

Abstract
Nucleosomes inhibit excision repair of DNA damage caused by ultraviolet (UV) light, and it has been generally assumed that repair inhibition is equivalent on both sides of the nucleosome dyad. Here, we use genome-wide repair data to show that repair of UV damage in nucleosomes is asymmetric. In yeast, nucleosomes inhibit nucleotide excision repair (NER) of the nontranscribed strand (NTS) of genes in an asymmetric manner, with faster repair of UV damage occurring on the 5′ side of the nucleosomal DNA. Analysis of genomic repair data from UV-irradiated human cells indicates that NER activity along the NTS is also elevated on the 5′ side of nucleosomes, consistent with the repair asymmetry observed in yeast nucleosomes. Among intergenic nucleosomes, repair activity is elevated on the 5′ side of both DNA strands. The distribution of somatic mutations in nucleosomes shows the opposite asymmetry in NER-proficient skin cancers, but not in NER-deficient cancers, indicating that asymmetric repair of nucleosomal DNA imposes a strand polarity on UV mutagenesis. Somatic mutations are enriched on the relatively slow-repairing 3′ side of the nucleosomal DNA, particularly at positions where the DNA minor groove faces away from the histone octamer. Asymmetric repair and mutagenesis are likely caused by differential accessibility of the nucleosomal DNA, a consequence of its left-handed wrapping around the histone octamer.
Efficient nucleotide excision repair (NER) is critical for preventing skin cancer and other diseases, because it removes mutagenic DNA lesions, including ultraviolet (UV)-induced cyclobutane pyrimidine dimers (CPDs), from the genome (Scharer 2013). Genetic defects that reduce NER activity (e.g., in xeroderma pigmentosum [XP] patients) result in significantly elevated rates of UV-induced mutations and up to about 10,000-fold higher rate of skin cancers, including squamous cell carcinomas (SCC) and melanomas (DiGiovanna and Kraemer 2012). The primary NER pathway, known as global genomic-NER (GG-NER), initiates when the XPC protein (and its cofactors, such as UV-DDB) recognize a bulky, helix-distorting DNA lesion (Scharer 2013). A second NER pathway, known as transcription coupled-NER (TC-NER), differs only in that lesion recognition occurs when RNA polymerase II stalls at the site of damage and is therefore confined to repairing the transcribed strand (TS) of expressed genes (Hanawalt and Spivak 2008; Marteijn et al. 2014).
In eukaryotic cells, the NER machinery must recognize and repair DNA lesions that are resident in chromatin, which significantly modulates NER efficiency. For example, regions of compact heterochromatin are associated with lower NER activity and concomitantly higher mutation rates in UV-exposed skin cancers (Adar et al. 2016; Hu et al. 2017b). NER activity is also modulated by individual nucleosomes, which comprise the basic building blocks of chromatin. NER is inhibited when a CPD lesion is packaged into a nucleosome in vitro (Hara et al. 2000; Liu and Smerdon 2000), which is consistent with in vivo studies indicating that NER activity is inhibited near the central dyad axis of nucleosomes (Smerdon and Thoma 1990; Wellinger and Thoma 1997; Tijsterman et al. 1999; Mao et al. 2016; Brown et al. 2018).
Because the canonical nucleosome structure is symmetric on both sides of the central dyad axis, with similar histone content, DNA structure, and histone-DNA contacts (Luger et al. 1997), it is generally assumed that repair of DNA lesions by the NER pathway is equally efficient on both sides of the nucleosome dyad axis. However, recent reports of the existence of asymmetric subnucleosome structures in gene coding regions (Rhee et al. 2014; Ramachandran et al. 2015, 2017), reports of asymmetric accessibility of nucleosomal DNA (Lutter 1978; Zhong et al. 2016), as well as the intrinsic asymmetry of the NER incision reaction (Huang et al. 1992), suggest that further study is warranted. Moreover, if the repair of UV lesions does occur asymmetrically in nucleosomes, what effect this might have on the genomic distribution of UV-induced somatic mutations in skin cancers, particularly in cancer driver genes, is currently unclear.
To address these issues, we analyzed repair of UV damage in nucleosomes at high resolution across the yeast and human genomes. In parallel, we characterized the nucleosome distribution of somatic mutations in repair-proficient and repair-deficient skin cancers, to discern how intranucleosomal patterns of repair affect UV mutagenesis in human skin cancers.
Results
Repair of the nontranscribed strand (NTS) is modulated by the nucleosome organization in yeast genes
To study how chromatin affects the activity of the GG-NER pathway at single-nucleotide resolution across the yeast genome, we analyzed CPD repair profiles using CPD-seq data for wild-type (Mao et al. 2016) and rad16Δ mutant cells. The rad16Δ mutant cells were used as a control for these experiments, because Rad16 is specifically required for GG-NER in yeast (Verhage et al. 1994). CPD-seq uses repair enzymes T4 endonuclease V and AP endonuclease (Ape1) to specifically cleave at the CPD damage and create a new ligatable 3′-OH group, which is subsequently ligated to a second adapter, purified, and sequenced (Supplemental Fig. S1A). Initial analysis of the CPD-seq data revealed significant enrichment of dipyrimidine-associated sequencing reads (i.e., TT, TC, CT, and CC) in UV-treated rad16Δ cells, but not in the “No UV” control (Supplemental Fig. S1B), consistent with previous data for UV-irradiated wild-type (WT) yeast and human cells (Mao et al. 2016, 2018). Analysis of CPD-seq data following 2 h of repair revealed more rapid repair of the transcribed strand (TS) along the transcribed regions of yeast genes in both the WT and rad16Δ strains (Supplemental Fig. S1C), consistent with previous results (Mao et al. 2016). Deletion of RAD16 results in a greater fraction of unrepaired CPDs along the nontranscribed strand (NTS), but has little impact on repair of the TS (Supplemental Fig. S1C), consistent with its role specifically in GG-NER.
To determine how nucleosome positioning in genes influences GG-NER, we aligned the previously mapped transcription start sites (TSS) of about 5200 yeast genes (Park et al. 2014) and analyzed CPD repair at each nucleotide position around the TSS (i.e., from −200 bp to +640 bp relative to the TSS). Regions near the TSS were chosen because highly phased nucleosomes are enriched toward the 5′ end of genes, beginning with the +1 nucleosome, which is nearest the TSS (Jiang and Pugh 2009). High-resolution analysis of the CPD-seq data in yeast genes revealed periodic peaks of unrepaired CPDs in the NTS of yeast genes following 2 h of repair in WT cells (Fig. 1A). The fraction of unrepaired CPD lesions, which accounts for variability in the initial CPD distribution (Supplemental Fig. S2A), shows a similar periodicity along the NTS (Fig. 1B). This indicates that the observed periodicity is caused by systematic variations in repair efficiency, not CPD formation. To investigate if peaks of unrepaired CPDs in the NTS correlate with nucleosome organization, we analyzed the average nucleosome occupancy around the TSS, using published nucleosome maps generated by MNase-seq (Weiner et al. 2015) or a chemical cleavage method (Brogaard et al. 2012). Regardless of which nucleosome map is used, peaks of unrepaired CPDs (indicative of slow repair) are associated with the principal dyad locations of the +1, +2, +3, and +4 nucleosomes (Fig. 1A,B; Supplemental Fig. S2B), whereas low levels of unrepaired CPDs (indicative of fast repair) are associated with linker DNA between these nucleosome positions, as well as the nucleosome-depleted region (NDR) upstream of the TSS. A similar but less pronounced periodicity in repair of the NTS is also apparent following 1 h repair in WT cells (Supplemental Fig. S2C). In contrast, nucleosome positioning has no effect on the repair of the TS (Fig. 1B), indicating that TC-NER functions in a nucleosome-independent manner in WT cells, with similar efficiency of repair near the central nucleosome dyad axis as in linker DNA. A rad16 deletion eliminates the periodicity of unrepaired CPD lesions in the NTS (Fig. 1C; Supplemental Fig. S2D), indicating the observed repair pattern is due to GG-NER.
Figure 1.

Repair of the NTS is modulated by the nucleosome organization downstream from the transcription start sites (TSSs) for nearly all yeast genes. (A) High-resolution analysis of normalized CPD levels around the transcription start site (TSS) in wild-type (WT) cells following 2-h repair. The TSSs for 5205 yeast genes were aligned, and the number of CPDs associated with either the TS or NTS from 200 bp upstream of to 640 bp downstream from the TSS (indicated by the dotted line/arrow) in WT cells following 2-h repair was counted. CPD counts were divided by the strand-specific dipyrimidine frequencies associated with each position to calculate the normalized CPD levels. The average nucleosome coverage associated with each nucleosome dyad position, derived from a published yeast MNase-seq nucleosome map (Weiner et al. 2015), was plotted in gray. NDR denotes the nucleosome-depleted region upstream of the TSS, and the highly phased nucleosomes downstream from the TSS are labeled as +1, +2, +3, and +4. (B) Same as A, except CPD counts at 2 h were divided by the counts at 0 h at each position to analyze the fraction of remaining CPDs in the WT 2-h sample (i.e., WT-2 h/WT-0 h). (C) Same as B, except CPD-seq data in the rad16Δ mutant were analyzed. (D) Gene cluster plot of normalized CPDs in WT cells after 2-h repair from 500 bp upstream of to 640 bp downstream from the TSS, across 4557 yeast genes. The TS is depicted in the left panel, and NTS is depicted in the middle panel. Each column represents a 30-bp window, and the rows correspond to the CPD data for yeast genes. To display the plot in a compact manner, each row/pixel corresponds to approximately 16 yeast genes. Median levels of CPDs are depicted in black, whereas blue and yellow indicate low and high levels of CPDs, respectively (see color bar). Rows were sorted by transcriptional frequency (Holstege et al. 1998), from lowest to highest. Nucleosome coverage associated with each dyad position, based on a published yeast MNase-seq nucleosome map (Weiner et al. 2015), is depicted in the right panel. (E) Same as D, except the fraction of CPDs remaining was plotted, by calculating the log2ratio of the CPDs in the WT 2-h sample relative to the WT 0-h sample for each 30-bp window. (F) Same as E, except CPD-seq data in the rad16Δ mutant were analyzed.
The periodic repair pattern along the NTS could reflect variations of GG-NER efficiency occurring at most yeast genes, or alternatively, a phenotype dictated by a subset of genes. To gain insights into repair patterns for genes across the yeast genome, we visualized the normalized CPD levels following 2-h repair around the TSS for all yeast genes, sorted by transcription frequency (Holstege et al. 1998). This analysis revealed that for most yeast genes, the level of unrepaired CPDs in the NTS after 2-h repair correlates with nucleosome positioning downstream from the TSS (Fig. 1D). A similar pattern is apparent across yeast genes if the fraction of unrepaired CPDs is analyzed (Fig. 1E), thereby normalizing for potential differences in initial damage levels. Additionally, there are consistently low levels of unrepaired CPDs at 2 h in the TS, indicating that TC-NER efficiently repairs CPD lesions at nearly all genes in WT cells, even those with low transcription frequency (Fig. 1D,E). In the rad16Δ mutant, the NTS repair pattern is uniformly abolished across all yeast genes (Fig. 1F), indicating that Rad16 is generally required for repair of the NTS and that slower repair of nucleosomal DNA in the NTS is attributable to the GG-NER pathway.
Repair of CPD lesions along the NTS is asymmetric in yeast nucleosomes
A careful examination of the repair data for the NTS in WT cells (Fig. 1A,B) indicates that the peaks of unrepaired CPD lesions are slightly shifted relative to the nucleosome centers. The peak of unrepaired CPD lesions in the NTS consistently occurs on the TSS-distal side of the nucleosome dyad peak in WT cells, both at the 1-h (Supplemental Fig. S2C) and 2-h (Fig. 1A,B) time points. To further investigate this finding, we obtained the dyad positions of the +1, +2, and +3 nucleosomes for each yeast gene (Weiner et al. 2015). We oriented the nucleosomal DNA based on the transcriptional direction of their associated genes, so that the NTS was always oriented in the 5′-to-3′ direction (Fig. 2A), and analyzed CPD repair at each nucleotide position relative to the central dyad axis. The fraction of unrepaired CPDs is higher near the central nucleosome dyad axis following 2-h repair in WT cells (Fig. 2B), but not in a rad16Δ mutant (Fig. 2C), indicating that repair by the GG-NER pathway is inhibited near the nucleosome dyad, consistent with our previous results (Mao et al. 2016; Brown et al. 2018). However, the peak of unrepaired CPD lesions on the NTS is shifted ∼10–20 nt to the 3′ side of the nucleosome dyad (Fig. 2B), consistent with the shift we observed between the CPD and nucleosome peaks downstream from the TSS (e.g., Fig. 1A,B). This shift is unlikely to result from a general UV-induced change in nucleosome positioning, because a recent study has shown that following UV irradiation, nucleosome positioning is essentially unchanged across nearly all yeast genes (van Eijk et al. 2019).
Figure 2.

Repair of the NTS is asymmetric in yeast nucleosomes. (A) Diagram depicting how the NTS associated with each intragenic nucleosome (i.e., +1, +2, and +3) was oriented in the 5′-to-3′ direction, so that the 5′ side (i.e., TSS-proximal side) is consistently to the left of the dyad axis, whereas the 3′ side (i.e., TSS-distal side) is to the right of the dyad axis. Arrow indicates the TSS and direction of transcription. Dashed lines indicate the dyad axis of the +1, +2, and +3 nucleosomes. (B) Repair of CPD lesions in the +1 (left), +2 (middle), and +3 nucleosomes (right) in yeast genes. The fraction of CPDs remaining at each position in the nucleosome (−73 to +73 bp relative to the dyad axis) is plotted for the WT 2-h repair sample, relative to WT 0-h sample. “Observed” plots the actual CPD repair data, whereas “Expected” plots the data from the 5′ side of the nucleosome on the 3′ side (i.e., the expected fraction of CPDs remaining if repair was symmetric across the nucleosome dyad). The relative difference between the observed and expected curves quantifies the degree of asymmetry (Asym.) in repair, which is expressed as percent value (Methods). Nucleosome dyad positions were obtained from Weiner et al. (2015). (C) Same as B, left panel (+1 nucleosome), except the CPD-seq data for the rad16Δ strain were analyzed. (D) Same as B, except the NTS was not oriented in a 5′-to-3′ direction. (E) Same as B, except the +1 nucleosome dyad positions were obtained from Chereji et al. (2018).
The fraction of unrepaired CPDs in the NTS is generally lower on the TSS-proximal (5′) side of the nucleosome dyad relative to the TSS-distal (3′) side (Fig. 2B). To highlight this asymmetry, the fraction of CPDs remaining on the 5′ side of the nucleosomal DNA is plotted on the 3′ side of nucleosome (Fig. 2B, “Expected”). The difference between the actual fraction of CPDs remaining (i.e., “Observed”) and Expected (assuming symmetric repair on both sides of the dyad) reflects the degree of asymmetry in repair. We quantified the degree of repair asymmetry by calculating the relative difference in the fraction of CPDs remaining between the 3′ (distal) side of the nucleosome (Observed) and the mirrored position derived from the 5′ (proximal) side of the nucleosome (Expected) (Supplemental Fig. S3A). In WT cells, there is 12%, 9%, and 7% asymmetry in repair in the +1, +2, and +3 nucleosomes, respectively, indicating significantly higher levels of unrepaired lesions on the 3′ side of the nucleosomal DNA (P < 0.0001) (Fig. 2B). These positive asymmetry values reflect consistently slower repair of the 3′ side of the nucleosome, which is already evident in the +1 and +2 nucleosomes 1 h post-irradiation (Supplemental Fig. S3B). In contrast, there is no asymmetry in repair of the +1 nucleosomes when the NTS is randomly oriented (i.e., in the 5′-to-3′ or 3′-to-5′ orientation, depending on the direction of transcription; P > 0.05) (Fig. 2D). Similar analysis of repair using a different yeast nucleosome map (i.e., generated by a high-resolution chemical cleavage method) (Chereji et al. 2018) showed a similar degree of asymmetry in repair of the NTS (P < 0.0001) (Fig. 2E). Taken together, these findings indicate that repair of the NTS by the GG-NER pathway is asymmetric in nucleosomes, with more rapid repair occurring in the TSS-proximal 5′ side of the nucleosomal DNA, and slower repair in the distal 3′ side.
Repair of CPD lesions along the NTS is asymmetric in human nucleosomes
To test whether repair of UV lesions on the NTS also occurs asymmetrically in human nucleosomes, we analyzed published XR-seq data derived from UV-irradiated human fibroblasts (Adar et al. 2016). The XR-seq method sequences CPD-containing DNA fragments excised by the NER machinery and is thus a direct measure of NER activity (Hu et al. 2015, 2017b; Adar et al. 2016). We analyzed the XR-seq repair data using a map of strongly positioned human nucleosomes derived from published DNase-seq and MNase-seq data sets (Methods). Only strongly positioned nucleosomes located in human protein-coding genes were analyzed (a total of 553,447 intragenic nucleosomes), and the NTS was aligned in the direction of transcription of the associated gene, so that it was always oriented in the 5′-to-3′ direction (Fig. 3A). At early repair time points, there are generally fewer XR-seq reads associated with the NTS in intragenic nucleosomes (i.e., nucleosomes in human protein-coding genes) than the TS (Supplemental Fig. S4A), consistent with more rapid repair of the TS by the TC-NER pathway. To account for potential differences in initial UV damage levels, we normalized the CPD XR-seq data using a cellular HS-Damage-seq data set (Hu et al. 2017a), which measured initial CPD levels in human fibroblasts immediately following UV irradiation (i.e., 0 h). We focused our analysis on the central 121 bp of the nucleosomal DNA (Fig. 3B), similar to a previous study (Pich et al. 2018). At the nucleosomal DNA ends (i.e., more than 60 nt from the central dyad axis), normalized repair activity is elevated at both 5′ and 3′ ends (Supplemental Fig. S4B), presumably reflecting increased lesion accessibility attributable to transient unwrapping of the nucleosomal DNA (Zhou et al. 2019).
Figure 3.

Repair of the NTS is asymmetric in human intragenic nucleosomes. (A) Diagram depicting the nontranscribed strand (NTS) associated with human intragenic nucleosomes oriented in the 5′-to-3′ direction, so that the 5′ side (i.e., TSS-proximal side) is consistently to the left of the dyad axis, whereas the 3′ side (i.e., TSS-distal side) is to the right of the dyad axis. Arrow indicates the direction of transcription. Dashed line indicates the aligned dyad axes of the intragenic nucleosomes. (B) The density of CPD excision repair-sequencing (XR-seq) reads (Adar et al. 2016) from UV-irradiated fibroblasts (4-h repair) along the NTS was calculated at each position in intragenic nucleosomes (from −73 to +73 bp from the dyad axis) and normalized using CPD HS-Damage-seq data (Hu et al. 2017a) from UV-irradiated fibroblasts (0-h repair). The normalized (Norm.) XR-seq read density was plotted for the nucleosome core, consisting of −60 to +60 bp from the dyad (lower). The “Expected” data are mirrored from the 5′ side of the dyad axis (i.e., expected if symmetric repair across the nucleosome dyad). The relative difference between the observed and expected curves quantifies the degree of asymmetry (Asym.) in repair, which is expressed as percent value, as in Figure 2. (C) Plot of normalized XR-seq reads along the NTS in intragenic nucleosomes, except the NTS was randomly oriented (i.e., 5′-to-3′ or 3′-to-5′). (D) Same as B, lower panel, except for different repair time points (1–48 h). XR-seq data were obtained from Adar et al. (2016). (**) P < 0.0001; (*) P < 0.01.
Our analysis revealed that the distribution of XR-seq reads along the NTS of strongly positioned nucleosomes shows a significant degree of asymmetry, even after normalizing for initial damage levels (Fig. 3B). At the 4-h time point, there is −9% asymmetry in the distribution of normalized XR-seq reads (P < 0.0001), the negative asymmetry reflecting less NER activity on the 3′ side of the nucleosome (cf. observed and expected curves in the bottom panel of Fig. 3B). The negative asymmetry in repair activity in human cells is equivalent to a positive asymmetry in unrepaired CPDs observed in yeast, indicating that repair is consistently slower on the 3′ side of nucleosomal DNA in both yeast and human cells. The un-normalized XR-seq read distribution at 4 h has a similar degree of asymmetry (−9%) (Supplemental Fig. S4C), whereas there is no asymmetry in the initial distribution of CPDs in the HS-Damage-seq data (Supplemental Fig. S4C), indicating that differences in repair activity is the source of the asymmetry in intragenic nucleosomes. Repair asymmetry is dependent on DNA strand orientation, because the distribution of normalized XR-seq reads is symmetric (i.e., 0% asymmetry) if the NTS in nucleosomes is randomly oriented (Fig. 3C). Significant asymmetry in repair activity is also apparent for the 1- and 8-h time points (−12% and −7%, respectively), but not at the 16- and 48-h time points (Fig. 3D). In general, the degree of repair asymmetry is greatest in magnitude at the earliest time point (1 h), and its magnitude is reduced at later repair time points, consistent with CPD lesions on the promoter-proximal 5′ side of the nucleosome being repaired more rapidly than CPDs located on the 3′ side.
Somatic mutations in human melanomas have an asymmetric distribution in intragenic nucleosomes
Previous studies have indicated that decreased NER activity in regions of heterochromatin (Adar et al. 2016; Roberts et al. 2019) and near the central dyad axis of the nucleosome (Sabarinathan et al. 2016; Brown et al. 2018; Pich et al. 2018) is associated with elevated somatic mutation rates in skin cancers. We wondered whether lower NER activity on the 3′ side of strongly positioned intragenic nucleosomes is associated with increased somatic mutation rates in UV-exposed cancers. To test this hypothesis, we analyzed a published set of somatic mutations derived from 183 melanomas, sequenced by the International Cancer Genome Consortium (ICGC) (Hayward et al. 2017). To analyze strand-specific mutation frequencies, we assigned each mutation to the DNA strand containing the pyrimidine (C or T) base, because UV lesions at dipyrimidine sequences are the origin of the vast majority of these somatic mutations. Analysis of strand-specific mutation frequencies in intragenic nucleosomes revealed that the somatic mutation density (and enrichment) is elevated on the NTS relative to the TS (Supplemental Fig. S5A), consistent with previous results (Brown et al. 2018).
More detailed analysis of the NTS (Fig. 4A) revealed that the distribution of somatic mutations in nucleosomes is asymmetric. Mutation density (i.e., mutations per nucleosome) is elevated on the 3′ side of intragenic nucleosomes (Supplemental Fig. S5B,C), even after normalizing for the expected mutation frequency derived from the DNA sequence context of the NTS (“Mutation Enrichment”; P < 0.01) (Fig. 4B). Furthermore, no asymmetry is present when the NTS was randomly oriented (i.e., in either the 5′-to-3′ or 3′-to-5′ orientation) (Supplemental Fig. S5D). Mutation enrichment also shows a ∼10 bp periodicity (consistent with previous results) (Brown et al. 2018; Pich et al. 2018), but this periodicity is dampened on the relatively fast-repairing 5′ side of the nucleosome (see below). Taken together, these results indicate that somatic mutations in melanoma are asymmetrically distributed in intragenic nucleosomes, being enriched on the slow-repairing 3′ side of the NTS.
Figure 4.

Somatic mutations in skin cancers are asymmetrically distributed in intragenic nucleosomes. (A) Diagram depicting how the NTS associated with human intragenic nucleosomes was oriented in the 5′-to-3′ direction. Arrow indicates the direction of transcription. Dashed line indicates the aligned dyad axes of the intragenic nucleosomes. (B) Enrichment of somatic mutations in 183 melanoma tumors along the NTS was plotted at positions from −60 to +60 bp from dyad axis of intragenic nucleosomes. Mutation enrichment was calculated by dividing the actual density of mutations along the NTS by the expected mutation density, based on the trinucleotide sequence context (Methods). The average relative difference between the mutation enrichment on the 5′ and 3′ sides of the nucleosome dyad was used to quantify the degree of asymmetry (Asym.) in mutagenesis, which is expressed as percent value. (C) Same as B, except for cutaneous squamous cell carcinomas (cSCCs) from NER competent (WT) patients. (D) Same as B, except for cSCCs from GG-NER-deficient patients with mutations in XPC. (E) Comparison of melanoma mutation enrichment among intragenic nucleosomes (top) with CPD enrichment (second from top), normalized XR-seq reads (third from top), and DNase-seq reads (bottom). “Out” rotational settings in the intragenic nucleosomes are indicated with vertical dashed lines. CPD enrichment was calculated from the scaled ratio of CPD lesions in UV-irradiated cells relative to CPD lesions in UV-irradiated naked DNA, using data from Brown et al. (2018) and Mao et al. (2018). Normalized XR-seq data for the 4-h repair time point are same as those depicted in Figure 3B, lower panel. DNase-seq data measure nucleosomal DNA accessibility and were obtained from Degner et al. (2012).
To further explore how asymmetric repair impacts UV mutagenesis in skin cancers, we analyzed somatic mutations in cutaneous squamous cell carcinomas (cSCC), derived from both NER-proficient patients (WT cSCC) and GG-NER-deficient patients (Zheng et al. 2014), who have germline mutations in the XPC gene (XPC−/− cSCC). Although the somatic mutation frequency is elevated on the NTS relative to the TS in nucleosomes in WT cSCCs, this strand disparity is magnified in the XPC−/− cSCCs (Supplemental Fig. S5E), consistent with a defect in GG-NER of the NTS in the XPC−/− cells. There is significant asymmetry in the mutation distribution in WT cSCCs along the NTS in intragenic nucleosomes (6%, P < 0.01) (Fig. 4C). Mutation density is elevated on the 3′ side of the nucleosome, particularly at “out” positions (see below). The trend in the WT cSCC data is somewhat noisier, likely because the WT cSCC data have only ∼5% as many somatic mutations as the melanoma data set. Analysis of the mutational distribution among XPC−/− cSCCs revealed no significant asymmetry in the mutational distribution in intragenic nucleosomes (P > 0.05) (Fig. 4D). Indeed, mutation density was slightly higher on the 5′ side of the nucleosome relative to the 3′ side in these tumors. These analyses indicate that the nucleosome asymmetry in somatic mutations requires a functional GG-NER pathway.
Mutation enrichment in melanoma has a ∼10-bp periodicity in nucleosomes along the NTS, with peaks at “out” positions (dashed lines in Fig. 4E), where the minor groove of the nucleosomal DNA faces away from the histone octamer. A very similar periodicity is observed in CPD enrichment (i.e., CPD levels in UV-irradiated cells relative to UV-irradiated naked DNA) (Fig. 4E, second panel from top), consistent with previous reports that elevated CPD formation at “out” positions in nucleosomal DNA promote increased somatic mutagenesis in melanoma (Brown et al. 2018; Pich et al. 2018). However, the periodicity in mutation enrichment differs across the nucleosome dyad, with the amplitude in periodicity greater on the 3′ side of the nucleosome (Fig. 4E, top). This appears to be primarily caused by differences in mutation enrichment at “out” positions, as mutation enrichment is higher at “out” positions on the 3′ side of the nucleosome dyad than on the 5′ side (12% asymmetry for “out” positions, P < 0.001). This asymmetry cannot be explained by elevated levels of CPD formation at 3′ out positions, because CPD enrichment is slightly lower at 3′ out positions relative to 5′ out positions (−2% asymmetry, P < 0.05) (Fig. 4E, second panel). Instead, it appears that repair of CPD lesions at “out” positions along the 3′ side of the nucleosome is inhibited compared to the 5′ side (−11% asymmetry, P < 0.05) (Fig. 4E, third panel).
Asymmetric repair of nucleosomal DNA could arise from differences in DNA accessibility on opposite sides of the dyad. To test this hypothesis, we analyzed published human DNase-seq data (Degner et al. 2012), because the frequency of cleavage by DNase I is a common measure of DNA accessibility in nucleosomes. “Out” positions in nucleosomal DNA are more efficiently cleaved by DNase I, because these DNA locations are more accessible to the nuclease (Noll 1974; Lutter 1978; Zhong et al. 2016), leading to the expected periodic pattern of DNase I cleavage sites (Fig. 4E, bottom). This analysis revealed asymmetry in the DNase-seq cleavage pattern along the NTS in intragenic nucleosomes, with a higher density of DNase-seq reads at “out” positions on the 5′ side of the nucleosome dyad (−9% asymmetry, P < 0.05) (Fig. 4E, bottom). Increased DNA accessibility at 5′ “out” positions, as measured by DNase-seq, is associated with elevated repair activity at these same locations (Fig. 4E). A similar asymmetry in DNase I cleavage has been previously observed in nucleosomes in vitro (Lutter 1978) and across the yeast genome (Zhong et al. 2016). These results suggest that increased DNA accessibility at “out” positions on the 5′ side of nucleosomes promotes more efficient repair of UV damage, whereas the reduced accessibility of “out” positions on the 3′ side of intragenic nucleosomes inhibits repair and promotes somatic mutations at these locations in melanoma.
Asymmetric repair and mutagenesis in intergenic nucleosomes
The results so far indicate that mutations in skin cancers are asymmetrically distributed in intragenic nucleosomes along the NTS, because of differences in lesion accessibility and repair between the 5′ and 3′ side of the nucleosomal DNA. These differences could arise because of ongoing transcription through these intragenic nucleosomes, which has been associated with the formation of asymmetric nucleosome structures (Rhee et al. 2014; Ramachandran et al. 2015, 2017), or could arise from structural features of canonical nucleosomes, independent of transcription. To distinguish between these two hypotheses, we analyzed repair and mutagenesis in strongly positioned nucleosomes located in nontranscribed (i.e., intergenic) regions. Repair and mutagenesis were analyzed along both DNA strands in their normal antiparallel orientation (Fig. 5A, upper).
Figure 5.

Asymmetric repair of human intergenic nucleosomes promotes a DNA strand polarity in somatic mutations in melanoma. (A) Normalized NER activity is elevated on the 5′ side of both DNA strands in intergenic nucleosomes. Normalized XR-seq reads for the 4-h repair time point were plotted for both DNA strands in their normal antiparallel orientation (top) at positions from −60 to +60 bp from the dyad axis of intragenic nucleosomes. (B) Same as A, except the plus and minus DNA strands were both aligned in the 5′-to-3′ direction, and the average of both aligned DNA strands is plotted. Data for the 5′ side of the dyad axis are depicted in orange, and data for the 3′ side of the dyad are depicted in purple. (C,D) Same as A and B, except DNase-seq read density (Degner et al. 2012) was plotted. (E,F) Same as A and B, except mutation enrichment in melanoma tumors was plotted. (G) Same as A, except CPD enrichment was plotted. (H) Structural model showing that at “out” positions (indicated with arrows), the 5′ side of the nucleosomal DNA (orange/gold) faces the solvent, whereas the 3′ side of the nucleosomal DNA (light purple/blue) faces the other DNA gyre, and is thereby less accessible for repair. PyMol (https://pymol.org/2/) was used to visualize the nucleosome structure (PDB ID: 1KX5) from Davey et al. (2002).
Analysis of NER activity in UV-irradiated fibroblasts revealed that repair activity (i.e., normalized XR-seq reads) is also asymmetric in intergenic nucleosomes (Fig. 5A, lower). Along both DNA strands, repair activity is elevated on the 5′ side of the nucleosomal DNA, which is apparent upon aligning both strands in a 5′-to-3′ orientation (Fig. 5B). Elevated repair on the 5′ side of each nucleosomal DNA strand is also apparent at other early repair time points (e.g., 1 h and 8 h) (Supplemental Fig. S6), but not at later time points (Supplemental Fig. S6). Analysis of DNase-seq data revealed that DNA accessibility is elevated at “out” positions along the 5′ side of both DNA strands in intergenic nucleosomes (P < 0.05) (Fig. 5C,D), similar to our analysis of intragenic nucleosomes. These findings suggest that asymmetric DNA accessibility and repair is an inherent feature of canonical nucleosomes (see below), even in the absence of transcription.
We also examined the distribution of somatic mutations in melanoma in intergenic nucleosomes. Somatic mutations are elevated at “out” positions, as expected (Brown et al. 2018; Pich et al. 2018), but the amplitude of the periodicity is diminished along the 5′ side of both DNA strands (Fig. 5E,F). This is primarily because of decreased mutation density at “out” positions on the 5′ side of the nucleosome dyad, consistent with our previous analysis of intragenic nucleosomes. This is not caused by potential differences in CPD formation, because CPD enrichment is largely symmetric across the nucleosome dyad along both DNA strands (Fig. 5G). Instead, faster repair of the 5′ side of the nucleosome, particularly at “out” positions, appears to suppress UV mutagenesis in intergenic nucleosomes.
Analysis of the canonical nucleosome structure indicates that “out” positions along the nucleosomal DNA have differing degrees of solvent accessibility, consistent with previous studies (Lutter 1978; Tanaka et al. 1996; Zhong et al. 2016; Mao et al. 2017). For both DNA strands, “out” positions along the 5′ half of the nucleosomal DNA face the solvent (orange highlights in Fig. 5H), and thus are readily accessible to DNase I cleavage and the NER machinery. In contrast, “out” positions along the 3′ half of the nucleosomal DNA face the other DNA gyre (blue highlights in Fig. 5H), which can explain the reduced accessibility of these “out” positions to DNase I and the NER machinery.
Discussion
Here, we have shown that the repair of UV damage in nucleosomes is asymmetric. High-resolution genomic repair maps in both yeast and human cells indicate that CPD lesions on the 5′ side of the central dyad axis are more efficiently repaired than those on the 3′ side. Although it has previously been established that nucleosomes inhibit NER, particularly for lesions near the dyad center of the nucleosome, our data indicate that the degree of this inhibition is dissimilar on opposite sides of the dyad axis. We have further shown that UV-induced somatic mutations in skin cancers have the opposite polarity, resulting in a lopsided distribution of mutations along each DNA strand. This is primarily attributed to elevated mutation rates at “out” positions along the slow-repairing 3′ side of the nucleosomal DNA. This strand polarity in UV mutagenesis is dependent on the GG-NER pathway, because it is absent in XPC−/− cutaneous squamous cell carcinomas, which are deficient in GG-NER activity.
Our analysis indicates that asymmetric repair activity in human cells is likely a consequence of the left-handed wrapping of the DNA around the histone octamer. Analysis of DNase I cleavage sites in nucleosomal DNA indicates that “out” positions along the 3′ side of the dyad axis are less accessible to repair enzymes, owing to the proximity of the neighboring DNA gyre (Fig. 6). In contrast, “out” positions along the 5′ side are more amenable to repair, because the DNA backbone at these positions faces the solvent (Fig. 6). This strand polarity in DNA accessibility, which is a consequence of the left-handed wrapping of nucleosome DNA around the histone octamer, was discovered more than 40 yr ago (Lutter 1978). There have been hints that this asymmetry in nucleosomal DNA accessibility could affect repair at individual yeast nucleosomes (Wellinger and Thoma 1997; Tijsterman and Brouwer 1999); however, its impact on the repair and mutagenesis of UV damage across the genome has not previously been investigated. It is known that the DDB2 subunit of the UV-DDB complex, which is critical for recognizing CPD lesions in human cells, can bind to CPD lesions resident in nucleosomes to initiate GG-NER (Fischer et al. 2011). Moreover, a recent study suggested that UV-DDB shows strand asymmetry in the recognition of a model lesion resident in a positioned nucleosome (Matsumoto et al. 2019). We hypothesize that UV-DDB, XPC, or possibly downstream NER factors (e.g., TFIIH) may be preferentially inhibited from initiating repair at “out” positions along the 3′ side of the nucleosome, owing to the interference of the neighboring DNA gyre, thereby promoting asymmetric repair and mutagenesis in nucleosomes (Fig. 6).
Figure 6.
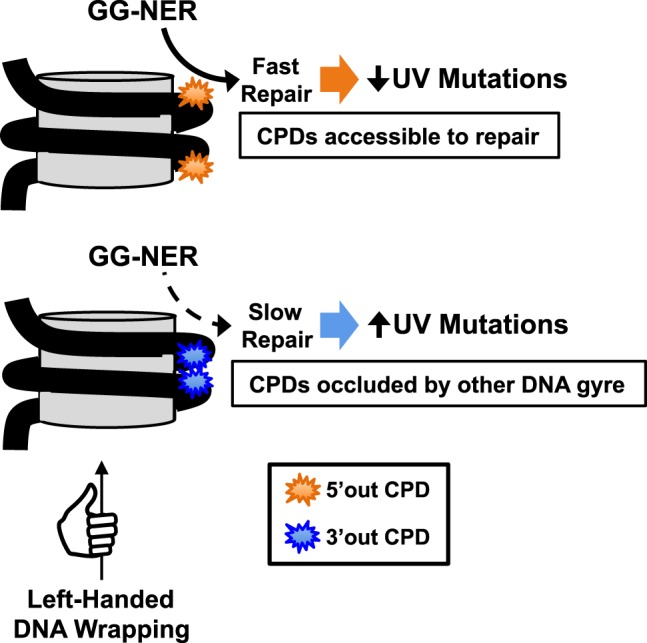
Molecular mechanism promoting asymmetric repair and mutagenesis in nucleosomes. Schematic showing how the left-handed wrapping of DNA (black line, depicting antiparallel DNA double helix) around the histone octamer (gray cylinder) promotes the accessibility and repair of CPD lesions on the 5′ side of the nucleosome dyad (orange), particularly at “out” rotational settings, but inhibits the repair of CPD lesions at “out” positions on the 3′ side of the nucleosome dyad (blue), because of the proximity of the neighboring DNA gyre. Two CPD lesions are shown in each nucleosome to highlight the asymmetry in solvent accessibility of the 5′ and 3′ sides of the nucleosome; however, typically only a single CPD lesion will be resident in an individual nucleosome.
Our data indicate that there is also asymmetric repair of CPD lesions in yeast nucleosomes, with faster repair on the 5′ side of the nucleosomal DNA along the NTS of yeast genes. Although this is essentially the same pattern observed in human nucleosomes, there are some subtle differences in the repair patterns between yeast and human cells. In human cells, asymmetry in repair activity is most apparent at “out” positions; however, in yeast nucleosomes there does not appear to be elevated repair activity or asymmetry at “out” rotational settings. This could result from species–specific differences in the GG-NER pathway (e.g., yeast lack the UV-DDB complex) or nucleosome organization. In yeast, the repair asymmetry is highest in the +1 nucleosome, which is positioned closest to the TSS. This could be a consequence of the formation of asymmetric subnucleosome structures, which are highly enriched at the +1 nucleosome (Rhee et al. 2014; Ramachandran et al. 2015, 2017). Asymmetric repair in yeast nucleosomes may also be modulated by histone post-translational modifications (PTMs) and variants, because certain histone PTMs and histone variants are preferentially enriched on one half of the nucleosome (Rhee et al. 2014). Because histone PTMs and variants can promote NER in chromatin (Waters et al. 2015; Mao and Wyrick 2016), their asymmetric distribution in yeast (or human) nucleosomes could promote asymmetric repair.
It will be interesting to determine whether other repair pathways show a similar degree of asymmetry in nucleosomes. Previously, we showed that repair of DNA alkylation damage by the base excision repair (BER) pathway is asymmetric in yeast nucleosomes (Mao et al. 2017). Alkylation damage is repaired more rapidly at “out” positions on the 5′ side of nucleosome dyad than at “out” positions on the 3′ side of the dyad, consistent with the human NER data presented here. Here, we have further shown that asymmetric NER activity in nucleosomes is associated with a strand polarity in somatic mutations in skin cancers. It will be interesting to determine whether asymmetric BER activity in nucleosomes impacts the distribution of mutations arising from DNA base lesions in other cancers.
Recently, it was shown that somatic mutations in skin cancers have a periodicity in nucleosomal DNA, with elevated mutation rates at “out” positions in nucleosomes attributed to elevated CPD formation (Brown et al. 2018; Pich et al. 2018). Our strand-specific analysis indicates that this mutational periodicity is primarily associated with the 3′ side of the nucleosome dyad. Although CPD formation is largely symmetric across the nucleosome dyad, faster repair of CPD lesions at “out” positions on the 5′ side of the dyad acts to suppress this periodicity. In contrast, slower repair on the 3′ side of the dyad acts to enhance mutation rates at “out” positions, thereby enhancing mutation periodicity. Because G/C dinucleotides are generally favored at “out” positions in nucleosomes (Struhl and Segal 2013; Lai and Pugh 2017), elevated rates of UV-induced C > T mutations at “out” rotational settings would presumably weaken nucleosome positioning, particularly on the 3′ side of the dyad. Asymmetry in UV mutagenesis could serve as a novel mechanism to alter nucleosome positions and potentially promote the formation of asymmetric subnucleosome structures in cells (or organisms) exposed to UV light over many generations. This mechanism should be especially active in transcribed nucleosomes, because asymmetric repair in nucleosomes should impact only one of the DNA strands (i.e., the NTS), with the other strand (i.e., the TS) being primarily repaired by the TC-NER pathway.
In summary, we have shown that the repair of UV damage in nucleosomes is asymmetric, in part because of the intrinsic difficulty of accessing and repairing DNA lesions on the 3′ side of each nucleosomal DNA strand. We have further shown that this repair asymmetry modulates UV mutagenesis in skin cancers. Because nucleosome-bound DNA comprises the vast majority of the human genome, this mutational asymmetry may have significant ramifications in carcinogenesis. For example, our preliminary analysis indicates that a number of protein-altering mutations in melanoma driver genes are associated with the 3′ side of strongly positioned nucleosomes (highlighted in bold in Supplemental Table S1). Moreover, previous studies have exploited DNA strand asymmetries in replication (R-class asymmetry) and transcription (T-class asymmetry) to elucidate specific DNA lesions or mutational processes causing different mutation signatures in tumors (Haradhvala et al. 2016). However, many classes of DNA lesions show neither T-class nor R-class asymmetry in human tumors. We propose that mutational asymmetries in nucleosomal DNA, which we term N-class asymmetry, could be used in a complementary fashion to investigate mutational processes in skin cancers and potentially other tumor types.
Methods
Yeast CPD-seq experiments and analysis
Yeast UV irradiation, genomic DNA isolation, and CPD-seq library preparation and analysis were performed as previously described (Mao et al. 2016). A detailed description of these methods, as well as the method used to calculate repair asymmetry in yeast nucleosomes, is provided in the Supplemental Methods. CPD-seq data for the WT 0-, 1-, and 2-h repair time points were originally published in Mao et al. (2016).
Human nucleosome map
Nucleosome positions were called using a new algorithm that combines both DNase-seq (Degner et al. 2012; Zhong et al. 2016) and MNase-seq data (Gaffney et al. 2012). Nucleosome scores derived from DNase-seq data (Zhong et al. 2016) were used to identify potential nucleosome dyad positions using a greedy algorithm, as previously described (Brown et al. 2018). A detailed description of the nucleosome calling algorithm is provided in the Supplemental Methods.
Analysis of CPD-seq, HS-Damage-seq, DNase-seq, and XR-seq data
Human CPD-seq analysis was performed as previously described using cellular (UV 0 h) and in vitro irradiated (naked DNA) CPD-seq data (Brown et al. 2018; Mao et al. 2018). Human HS-Damage-seq, DNase-seq, and XR-seq reads was obtained from published sources (Degner et al. 2012; Adar et al. 2016; Hu et al. 2017a). A detailed description of the data processing and analysis is included in the Supplemental Methods.
Analysis of melanoma and squamous cell carcinoma mutation data
Melanoma mutation data were from the ICGC data portal (data release 20), and squamous cell carcinoma mutation data were from Zheng et al. (2014). Expected mutation frequencies were determined using the mutation frequency of each trinucleotide sequence (e.g., TCG, in which the underlined C is mutated), essentially as previously described (Brown et al. 2018; Mao et al. 2018). Expected mutation frequencies were determined for each DNA strand separately, in that the expected mutation frequency at each position was assigned to the pyrimidine-containing strand. Additional details of mutation data sets and analysis are provided in the Supplemental Methods.
Calculating repair and mutational asymmetry in human nucleosomes
Repair asymmetry was analyzed for the NTS in intragenic nucleosomes (i.e., nucleosomes occurring in protein-coding gene sequences) and for both DNA strands in intergenic nucleosomes using the called nucleosome dyad positions. A detailed description of the repair asymmetry analysis is provided in the Supplemental Methods. Moreover, the key Perl and C++ scripts used to analyze these and other data are included in the Supplemental Code.
Data access
The rad16Δ CPD-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE131101.
Supplementary Material
Acknowledgments
We thank Weiwei Du and Mark Wildung for Ion Proton sequencing and Alexander Brown for bioinformatics support. This study was supported by National Institute of Environmental Health Sciences Grants R21ES027937 (to S.A.R. and J.J.W.), R03ES027945 (to P.M.), R21ES029302 (to P.M. and J.J.W.), and R01ES028698 (to J.J.W. and M.J.S.). We are grateful to the International Cancer Genome Consortium (ICGC) for identifying somatic mutations in melanoma tumors and making the data publicly available.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.253146.119.
References
- Adar S, Hu J, Lieb JD, Sancar A. 2016. Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc Natl Acad Sci 113: E2124–E2133. 10.1073/pnas.1603388113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang JP, Widom J. 2012. A map of nucleosome positions in yeast at base-pair resolution. Nature 486: 496–501. 10.1038/nature11142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AJ, Mao P, Smerdon MJ, Wyrick JJ, Roberts SA. 2018. Nucleosome positions establish an extended mutation signature in melanoma. PLoS Genet 14: e1007823 10.1371/journal.pgen.1007823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereji RV, Ramachandran S, Bryson TD, Henikoff S. 2018. Precise genome-wide mapping of single nucleosomes and linkers in vivo. Genome Biol 19: 19 10.1186/s13059-018-1398-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. 2002. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J Mol Biol 319: 1097–1113. 10.1016/S0022-2836(02)00386-8 [DOI] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. 2012. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482: 390–394. 10.1038/nature10808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna JJ, Kraemer KH. 2012. Shining a light on xeroderma pigmentosum. J Invest Dermatol 132: 785–796. 10.1038/jid.2011.426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Scrima A, Böhm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, et al. 2011. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 147: 1024–1039. 10.1016/j.cell.2011.10.035 [DOI] [PubMed] [Google Scholar]
- Gaffney DJ, McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, Pritchard JK. 2012. Controls of nucleosome positioning in the human genome. PLoS Genet 8: e1003036 10.1371/journal.pgen.1003036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G. 2008. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9: 958–970. 10.1038/nrm2549 [DOI] [PubMed] [Google Scholar]
- Hara R, Mo J, Sancar A. 2000. DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol Cell Biol 20: 9173–9181. 10.1128/MCB.20.24.9173-9181.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, Rheinbay E, Kim J, Maruvka YE, Braunstein LZ, et al. 2016. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 164: 538–549. 10.1016/j.cell.2015.12.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch AM, Kakavand H, Alexandrov LB, Burke H, et al. 2017. Whole-genome landscapes of major melanoma subtypes. Nature 545: 175–180. 10.1038/nature22071 [DOI] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95: 717–728. 10.1016/S0092-8674(00)81641-4 [DOI] [PubMed] [Google Scholar]
- Hu J, Adar S, Selby CP, Lieb JD, Sancar A. 2015. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev 29: 948–960. 10.1101/gad.261271.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Adebali O, Adar S, Sancar A. 2017a. Dynamic maps of UV damage formation and repair for the human genome. Proc Natl Acad Sci 114: 6758–6763. 10.1073/pnas.1706522114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Selby CP, Adar S, Adebali O, Sancar A. 2017b. Molecular mechanisms and genomic maps of DNA excision repair in Escherichia coli and humans. J Biol Chem 292: 15588–15597. 10.1074/jbc.R117.807453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JC, Svoboda DL, Reardon JT, Sancar A. 1992. Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc Natl Acad Sci 89: 3664–3668. 10.1073/pnas.89.8.3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10: 161–172. 10.1038/nrg2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WKM, Pugh BF. 2017. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol 18: 548–562. 10.1038/nrm.2017.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Smerdon MJ. 2000. Nucleotide excision repair of the 5 S ribosomal RNA gene assembled into a nucleosome. J Biol Chem 275: 23729–23735. 10.1074/jbc.M002206200 [DOI] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389: 251–260. 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- Lutter LC. 1978. Kinetic analysis of deoxyribonuclease I cleavages in the nucleosome core: evidence for a DNA superhelix. J Mol Biol 124: 391–420. 10.1016/0022-2836(78)90306-6 [DOI] [PubMed] [Google Scholar]
- Mao P, Wyrick JJ. 2016. Emerging roles for histone modifications in DNA excision repair. FEMS Yeast Res 16: fow090 10.1093/femsyr/fow090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Smerdon MJ, Roberts SA, Wyrick JJ. 2016. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc Natl Acad Sci 113: 9057–9062. 10.1073/pnas.1606667113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Brown AJ, Malc EP, Mieczkowski PA, Smerdon MJ, Roberts SA, Wyrick JJ. 2017. Genome-wide maps of alkylation damage, repair, and mutagenesis in yeast reveal mechanisms of mutational heterogeneity. Genome Res 27: 1674–1684. 10.1101/gr.225771.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Brown AJ, Esaki S, Lockwood S, Poon GMK, Smerdon MJ, Roberts SA, Wyrick JJ. 2018. ETS transcription factors induce a unique UV damage signature that drives recurrent mutagenesis in melanoma. Nat Commun 9: 2626 10.1038/s41467-018-05064-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. 2014. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15: 465–481. 10.1038/nrm3822 [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Cavadini S, Bunker RD, Grand RS, Potenza A, Rabl J, Yamamoto J, Schenk AD, Schübeler D, Iwai S, et al. 2019. DNA damage detection in nucleosomes involves DNA register shifting. Nature 571: 79–84. 10.1038/s41586-019-1259-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll M. 1974. Internal structure of the chromatin subunit. Nucleic Acids Res 1: 1573–1578. 10.1093/nar/1.11.1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Morris AR, Battenhouse A, Iyer VR. 2014. Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res 42: 3736–3749. 10.1093/nar/gkt1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pich O, Muiños F, Sabarinathan R, Reyes-Salazar I, Gonzalez-Perez A, Lopez-Bigas N. 2018. Somatic and germline mutation periodicity follow the orientation of the DNA minor groove around nucleosomes. Cell 175: 1074–1087.e18. 10.1016/j.cell.2018.10.004 [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Zentner GE, Henikoff S. 2015. Asymmetric nucleosomes flank promoters in the budding yeast genome. Genome Res 25: 381–390. 10.1101/gr.182618.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Ahmad K, Henikoff S. 2017. Transcription and remodeling produce asymmetrically unwrapped nucleosomal intermediates. Mol Cell 68: 1038–1053.e4. 10.1016/j.molcel.2017.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Bataille AR, Zhang L, Pugh BF. 2014. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 159: 1377–1388. 10.1016/j.cell.2014.10.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Brown AJ, Wyrick JJ. 2019. Recurrent noncoding mutations in skin cancers: UV damage susceptibility or repair inhibition as primary driver? Bioessays 41: 1800152 10.1002/bies.201800152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabarinathan R, Mularoni L, Deu-Pons J, Gonzalez-Perez A, López-Bigas N. 2016. Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature 532: 264–267. 10.1038/nature17661 [DOI] [PubMed] [Google Scholar]
- Scharer OD. 2013. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 5: a012609 10.1101/cshperspect.a012609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerdon MJ, Thoma F. 1990. Site-specific DNA repair at the nucleosome level in a yeast minichromosome. Cell 61: 675–684. 10.1016/0092-8674(90)90479-X [DOI] [PubMed] [Google Scholar]
- Struhl K, Segal E. 2013. Determinants of nucleosome positioning. Nat Struct Mol Biol 20: 267–273. 10.1038/nsmb.2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Livingstone-Zatchej M, Thoma F. 1996. Chromatin structure of the yeast URA3 gene at high resolution provides insight into structure and positioning of nucleosomes in the chromosomal context. J Mol Biol 257: 919–934. 10.1006/jmbi.1996.0212 [DOI] [PubMed] [Google Scholar]
- Tijsterman M, Brouwer J. 1999. Rad26, the yeast homolog of the cockayne syndrome B gene product, counteracts inhibition of DNA repair due to RNA polymerase II transcription. J Biol Chem 274: 1199–1202. 10.1074/jbc.274.3.1199 [DOI] [PubMed] [Google Scholar]
- Tijsterman M, de Pril R, Tasseron-de Jong JG, Brouwer J. 1999. RNA polymerase II transcription suppresses nucleosomal modulation of UV-induced (6-4) photoproduct and cyclobutane pyrimidine dimer repair in yeast. Mol Cell Biol 19: 934–940. 10.1128/MCB.19.1.934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eijk P, Nandi SP, Yu S, Bennett M, Leadbitter M, Teng Y, Reed SH. 2019. Nucleosome remodeling at origins of global genome–nucleotide excision repair occurs at the boundaries of higher-order chromatin structure. Genome Res 29: 74–84. 10.1101/gr.237198.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage R, Zeeman AM, de Groot N, Gleig F, Bang DD, van de Putte P, Brouwer J. 1994. The RAD7 and RAD16 genes, which are essential for pyrimidine dimer removal from the silent mating type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Mol Cell Biol 14: 6135–6142. 10.1128/MCB.14.9.6135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters R, van Eijk P, Reed S. 2015. Histone modification and chromatin remodeling during NER. DNA Repair (Amst) 36: 105–113. 10.1016/j.dnarep.2015.09.013 [DOI] [PubMed] [Google Scholar]
- Weiner A, Hsieh TH, Appleboim A, Chen HV, Rahat A, Amit I, Rando OJ, Friedman N. 2015. High-resolution chromatin dynamics during a yeast stress response. Mol Cell 58: 371–386. 10.1016/j.molcel.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger RE, Thoma F. 1997. Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J 16: 5046–5056. 10.1093/emboj/16.16.5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng CL, Wang NJ, Chung J, Moslehi H, Sanborn JZ, Hur JS, Collisson EA, Vemula SS, Naujokas A, Chiotti KE, et al. 2014. Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes. Cell Rep 9: 1228–1234. 10.1016/j.celrep.2014.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Luo K, Winter PS, Crawford GE, Iversen ES, Hartemink AJ. 2016. Mapping nucleosome positions using DNase-seq. Genome Res 26: 351–364. 10.1101/gr.195602.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Gaullier G, Luger K. 2019. Nucleosome structure and dynamics are coming of age. Nat Struct Mol Biol 26: 3–13. 10.1038/s41594-018-0166-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.