ∼10% of accuracy drops when CTCF-MP is trained without DNase I and ChIP-seq window features and F1-score drops ∼2%–4% when EP2vec is trained without TargetFinder-derived window features.
In the recent preprint describing a tool for HiC-data prediction, HiC-Reg (Zhang et al. 2019), the authors also show that sharing genomic regions between training and validation data sets improves prediction scores. However, the authors connect this observation to a chromosome-specific biological mechanism, which cannot be modeled when overlapping data are omitted from the validation set. Whereas chromosome- and even region-specific mechanisms of DNA-packaging indeed exist (Jiang et al. 2017)—and we have also shown that prediction is better when multiple chromosomes are used to train the model—better results of intrachromosomal cross-validation are likely to originate from existence of overlapping regions. One should note that, although pairs with overlapping left or right anchors are not shared between training and validation data sets in HiC-Reg (so-called easy samples), authors do not exclude regions, which share a part of the window between interacting anchors.
We next raised the question of definition of promoter–enhancer interaction. Currently, most of the studies use all 3C-interactions, which differ statistically from distance-adjusted background as functional EP interactions (Whalen et al. 2016; Mishra and Hawkins 2017). According to our results, functional interactions of promoters and enhancers do not fully overlap with Hi-C loops and probably do not overlap completely with any other set of enriched interactions. Whereas spatial proximity is required for EP communication, it is not clear which spatial distance is necessary and sufficient to achieve functional interaction. For example, the recent study of the Shh-ZRS TAD showed that nearly the entire ∼900-kb intra-TAD region can be activated by the ZRS enhancer, although pronounced looping was observed only between the Shh promoter and ZRS enhancer (Symmons et al. 2016). Removing the Shh-ZRS TAD boundary reduces intra-TAD contact frequencies to the background level and disturbs Shh expression in the developing limbs; however, relocating the enhancer closer to the Shh promoter region restores the expression pattern. These results indicate that background-level interactions within the TAD might be sufficient to establish functional connections of the promoter and enhancer. Moreover, a recent paper reports that intra-TAD promoter regions often show a significant level of interaction with TAD boundaries, and disruption of these interactions does not lead to changes of expression levels (Sun et al. 2019). To sum up, our view is that a statistical increase of spatial contact frequencies, i.e., formation of loops, is an important indicator of promoter–enhancer connectivity but cannot be solely used to distinguish functional interactions. In accord with this, a recent large-scale CRISPR assay of promoter–enhancer connections (Fulco et al. 2019) suggested a quantitative “contact-by-activity” model of EP interaction. In this model, enhancer impact is quantitative and proportional to both promoter–enhancer proximity and enhancer activity. Whereas the latter can be estimated using DNase I or ATAC-seq data available for many cell types, here, we described 3DPredictor, which can be used to quantitatively predict the spatial architecture of chromatin, including enhancer–promoter interactions, to supplement this ATAC-seq or DNA-seq data.
There are many methods published previously for prediction of TAD boundaries, Hi-C interactions, and enhancer–promoter interactions (Xu et al. 2018). CITD (Chen et al. 2016), MEGABASE (Di Pierro et al. 2017), EpiTensor (Zhu et al. 2016), and the model described in Qi and Zhang (2019) can predict 3D-interactions, including EP interactions, at various resolutions based on epigenetic data. However, these tools require a large amount of epigenetic information: 5–10 patterns of histone modifications for CITD; 11 for MEGABASE; 12 for Qi and Zhang (2019); and 16 histone modifications and additional data for EpiTensor. CISD (Zhang et al. 2017) and PRISMR (Bianco et al. 2018) are able to infer chromatin contacts genome-wide as well but require Hi-C data as an input. In contrast, 3DPredictor could make predictions when supplied with CTCF and RNA-seq data only.
Chromatin simulations proposed by Rowley et al. (2017) also utilize CTCF and transcription data only; however, they require manual selection of interacting CTCF sites based on Hi-C data, thus making it impossible to predict 3D-interactions from epigenetic data alone.
3DEpiLoop (Al Bkhetan and Plewczynski 2018b), Lollipop (Kai et al. 2018), CTCF-MP (Zhang et al. 2018), the BART model (Huang et al. 2015), and other tools (Fortin and Hansen 2015; Jenkinson et al. 2017; Al Bkhetan and Plewczynski 2018a) could predict specific chromatin features, such as TAD boundaries, A/B-compartments, and CTCF-interactions or loops. However, in contrast to 3DPredictor, these approaches (1) do not infer EP interactions, (2) perform qualitative, rather than quantitative, prediction (i.e., classification), and (3) for most of them, significantly more input information is required than for 3DPredictor.
There are multiple computation tools designed specifically to infer EP interactions (for example, Whalen et al. 2016; Cao et al. 2017; O'Connor et al. 2017; Hait et al. 2018; Zeng et al. 2018; Li et al. 2019). However, all these tools are fundamentally different from 3DPredictor, as they consider EP interaction as qualitative, rather than quantitative. Moreover, most of them require a large amount of epigenetic data to make predictions, and performance of some of them (Whalen et al. 2016; Zeng et al. 2018) might be overestimated, as discussed above.
To sum up, 3DPredictor is a unique tool, which allows predicting a large set of interactions, including EP interactions, quantitatively, using only a small amount of input epigenetic data.
It is essential that our model not only predicts chromatin interactions in the normal genome but could also capture ectopic interactions, which are formed as a result of chromosomal rearrangements. Currently, both the experimental data describing 3D genome alterations associated with known rearrangements and tools modeling the spatial landscape of novel variants are limited. At the same time, we (Gridina et al. 2018) and others (Lupiáñez et al. 2015; Franke et al. 2016; Redin et al. 2017; Zepeda-Mendoza et al. 2017) have recently reported novel variations with unexpected pathological phenotypes, which might be explained, at least partially, by changes of chromatin organization (Fishman et al. 2018; Spielmann et al. 2018). Future development and validation of models predicting chromatin contacts in a rearranged genome is essential for a better understanding of the biological consequences of these rearrangements. Moreover, integrating chromatin interactions, derived from 3DPredictor, with enhancer activity information using the “activity-by-contact” model may allow precise estimation of transcriptional changes caused by structural variations.
Methods
Hi-C data processing
Hi-C data for mouse hepatocytes (GSE95116) and cardiomyocytes (SRX2658510) were downloaded from NCBI and processed using Juicer (Durand et al. 2016b). Resulting .hic-files are deposited at the genedev hic-file server (http://genedev.bionet.nsc.ru/site/hic_out/) under accessions “Hepat” and “CardioMyo”. Hi-C data for mouse ES cells, NPC, cortex (Bonev et al. 2017), CH12.LX lymphocytes (Rao et al. 2014), and human GM12878, K562, IMR-90, NHEK (Rao et al. 2014), macrophages, and monocytes (Phanstiel et al. 2017) are available at the AidenLab hic-file server via Juicebox and Juicer Tools (Durand et al. 2016a,b). All data sets were KR-normalized. For each Hi-C data set, contacts were obtained at 25- or 5-kb resolutions using the Juicer Tools dump command. To be able to perform comparisons between cell types, we normalized data sets, dividing each contact by normalization coefficient Coef, which reflects average bin coverage
where Ci,j is the contacts between the ith and jth bins, K is the number of bins on Chromosome 1, and N is the number of bins in the genome. To speed up Coef computation, we only used bins of Chromosome 1, although this should not affect results, as we use KR-normalized matrices where coverage of all bins is roughly equal.
Loops were called by the Juicer Tools HiCCUPS command with default parameters using heat maps at 25- or 5-kb resolution. K562 loops presented in Figure 2 are from Rao et al. (2014).
First eigenvector (E1) values of Hi-C matrixes were obtained using the Juicer Tools eigenvector module.
5C data describing the three-dimensional organization of the wild-type and mutated mouse Epha4 locus in distal limb buds were downloaded from GEO: GSE92291. Data were processed by the HiC-Pro (Servant et al. 2015) pipeline using the mm10 genome.
The relative error of Hi-C contact counts shown in Supplemental Figure S3 was estimated based on a binomial distribution
where N is a number of Hi-C reads between contacted loci. The average and standard deviation of relative errors were independently calculated for each genomic distance.
To estimate correlation of contact counts on different resolutions for Supplemental Figure S9, we used data for Chromosome 10 of the GM12878 cell line. We randomly choose 1000 loci pairs and calculated Pearson's correlation between KR-normalized contact frequencies on different resolutions. We aggregated calculations of 100 independent samplings by averaging to obtain final results.
Definition of promoters and enhancers
For human macrophages and monocytes, enhancers were defined using the SlideBase (http://slidebase.binf.ku.dk) database. This database is supported by the FANTOM5 consortium (http://fantom.gsc.riken.jp/data/) (Andersson et al. 2014) and represents a map of human regulatory elements of each cellular state. It contains levels of enhancer expression based on CAGE sequencing of RNA isolated from every major human organ, over 200 cancer cell lines, 30 time courses of cellular differentiation, mouse developmental time courses, and over 200 primary cell types. Thereby, an enhancer can be specific to a set of primary cells and organs (tissue samples) or can be broadly (or ubiquitously) expressed. We took into account enhancers displayed in >25% of samples related to the target cell line.
Using the GeneHancer database (https://www.genecards.org), we defined gene promoters regulated by a given enhancer. GeneHancer is a database of genome-wide enhancer-to-gene and promoter-to-gene associations, embedded in GeneCards. GeneHancer EP associations were generated using the following information:
eQTLs (expression quantitative trait loci) from GTEx;
capture Hi-C EP long-range interactions;
expression correlations between eRNAs and candidate target genes from FANTOM5;
cross-tissue expression correlations between a transcription factor interacting with an enhancer and a candidate target gene; and
GeneHancer-gene distance-based associations, scored utilizing inferred distance distributions. Associations include several approaches: (a) nearest neighbors, where each GeneHancer is associated with its two proximal genes; (b) overlaps with the gene territory (intragenic); (c) proximity to the gene TSS (<2 kb). TSS proximity scores are boosted to elevate GeneHancer associations in the vicinity of the gene TSS.
The “true” interacting EP pairs of human monocytes and macrophages were calculated by combining the list of cell type–specific enhancers from SlideBase and a list of enhancer-gene associations from GeneHancer.
When using the TargetFinder pipeline on human data, we used the authors’ definition of active promoters and enhancers and obtained coordinates from https://github.com/shwhalen/targetfinder. For mouse data, we first obtained promoters using TSSs (transcription start sites) downloaded from UCSC and active enhancers based on annotations from Bogu et al. (2016). Next, we defined interacting pairs as promoters and enhancers located within the anchors of one loop.
ChIP-seq data processing
All ChIP-seq data for human GM12878 and K562 cell lines were downloaded from https://github.com/shwhalen/targetfinder. ChIP-seq data for mouse hepatocytes (NCBI SRX2578761–SRX2578762), mouse NPC (NCBI SRX2636706–SRX2636707, ENCODE ChIP-seq data for forebrain embryo 13.5, GSE96107, GSE96107), mouse cortex (ENCODE ChIP-seq data for forebrain embryo 13.5, GSE96107, GSE96107), and human monocytes (ENCODE ENCSR000ATN) were downloaded from NCBI or ENCODE and processed using the aquas pipeline (https://github.com/kundajelab/chipseq_pipeline). CTCF motif orientation was defined using GimmeMotifs (van Heeringen and Veenstra 2011) software.
RNA-seq data processing
RNA-seq data for human GM12878 (ENCODE ENCFF212CQQ) and human K562 (ENCODE ENCFF026BMH) cell lines were downloaded from ENCODE. RNA-seq data for mouse hepatocytes (NCBI GSE95111) and mouse NPC (NCBI GSM2533845) were downloaded from NCBI. Data for human monocytes (NCBI SRX2785183) were downloaded from NCBI and processed using standard protocols with HISAT2 and StringTie (Pertea et al. 2016).
Prediction of three-dimensional interaction frequencies
For training purposes, all data were split into nonoverlapping genomic intervals. Usually, we use one or several chromosomes for training and other chromosomes for validation. To perform prediction genome-wide, we first used odd chromosomes for training and made predictions for contacts on even chromosomes, and then used even chromosomes for training and predicted contacts on odd chromosomes. Unless otherwise mentioned, we used only CTCF and RNA-seq data for predictions. For all results except those described in the Supplemental Note, we used Gradient Boosting with parameters n_estimators = 100, max_depth = 9, subsample = 0.7. Predictors parametrization and other details are explained in detail in the Supplemental Note.
Estimating predictions efficiency
We used several metrics to choose the best model. Pearson's correlation is the most common metric; however, Pearson's correlation is dominated by dependence of contact frequencies from genomic distance. Thus, we also used the SCC metric (Yang et al. 2017a) which measures correlation of contact frequencies on each diagonal of the Hi-C matrix independently, thereby neglecting the factor of genomic distance. To reduce the effect of random noise, we smoothed the Hi-C matrices before calculating SCC, as was suggested by Yang et al. (2017a). All comparisons were carried out with the same noise smoothing parameter h = 2 (see Supplemental Note; Supplemental Table S4 for justification of the h value). In addition, to evaluate the model's quality, we used other metrics such as MSE, MAE, and MRE.
To benchmark model predictions against transfer of contact counts from another cell type, we performed pairwise comparisons of mouse Hi-C data (CH12.LX lymphocytes, cortical neurons, cardiomyocytes cells, and hepatocytes) using the same metrics as described above. Similarly, we compared human NHEK, K562, IMR-90, GM12878 to benchmark predictions of human data.
Comparing 3DPredictor with other models
Chromatin interactions for GM12878 cells predicted by MEGABASE + MiChroM were downloaded from the Juicebox server (https://s3.amazonaws.com/hicfiles/external/ctbp_8_4_17/all_intra_megabase_michrom.hic). As these interactions were at 50-kb resolution, we predicted the same regions at 5-kb resolution and averaged data to obtain contacts at 50 kb.
Interaction frequencies for a 53- to 75-Mb region on Chromоsome 4 of GM12878 cells, predicted by Rowley et al. (2017), as well as CTCF loop anchors were provided by VG Corces, MJ Rowley, and MH Nichols (pers. comm.).
Interaction frequencies for a 20- to 45-Mb region on Chromоsome 1 of GM12878 cells, predicted by Qi and Zhang (2019), were provided by Y Qi and B Zhang (pers. comm.).
Note that we used here a SCC smoothing parameter of 2 for all comparisons, whereas Qi and Zhang (2019) used SCC smoothing parameter values >5. Note that changing the smoothing parameter does not affect results of the 3DPredictor benchmark (see Supplemental Fig. E in Qi and Zhang 2019 and Supplemental Note; Supplemental Tables S4, S7 in this paper for details of the SCC smoothing parameter effect).
Defining cell type–specific TADs and cell type–specific EP interactions
To define cell type–specific TAD boundaries, we utilized an insulation-based score, which reflects depletion of contacts spanning the putative TAD boundary, similar to the approach used in Sexton et al. (2012), Vietri Rudan et al. (2015), and Fishman et al. (2019) but with some modifications. The schematic representation of the current approach is shown in Supplemental Figure S8. In particular, for each bin i of the Hi-C matrix A, we define four vectors aL, bL, aR, bR , each containing N elements
where Ai,j is the number of contacts between bins i and j, and N is empirical constant which was equal to 5 in this study. Thus, for two bins a and b, surrounding the bin i, vectors aL, bL, aR, bR describe local (± N bins) contacts.
Then, the insulation score Si of bin i was computed by dividing the frequency of contacts crossing bin i to the frequency of distance-matched contacts located downstream from or upstream of i (Supplemental Fig. S8) and summing obtained ratios
If, for a bin i, we observed a high (above the empirically defined upper threshold) insulation-based score in one cell type and low (under the empirically defined lower threshold) score in another cell type, then we considered the bin i as a center of the cell type–specific region. We defined the upper and lower thresholds based on the distribution of insulation scores (Supplemental Fig. S18) so that the upper threshold corresponded to the strong insulation and the lower threshold was close to the natural noise of insulation in Hi-C data. In particular, we used the following parameters to compare NPC and hepatocytes: bin size = 25 kb; N = five, which means that we used the interval ±100 kb around the putative boundary to calculate the insulation score; the upper threshold = 3 × N = 15; the lower threshold = 2.4 × N = 12. With these parameters, we obtained 88 cell type–specific regions.
To estimate prediction accuracy for cell type–specific EP interactions, we compared differences between predicted and control data with differences between cell types and replicates. We characterized contacts of EP pairs by an observed-to-expected contacts ratio (OE). The EP interactions were referred to as cell type–specific, if the OE differs between replicates less than two times and differs between cell types more than two times:
To measure the similarity of cell type–specific interactions in two samples (i.e., predicted and experimental data or two experimental samples), we calculated the mean difference of OE values for corresponding interactions:
The cell type–specific interactions were obtained comparing mouse hepatocytes (combined data and replicates) with NPC (only combined data) and human K562 cell (combined data and by replicates) with monocytes (only combined data) on 5-kb and 25-kb resolution.
Analysis of looping contacts
For quantitative comparison of interactions in loop anchors shown in Supplemental Figure S17, we used loops derived from the experimental NPC Hi-C data using HiCCUPS. We next aimed to call loop anchors based on 3DPredictor data using HiCCUPS. However, HiCCUPS required normalization vectors to be provided in .hic-files, and since these vectors were not available for predicted data, we failed to annotate the loops automatically. Thus, we manually annotated all loops for both experimental and predicted Hi-C maps of Chromosome 5 of NPC cells (Supplemental Tables S8, S9 show coordinates of the manually annotated loop anchors) and compared obtained data to estimate the number of correctly predicted loops.
Modeling chromosomal rearrangements
To model chromosomal rearrangements, we used 3DPredictor trained on mouse hepatocyte cells. To generate predictors, we obtained CTCF (NCBI SRX1975285–SRX1975286) and RNA-seq (NCBI SRX1975216–SRX1975217) data from wild-type mouse hindlimb E11.5 cells. Next, we deleted all CTCF peaks and genes from the region [mm10]: Chr 1: 76,392,403–78,064,264, which corresponds to the deletion coordinates described in Bianco et al. (2018). The resulting set of predictors was used to model all chromatin contacts within the region [mm10]: Chr 1: 70,950,000–81,000,000. To compare contact frequencies predicted by the model with experimental data, we defined ectopic interactions as described in Bianco et al. (2018). We first generated a normalized difference matrix between mutated and WT matrices. For this, we multiplied the mutant matrix by a coefficient that equalizes the coverage of regions that are not involved in the mutation. Next, we subtracted the WT matrix from the mutated matrix. We normalized the difference matrix by dividing each subdiagonal by the average number of reads observed at the corresponding genomic distance in WT data. After we get the normalized difference matrix, we found ectopic interactions for each subdiagonal. Specifically, we filtered out the subdiagonal elements, which were above the 96th percentile of all subdiagonal values, and calculated the standard deviation of the remaining values. All points that differ from zero by more than three standard deviations were considered ectopic.
Software availability
3DPredictor source code (https://github.com/labdevgen/3DPredictor) and Jupiter Notebook with the code used to reproduce TargetFinder results (https://github.com/labdevgen/targetFinderTests) are both freely available on GitHub and in Supplemental Code.
Supplementary Material
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (RFBR) grant #18-29-13021 and Russian Science Foundation grant #17-74-10143. All computations were performed with support from the Computational Cluster of the Novosibirsk State University and Computational Nodes of the Institute of Cytology and Genetics (Budget Project #0324-2019-0041). We thank Dr. Nariman Battulin and Emil Valeev for scientific discussions. We also thank V.G. Corces, M.J. Rowley, and M.H. Nichols, and Y. Qi and B. Zhang, who provided their predictions of GM12878 contacts. We also thank Michele Di Pierro, who pointed us to the MEGABASE + MiChroM predicted contacts on the Juicebox web-server.
Author contributions: V.F. proposed the study. P.S.B., D.F., and V.F. benchmarked the TargetFinder algorithm. V.F. and P.S.B. wrote the main parts of the 3DPredictor code. E.A.M. implemented the metrics of algorithm performance and cell type–specific TADs comparisons. V.F., P.S.B., E.A.M., and M.A.N. developed different predictor parameterizations and compared training parameters. M.A.N. prepared training and validation data and identified and analyzed cell type–specific EP interactions. P.S.B. performed Epha4 locus modeling. All authors contributed to manuscript preparation.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.249367.119.
References
- Al Bkhetan Z, Plewczynski D. 2018a. Three-dimensional epigenome statistical model: genome-wide chromatin looping prediction. Sci Rep 8: 5217 10.1038/s41598-018-23276-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Bkhetan Z, Plewczynski D. 2018b. Three-dimensional epigenome statistical model: genome-wide chromatin looping prediction. Sci Rep 8: 5217 10.1038/s41598-018-23276-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. 2014. An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461. 10.1038/nature12787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlasi Y, Stunnenberg HG. 2017. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet 18: 643–658. 10.1038/nrg.2017.57 [DOI] [PubMed] [Google Scholar]
- Battulin N, Fishman VS, Mazur AM, Pomaznoy M, Khabarova AA, Afonnikov DA, Prokhortchouk EB, Serov OL. 2015. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol 16: 77 10.1186/s13059-015-0642-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco S, Lupiáñez DG, Chiariello AM, Annunziatella C, Kraft K, Schöpflin R, Wittler L, Andrey G, Vingron M, Pombo A, et al. 2018. Polymer physics predicts the effects of structural variants on chromatin architecture. Nat Genet 50: 662–667. 10.1038/s41588-018-0098-8 [DOI] [PubMed] [Google Scholar]
- Bogu GK, Vizán P, Stanton LW, Beato M, Di Croce L, Marti-Renom MA. 2016. Chromatin and RNA maps reveal regulatory long noncoding RNAs in mouse. Mol Cell Biol 36: 809–819. 10.1128/mcb.00955-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, et al. 2017. Multiscale 3D genome rewiring during mouse neural development. Cell 171: 557–572.e24. 10.1016/j.cell.2017.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckle A, Brackley CA, Boyle S, Marenduzzo D, Gilbert N. 2018. Polymer simulations of heteromorphic chromatin predict the 3D folding of complex genomic loci. Mol Cell 72: 786–797.e11. 10.1016/j.molcel.2018.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Anyansi C, Hu X, Xu L, Xiong L, Tang W, Mok MTS, Cheng C, Fan X, Gerstein M, et al. 2017. Reconstruction of enhancer–target networks in 935 samples of human primary cells, tissues and cell lines. Nat Genet 49: 1428–1436. 10.1038/ng.3950 [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang Y, Xuan Z, Chen M, Zhang MQ. 2016. De novo deciphering three-dimensional chromatin interaction and topological domains by wavelet transformation of epigenetic profiles. Nucleic Acids Res 44: e106 10.1093/nar/gkw225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiariello AM, Annunziatella C, Bianco S, Esposito A, Nicodemi M. 2016. Polymer physics of chromosome large-scale 3D organisation. Sci Rep 6: 29775 10.1038/srep29775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pierro M, Cheng RR, Lieberman Aiden E, Wolynes PG, Onuchic JN. 2017. De novo prediction of human chromosome structures: epigenetic marking patterns encode genome architecture. Proc Natl Acad Sci 114: 12126–12131. 10.1073/pnas.1714980114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. 2016a. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3: 99–101. 10.1016/j.cels.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, Aiden EL. 2016b. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3: 95–98. 10.1016/j.cels.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman VS, Salnikov PA, Battulin NR. 2018. Interpreting chromosomal rearrangements in the context of 3-dimensional genome organization: a practical guide for medical genetics. Biochemistry (Mosc) 83: 393–401. 10.1134/S0006297918040107 [DOI] [PubMed] [Google Scholar]
- Fishman V, Battulin N, Nuriddinov M, Maslova A, Zlotina A, Strunov A, Chervyakova D, Korablev A, Serov O, Krasikova A. 2019. 3D organization of chicken genome demonstrates evolutionary conservation of topologically associated domains and highlights unique architecture of erythrocytes’ chromatin. Nucleic Acids Res 47: 648–665. 10.1093/nar/gky1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J-P, Hansen KD. 2015. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol 16: 180 10.1186/s13059-015-0741-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schöpflin R, Kraft K, Kempfer R, Jerković I, Chan W-L, et al. 2016. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538: 265–269. 10.1038/nature19800 [DOI] [PubMed] [Google Scholar]
- Fudenberg G, Abdennur N, Imakaev M, Goloborodko A, Mirny LA. 2017. Emerging evidence of chromosome folding by loop extrusion. Cold Spring Harb Symp Quant Biol 82: 45–55. 10.1101/sqb.2017.82.034710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, Engreitz JM. 2016. Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 354: 769–773. 10.1126/science.aag2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. 2019. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat Genet 51: 1664–1669. 10.1038/s41588-019-0538-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong EEM, Levine M. 2018. Developmental enhancers and chromosome topology. Science 361: 1341–1345. 10.1126/science.aau0320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gridina MM, Matveeva NM, Fishman VS, Menzorov AG, Kizilova HA, Beregovoy NA, Kovrigin II, Pristyazhnyuk IE, Oscorbin IP, Filipenko ML, et al. 2018. Allele-specific biased expression of the CNTN6 gene in iPS cell-derived neurons from a patient with intellectual disability and 3p26.3 microduplication involving the CNTN6 gene. Mol Neurobiol 55: 6533–6546. 10.1007/s12035-017-0851-5 [DOI] [PubMed] [Google Scholar]
- Gudmundsson J, Thorleifsson G, Sigurdsson JK, Stefansdottir L, Jonasson JG, Gudjonsson SA, Gudbjartsson DF, Masson G, Johannsdottir H, Halldorsson GH, et al. 2017. A genome-wide association study yields five novel thyroid cancer risk loci. Nat Commun 8: 14517 10.1038/ncomms14517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait TA, Amar D, Shamir R, Elkon R. 2018. FOCS: a novel method for analyzing enhancer and gene activity patterns infers an extensive enhancer–promoter map. Genome Biol 19: 56 10.1186/s13059-018-1432-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Marco E, Pinello L, Yuan G. 2015. Predicting chromatin organization using histone marks. Genome Biol 16: 162 10.1186/s13059-015-0740-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibn-Salem J, Andrade-Navarro MA. 2019. 7C: Computational Chromosome Conformation Capture by Correlation of ChIP-seq at CTCF motifs. BMC Genomics 20: 777 10.1186/s12864-019-6088-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson G, Pujadas E, Goutsias J, Feinberg AP. 2017. Potential energy landscapes identify the information-theoretic nature of the epigenome. Nat Genet 49: 719–729. 10.1038/ng.3811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Loh YHE, Rajarajan P, Hirayama T, Liao W, Kassim BS, Javidfar B, Hartley BJ, Kleofas L, Park RB, et al. 2017. The methyltransferase SETDB1 regulates a large neuron-specific topological chromatin domain. Nat Genet 49: 1239–1250. 10.1038/ng.3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai Y, Andricovich J, Zeng Z, Zhu J, Tzatsos A, Peng W. 2018. Predicting CTCF-mediated chromatin interactions by integrating genomic and epigenomic features. Nat Commun 9: 4221 10.1038/s41467-018-06664-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wong WH, Jiang R. 2019. DeepTACT: predicting 3D chromatin contacts via bootstrapping deep learning. Nucleic Acids Res 47: e60 10.1093/nar/gkz167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. 2015. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161: 1012–1025. 10.1016/j.cell.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Hawkins RD. 2017. Three-dimensional genome architecture and emerging technologies: looping in disease. Genome Med 9: 87 10.1186/s13073-017-0477-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BL, Aitken S, Semple CA. 2015. Integrative modeling reveals the principles of multi-scale chromatin boundary formation in human nuclear organization. Genome Biol 16: 110 10.1186/s13059-015-0661-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy SD, Davidson S, Shchuka VM, Singh G, Malek-Gilani N, Langroudi L, Martchenko A, So V, Macpherson NN, Mitchell JA. 2017. Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res 27: 246–258. 10.1101/gr.210930.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor T, Bodén M, Bailey TL. 2017. CisMapper: predicting regulatory interactions from transcription factor ChIP-seq data. Nucleic Acids Res 45: e19 10.1093/nar/gkw956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. 2016. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11: 1650–1667. 10.1038/nprot.2016.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Van Bortle K, Spacek D, Hess GT, Shamim MS, Machol I, Love MI, Aiden EL, Bassik MC, Snyder MP. 2017. Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol Cell 67: 1037–1048.e6. 10.1016/j.molcel.2017.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Zhang B. 2019. Predicting three-dimensional genome organization with chromatin states. PLoS Comput Biol 15: e1007024 10.1371/journal.pcbi.1007024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680. 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, Hanscom C, Pillalamarri V, Seabra CM, Abbott MA, et al. 2017. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet 49: 36–45. 10.1038/ng.3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, Corces VG. 2017. Evolutionarily conserved principles predict 3D chromatin organization. Mol Cell 67: 837–852.e7. 10.1016/j.molcel.2017.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, Heard E, Dekker J, Barillot E. 2015. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16: 259 10.1186/s13059-015-0831-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. 2012. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148: 458–472. 10.1016/j.cell.2012.01.010 [DOI] [PubMed] [Google Scholar]
- Spielmann M, Lupiáñez DG, Mundlos S. 2018. Structural variation in the 3D genome. Nat Rev Genet 19: 453–467. 10.1038/s41576-018-0007-0 [DOI] [PubMed] [Google Scholar]
- Stricker SH, Köferle A, Beck S. 2017. From profiles to function in epigenomics. Nat Rev Genet 18: 51–66. 10.1038/nrg.2016.138 [DOI] [PubMed] [Google Scholar]
- Sun F, Chronis C, Kronenberg M, Chen XF, Su T, Lay FD, Plath K, Kurdistani SK, Carey MF. 2019. Promoter-enhancer communication occurs primarily within insulated neighborhoods. Mol Cell 73: 250–263.e5. 10.1016/j.molcel.2018.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons O, Pan L, Remeseiro S, Aktas T, Klein F, Huber W, Spitz F. 2016. The Shh topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Dev Cell 39: 529–543. 10.1016/j.devcel.2016.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. 2015. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163: 1611–1627. 10.1016/j.cell.2015.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heeringen SJ, Veenstra GJC. 2011. GimmeMotifs: a de novo motif prediction pipeline for ChIP-sequencing experiments. Bioinformatics 27: 270–271. 10.1093/bioinformatics/btq636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietri Rudan M, Barrington C, Henderson S, Ernst C, Odom DT, Tanay A, Hadjur S. 2015. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep 10: 1297–1309. 10.1016/j.celrep.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen S, Truty RM, Pollard KS. 2016. Enhancer–promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet 48: 488–496. 10.1038/ng.3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zeng J, Zhang F, Zhu Z, Qi T, Zheng Z, Lloyd-Jones LR, Marioni RE, Martin NG, Montgomery GW, et al. 2018. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun 9: 918 10.1038/s41467-018-03371-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi W, Beer MA. 2018. Local epigenomic state cannot discriminate interacting and non-interacting enhancer–promoter pairs with high accuracy. PLoS Comput Biol 14: e1006625 10.1371/journal.pcbi.1006625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Zheng X, Li B, Jin P, Qin Z, Wu H. 2018. A comprehensive review of computational prediction of genome-wide features. Brief Bioinform 10.1093/bib/bby110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Zhang F, Yardımcı GG, Song F, Hardison RC, Noble WS, Yue F, Li Q. 2017a. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res 27: 1939–1949. 10.1101/gr.220640.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhang R, Singh S, Ma J. 2017b. Exploiting sequence-based features for predicting enhancer–promoter interactions. Bioinformatics 33: i252–i260. 10.1093/bioinformatics/btx257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Wu M, Jiang R. 2018. Prediction of enhancer-promoter interactions via natural language processing. BMC Genomics 19: 84 10.1186/s12864-018-4459-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepeda-Mendoza CJ, Ibn-Salem J, Kammin T, Harris DJ, Rita D, Gripp KW, MacKenzie JJ, Gropman A, Graham B, Shaheen R, et al. 2017. Computational prediction of position effects of apparently balanced human chromosomal rearrangements. Am J Hum Genet 101: 206–217. 10.1016/j.ajhg.2017.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li F, Jia Y, Xu B, Zhang Y, Li X, Zhang Z. 2017. Characteristic arrangement of nucleosomes is predictive of chromatin interactions at kilobase resolution. Nucleic Acids Res 45: 12739–12751. 10.1093/nar/gkx885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Wang Y, Yang Y, Zhang Y, Ma J. 2018. Predicting CTCF-mediated chromatin loops using CTCF-MP. Bioinformatics 34: i133–i141. 10.1093/bioinformatics/bty248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chasman D, Knaack S, Roy S. 2019. In silico prediction of high-resolution Hi-C interaction matrices. Nat Commun 10: 5449 10.1038/s41467-019-13423-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Chen Z, Zhang K, Wang M, Medovoy D, Whitaker JW, Ding B, Li N, Zheng L, Wang W. 2016. Constructing 3D interaction maps from 1D epigenomes. Nat Commun 7: 10812 10.1038/ncomms10812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Deng W, Hu H, Ma R, Zhang S, Yang J, Peng J, Kaplan T, Zeng J. 2018. Reconstructing spatial organizations of chromosomes through manifold learning. Nucleic Acids Res 46: e50 10.1093/nar/gky065 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.