

Summary
We investigated the role of chemokines in regulating T-cell accumulation in solid tumors. CCL5 and CXCL9 overexpression was associated with CD8+ T-cell infiltration in solid tumors. T-cell infiltration required tumor cell-derived CCL5 and was amplified by IFNγ-inducible, myeloid cell-secreted CXCL9. CCL5 and CXCL9 co-expression revealed immunoreactive tumors with prolonged survival and response to checkpoint blockade. Loss of CCL5 expression in human tumors was associated with epigenetic silencing through DNA methylation. Reduction of CCL5 expression caused tumor-infiltrating lymphocytes (TILs) desertification whereas forced CCL5 expression prevented Cxcl9-expression and TILs loss and attenuated tumor growth in mice through IFNγ. The cooperation between tumor-derived CCL5 and IFNγ-inducible CXCR3 ligands secreted by myeloid cells is key for orchestrating T-cell infiltration in immunoreactive and immunoresponsive tumors.
Graphical Abstract
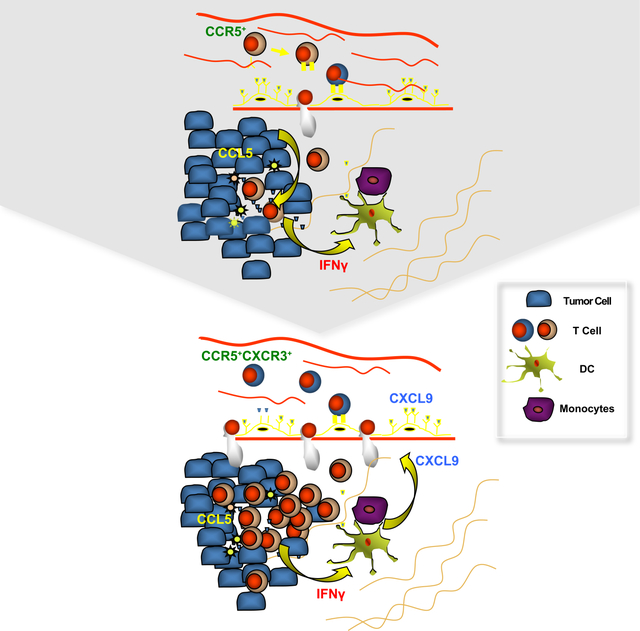
Dangaj et al. show that tumor cell-expressed CCL5 and macrophage- and DC-expressed CXCL9 are important for the infiltration of T cells into tumors, a process that also requires recognition of tumor antigens by T cells. CCL5 is often epigenetically silenced in tumor cells but can be reactivated by Decitabine.
Introduction
Immune recognition can lead to tumor suppression or outright rejection. T-cell inflamed tumors exhibit T-cell infiltration and molecular signatures of immune activation. The presence of intraepithelial tumor-infiltrating lymphocytes (ieTILs) is associated with longer survival in ovarian cancer (Hwang et al., 2012; Zhang et al., 2003), colon cancer (Galon et al., 2006), and most other solid tumor types (Fridman et al., 2012; Gooden et al., 2011). TILs detection prior to therapy has been associated with response to immune checkpoint blockade in melanoma (Tumeh et al., 2014). Moreover, TILs activation by immune modulation can lead to eradication of human tumors in the context of adoptive T-cell therapy (Dudley et al., 2002; Garon et al., 2015; Postow et al., 2015; Wolchok et al., 2013).
The molecular mechanisms governing the establishment of the T-cell inflamed phenotype are only partly understood. Lymphocyte migration into tumors is a carefully orchestrated process and includes adhesion to endothelium, trans-endothelial migration, infiltration through extracellular matrix and stroma, and ultimately engraftment in tumor islets. The tumor vasculature plays a key role in T-cell homing into tumors (Buckanovich et al., 2008; Motz et al., 2014). Chemotactic cytokines or chemokines determine the migratory behavior of leukocytes, but how they regulate immunoreactive tumors at the steady state is not well understood. Based on their expression, chemokines are recognized as constitutive (homeostatic) or inducible (inflammatory). Homeostatic chemokines regulate the physiological trafficking of immune cells through secondary lymphoid organs and tissues. Inducible chemokines are upregulated at sites of inflammation and recruit activated effector leukocytes to tissues in response to immunological stimuli (Griffith et al., 2014).
Chemokines regulate immune cell trafficking in tumors and are implicated in tumor development, progression and angiogenesis. Most tumors shape local chemokine networks to promote their growth by recruiting stromal cells like tumor-associated macrophages (TAMs) (Viola et al., 2012), myeloid-derived suppressor cells (MDSC) (Li et al., 2018), and regulatory T cells (Treg) (Curiel et al., 2004; Facciabene et al., 2011), all associated with poor patient prognosis. Several recent studies have investigated the involvement of chemokines in T-cell recruitment to tumors. For example, expression of CCL2, CCL3, CCL4, CCL5, CXCL9 and CXCL10 has been correlated with the presence of TILs in melanoma (Harlin et al., 2009; Spranger et al., 2015). Furthermore, interferon (IFN) γ-inducible chemokines CXCL9, CXCL10, and CXCL11 have been associated with activation of Th1 immunity within the tumor microenvironment (TME) and favorable response to chemotherapy and immunotherapy in melanoma (Harlin et al., 2009; Hong et al., 2011; Mikucki et al., 2015; Peng et al., 2015). CCR5 and CXCR3 expression on TILs was found to be important for their infiltration in tumor beds (Gonzalez-Martin et al., 2011; Mikucki et al., 2015) while expression of their cognate ligands was correlated with response to adoptive TILs therapy in human melanoma (Bedognetti et al., 2013). The naturally occurring CCR5∆32 polymorphism, which results in a non-functional CCR5 protein and reduced CCR5 surface expression in human heterozygotes, has been associated with poor efficacy of IFNα or interleukin-2 (IL2) immunotherapy in melanoma (Ugurel et al., 2008). Furthermore, activation of type-I IFN signaling in melanoma has been correlated with T-cell infiltration (Spranger et al., 2015), and CCL4 was as a key chemokine recruiting BATF3-expresssing dendritic cells (DCs), an important determinant of T-cell inflammation and response to immunotherapy in melanoma (Sanchez-Paulete et al., 2016; Spranger et al., 2015; Spranger et al., 2017). Tumors such as ovarian cancer express a variety of chemokines, which permit the homing of vaccine-primed and activated adoptively transferred T cells (Zsiros et al., 2015). Epigenetic silencing of Th1-type chemokines CXCL9 and CXCL10 in tumor cells has been identified as a reversible mechanism of immune escape in ovarian cancer (Peng et al., 2015).
Collectively, the existing evidence suggests that chemokine circuitries are key for T-cell homing in tumors, but how these mechanisms are orchestrated in the TME and whether hierarchies exist within such chemokine circuitries is not well understood. Our study aims to reveal associations of chemokines with T-cell infiltration across multiple solid tumor types and dissect these associations functionally.
Results
CCL5 and CXCL9 uniquely correlate with CD8+ TILs in human solid tumors
We took an unbiased approach to identify chemokines associated with T-cell infiltration in cancers. We found that CD8A expression significantly correlated to CD8+ T-cell infiltration and CD3E and CD3D expression across all solid tumors examined (Figure 1A, 1B; Figure S1A, S1B). Given the key role of CD8+ T cells in immune-mediated tumor rejection and in predicting clinical outcome in many solid tumors, we chose CD8A as a gene marker for quantifying TILs in cancer. Among all chemokines, only the expression of CCL5 and CXCL9 correlated consistently with that of CD8A across several cancer types (Figure 1C–1E). No other chemokine exhibited this universal correlation with CD8A across all tumor types. Matched scatterplots revealed a proportionality of expression between CXCL9 and CD8A, and CCL5 and CD8A, over a wide range of expression in 7 solid tumor types (Figure 1F). Concordant results were found analyzing TCGA data (Figure S1C–S1E). We confirmed by qPCR the positive correlation between CD8A and CCL5 or CD8A and CXCL9 in an independent set of 57 ovarian cancer specimens as well as the correlation between CD8A and CD3E and CD247, encoding CD3 subunits epsilon and zeta respectively (Figure S1F). We further studied associations of the expression of chemokine with the expression of known lineage markers for CD45+ leukocytes (PTPRC), CD4+ TILs (CD4), Treg cells (FOXP3), NK cells (KLRB1), CD11b+ monocytes (ITGAM), macrophages (CD68), myeloid DCs (ITGAX), and BATF3+ DCs (BATF3). No correlation was seen between CCL5 and CXCL9 with any of the above genes or of the above lineage markers with any chemokine (Figure S2A, S2B). Thus, analysis of over 9000 tumors reveals a specific and universal association of T-cell infiltration with CCL5 and CXCL9.
Figure 1. Identification of chemokines correlating with CD8A in solid tumors.

(A) IHC examples of advanced ovarian tumors with low and high levels of CD8+ TILs (left) and Pearson correlation plot of CD8A mRNA and CD8+ TILs in EOC samples (n=19) (right). (B) Pearson correlation plot of expressions of CD8A and CD3D (n=125). (C) Correlation analyses of CD8A expression with that of CCL and CXCL chemokine genes in the ExpO microarray dataset. Estimate (square) in a subset of 6 tumor types was plotted with 95% confidence intervals (CI) (lines) truncated on the left (n=1383). (D-E) Forest plots and meta-analytical estimation of the correlation between expressions of CD8A with CCL5 (D) or with CXCL9 (E) for 13 tumor types (n=1752). Estimates (squares) are drawn in proportion to n with 95% CI (lines). Average correlation r (diamond) to CD8A: r=0.86 and r=0.76, for CCL5 and CXCL9 respectively. (F) Scatterplots showing the range of associations (r) with 95% CI and proportionality of expression levels for CD8A and CCL5 or CXCL9 in seven solid tumor types. All lower bounds being higher than zero indicate highly significant associations. See also Figures S1, S2.
Constitutive expression of CCL5 by tumor cells is associated with ieCD8+ TILs and is epigenetically regulated
Next, we sought to decipher the role of each chemokine in T-cell engraftment. We used epithelial ovarian cancer (EOC) to characterize the association of CCL5 with TILs. In an EOC tissue microarray (Helsinki, n=522), ≈75% of tumors expressed CCL5 and 95% of tumors exhibiting ieCD8+ TILs displayed CCL5 expression (Figure 2A). In fact, CCL5+ tumors were more likely than CCL5− tumors to exhibit ieCD8+ TILs (54% vs. 8%, respectively, p=2.2×10−16). In a different cohort (UPenn, n=86), 79% of cases expressed CCL5 and the frequency of ieCD8+ TILs was higher in CCL5+ than CCL5− tumors (Figure 2B). In both cohorts (n=608), CCL5 immunolocalized in the tumor cell clusters (islets) and specifically within the tumor cells (Figure 2C). We confirmed tumor-cell CCL5 expression in situ by multispectral imaging microscopy (Figure 2D), where CCL5 colocalized with cytokeratin, and by detecting CCL5 mRNA in FACS-purified ovarian cancer cells (Figure 2E). The detection of CCL5 mRNA in numerous established ovarian cancer cell lines indicated constitutive expression of the chemokine in ovarian tumor cells (Figure S3A). However, unlike in other tumor types (Halama et al., 2016; Velasco-Velazquez et al., 2014), we could not demonstrate coexpression of CCL5 and any of its receptors (CCR1, CCR3, or CCR5) on ovarian tumors cell lines (Figure S3A). Consistent with the literature, CCL5 expression was also detected in sorted tumor leukocytes (Figure S3B) and specifically in T cells by immunostaining (Figure 2D).
Figure 2. CCL5 is intrinsically expressed by ovarian cancer cells and is associated with CD8+ T cells infiltration in tumors.

(A) Representative IHC images and summary of CCL5 protein expression and ieCD8+ TILs in the Helsinki EOC TMA and comparison of absolute number for CCL5+/− and CD8+/− categories (Fisher’s exact test p=2.2×10−16) (B) Quantification of ieCD8+ TILs in CCL5low and CCL5high tumors (UPenn cohort). P value was calculated with Mann-Whitney test. (C) CCL5 IHC staining in EOC. A tumor islet with cytoplasmic CCL5 is projected. (D) Multispectral immunofluorescence staining of CCL5− (upper) and CCL5+ (lower) EOC cases for CCL5 (red), keratin (grey), and CD8+ (green). (E) Representative FACS dot plot of FACS-sorted Epcam+CD45− ovarian cancer cells before (upper left) and after sorting (upper right) and relative quantification of CCL5 mRNA in FACS-sorted Epcam+CD45− EOC cells (lower bar graph). (F) FACS analysis of CCR5 expression in CD8+ and CD4+ T cells isolated from solid tumor, ascites and matched blood (PBL) of EOC patient. (G) FACS analysis of CCR5 in TILs or PBMCs from EOC patients. (H) Quantification of CCR5 expression in CD8+ and CD4+ T cells from PBMCs or TILs from EOC patients. P values were calculated with unpaired T test. (I) CCL5 methylation β values in CCL5high and CCL5low groups of five tumor types (TCGA datasets). Boxplots represent 25th and 75th percentiles with midline indicating the median; whiskers extend to maximally 1.5 interquartile (IQ) range beyond the box limits. Points indicate values for individual subjects beyond whiskers. (J) CCL5 expression in the indicated ovarian cancer cell lines 72 hr after 5’-aza-2’-deoxycytidine or DZNeP treatment. P values were calculated with unpaired T test. All bar-graph data are presented as mean ± SEM. See also Figure S3.
CCL5 exerts its chemotaxis effects on T cells through CCR5 (Gonzalez-Martin et al., 2011). We found CD8+ T cells within the islets and in proximity of tumor cells expressing CCL5 (Figure 2D). TILs and ascites lymphocytes were enriched for CCR5+ T cells relative to matched peripheral blood T cells (Figure 2F). Indeed, a large fraction of CD8+ and CD4+ TILs were CCR5+ (Figure 2G), and overall the frequency of CCR5+ lymphocytes was higher in TILs than in peripheral blood T cells (Figure 2H).
Next, we asked whether tumor-intrinsic oncogenic events could contribute to CCL5 loss. Absent or low CCL5 expression was associated with increased DNA methylation of the 5’UTR of CCL5 in several human tumor types (Figure 2I). Consistently, DNA methyltransferase inhibitor 5’-aza-2’-deoxycitine increased CCL5 expression in ovarian cancer cell lines (Figure 2J). Conversely, the global histone methylation inhibitor 3-Deazaneplanocin A (DZNep), which induces EZH2 protein depletion (Miranda et al., 2009), upregulated CCL5 in only 1 out of 4 cell lines tested. These results collectively indicate that constitutive expression of CCL5 by tumor cells may be implicated in the recruitment of ieTILs in ovarian cancer. Its expression by tumors cells is associated with successful T-cell engraftment in tumor islets, while its absence, partly due to epigenetic silencing, consistently correlates with lack of ieTILs.
CXCL9 is expressed by TAMs and DCs, and correlates with ieTILs
Although the majority of CCL5− tumors lacked ieCD8+ TILs, a fraction of CCL5+ tumors showed ieCD8+ TILs, suggesting that CCL5 is necessary but not sufficient for T-cell engraftment. We examined CXCL9, whose expression correlated well with CD8A expression. In the UPenn cohort we found that all ovarian tumors with ieCD8+ TILs expressed CXCL9 whereas tumors lacking ieCD8+ TILs did not (Figure 3A). By immunostaining we detected CD8+ TILs almost exclusively in tumors where CXCL9 was expressed (Figure 3B), while TILs density was commensurate with CXCL9 expression (Figure 3C). CXCL9+ cells were mostly detected around the tumor islets (Figure 3C) and only some were found within. CXCL9+ cells were also observed in tertiary lymphoid aggregates in the stroma (not shown). Using multispectral microscopy, we ascertained that CXCL9+ cells were CD68+ tumor-associated macrophages (TAMs) as well as CD11c+ DCs (Figure 3D, 3E). Intracellular CXCL9 was confirmed in CD11c+ cells (not shown) as well as in CD14+CD11b+HLADR+CD141+CD16−CD3−CD1c− TAMs by FACS, but not in circulating monocytes (Figure S3C), while CXCL9 mRNA was detected in sorted tumor leukocytes, but not in tumor cells (Figure 3F). Consistent with the correlation between CXCL9 and TILs, a large fraction of freshly isolated CD8+ and CD4+ TILs were CXCR3+ relative to matched peripheral blood (Figure 3G, 3H). Thus, CXCL9 is produced by TAMs and DCs, and may be implicated in the recruitment of TILs.
Figure 3. CXCL9 is expressed by tumor macrophages and dendritic cells and closely associates with ieCD8+ TILs.

(A) CXCL9 mRNA in ovarian tumors with or without ieCD8+ TILs (n=86). Data are represented as mean ± SEM. P value was calculated with unpaired T test. (B) Quantification of ieCD8+ TILs in tumor islets of EOC that are positive or negative for CXCL9 expression (n=86). Data are represented as mean ± SEM. P value was calculated with Mann-Whitney test. (C) Representative IHC of EOC with low or high CXCL9 in the tumor stroma and corresponding infiltration of CD8+ TILs in the same cases. S: stroma; T: tumor (D) Multispectral immunostaining of an EOC case for CXCL9 (green), CD11c (orange, DCs), and CD68 (red, macrophages). (E) Representative images of CXCL9 IHC (left) and multispectral immunostaining for CXCL9 (green) and CD68 (red) (right). (F) CXCL9 mRNA levels in sorted CD45+Epcam− TILs and CD45−Epcam+ ovarian cancer cells. Boxplots represent 25th and 75th percentiles with midline indicating the median; whiskers extend to the lowest/highest values. P value was calculated with Mann-Whitney test. (G) FACS histogram of CXCR3 in CD4+ and CD8+ T cells from matched tumor, ascites, and blood (PBL) of an EOC case. (H) FACS histogram CXCR3 surface expression in PBMCs and TILs from several EOC patients. See also Figure S3.
CXCL9 is specifically upregulated by IFNγ in TAMs and DCs
To understand more on CXCL9’s role, we asked how this chemokine is regulated in TME. We found that CD14+CD11b+ TAMs purified from EOC samples secreted high levels of CXCL9 responding to human recombinant IFNγ, but not type-I IFNs (IFNα or IFNβ) (Figure 4A). This behavior was similar to CD11b+ monocytes or CD11c+ DCs derived from peripheral blood mononuclear cells (PBMCs) of EOC patients and healthy donors (Figure S3D, S3E). In contrast, CXCL10 was induced at high levels by both type-I and type-II IFNs in TAMs (Figure 4B) and healthy donor monocytes (Figure S3F). TILs did not express CXCL9 or CXCL10 at baseline or under IFN stimulation (Figure 4A, 4B). Similarly, CXCL9 expression in sorted tumor cells was minimal, and CXCL10 was low even when tumor cells were stimulated with high-dose IFNγ (40 ng/ml, Figure 4A, 4B), in line with silencing of these chemokines in ovarian cancer cells (Peng et al., 2015). Thus, TAMs and DCs are the main contributors of CXCL9 in the TME, and its production is dependent on IFNγ and not on type-I IFNs. Ovarian cancers express IFNA1, IFNB1 and IFNG mRNA but only IFNG correlated positively with CD8A (Figure 4C). Corroborating that CXCL9 depends on IFNγ, we found that CXCL9 expression correlated only with IFNG, but not type-I IFNs’ expression whereas CXCL10 expression highly correlated with that of IFNG and of IFNB1 (Figures 4D, 4E; S3G, S3H; Table S1).
Figure 4. CXCL9 is only upregulated by IFNγ in TAMs and DCs whereas CXCL10 is upregulated by both type-I IFNs and IFNγ in tumor cells, TAMs and DCs.

(A, B) Cytokine bead array (CBA) quantification of CXCL9 (A) and CXCL10 (B) in EOC-derived T cells, TAMs and tumor cells after 72 hr stimulation with IFNα, IFNβ or IFNγ. P values of IFNγ vs. untreated conditions are shown. (C) Pearson correlation plots of expressions of CD8A with IFNA1 (left), IFNB1 (center), or IFNG (right) in the TCGA ovarian cancer dataset. (D) Pearson correlation plots of expressions of IFNG with CXCL9 (left) and CXCL10 (right) in the TCGA ovarian cancer dataset. (E) Correlation coefficient heatmaps summarizing associations between expressions of IFNA1, IFNB1 and IFNG with those of CD8A, CXCL9, CXCL10 and CXCL11 for 6 solid tumor types in TCGA. All p values are shown is Table S1. (F) Illustration of Ovarian TME co-cultures (left) and CXCL9 secretion under no blockade (medium alone or isotype control Abs), single or combined Ab blockade (right). P values were calculated with corrected T tests comparing each condition to medium only (n=4). (G) Pearson correlation of relative IFNγ and CXCL9 secretion in four patients. (H, I) Secretion of IFNγ (H) and CXCL9 (I) in mixed autologous tumor co-cultures, in the presence of HLA-ABC blocking Ab or control IgG (n=10). (J) Illustration of chemotaxis assay of autologous blood T cells or TILs towards supernatants derived from mixed autologous tumor co-cultures (left) and quantification of TILs migration in tumor-conditioned medium in the presence of anti-CCR5 and anti-CXCR3 Abs (right). (K) Autologous blood T cells migration towards supernatants from 2D co-cultures, purified tumor cells (CD45−) or purified tumor leukocytes (CD45+) in the presence of anti-CXCR3 Ab. (L) Illustration for the co-culture chemotaxis system using human tumor spheroids, monocytes and CD8+ T cells transduced with cognate TCR (left). Migration of TCR-transduced CD8+ T cells in the following tumor spheroids: OV79-NYESO1+ coexpressing or not CCL5 (NY/CCL5 and NY respectively) and enriched with CXCL9-producing monocytes (NY/CCL5/CXCL9), CCL5+ only expressing OV79 tumor cell spheroids (CCL5), or OV79 tumor spheroids coated with CXCL9+ monocytes (CXCL9) (right). (M) Spheroid infiltration by antigen-specific T cells in the presence of anti-CXCR3 Ab. NY: NY-ESO+ OV79 tumor cells, CCL5: CCL5+ OV79 tumor cells, CXCL9: CXCL9+ monocytes. (N) Spheroid infiltration by TCR-transduced or non-transduced CD8+ T cells infiltrating OV79-NYESO1+/CCL5/CXCL9 tumor spheroids. All bar-graph data are represented as mean ± SEM. All p values were calculated with multiple T tests unless specified otherwise. See also Figure S3 and Table S1.
To investigate further the regulation of CXCL9 in the TME, we generated whole primary tumor cultures derived from dissociated EOC samples with documented ieCD8+ TILs. In these 2D unsorted TME cultures, we confirmed that only neutralization of IFNγ – but not IFNα or IFNβ – reduced CXCL9 substantially (Figure 4F). Indeed, IFNγ and CXCL9 secreted levels were highly correlated (Figure 4G). Importantly, release of IFNγ as well as CXCL9 largely depended on antigen recognition by TILs, since MHC class I and/or II blockade attenuated both (Figure 4H, 4I; Figure S3I, S3J). Thus, CXCL9 expression in the ovarian TME is restricted to TAMs and DCs, and is induced specifically by IFNγ, dependent on tumor antigen recognition.
IFNγ-inducible chemokines from TAMs cooperate with tumor-derived CCL5 for the engraftment of tumor-reactive TILs
The above data indicate that CXCL9 is a reliable biomarker of T-cell engraftment and activation in the TME. It led us to hypothesize a cascade of events whereby patrolling TILs (initially recruited by constitutive tumor chemokines such as CCL5) get activated by tumor antigen and release IFNγ, which in turn activates TAMs and DCs to secrete CXCL9. To test this hypothesis, we collected the supernatants of primary TME cultures and performed chemotaxis assays with autologous TILs. CCR5 blockade attenuated the recruitment of TILs towards TME-conditioned media, which was further suppressed by CXCR3 blockade (Figure 4J). Furthermore, CXCR3-dependent recruitment of T cells was activated only by supernatants of primary autologous co-cultures that comprised both sort-purified tumor cells and leukocytes (Figure 4K), confirming that CXCR3-dependent recruitment of T cells requires interaction between tumor associated leukocytes and autologous tumor cells.
To further understand the interplay between CXCL9 and CCL5 in the recruitment of tumor-specific T cells, we developed an in vitro model of EOC TME spheroids using OV79 ovarian cancer cells ectopically expressing (or not) HLA-A*02 and NY-ESO-1 and/or CCL5. IFNγ-activated CD11b+ monocytes (γMs) adhered to tumor spheroids mimicking the juxta-epithelial CXCL9+ TAMs observed in EOC. CD3/CD28-stimulated NY-ESO-1 TCR transduced CD8+ T cells upregulated CXCR3 and CCR5 (Figure S3K). In the absence of CCL5 or γMs, tumor expression of cognate antigen alone enabled low-level engraftment of NY-ESO-1 TCR+ CD8+ T cells in the spheroids. Similarly, expression of CCL5 or CXCL9 alone (without tumor NY-ESO-1), or expression of NY-ESO-1 and CCL5 or CXCL9 resulted in low-level engraftment of T cells. However, T-cell infiltration markedly increased when tumors coexpressed antigen and CCL5 and were enriched with γMs (Figure 4L). T-cell recruitment was abrogated by CXCR3-neutralizing Ab (Figure 4M). Importantly lack of tumor antigen or lack of tumor-specific TCR by T cells abolished T-cell engraftment (Figure 4N). Collectively our data indicate that tumor CCL5 expression is necessary but not sufficient to enable TILs engraftment. Antigen recognition by tumor-reactive T cells and IFNγ-activated TAMs and DC are also required to establish a positive loop effectively amplifying T-cell recruitment through CXCL9.
CCL5/CXCL9 co-expression reveals immunoreactive tumors with longer survival and response to PD-1 inhibition
Given the above observations, we next asked whether coexpression of CCL5 and CXCL9 reveals immunoreactive tumors in a large EOC dataset (Ganzfried et al., 2013). We found again a strong correlation between CXCL9 and CCL5 expression and observed that overexpression of CXCL9 occurs mainly in the context of CCL5 overexpression (Figure 5A). Patients with CCL5hiCXCL9hi ovarian tumors survived longer than the other groups (Figure 5B). We further documented a strong correlation between CXCL9 and CCL5 expressions (where CXCL9 was expressed mainly in CCL5-overexpressing tumors, not shown) in triple-negative breast, colon, lung cancer and melanoma. Similarly, in each tumor type CCL5hiCXCL9hi tumors exhibited longer survival than the other groups (Figure 5B). Using established methods (Bindea et al., 2013), CCL5hiCXCL9hi tumors predicted to have higher infiltration of CD8+, cytotoxic and Th1 T cells as well as DCs and macrophages, while the CCL5loCXCL9lo tumor type displayed a general lack of immune infiltration (Figure 5C, 5D). The same observations were made across all tumor types examined (Figure S4A). By the CIBERSORT algorithm, CCL5hiCXCL9hi tumors exhibited higher percentage of activated CD8+ and CD4+ T cells and M1 macrophages, and higher immunophenoscores (Charoentong et al., 2017) than CCL5loCXCL9lo tumors in ovarian and breast cancer as well as melanoma (Figure S4A–S4C).
Figure 5. Co-expression of CCL5 and CXCL9 reveals immunoreactive tumors that respond to PD1 blockade.

(A) Scatter plot of expressions of CXCL9 and CCL5 in 2402 serous EOC cases and definition of four subgroups. (B) Kaplan-Meier survival analysis according to chemokine subgroups (as defined in A) in five solid tumor types. Due to its low frequency, the CXCL9-CCL5 Hi-Lo group for each cancer type is omitted. (C) Heatmap showing scoring of immune gene signatures (Bindea et al., 2013) in EOC according to CCL5 and CXCL9 expression subgroups. (D) Summary pie chart displaying the immune subset gene signatures of Hi-Hi and Lo-Lo groups in five cancer types computed as in (C). Heatmap displaying the significance level of the differences in immune subsets between Hi-Hi and Lo-Lo tumors for each cancer type (ANOVA followed by Tukey test) and the directionality of the difference (red: over- representation in Hi-Hi, light blue over-representation in Lo-Lo). (E) Hierarchical biclustering and gene expression heatmap (red, high; green, low) with names of strongest genes discriminating the Hi-Hi group (FDR cutoff =1×10−38). (F) Enrichment analysis heatmap for gene sets in BioCarta and Wikipathways in the top 202 differentially expressed genes characteristic for the Hi-Hi groups in five different cancer types. Colors in the heatmap represent the level of significance of the enrichment (-log10 of the adjusted p values). (G) Protein-protein interaction network (STRING) for the chemokine-derived 21-gene shared signature consisting of the included genes. Average local clustering coefficient=0.727 and protein-protein interaction (PPI) enrichment p<2.2e-16. (H) 21-gene signature score for pre-treatment samples are shown for responders (R) and non-responders (NR) (left) and ROC plot for 21-gene signature score vs. response (right) from the Nivolumab-Ipilimumab sequential treatment arm in (Rodig et al., 2018). Statistical comparison is based on one-sided Wilcoxon rank sum tests. (I) Variation of the 21-gene signature score for on-treatment samples from Chen et al. (Chen et al., 2016) defined by the treatment received, the response status of the patient classified and the time point of the profiling. All boxplots represent 25th and 75th percentiles with midline indicating the median; whiskers extend to maximally 1.5 IQ range beyond the box limits. Points indicate values for individual subjects beyond whiskers. See also Figures S4, S5 and Tables S2–S4.
In each tumor type, we could identify partly overlapping (among tumor types) individual ≈200-gene signatures that were overexpressed uniquely in the CCL5hiCXCL9hi tumors (FDR cutoff=1×10−38, Figure 5E depicts ovarian data) (Tables S2–S4). Their pathway analysis revealed enrichment in TCR signaling, T-cell activation, costimulation, and IFN signaling in CCL5hiCXCL9hi tumors regardless of histology (Figure 5F). We merged these to derive a final common 21-gene signature overexpressed in CCL5hiCXCL9hi tumors that was shared across all five-cancer types (Tables S3, S4). Protein-protein interaction network analysis confirmed functional attributes of immune activation to this 21-gene signature (Figure 5G). We reasoned that if this 21-gene signature revealed an ongoing tumor immune recognition, it should be present at baseline in melanoma lesions that respond to PD-1 blockade where pre-existing TILs are required (Tumeh et al., 2014). We analyzed pre-treatment biopsies of melanoma patients who received nivolumab followed by ipilimumab (Rodig et al., 2018). Overexpression of the signature was associated with response to nivolumab (Figure 5H), with an area under the curve (ROC) for predicted vs. best overall response by RECIST criteria of 78.7% (Figure 5H). On the contrary, this relationship was lost in the cohort treated with ipilimumab followed by nivolumab in the same study (Figure S5A, S5B). We also examined whether this signature overlapped with overexpressed genes found on-treatment with immune checkpoint blockade therapy (Chen et al., 2016). Fourteen genes of the signature (CCL5, CD247, CD3E, CD4, CD86, CD8A, CXCL9, GZMK, IL10RA, IL2RB, IRF1, PTPRC, STAT1 and TNFRSF) were included in the gene panel and found to be increased upon treatment in tumors that responded to immune checkpoint blockade therapy (Figure 5I). In addition, most of the genes of immune activation emerging during effective checkpoint blockade therapy (Chen et al., 2016) were included in our signatures and were strongly upregulated in CCL5hiCXCL9hi melanoma, breast, ovarian, colon and lung cancers at the steady state (Figure S5C).
Tumor immune desertification in vivo is due to tumor loss of CCL5 and associated with macrophage loss of CXCL9 in the mouse
Next, we used the ID8 syngeneic mouse ovarian cancer model to test if CXCL9 expression depends on prior CCL5 expression in tumors. We have previously shown that early orthotopic ID8 tumors exhibit an infiltration of CD8+ T cells, which is spontaneously lost within 6–7 weeks of tumor development (Figure 6A) (Duraiswamy et al., 2013). Principal component analysis showed that early (ieCD8+ T cells) and late (ieCD8− T cells). ID8 tumors were clearly separated based on immune-related genes (Figure 6B). Ccl5 and Cxcl9 were among the most highly upregulated genes in ieCD8+ relative to ieCD8− tumors (p<0.01, Figure 6C). Immune-related genes overexpressed in ID8 tumors correlated with human orthologs overexpressed in CCL5hiCXCL9hi human EOC (Figure 6D). As their human counterparts, Ccl5 and Cxcl9 correlated significantly with Cd3e and with each other in ID8 tumors (Figure 6E). Finally, early ID8 tumors expressing such chemokine gene signature were responsive to PD-1 blockade-based immunotherapy, while later tumors that lost the signature were not (Figure S6).
Figure 6. Epigenetic loss of Ccl5 drives tumor immune desertification in vivo and is associated with loss of Cxcl9 in TAMs.

(A) Schema of the experiment (left) and IHC analysis of cytokeratin 8 (CK8) and ieCD8+ TILs in early and late stage mouse ID8 tumors (right). (B) Principal component analysis of gene expression in early and late ID8 tumors. (C) Volcano plots displaying names of the genes that were significantly (FDR=0.05) and ≥2-fold downregulated (left) or upregulated (right) in late versus early ID8 tumors. (D) Correlation of mouse ID8 tumor genes with human ovarian cancer orthologs. (E) Correlation plots of expressions of indicated genes. (F) Relative mRNA quantification of Cxcl9 in FACS sorted CD45− tumor cells and CD45+CD3− CD11b+ TAMs. P value was calculated with unpaired T test. (G) Relative mRNA quantification of the top genes of the human chemokine signature genes by qPCR in early and late ID8 tumors. P values were calculated with multiple T tests. (H) Relative mRNA quantification of Ccl5 in the ID8 cell line and in ID8 cells purified from late ID8 tumors 3 days post-treatment with 5’-aza-2’-deoxycytidine or DZNep. Data are represented as mean ± SEM. P values were calculated with unpaired T test.
All boxplots represent 25th and 75th percentiles with midline indicating the median; whiskers extend to the lowest/highest values. See also Figure S6.
We asked which cells produced CXCL9 in early ID8 tumors. Recapitulating our human observations, Cxcl9 mRNA was only detected in sorted CD45+CD3−CD11b+ TAMs and not in tumor cells from early tumors (Figure 6F). Expression of Cxcl9 and ortholog genes from the shared 21-gene signature was lost in advanced ID8 tumors (Figure 6G). Further recapitulating our findings in human EOC, ID8 tumor cell line constitutively expressed Ccl5 in vitro whereas late tumors lacking TILs did not express Ccl5 in tumor cells in vivo. This was reversed by 5’-aza-2’-deoxycitine but not by DZNep (Figure 6H). Thus, like in the human, CCL5 and CXCL9 coexpression was associated with TILs engraftment and activation, and with response to PD-L1 blockade in the mouse. CXCL9 was produced by TAMs and loss of tumor CCL5 by DNA methylation in tumor cells was associated with loss of CXCL9 and TILs engraftment in advanced tumors.
CCL5 drives CXCL9 expression in tumor macrophages and TILs engraftment in ovarian cancer
To further test whether CCL5 expression by tumor cells can drive CXCL9 expression in the tumors, we first asked whether prevention of Ccl5 silencing would reverse the loss of Cxcl9 and the immune desertification of ID8 tumors. We generated ID8 tumor lines overexpressing CCL5 (ID8Luc CCL5OE) (Figure 7A; Figure S7A) and found that sustained expression of CCL5 by tumor cells indeed resulted in increased expression of Cxcl9, Cd8a, and Ifng in vivo in late ID8 tumors (Figure 7B). This also resulted in reduced tumor growth (Figure S7B) and significantly longer survival (Figure 7C).
Figure 7. IFNγ and CXCL9 enhance T-cell engraftment in CCL5-expressing tumors in vivo.

(A, B) Relative mRNA quantification of Ccl5 (A) and Cd8a, Ifng, and Cxcl9 (B) in ID8Luc CCL5OE tumors and ID8Luc control tumors. (C) Kaplan-Meier survival curves of mice injected with ID8Luc CCL5OE or ID8Luc control cells. P values were calculated with Log-rank (Mantel-Cox) test. (D) Relative mRNA quantification of Ccl5 in FACS sorted CD45− tumor cells derived from ID8Luc scr sh control tumors or ID8Luc CCL5KD tumors. (E) Relative mRNA quantification of indicated genes by qPCR in ID8Luc scr sh or ID8Luc CCL5KD tumors. (F) Kaplan-Meier curves showing survival of mice with intraperitoneal ID8Luc scr sh or ID8Luc CCL5KD tumors. (G) Relative mRNA quantification of Cxcl9 in FACS sorted CD45+CD3−CD11b+ TAMs derived from ID8Luc scr sh or ID8Luc CCL5KD tumors. (H) Relative mRNA quantification of indicated genes by qPCR in ID8Luc CCL5OE and treated in vivo with an Ab neutralizing IFNγ or an isotype control Ab. (I) Intraperitoneal growth of ID8Luc CCL5OE cancers as detected by bioluminescence imaging during treatment with IFNγ neutralizing or isotype control Abs. Data presented as mean ± SEM. P values were calculated with Two-way Anova T tests. (J) Relative mRNA quantification of indicated genes by qPCR in ID8Luc CCL5OE tumors and treated in vivo with an Ab neutralizing CXCL9 or an isotype control Ab. (K) Intraperitoneal tumor growth kinetics of ID8Luc CCL5OE cancers as detected by bioluminescence imaging during treatment with CXCL9 neutralizing or isotype control Abs. Data presented as mean ± SEM. P values were calculated with Two-way Anova T tests.
All boxplots represent 25th and 75th percentiles with midline indicating the median; whiskers extend to the lowest/highest values. Corresponding p values were calculated with Mann-Whitney tests. See also Figure S7.
We then stably knocked down CCL5 in ID8 cells (ID8Luc CCL5KD, Figure 7D; Figure S7C). ID8Luc CCL5KD tumors exhibited significantly lower expression of Cxcl9, Cd8a, Ifng, and Grzmb (Figure 7E) and reduced ieTILs (Figure S7D, S7E) relative to control ID8 tumors. In agreement, CCL5 knockdown resulted in accelerated tumor growth (Figure S7F) and shorter survival (Figure 7F). Confirming that tumor cell CCL5 drives CXCL9 expression in TAMs, tumor-infiltrating CD11b+ cells isolated from these early ID8Luc CCL5KD tumors exhibited significantly decreased Cxcl9 expression relative to TAMs from control ID8Luc tumors (Figure 7G). Importantly, ID8Luc CCL5KD tumors exhibited no difference in CD11b+ cell infiltration (Figure S7G) or in Batf3 expression (Figure S7H) relative to control tumors. These results confirm that tumor-intrinsic CCL5 expression drives CXCL9 expression in tumor myeloid cells and determines the tumor immunophenotype.
IFNγ and CXCL9 enhance T-cell engraftment in CCL5-expressing tumors in vivo
Next, we tested whether sustained tumor expression of CCL5 drives CXCL9 expression in TAMs through IFNγ. We blocked IFNγ using neutralizing Abs in the ID8Luc CCL5OE model and found that Cxcl9 expression was abrogated in CCL5OE tumors under IFNγ blockade (Figure 7H). Importantly, Ifnb levels remained unaltered in tumors (Figure 7H), confirming that CXCL9 regulation in TAMs is independent of type-I IFNs. IFNγ blockade significantly reduced CD8+ and total TILs infiltration (Figure S7I, S7J) and reduced the Cxcr3 expression (Figure 7H). IFNγ blockade also accelerated the growth of CCL5OE tumors (Figure 7I). Importantly, although the total frequency of CD11b+ TAMs was not altered (Figure 7H; Figure S7K), the frequency of M2-like CD206+ TAMs was significantly increased (Figure S7L), consistent with loss of IFNγ effects in the TME.
Finally, we tested whether CXCL9 contributes to T-cell engraftment in CCL5OE tumors. CXCL9 blockade in the ID8Luc CCL5OE model decreased Cd8a expression, indicating reduced CD8+ TILs, and Cxcr3 expression (Figure 7J, Figure S7M). Importantly, we observed that in contrast to Cxcl9, Cxcl10 was still expressed, and CD11b+ TAMs did not change significantly (Figure 7J, Figure S7N). Furthermore, inhibition of CXCL9 resulted in overall increased tumor growth (Figure 7K). The above results, in line with our in vitro human data, confirm that in the presence of CCL5 expression, IFNγ activation and CXCL9 expression are downstream mediators ensuring successful T-cell engraftment and immune attack in tumors.
Discussion
In this study, we identified a close correlation between CCL5 and CXCL9 expressions in human ovarian and other cancers and their coexpression correlated with ieTILs and an immunoreactive molecular phenotype. CCL5 loss in human and murine ovarian cancer led to significant reduction of CXCL9 expression in TAMs, abrogation of the entire pathway and loss of ieTILs. Although necessary, expression of CCL5 by tumor cells is insufficient. CXCL9 acted as an amplifier of T-cell engraftment in human TME co-cultures in vitro, and blockade of CXCL9 significantly reduced ieTILs in CCL5OE tumors in vivo. Our in vitro TME co-cultures revealed that cognate tumor antigen recognition by tumor-specific T cells is the catalyst triggering IFNγ release and thus, activation of TAMs and DCs to produce CXCL9 and CXCL10 in the TME. Although prior important work has revealed the key roles of chemokines in T-cell recruitment in tumors (Harlin et al., 2009; Peng et al., 2015; Spranger et al., 2015; Spranger et al., 2017), the present data reveals a hierarchical cascade, where tumor-intrinsic constitutive chemokines, e.g. CCL5, enable T-cell trafficking in tumors and cooperate with IFNγ-inducible chemokines including CXCL9 that amplify engraftment of TILs and help establish the “hot” tumor immunophenotype. Our findings are supported by previous reports showing that TILs in ovarian cancer recognize autologous antigen and produce markers of TCR-dependent activation including IFNγ (Westergaard et al., 2019 2018; Ye et al., 2014), and explain observations where loss of the antigen presentation machinery is associated with TILs loss in cancers (Angelova et al., 2018; Zaretsky et al., 2016).
Since constitutive CCL5 expression enables tumor immune recognition, it is not surprising that tumor evolutionary pressure may lead to its silencing. We report that DNA methylation is a principal epigenetic mechanism negatively regulating CCL5, in agreement with prior evidence in lung and colon cancer (Li et al., 2014). It should be noted that methylation may not be the only mechanism downregulating CCL5 expression in tumors. In fact, we also identified a reverse association between the NOTCH, MYC or WNT pathways and CCL5 mRNA levels across tumor types (not shown), while a previously described WNT/β-catenin gene signature (Spranger et al., 2015) was upregulated specifically in CCL5low tumors in ovarian, breast, lung and melanoma datasets (not shown). These pathways may be interdigitated. Indeed, DNMT1 inhibition decreased MYC and increased CCL5 expression in lung cancer (Topper et al., 2017). These mechanisms may affect additional constitutive chemokines with overlapping roles. For example, WNT/β-catenin-driven suppression of CCL4 leads to “cold” tumor phenotypes in melanoma (Spranger et al., 2015; Spranger et al., 2017).
CXCL9 emerged as a biomarker of tumor immune recognition and local IFNγ production, explaining its close correlation with TILs engraftment. Through this association we uncovered the important role of TAMs and DCs, which in response to local IFNγ produce chemokines to further enhance T-cell recruitment. In agreement with older literature (Farber, 1990; Farber, 1993), CXCL9 was induced only by IFNγ, whereas CXCL10 was also induced by type-I IFNs in tumors. This suggests that while CXCL9 is expressed only under conditions of sufficient IFNγ expression (i.e. by tumor-specific T cells), CXCL10 may in addition be sensitive to innate inflammatory conditions such as type-I IFN signaling, NF-κB signaling (Burke et al., 2013), or hypoxia-reperfusion (Zhai et al., 2008), and therefore its expression may not necessarily translate to effective T-cell infiltration (Kunz et al., 1999). This explains the close correlation of CXCL9 (and IFNγ) – but not CXCL10 (or type-I IFNs) – with CD8+ T-cell infiltration in gene expression data sets. However, our data do not exclude a complementary role for CXCL10 in the dynamic interplay with constitutive chemokines, since CXCL10 is also induced in macrophages by IFNγ. Furthermore, our data do not contradict the well-established role of innate immune pathways, including type-I IFN signaling, in the establishment of the immunoreactive phenotype in tumors. Type-I IFN signals can sensitize tumor macrophages to IFNγ through increased STAT1 signaling (Karonitsch et al., 2012). It is possible that in so-called inflamed tumors, type-I IFNs “prime” macrophages and DCs in the TME, lowering the threshold of local IFNγ required for enabling the dynamic circuit between tumor cell-expressed CCL5 and macrophage-expressed CXCR3 ligands, which can effectively amplify T-cell infiltration. Additionally, the tonic expression of CXCL10, induced by type-I IFNs, could lower the requirements for de novo production of CXCR3 ligands by IFNγ-activated TAMs for attaining effective TILs engraftment. In fact, we found that blockade of type-I IFN signaling through an IFNAR1-neutralizing Ab significantly decreased the levels of CXCL9 and largely attenuated T-cell infiltration in CCL5OE ID8 tumors (not shown).
Pre-existing T-cell infiltration has been established as a prerequisite for responding to PD-1/PD-L1 blockade therapy in melanoma (Tumeh et al., 2014). In agreement, we found that the expression levels of our CCL5hiCXCL9hi-derived 21-gene signature was significantly higher in melanomas that responded to PD-1 followed by CTLA-4 blockade. Interestingly, there was no difference in gene expression among responders and non-responders to CTLA-4 followed by PD-1 blockade, indicating that the inverse sequence has different requirements in terms of pre-existing immune activation (Rodig et al., 2018). Similarly, we found that mouse ovarian Ccl5hiCxcl9hi ID8 tumors were infiltrated by T cells and responded to PD-L1 blockade therapy, while tumors that lost Ccl5 and Cxcl9 expression became unresponsive.
Collectively, our results reveal a hierarchy in how chemokines determine the immunophenotype of tumors. The cooperation between constitutive chemokines such as CCL5 and IFNγ-inducible chemokines such as CXCL9 plays a key and universal role in the orchestration of T-cell responses in tumors and enables the establishment of the T-cell inflamed phenotype and response to checkpoint immunotherapy. Our results also suggest that loss of tumor-intrinsic chemokines supporting T-cell recruitment is a common mechanism of immune desertification and escape, with CCL5 being a prevalent chemokine target. Together with loss of tumor antigen presentation, silencing of CCL5 driven by oncogenic pathways should be added to the molecular alterations that lead to tumor immune escape and resistance to immune checkpoint blockade.
STAR methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, George Coukos (george.coukos@chuv.ch)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human tumor and blood specimens
An EOC tissue microarray derived from 522 treatment-naïve (International Federation of Gynecologists and Obstetricians (FIGO) stage-III) EOC specimens, collected under institutional (University of Helsinki, Finland) approved protocol, was provided by Dr Ralf Bützow and was used for studying expression of CCL5 and TILs. A cohort of 57 snap-frozen treatment-naïve and advanced-stage EOC samples, collected under institutional approved protocol, was kindly provided by Dr Katsaros at the University of Turin, Italy and was used to confirm CCL5, CXCL9 and CD8A correlations. The rest of the EOC samples were collected and studied under a protocol approved by the University of Pennsylvania Institutional Review Board: 86 matched snap-frozen and paraffin-embedded samples provided by the Tumor Tissue and Biospecimen Bank (TTAB), Department of Pathology, at the University of Pennsylvania, Philadelphia, USA, and were used to study expression of CCL5 and CXCL9, and TILs; 20 snap-frozen and 25 fresh tumor samples collected by the Ovarian Cancer Center, Department of Obstetrics & Gynecology, University of Pennsylvania, Philadelphia, USA. Those samples were used for laser capture microdissection and cell culture functional assays, respectively. Peripheral blood of healthy donors was obtained from the human immunology core at the University of Pennsylvania and collected by the Ovarian Cancer Center, Department of Obstetrics & Gynecology, University of Pennsylvania, Philadelphia, USA. Informed consent was obtained from all human subjects included in this study.
Mouse models
Six to eight-week old female C57BL/6mice were purchased from Envigo and maintained in pathogen-free conditions. Animal experimentation procedures were performed according to the protocols approved by the Veterinary Authorities of the Canton Vaud according to Swiss law.
Cell Cultures
Human ovarian cancer cell lines OVCAR5, OVCAR3, A1847, A2780/C30 were cultured in RPMI Glutamax supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptavidin (P/S). Ovarian cancer cell line OV79 was grown and propagated in RPMI media supplemented with 10% FBS and 1% P/S. The human primary cultures were propagated in RPMI enriched with 10% (human/fetal bovine) serum supplemented with 20 IU/ml recombinant human (rh) IL-2 and P/S at a concentration of 100 IU/ml. Mouse ovarian cancer cell line ID8 was cultured in DMEM Glutamax supplemented with 10% FBS and 1% P/S. All cell lines were cultured at 37°C in 5% CO2 atmosphere.
METHOD DETAILS
Human tumor dissociation
Fresh tumor specimens were processed by sterile mechanical dissociation and the tissue was filtered through a wire grid to yield a single cell suspension. Cells were separated on a Percoll density gradient for 30 min at 1500xg at room temperature. In some experiments, all cells recovered from tumors were used directly to generate mixed primary co-cultures. For other experiments, a >95% enriched population of tumor-derived CD45+ leukocytes was procured by magnetically activated sorting (MACS) using nanoparticles conjugated to streptavidin and a biotinylated anti-CD45 Ab. The remaining leukocyte-depleted cells were plated on plastic for 30 min, to further eliminate fast adhering cells (mostly monocytes), and floating cells were replated on plastic. Two hours later floating cells (containing most lymphocytes) were removed to leave an adherent fraction highly enriched in tumor cells (>90%), as previously described.
Peripheral blood lymphocytes isolation
Peripheral blood lymphocytes (PBL) were isolated from patients with advanced ovarian carcinoma and normal healthy donors by elutriation. T cells were stimulated with anti-CD3/anti-CD28-coated beads (Invitrogen) at a 3:1 bead:cell ratio at a final concentration of 1 × 106/ml for 48 hr in the presence of 20 IU/ml rhIL-2. A population enriched in human peripheral blood monocytes was derived from PBMCs of patients with advanced EOC through elutriation. Immature dendritic cells were generated from PBMCs of patients with advanced EOC using IL-4 and GM-CSF as previously described (Schlienger et al., 2000). In some experiments, cells were stimulated with 40 ng/ml rhIFNγ.
IFNα, β, γ stimulation of tumor infiltrating T cells, monocytes and tumor cells isolated from human ovarian cancer dissociates
CXCL9 expression upon IFNα, β, γ stimulation in human monocytes was evaluated in vitro using PBMC-derived monocytes (n=2) or tumor dissociates of EOC patients (UPENN Cohort) (n=3). EOC dissociates were washed in PBS and stained for Fluorescence-Activated Cell Sorting based on their expression of CD45, CD3, CD11b and CD14 markers. Isolated T lymphocytes (CD3+CD45+ live cells), monocytes (CD14+CD11b+CD45+ live cells) and tumor cells (CD45− live cells) were plated in round bottom 96 well-plate at 100 000 cells per well. Cells were stimulated with human recombinant IFNα, IFNβ or IFNγ at 40 ng/ml in full media (RPMI + 10% FBS + 1% P/S) and supernatants were harvested after 72 hr for quantification of CXCL9 and CXCL10 expression by BD™ Cytometric Bead Array System. All Abs are listed in the Key Resource Table.
KEY RESOURCES Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMAb anti-mouse IFNγ clone R4–6A2 | BioXCell | BE0054, RRID: AB_1107692 |
| InVivoMAb rat IgG1 Isotype control, anti-trinitrophenol, clone TNP6A7 | BioXCell | BE0290, RRID: AB_2687813 |
| InVivoMAb anti-mouse CXCL9 (MIG), clone MIG-2F5.5 | BioXCell | BE0309, RRID: AB_2736989 |
| InVivoMAb polyclonal Armenian hamster IgG | BioXCell | BE0091, RRID: AB_1107773 |
| anti-Human CXCR3 Antibody, clone 49801 | R&D Systems | MAB160–100 |
| anti-Human CCR5 Antibody, clone 45531 | R&D Systems | MAB182–100 |
| Ultra-LEAF™ Purified anti-human HLA-A,B,C Antibody | Biolegend | 311427, RRID: AB_2561492 |
| Purified anti-human HLA-DR, DP, DQ Antibody | Biolegend | 361702, RRID: AB_2563139 |
| anti-human IFNα | R&D systems | 21100–2 |
| Purified anti-human IFN-β Antibody | Biolegend | 514002, RRID: AB_2122765 |
| LEAF™ Purified anti-human IFN-γ Antibody | Biolegend | 506513, RRID: AB_315446 |
| anti-mouse CD45-BV650, clone 30F11 | Biolegend | 103151, RRID: AB_2565884 |
| anti-mouse CD3e PECy5.5, clone 145–2C11 | Invitrogen | 35–0031-80, RRID: AB_11218085 |
| anti-mouse CD3 eFluor 660, clone 17A2 | eBioscience | 50–0032-82, RRID: AB_10598657 |
| anti-mouse CD3 eFluor450 | Home made | |
| anti-mouse CD4 PB, clone GK1.5 | Biolegend | 100428, RRID: AB_493647 |
| anti-mouse CD8 BV711, clone 53.6.7 | Biolegend | 100748, RRID: AB_2562100 |
| anti-mouse CD11b PECy7, clone M1/70 | eBioscience | 25–0112-81, RRID: AB_469587 |
| anti-mouse CD11c BV605, clone N418 | Biolegend | 117334, RRID: AB_2562415 |
| anti-mouse CD206 PE/Dazzle™ 594, clone C068C2 | Biolegend | 141732, RRID: AB_2565932 |
| anti- mouse CD16/CD32, clone 2.4G2 | BD Pharmigen | 553142 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain | Invitrogen | L34966 |
| anti-human CD45 APC, clone HI30 | Biolegend | 304012, RRID: AB_314400 |
| anti-human CD3 BV510, clone UCHT1 | Biolegend | 300448, RRID: AB_2563468 |
| anti-human CD11b BB515, clone ICRF44 | BD | 564517 |
| anti-human CD14 BV605, clone M5E2 | BD | 564054, RRID: AB_2564142 |
| anti-human CD19 PE, clone HIB19 | Biolegend | 302254 |
| anti-human CD56 PE, clone HCD56 | Biolegend | 318305, RRID: AB_604093 |
| anti-human EPCAM PeCy7, clone 9C4 | Biolegend | 324222, RRID: AB_2561506 |
| Zombie UV | Biolegend | 423108 |
| anti-human CD11c (rabbit monoclonal, clone EP1347Y) | Abcam | ab52632 |
| anti-human CD68 (mouse monoclonal, clone PG-M1) | Dako | M0876 |
| anti-human CD8 (rabbit monoclonal, clone SP16) | Thermo Scientific | MA5–14548 |
| anti-human Keratin | Dako | M3515 |
| anti-human CCL5/RANTES (rabbit polyclonal) | Abcam | ab9679 |
| anti-human CXCL9/MIG (rabbit polyclonal) | Abcam | ab9720 |
| anti-mouse Immunoglobulins/HRP (goat polyclonal) | Dako | P0447 |
| anti-rabbit Immunoglobulins/HRP (goat polyclonal) | Dako | P0448 |
| anti-mouse Cytokeratin 8+18 (clone EP1628Y) | Abcam | ab53280 |
| anti-mouse CD8, clone 4SM15 | eBioscience | 14–0808-82 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human IFN-alpha I (alpha 17) Protein | R&D Systems | 11150–1 |
| Animal-Free Recombinant Human IFN-β | Peprotech | AF-300–02B |
| Animal-Free Recombinant Human IFN-γ | Peprotech | AF-300–02 |
| D-luciferin | Biosynth | L-8220 |
| Liberase TL | Roche | 540102001 |
| Dnase I | Sigma Aldrich | D4527 |
| 5’-aza-2’-deoxycitine | Sigma Aldrich | A3656 |
| DZNeP | Selleckchem | S7120 |
| CFSE | Invitrogen | C34554 |
| Critical Commercial Assays | ||
| Mouse RANTES (CCL5) Flex Set | BD | 558345 |
| Human MIG (CXCL9) Flex Set | BD | 558286 |
| Human CXCL10 | BD | 558280 |
| Dynabeads™ Human T-Activator CD3/CD28 for T Cell Expansion and Activation | Invitrogen | 11131D |
| 561 mRNA targets on the nCounter GX Immunology panel (mouse) | NanoString Technologies | NS_IMMUNOLOGY_MM_C2269 |
| Deposited Data | ||
| CuratedOvarianData Package | (Ganzfried et al., 2013) | https://bioconductor.org/packages/release/data/experiment/html/curatedOvarianData.html |
| METABRIC | (Curtis et al., 2012) | https://www.ebi.ac.uk/ega/studies/EGAS00000000083 |
| NKI | (van ‘t Veer et al., 2002) | https://doi.org/10.1038/415530a |
| Uppsala | (Calza et al., 2006; Viola et al., 2012) | GSE4922 |
| Stockholm | (Calza et al., 2006) | GSE1456 |
| PETACC-3 | (Popovici et al., 2012) | E-MTAB-990 |
| ALMAC | (Kennedy et al., 2011) | E-MTAB-863, E-MTAB-864 |
| CIT | (Marisa et al., 2013) | GSE39582 |
| DFCI | (Director’s Challenge Consortium for the Molecular Classification of Lung et al., 2008) | GSE68465 |
| HLMCC | (Director’s Challenge Consortium for the Molecular Classification of Lung et al., 2008) | GSE68465 |
| UMCC | (Director’s Challenge Consortium for the Molecular Classification of Lung et al., 2008) | GSE68465 |
| MSKCC | (Director’s Challenge Consortium for the Molecular Classification of Lung et al., 2008) | GSE68465 |
| DKFZ | (Kuner et al., 2009) | GSE10245 |
| Sato | (Sato et al., 2013) | GSE41271 |
| NCCH | (Wilkerson et al., 2010) | GSE1771 |
| Expression Project for Oncology (ExpO) | http://www.intgen.org/, GEO GSE2109 | |
| ConsensusPathDB interface | http://cpdb.molgen.mpg.de | |
| STRING database | http://string-db.org | |
| MSigDB | http://software.broadinstitute.org/gsea/msigdb | |
| Experimental Models: Cell Lines | ||
| ID8 | our own laboratory | |
| OVCAR3 | our own laboratory | |
| OVCAR5 | our own laboratory | |
| A1847 | our own laboratory | |
| A2780/C30 | our own laboratory | |
| OV79 | our own laboratory | |
| Experimental Models: Organisms/Strains | ||
| C57/BL6 mice | Envigo (former Harlan Laboratories) | |
| Oligonucleotides | ||
| CCL5 | Applied Biosystems | Hs00982282_m1 |
| CXCL9 | Applied Biosystems | Hs00171065_m1 |
| CXCL10 | Applied Biosystems | Hs01124251_g1 |
| CD8A | Applied Biosystems | Hs00233520_m1 |
| IFNB1 | Applied Biosystems | Hs01077958_s1 |
| IFNA1 | Applied Biosystems | Hs00256882_s1 |
| IFNG | Applied Biosystems | Hs00989291_m1 |
| GAPDH | Applied Biosystems | Hs03929097_g1 |
| Cd8a | Applied Biosystems | Mm01188922_m1 |
| Ccl5 | Applied Biosystems | Mm01302427_m1 |
| Cxcl9 | Applied Biosystems | Mm00434946_m1 |
| Stat1 | Applied Biosystems | Mm01257286_m1 |
| Cxcl10 | Applied Biosystems | Mm00445235_m1 |
| Cxcr3 | Applied Biosystems | Mm99999054_s1 |
| Gzmk | Applied Biosystems | Mm00492530_m1 |
| Gzmb | Applied Biosystems | Mm00442837_m1 |
| Ifng | Applied Biosystems | Mm01168134_m1 |
| Ifnb1 | Applied Biosystems | Mm00439552_s1 |
| Gapdh | Applied Biosystems | Mm99999915_g1 |
| CD3e | Applied Biosystems | Mm01179194_m1 |
| Cd2 | Applied Biosystems | Mm00488928_m1 |
| Cd53 | Applied Biosystems | Mm00514262_m1 |
| Batf3 | Applied Biosystems | Mm01318274_m1 |
| Gbp2 | Applied Biosystems | Mm00494575_m1 |
| Il2rb | Applied Biosystems | Mm00434268_m1 |
| CCL5 cDNA amplification primer 5’-ACGCGCGATCGCCATGAAGATCTCTGCAGCTGCCCTC -3’ | Microsynth | Custom made |
| CCL5 cDNA amplification primer 5’-ACGCCAATTGTCACTAGCTCATCTCCAAATAGTTGATG -3’ | Microsynth | Custom made |
| Recombinant DNA | ||
| MISSION pLKO.1-puro non-target shRNA control | Sigma | SHC016 |
| CCL5 MISSION shRNA Bacterial Glycerol Stock chemokine (C-C motif) ligand 5 | Sigma | SHCLNG-NM_013653 |
| MSCV Luciferase PGK-hygro construct | Addgene | 18782 |
| MSGV1-puromycin-hPGK | Home made | N/A |
| pMD18-T-CCL5 vector | Sino biological | MG50022-M |
| NYESO-1 plasmid | Home made | |
| NY-ESO-1/CCL5 plasmid | Home made | |
| Software and Algorithms | ||
| R Bioconductor package EdgeR (version 3.6.8) | R Project for Statistical Computing | |
| R Bioconductor package limma (version 3.20.9) | R Project for Statistical Computing | |
| RTCGAToolbox R package | R Project for Statistical Computing | |
| R version 3.3.2 with ggplot2 version 2.2.1 | R Project for Statistical Computing | |
| R version 3.3.2 with pROC version 1.9.1 | R Project for Statistical Computing | |
| Other | ||
Ex vivo Mixed Tumor Co-cultures
Requirement of IFNα, β, γ or HLA signaling for CXCL9 and IFN γ production in human ovarian cancer was evaluated using an ex vivo mixed tumor co-culture model. EOC dissociated from 5 different patients (UPENN Cohort) were washed in full media and plated at a concentration of 200 000 cells/well in a 96 flat well-plate. Fc Receptors were blocked through incubation for 30min at 37°c in Fc blocking solution diluted 1:4 in full media. Blocking Abs were subsequently added at specific concentrations mentioned below at t= 0 and 48 hr after the initiation of the ex vivo culture: anti-HLA-A,B,C (10 µg/ml), anti-HLA-DR, DP, DQ (20 µg/ml), anti-IFNα (10 µg/ml), anti-IFNβ (10 µg/ml), anti-IFNγ (10 µg/ml). Supernatants were harvested after 72 hr of co-culture for quantification of CXCL9 expression by BD™ Cytometric Bead Array System and IFNγ by Mesoscale Discovery. All Abs are listed in the Key Resource Table.
Chemotaxis assay
Lymphocyte migration using tumor infiltrating lymphocytes from an EOC patient were isolated using a PAN T cell isolation kit and CFSE labeled for 6 min at RT in 1uM CFSE. 150 µl/well of conditioned media from ex vivo autologous tumor co-culture assays (medium control group) were loaded in the bottom part of a Transwell migration chamber and 50 000 CFSE-labeled TILs were added on the top chamber in media containing or not blocking Abs for CXCR3 and/or CCR5 at 10 µg/ml. Migration was evaluated after 24 hr by quantification of the number of migrated cells in the bottom chamber using FACS analysis.
Lymphocyte migration using peripheral blood lymphocytes was assessed as follows: PBL were activated with beads coated with Abs against CD3 and CD28 for 48 hr labeled with CFSE and then placed (1 × 106 cells/ml) on the top Transwell migration chamber. 150 µl/well of conditioned media from ex vivo autologous tumor co-culture assays of either purified (CD45− depleted) tumor cells, purified CD45+ leukocytes or unprocessed mixed leukocytes and tumor cells derived from fresh EOC solid specimens were plated in the bottom part of a Transwell migration chamber. Migration was evaluated after 24 hr as described above. Results are presented as chemotactic index, defined as the fold increase in cell migration towards media of tumor-derived CD45+ cells, tumor cells or admixed co-cultures in the presence or absence of 50 µg/ml anti-human CXCR3 compared to media alone. Each experiment was performed separately with tumors from eight consecutive patients and in triplicate.
Transfection of human cell lines
For transfections, the OV79 cell line was seeded in a six well plate at a density of 5×105 cells per well and transfected first with an HLA-A2 plasmid. Cells were sorted for HLA-A2 expression and subsequently transfected with NYESO-1 or NY-ESO-1/CCL5 plasmids and selected by antibiotic resistance (5 mg/ml of gentamycin) for the transgenes’ expression.
Transduction of primary T cells and isolation of positive cells
Primary CD8+ T cells were obtained from healthy donors and activated with CD3/CD28 Dynabeads for 24 hr. Post activation, lentiviral particles encoding for the NYESO-1 T-cell receptor were added to the culture along with Polybrene (8 µg/ml). Cells were spin-inoculated for 90 min at 2500 rpm. After the transduction the T cells were expanded for 4 days and the media was changed every two days. On the fifth day, the Dynabeads were removed and the cells were rested for an additional five-six days. The efficiency of transduction was detected by staining the cells with Vbeta-13.1 Ab (Becton Dickinson) and by using a NYESO-1 tetramer (a kind gift from the Ludwig Institute, Lausanne). For further experiments the transduced T cells were stained with Vbeta-13.1 and positively selected using the PE-selection kit according to manufacturer’s instructions.
Three-dimensional culture system to study tumor-T cell interaction
To further understand the interplay between CXCL9 and CCL5 in the recruitment of tumor-specific T cells, we developed in vitro human EOC cell spheroids. To reproduce ovarian cancer islet conditions, we transduced OV79 tumor cells, which express neither CCL5 nor CXCL9, with HLA-A*02 and NY-ESO-1 (to force expression of relevant antigen) and/or CCL5, and allowed them to form spheroids in vitro. Control tumor cells were transduced with empty vector. The OV79 cell line was used to make spheroids by seeding 104 cells on flat 24-well plates coated with agar. Spheroid plates were continuously rotated using a shaker at 150 rpm for 4–6 days. Once the spheroids were formed, they were transferred to the lower well of the 24-well chemotaxis chamber of plate that had been pre-coated with 1 % agar and contained 10 % RPMI.
CXCL9-producing monocytes were developed from donor PBMC-derived CD11b+ cells exposed to recombinant human IFNγ (40 µg/ml for 48 hr), and were seeded and allowed to adhere on the surface of already formed tumor spheroids (at a concentration of 104-105 to the lower chamber along with the spheroids), to reproduce the juxta-epithelial CXCL9-producing tumor monocytes observed in EOC. Monocyte-coated or “naked” spheroids were placed in the bottom compartment of chemotaxis chambers. To develop tumor-specific T cells expressing CXCR3 and CCR5, autologous donor CD8+ cells were transduced (or not) with HLA-A*02-restricted NY-ESO-1 TCR linked with GFP, and were CD3/CD28 costimulated to upregulate the two receptors. T cells were then placed in a trans-well plate and allowed to migrate towards the tumor spheroids. After 24 hr, spheroids were thoroughly rinsed and analyzed by confocal microscopy to detect intraepithelial GFP+ TILs and exclude the presence of surface T-cell aggregate contaminants or dissociated and analyzed by FACS to count TCR-transduced T cells via the Vβ13.1 tetramer. For blocking experiments, CD8+ T cells were pretreated with anti-human CXCR3 Ab before adding them to the chemotaxis chamber.
Transduction of mouse cell lines
For transduction, the ID8 mouse ovarian cancer cell line was seeded in a six-well plate at a density of 5×105 cells per well and incubated with retroviruses carrying the Luciferase gene and selected with hygromycin (400 µg/ml final concentration). MSCV Luciferase PGK-hygro construct was a gift from Scott Lowe (Addgene plasmid # 18782).
CCL5 overexpressing ID8 (ID8Luc CCL5OE)
The retroviral vector MSGV1-puromycin-hPGK was used as a backbone to generate a vector where CCL5 expression is driven by the human PGK promoter (designated MSGV1-puromycin-hPGK-ccl5). The mouse CCL5 CDNA was amplified from the pMD18-T-CCL5 vector and cloned in the MSGV1-puromycin-PGK vector by AsiSI and MfeI digestion of both the PCR product and the parental vector, gel purification and ligation. The final construct was fully sequenced by Microsynth AG.
For the propagation of retroviral particles Phoenix Eco cells were seeded at 5 × 106 per T-75 tissue culture flask in RPMI-10+10% FBS medium 24 hr before transfection. Cells were then transfected with 21.4 µg gene expression plasmid and 14.4 µg of pCL-Eco plasmid using 107.4 µl turbofect. The viral supernatant was harvested at 24 and 48 hr post-transfection. MSGV1-puromycin-hPGK retroviral particles were also produced and used as control. For the transduction of ID8Luc with MSGV1-puromycin-hPGK-ccl5 or MSGV1-puromycin-hPGK retroviral particles, 1×105 tumor cells were seeded in a six-well plate one day prior their transduction. Next day, the medium was removed and the viral supernatant was added when the cells reached a confluence of about 30–40%. Protamine sulfate was also added at a concentration of 10 µg/ml and the plate was centrifuged for 20 min at 2500 rpm. The medium was replaced 24 hr after transduction with fresh DMEM −10 % FBS medium. Transduced cells when then selected with puromycin (2 µg/ml). Secretion of CCL5 was assessed using the BD cytokine bead array as per manufacturer’s instructions. Luciferase expression was also assessed post retroviral infection and ID8Luc CCL5OE expressed luciferase 2-fold higher than ID8Luc control cells (data not shown).
CCL5 knockdown ID8 (ID8Luc CCL5KD)
In order to generate an ID8Luc CCL5 knockdown cell line, the lentiviral vector pLKO.1-puro was used. For the propagation of retroviral particles 293T cells were seeded at 6 × 106 per T75 tissue culture flask in RPMI-10+10% FBS medium 24 hr before transfection. Cells were then transfected with 10 µg pLKO.1-puro CCL5 short hairpin plasmid, 1.58 µg pCMV-PAX2 plasmid and 3.125 µg of pMD2G plasmid using 59.175 µl Turbofect (Fisher Scientific). The viral supernatant was harvested at 24 and 48 hr post-transfection. PLKO.1-puromycin non-specific targeting lentiviral particles were also produced and used as control. The transduction of ID8Luc cells with lentiviruses was carried out as described above. The infected cells were then selected with puromycin (2 µg/ml). Secretion of CCL5 was assessed using the BD cytokine bead array as per manufacturer’s instructions. Luciferase expression was also assessed post retroviral infection and ID8Luc CCL5KD expressed luciferase 1.4-fold higher than ID8Luc scr sh cells (data not shown).
In vivo experiments
ID8 tumor cells (5 × 106) expressing luciferase were injected intraperitoneally in six to eight-week old C57/BL6 female mice. In order to normalize for different expression levels of the luciferase between the two cell lines we calculated the fold of luciferase-based tumor growth. Specifically, mice were injected with 5×106 ID8Luc CCL5OE or 5×106 ID8Luc control cells and fold of luciferase-based tumor growth was calculated by normalizing the absolute Luciferase flux (photons/sec) at week 8 to the absolute Luciferase flux (photons/sec) of day1 for each mouse. In vivo experiments with ID8Luc CCL5KD or ID8Luc scr sh were carried as described above. Tumors were harvested at early (four weeks) or late stages (six to seven weeks) post i.p. injection for RNA extractions. Tumor volume was measured by bioluminescence imaging quantification of luciferase activity (photons/sec).
In vivo blockade experiments
ID8Luc CCL5OE or ID8Luc control tumor cells (5×106) were injected intraperitoneally in 8-week-old C57/BL6 female mice. One or three weeks post tumor inoculation, mice were first standardized in groups with equal average of luciferase signals and then divided into treated and control groups. Mice were inoculated i.p. with α-IFNAR1 mAb (1 mg and then 250 µg), α-IFNγ (250 µg), α-CXCL9 (250 µg), rat IgG1 (250 µg), polyclonal Armenian hamster IgG (500 µg) or mouse IgG1 (1 mg and then 250 µg) isotype control twice a week.
Immunohistochemistry (IHC) staining and evaluation
IHC was performed on 4-µm paraffin sections on the Ventana Discovery Ultra staining module (Ventana, Roche). The following primary and secondary Abs were used: anti-CD8 (dilution 1:100), anti-CD11c (1:800), anti-CD68 (1:100), anti-CCL5/RANTES (1:125), anti-CXCL9/MIG (1:200), anti-mouse Immunoglobulins/HRP (1:200) and anti-rabbit Immunoglobulins/HRP (1:200). For the singleplex assays (CD8, CCL5, CXCL9), the slides were placed on the staining module for deparaffinization, epitope retrieval and endogenous peroxidase quenching and were incubated with the primary Abs for 1 h at room temperature. Detection of each primary Ab was carried out with the Discovery OmniMap anti-Ms or anti-Rb HRP (Ventana) and the immunoreaction was visualized with the Discovery ChromoMap DAB kit (Ventana) whereas hematoxylin was used as counterstain.
For triplexed immunofluorescent staining an iterative staining method was used, with 3 rounds of staining. Each round includes a three-step staining protocol i.e. primary Ab - secondary Ab HRP-conjugated – TSA-fluorophore (FITC, Cy3, Cy5.5) and was followed by an Ab denaturation (stripping) step and in turn by the next round of staining. The denaturation step removes both the primary and the HRP-conjugated secondary Ab of the previous round, whereas through covalent binding to tyrosine residues of the targeted epitope, the TSA-fluorophore remains, allowing for sequential steps of staining. Nuclei were visualized with DAPI.
The expression of CCL5 in tumor islets was assessed blindly by a qualified pathologist (PGF) using the H-score, a continuous variable that ranges from 1 to 300 which is based both on the staining intensity of the CCL5 protein expression (semiquantitatively on a 0–3+ scale) and the percentage of tumor cells with a given staining intensity, using the following formula: H-Score = (% at 0) * 0 + (% at 1+) * 1 + (% at 2+) * 2 + (% at 3+) * 3.
CXCL9 expression in the stroma was classified as low (0, rare positive cells) or high (1, numerous or aggregates of positive cells). Stained slides for CD8 were loaded onto PerkinElmer Vectra microscope and a scanning protocol was created in which, twenty random high-power fields (20x) were selected for multispectral imaging. Single color control slides i.e. DAB only and hematoxylin only were used to create the spectral libraries using the Nuance software, necessary for the unmixing of signals and accurate measurement of expression. Multispectral images from each case were loaded to InForm software, channels were unmixed using the single color spectral library and tissue segmentation (tumor/stroma) and cell segmentation algorithms were created. All images were processed and scored in a batch mode and merged data were processed in Excel. For statistical analysis the GraphPad Prism 6 software was used.
RNA Isolation, RT-PCR and Quantitative Real-time (q)PCR
Total RNA was isolated from 100 to 500 mg of frozen tissue or 1 × 106 cultured cells with TRIzol reagent (Invitrogen, Carlsbad, CA). After treatment with RNase-free DNase (Invitrogen), total RNA was reverse-transcribed using Superscript First-Strand Synthesis Kit for RT-PCR (Invitrogen) under conditions defined by the supplier. For qPCR cDNA was quantified on the ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster City, CA). Quantitative RT-PCR was performed using Fast Taqman reagents (Applied Biosystems) according to the manufacturer’s instructions. PCR amplification of the housekeeping gene, GAPDH, was performed for each sample as control for sample loading and to allow normalization among samples. A standard curve was constructed with PCR-II TOPO cloning vector (Invitrogen) containing the same inserted fragment and amplified by the qPCR. Each sample was run in duplicate and each PCR experiment included two non-template control wells. PCR products were confirmed as single bands using gel electrophoresis.
Nanostring Analysis for mouse experiment
100 ng total RNA was used for measuring 561 mRNA targets on the nCounter GX Immunology panel (mouse) on a nCounter® MAX Analysis System (NanoString Technologies). Gene expression was normalized with predefined reference genes included on the panel using the R Bioconductor packages EdgeR (version 3.6.8) and limma (version 3.20.9) and statistical analysis was performed using the R Bioconductor package limma (version 3.20.9).
Evaluation of methylation status
To evaluate the impact of DNA and histone methylation states on CCL5 expression in human and mouse ovarian cancer cells, the tumor cell lines OVCAR5, OVCAR3, A1847, A2780/C30 and ID8 were plated at 200 000 cells/well in 48 well-plate and stimulated with 5 µM of either 5-Aza-2′-deoxycytidine or DZNep. After 72 hr in vitro culture, cells were washed with PBS and lysed in TRIzol reagent for RNA extraction and CCL5 or GAPDH were quantified by qPCR using Taqman primers listed in the Key Resource Table.
Quantitative RT-PCR primers for cell lines and tumor tissue analyses
Human Taqman probes were used to quantify the expression of CCL5, CXCL9 and GAPDH (housekeeping). Mouse Taqman probes were used to quantify the expression of Cd8a, Ccl5, Cxcl9, Stat1, Gzmk, Cd2, Cd53, Gbp2, Il2rb, Ifng, Grzmb, Foxp3, Batf3 and Gapdh (housekeeping). P values were calculated using Mann–Whitney–Wilcoxon tests. All probes are listed in the Key Resource Table.
FACS and ELISA of human samples
Cells were subjected to six-color FACS on a FACSCanto flow cytometer using CellQuest 3.2.1f1 software (Becton Dickinson, San Jose, CA), utilizing monoclonal Abs against CD45, CD3, CD4, CD8, CD14, CD11c, CCR5, CXCR3, Vbeta- 13.1. Data representing 10 000 to 30 000 events were recorded and analyzed with CellQuest software (Becton Dickinson). Intracellular cytokine staining was performed as following. Briefly, 2 × 106 freshly isolated tumor-derived cells in 1 ml RPMI 10% FCS were incubated with brefeldin A (1 µg/ml) at 37°C for 6 hr. Cells were washed; stained with surface Abs; fixed, permeabilized; and incubated with the CXCL9 or CCL5 Ab. Cells were fixed with 1% paraformaldehyde solution in PBS and analyzed on a FACS-Canto flow cytometer, using Diva software (Becton Dickinson). IFN-γ ELISA was performed using NIB42 as capture Ab and biotinylated 4S.B3 as detection Ab (source) in the concentrations described by the manufacturer. For detection of CXCL9, ELISA was done using cell supernatants as per the instructions from the supplier.
FACS and analysis of murine samples
At the time of sacrifice, i.p. cancers were dissected. Tumors were digested in 200µg/ml Liberase TL and 5 units/ml DNase I in DMEM for 1 hour at 37°C, with rotation. For ex vivo staining, 1–2×106 cells were stained with LIVE/DEADTM Fixable Aqua Dead Cell Stain (1:500). Fc receptors were blocked for 10 min at 4°C with 5µg/ml Mouse BD FC Block. Cells were fluorescently labelled with Abs for 30 min at 4°C, washed and resuspended in fixation buffer (1% formaldehyde in PBS) or intracellularly stained according to the manufacturer’s protocol (eBiosciences). Fluorescently labeled anti-mouse monoclonal Abs used for FACS were: CD45-BV650 (1:400), CD3e-PECy5.5 (1:100), CD3-eFluor450 (1:200), CD3-eFluor 660 (1:100), CD4-PB (1:100), CD8-BV711 (1:100), CD11b-PECy7 (1:200), CD11c-BV605 (1:100), CD86-APCCy7 (1:100), CD206-PE/DazzleTM 594 (1:100). Flow cytometric analysis was performed on a LSR II flow cytometer and analyzed using FlowJo software. All Abs are listed in the Key Resource Table.
FACS sorting
Samples were prepared as described above. Fluorescently labeled anti-mouse monoclonal Abs used for FACS were: CD45-BV650 (1:400), CD3-eFluor 660 (1:100), CD11b-PECy7 (1:200). CD45+CD3−CD11b+ and CD45− cells were sorted using BD Aria II or FACS Aria III FACS sorters.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bioinformatics and Statistical Analyses
Computations were performed with the R software system for statistics.
For the multi-tumor CD8A correlation analysis (see Figure 1 and Figure S1, S2) we used the multi-tumor gene-expression microarray dataset from the Expression Project for Oncology (ExpO) (http://www.intgen.org/, GEO GSE2109) and the multi-tumor gene-expression data as prepared by The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov) using the RTCGAToolbox R package. Confidence intervals for the Pearson’s correlation coefficients were computed using the normal approximation given by the Fisher Z-transformation. Forest plots were generated using the correlation meta-analysis methods in the metafor package (https://www.jstatsoft.org/v036i03) for R using the weighted fixed effect model.
For survival analyses in melanoma, ovarian, breast, colon and lung tumor types, we used a collection of different datasets (see Key Resources Table). For melanoma we obtained the gene expression and patient data from the TCGA database as described above. For serous type ovarian cancer, we applied the curatedOvarianData R package in June 2016, which collected data and eliminated duplicates from 16 sources (identified as E.MTAB.386, GSE13876, GSE14764, GSE17260, GSE18520, GSE19829.GPL8300, GSE26193, GSE26712, GSE30161, GSE32062.GPL648, GSE49997, GSE51088, GSE9891, PMID17290060, TCGA and TCGA.RNASeqV2). To improve commensurability across all samples we performed sample-wise centering and gene-wise centering and rescaling to unit standard deviation in each of the 16 studies. We obtained 2402 cases selected to have patient survival available with positive survival times and expression data for the genes CCL5 and CXCL9. The same standardization and case selection were performed for the three cancer types described below (see also Key Resources Table). For lung cancer we assembled a data collection from seven studies and obtained initially 832 cases of which 720 were suitable for survival analysis. For colon cancer, we obtained 1630 cases from three studies. For breast cancer, we first pooled 2739 cases from four studies and present an analysis for the 420 cases thereof that we assigned to the triple negative subgroup, defined by manually setting a cutoff separating high and low expressers of ER (ESR1 gene) and HER2 (ERBB2 gene) in the gene expression data. When available for most patients of a tumor type, we used as endpoint relapse-free survival (colon, lung), otherwise overall survival when this was more complete (ovarian, melanoma, breast).
In each of the five tumor types the genes CCL5 and CXCL9 were associated with longer survival by Cox regression analysis. To define the physiologically most adequate cutoffs for classifying tumors as CCL5hiCXCL9hi we investigated by visual inspection curves of estimated survival in dependence of the gene level using the survfit function in R (package « survival »). This classification was used for differential expression analysis and QQplots. We selected approx. 200 genes more highly expressed in CCL5hiCXCL9hi group in EOC (FDR cutoff 10–38, 202 genes) and approx. 100 genes with lower expression (FDR cutoff 10–8, 104 genes) for heatmap visualization. The cutoffs differ, as the FDR values were much lower for the “higher” gene class. For intersection analysis and pathway analysis we took the top 202 highly differentially expressed genes in each of the five tumor types. The pathway-based gene set enrichment analysis was performed with the ConsensusPathDB interface at http://cpdb.molgen.mpg.de, limiting the query to the gene sets from Wikipathways and BioCarta (Herwig et al., 2016; Kamburov et al., 2013). The obtained adjusted p values were transformed as minus log10 of the p values and plotted as heatmaps using the pheatmap R package. The gene interaction network is based on gene-gene interaction information of the STRING database (http://string-db.org) and was generated via the query interface on their webpage.
The average of the expression of the set of the 21 common genes was used to score tumors with the same method in all five-tumor types. Cutpoint-free Cox’s proportional hazards survival regression analysis was performed with the signature scores rescaled to unit interquartile range. Follow-up was right-censored at 5 years, 15 years for melanoma.
To visualize the survival pattern with Kaplan-Meier curves, the cohorts were subdivided using population percentiles (top 20%, except 15% for ovarian, and median cut of the rest).
Immune pathway analyses were performed as follows. The signature scores for the immune subsets taken from Bindea et al (Bindea et al., 2013) were computed as the median of the expression of the genes used to define each subset. These scores were then plotted as heatmaps for each cancer type. The relative composition of the immune component was assessed using the CIBERSORT tool (Newman et al., 2015) and the averages for each immune subset ratio were plotted as pie charts using the ggplot2 R package. ANOVA were performed for each cancer type in order to find which immune subsets were differentially represented between CCL5hiCXCL9hi and CCL5loCXCL9lo groups and the p values were plotted as a heatmap after minus log10 transformation. The immunophenoscores as defined in Charoentong et al. (Charoentong et al., 2017) for TCGA patients were taken from the Cancer Immunome Atlas (https://tcia.at/home) and were plotted as pie charts. Fisher’s exact tests were performed to assess statistical differences between CCL5hiCXCL9hi and CCL5loCXCL9lo groups by considering IPS between 8 and 10 as high and below 8 as low.
Pathway analyses for chemokine subtype characterization were performed as follows: The Hallmark geneset collection from MSigDB (http://software.broadinstitute.org/gsea/msigdb) was used. For each geneset, we computed the median of the gene expression in each patient and averaged the signature scores per chemokine subtype. We then plotted these averaged signature scores as a heatmap. Only pathways displaying significant differences (ANOVA followed by Tukey test for multiple comparisons) between CCL5hiCXCL9hi and CCL5loCXCL9lo were shown.
For analyses involving the immunophenoscore and CCL5 regulation (DNA methylation and pathway analysis), only TCGA data were used, taken from the firehose interface (https://gdac.broadinstitute.org/). The methylation data were derived from probe cg10315334, which recognized the 5’ UTR locus of CCL5, between the TSS and the ATG start site (genomic region 34,207,332 on chromosome 17, genome build =17). Methylation data were used as preprocessed β-values; RSEM-normalized RNA sequencing data (level 3) were transformed as log2(1+RSEM).
Signature scores were calculated for RNA-seq transcriptome profiles of pre-treatment samples from melanoma trial Checkmate 064 (Rodig et al., 2018). Response was assessed using modified RECIST 1.1 criteria; patients with best overall response (BOR) of complete or partial response were designated as responders (R) and patients with best overall response progressive disease, stable disease or who were not evaluable due to disease progression were designated as non-responders (NR) Signature score vs. response plots and AUC plots were generated using R version 3.3.2 with ggplot2 version 2.2.1 and pROC version 1.9.1. P values for R vs. NR comparisons were calculated using a generalized linear model and multcomp version 1.4–6.
The gene expression data in melanoma under checkpoint blockade treatment were extracted from the Supplementary Table 6 of Chen et al. (Chen et al., 2016), the list of genes that changed under treatment from their Supplementary Table 10. The number of genes in common between the CCL5hiCXCL9hi gene signatures of each cancer type we studied and the Chen et al. gene signature was as follows: ovarian 59 in common; colon 87 in common; melanoma 107 in common; breast 72 in common lung 71 in common.
Supplementary Material
Table S4 related to Figure 5: Gene sets I, II and III show the genes that were common in 5, 4 and 3 cancer types, respectively.
Table S1 related to Figure 4: Associations of IFNA1, IFNB1 and IFNG with CD8A, CXCL9, CXCL10 and CXCL11
Table S2 related Figure 5: Differentially expressed genes in CCL5hiCXCL9hi ovarian cancer group
Table S3 related to Figure 5: CCL5hiCXCL9hi -derived genes and their overlap in solid tumors
Significance.
Improving T cell-mediated anti-tumor immunity and T-cell trafficking to the tumor site is essential for effective cancer therapy. We report the significance of the synergy between CCL5, constitutively expressed by tumor cell and CXCL9, expressed by macrophages and dendritic cells upon IFNγ stimulation. Co-expression of CCL5 and CXCL9 dictates immunoreactive and immunoresponsive tumors with increased cytotoxic T-cell infiltration. Furthermore, while stable CCL5 expression can sustain T-cell infiltration and CXCL9 expression, its downregulation is dependent on DNA methylation. Our study concludes that the cooperation between CCL5 and CXCL9 represents an important mechanism that orchestrates successful tumor-rejecting T cell responses.
Highlights.
CD8+ T-cell infiltration in tumors is associated with CCL5 and CXCL9 coexpression
CCL5 is expressed in tumor cells and CXCL9 is induced in APCs in response to IFNγ
CCL5hiCXCL9hi tumors are immunoreactive and respond to checkpoint blockade
Cancer cells negatively regulate CCL5 expression by epigenetic silencing mechanisms
Acknowledgments
This work was supported by grants P50 CA083638 SPORE in Ovarian Cancer, R01-CA098951, R01-CA116779-ARRA, a grant from the Sidney Kimmel Foundation, the Emma Mouschamp Foundation, the Ludwig Institute for Cancer Research and the Swiss Medic Foundation. We thank Dr Ralf Bützow from the Department of Obstetrics and Gynecology, University of Helsinki, Helsinki, Finland, for providing the ovarian TMA and Dr Michael Feldman and the staff of TTAB at the University of Pennsylvania Health System for providing ovarian tumors. We thank Pinelopi Chatziemmanouil and Esther Danenberg for IHC technical support; Romain Bedel for supporting with FACS sorting; Corinne Peter and Leonore Wigger, for Nanostring experiments and analysis.
Declaration of Interest
J.C-G is a member of the advisory board of Compass Therapeutics and Anixa Biosciences and has stock options from both. D.J.P.Jr is or has provided consultation for Iovance Biotherapeutics, Bellicum Pharmaceuticals, Neon Therapeutics and Tmunity Therapeutics, and holds patents in the areas of tumor-infiltrating lymphocytes and gene-engineered T cells. D.G.J is an employee and shareholder of Bristol-Myers Squibb. J.S.W has stock or other ownership from Altar BioScience, Biond, CytomX Therapeutics, Protean Biodiagnostics; honoraria from Bristol-Myers Squibb, Merck, Genentech, AbbVie, AstraZeneca, Daiichi Sankyo, GlaxoSmithKline, Eisai, Altar BioScience, Amgen, Roche, lchor Medical Systems, Celldex, CytomX Therapeutics, Nektar, Novartis, Sellas, WindMIL, Takeda, Protean Biodiagnostics. J.S.W has a consulting or advisory role at Celldex, lchor Medical Systems, Biond, Altar BioScience, Bristol-Myers Squibb, Merck, Genentech, Roche, Amgen, AstraZeneca, GlaxoSmithKline, Daiichi Sankyo, AbbVie, Eisai, CytomX Therapeutics, Nektar, Novartis, Sellas, Wind MIL, Takeda. J.S.W has received research funding (to the Institution) from Bristol-Myers Squibb, Merck, GlaxoSmithKline, Genentech, Astellas Pharma, lncyte, Roche, Novartis and declares travel, accommodations, expenses from Bristol-Myers Squibb, GlaxoSmithKline, Daiichi Sankyo, Roche, Celldex, Amgen, Merck, AstraZeneca, Genentech, Novartis, WindMIL, Takeda. S.J.R has received research funding from Bristol-Myers-Squibb, Merck, and KITE/Gilead Pharmaceuticals. During the conduct of the study, S.F.H reports that (other from Bristol-Myers Squibb to institution) has received: grants, personal fees and other from Bristol-Myers Squibb, grants and personal fees from Novartis, personal fees from MercK, EMD Serono, Takeda, Surface, Genentech/Roche, Compass Therapeutics, Apricity, Bayer, Aduro, Partners Therapeutics, Sanofi, Pfizer, Pionyr, from 7 Hills Pharma, Verastem, Rheos, and other from Torque, personal fees from outside the submitted work. S.F.H holds a patent method for treating MICA-related disorders (#20100111973). G.C has received grants, research support or is coinvestigator in clinical trials by BMS, Celgene, Boehringer Ingelheim, Roche, Iovance and Kite. G.C has received honoraria for consultations or presentations by Roche, Genentech, BMS, AstraZeneca, Sanofi-Aventis, Nextcure and GeneosTx. The other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angelova M, Mlecnik B, Vasaturo A, Bindea G, Fredriksen T, Lafontaine L, Buttard B, Morgand E, Bruni D, Jouret-Mourin A, et al. (2018). Evolution of Metastases in Space and Time under Immune Selection. Cell 175, 751–765 e716. [DOI] [PubMed] [Google Scholar]
- Bedognetti D, Spivey TL, Zhao Y, Uccellini L, Tomei S, Dudley ME, Ascierto ML, De Giorgi V, Liu Q, Delogu LG, et al. (2013). CXCR3/CCR5 pathways in metastatic melanoma patients treated with adoptive therapy and interleukin-2. Br J Cancer 109, 2412–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. [DOI] [PubMed] [Google Scholar]
- Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O’Brien-Jenkins A, Gimotty PA, and Coukos G (2008). Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med 14, 28–36. [DOI] [PubMed] [Google Scholar]
- Burke SJ, Goff MR, Lu D, Proud D, Karlstad MD, and Collier JJ (2013). Synergistic expression of the CXCL10 gene in response to IL-1beta and IFN-gamma involves NF-kappaB, phosphorylation of STAT1 at Tyr701, and acetylation of histones H3 and H4. J Immunol 191, 323–336. [DOI] [PubMed] [Google Scholar]
- Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, and Trajanoski Z (2017). Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell reports 18, 248–262. [DOI] [PubMed] [Google Scholar]
- Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. (2016). Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov 6, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. (2004). Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10, 942–949. [DOI] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. (2002). Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298, 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraiswamy J, Freeman GJ, and Coukos G (2013). Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res 73, 6900–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L, and Coukos G (2011). Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475, 226–230. [DOI] [PubMed] [Google Scholar]
- Farber JM (1990). A macrophage mRNA selectively induced by gamma-interferon encodes a member of the platelet factor 4 family of cytokines. Proc Natl Acad Sci U S A 87, 5238–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber JM (1993). HuMig: a new human member of the chemokine family of cytokines. Biochem Biophys Res Commun 192, 223–230. [DOI] [PubMed] [Google Scholar]
- Fridman WH, Pages F, Sautes-Fridman C, and Galon J (2012). The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12, 298–306. [DOI] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. [DOI] [PubMed] [Google Scholar]
- Ganzfried BF, Riester M, Haibe-Kains B, Risch T, Tyekucheva S, Jazic I, Wang XV, Ahmadifar M, Birrer MJ, Parmigiani G, et al. (2013). curatedOvarianData: clinically annotated data for the ovarian cancer transcriptome. Database (Oxford) 2013, bat013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. (2015). Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Martin A, Gomez L, Lustgarten J, Mira E, and Manes S (2011). Maximal T cell-mediated antitumor responses rely upon CCR5 expression in both CD4(+) and CD8(+) T cells. Cancer Res 71, 5455–5466. [DOI] [PubMed] [Google Scholar]
- Gooden MJ, de Bock GH, Leffers N, Daemen T, and Nijman HW (2011). The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer 105, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JW, Sokol CL, and Luster AD (2014). Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 32, 659–702. [DOI] [PubMed] [Google Scholar]
- Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, Suetterlin T, Brand K, Krauss J, Lasitschka F, et al. (2016). Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 29, 587–601. [DOI] [PubMed] [Google Scholar]
- Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, and Gajewski TF (2009). Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69, 3077–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herwig R, Hardt C, Lienhard M, and Kamburov A (2016). Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc 11, 1889–1907. [DOI] [PubMed] [Google Scholar]
- Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, Kato M, Prevost-Blondel A, Avril MF, Nardin A, and Abastado JP (2011). Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res 71, 6997–7009. [DOI] [PubMed] [Google Scholar]
- Hwang WT, Adams SF, Tahirovic E, Hagemann IS, and Coukos G (2012). Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol 124, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A, Stelzl U, Lehrach H, and Herwig R (2013). The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res 41, D793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karonitsch T, von Dalwigk K, Steiner CW, Bluml S, Steiner G, Kiener HP, Smolen JS, and Aringer M (2012). Interferon signals and monocytic sensitization of the interferon-gamma signaling pathway in the peripheral blood of patients with rheumatoid arthritis. Arthritis Rheum 64, 400–408. [DOI] [PubMed] [Google Scholar]
- Kunz M, Toksoy A, Goebeler M, Engelhardt E, Brocker E, and Gillitzer R (1999). Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. J Pathol 189, 552–558. [DOI] [PubMed] [Google Scholar]
- Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, Topper MJ, Luo J, Connolly RM, Azad NS, et al. (2014). Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 5, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. (2018). Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 49, 178–193 e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. (2015). Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun 6, 7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, and Jones PA (2009). DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther 8, 1579–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, and Coukos G (2014). Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 20, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nature methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. (2015). Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372, 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, Chang H, Lovitch SB, Horak C, Weber JS, et al. (2018). MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med 10. [DOI] [PubMed] [Google Scholar]
- Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, et al. (2016). Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov 6, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlienger K, Craighead N, Lee KP, Levine BL, and June CH (2000). Efficient priming of protein antigen-specific human CD4(+) T cells by monocyte-derived dendritic cells. Blood 96, 3490–3498. [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Spranger S, Dai D, Horton B, and Gajewski TF (2017). Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31, 711–723 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, Wenzel A, Hicks J, Ballew M, Stone M, et al. (2017). Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 171, 1284–1300 e1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugurel S, Schrama D, Keller G, Schadendorf D, Brocker EB, Houben R, Zapatka M, Fink W, Kaufman HL, and Becker JC (2008). Impact of the CCR5 gene polymorphism on the survival of metastatic melanoma patients receiving immunotherapy. Cancer Immunol Immunother 57, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Velazquez M, Xolalpa W, and Pestell RG (2014). The potential to target CCL5/CCR5 in breast cancer. Expert Opin Ther Targets 18, 1265–1275. [DOI] [PubMed] [Google Scholar]
- Viola A, Sarukhan A, Bronte V, and Molon B (2012). The pros and cons of chemokines in tumor immunology. Trends Immunol 33, 496–504. [DOI] [PubMed] [Google Scholar]
- Westergaard MCW, Andersen R, Chong C, Kjeldsen JW, Pedersen M, Friese C, Hasselager T, Lajer H, Coukos G, Bassani-Sternberg M, et al. (2019). Tumour-reactive T cell subsets in the microenvironment of ovarian cancer. Br J Cancer [DOI] [PMC free article] [PubMed]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. (2013). Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, Coukos G, and Powell DJ Jr. (2014). CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res 20, 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, Luster AD, Busuttil RW, and Kupiec-Weglinski JW (2008). CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology 47, 207–214. [DOI] [PubMed] [Google Scholar]
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al. (2003). Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348, 203–213. [DOI] [PubMed] [Google Scholar]
- Zsiros E, Duttagupta P, Dangaj D, Li H, Frank R, Garrabrant T, Hagemann IS, Levine BL, June CH, Zhang L, et al. (2015). The Ovarian Cancer Chemokine Landscape Is Conducive to Homing of Vaccine-Primed and CD3/CD28-Costimulated T Cells Prepared for Adoptive Therapy. Clin Cancer Res 21, 2840–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4 related to Figure 5: Gene sets I, II and III show the genes that were common in 5, 4 and 3 cancer types, respectively.
Table S1 related to Figure 4: Associations of IFNA1, IFNB1 and IFNG with CD8A, CXCL9, CXCL10 and CXCL11
Table S2 related Figure 5: Differentially expressed genes in CCL5hiCXCL9hi ovarian cancer group
Table S3 related to Figure 5: CCL5hiCXCL9hi -derived genes and their overlap in solid tumors