

ABSTRACT
Dysfunctional mitochondria have been implicated in a variety of human pathophysiological conditions such as cancer, neurodegeneration, and aging. However, the precise role of mitochondrial-generated reactive oxygen species (ROS) in these maladies is unclear. Using a light-activated mitochondrially targeted approach, we recently reported direct evidence that damaged mitochondria produce a wave of secondary ROS, causing rapid and preferential telomere dysfunction but not gross nuclear DNA damage (Fig 1).
KEYWORDS: Mitochondria, ROS, telomeres
Oxidative damage initiated in the mitochondria can result in a vicious cycle of reactive oxygen species (ROS) generation, inducing further mitochondrial damage and subsequent alteration in cellular metabolism and survival.1 Although ROS is believed to play a role in molecular communication between mitochondria and the nucleus,2 the relationship between dysfunctional mitochondria, subsequent nuclear alterations, and diseases remains unresolved.3 In order to fully address this research question, the field needs new tools to selectively damage the mitochondria with high temporal and spatial precision. Previously developed chromophore-assisted light inactivation (CALI) technologies that combine a genetically targeted fluorescent moiety (such as miniSOG and KillerRed) and light to generate ROS4,5 are difficult to control, as the fluorescent moiety, once synthesized, is always “on” and thus prone to activation by spurious light. Also, the wavelengths of light at the power required to produce sufficient flux of ROS in the CALI systems are capable of directly causing phototoxicity-mediated cellular damage.
To this end, we developed and validated a chemoptogenetic approach that uses three separate components to maintain rigid control over light-induced damage6 as shown in Figure 1. The basis of this new technology is a photosensitizer dye MG-2I (iodine-substituted malachite green analog) that can only be light-activated to produce singlet oxygen (1O2) when bound by a mitochondrial-targeted fluorogen-activating peptide (Mito-FAP). Exposure of this protein-dye complex to innocuous near-infrared (NIR) 660 nm light produces singlet oxygen with high quantum yield,6 attacking macromolecules that are in close proximity, including DNA, RNA, proteins and lipids, exclusively in mitochondria. By using HEK293 (human embryonic kidney) cells stably expressing Mito-FAP, we were able to show mitochondrial damage and dysfunction that were both light and dye dependent.7 We observed a significant decrease in the mitochondrial basal oxygen consumption rate (OCR) and a compensatory increase of the extracellular acidification rate (ECAR) within 4 h of treatment. Our data indicate that a brief pulse of singlet oxygen through the FAP system results in damage to the mitochondrial electron transport chain complexes (ETC I, III, and IV), producing a long-lived secondary wave of superoxide (O2•–) and hydrogen peroxide (H2O2) generation in mitochondria, which then leads to mitochondrial DNA damage and mitochondrial fragmentation.
Figure 1.
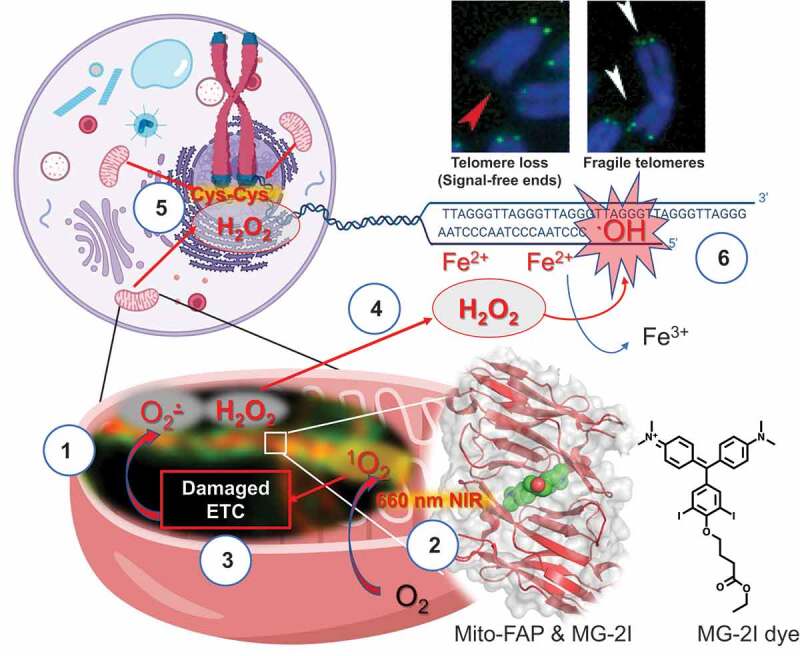
Dysfunctional mitochondria lead to preferential nuclear DNA damage at telomeres.
(1) Super resolution stimulated emission depletion (STED) image of mitochondrial-targeted fluorogen-activating peptide (Mito-FAP, orange), which is shown localized in the mitochondrial intermembrane space and surrounded by mNEON-TOM20 (green), an outer membrane marker. (2) Upon binding to MG-2I photosensitizer and irradiation with near-infrared (NIR) light (660 nm), Mito-FAP system produces singlet oxygen (1O2) with high quantum yield in the mitochondrial intermembrane space. (3) Singlet oxygen induces direct oxidative damage to the components in the intermembrane space, including mitochondrial electron transport chain (ETC). Oxidative damage to ETC initiates a persistent secondary wave of a generation of superoxide (O2·−), which can then be converted to hydrogen peroxide (H2O2) by manganese superoxide dismutase (MnSOD) inside mitochondria. The continuous generation of reactive oxygen species thus amplifies mitochondrial damage. (4) Due to the prolonged generation of hydrogen peroxide and its highly diffusible nature, (5) hydrogen peroxide can reach the nucleus causing oxidation of nuclear proteins at cysteine residues (Cys-Cys). (6) Since telomere DNA has a unique iron (Fe2+)-binding propensity, hydrogen peroxide can be converted to highly reactive hydroxyl radicals (.OH) preferentially at the telomeres causing telomere loss (red arrow) and fragile sites (white arrows), but not gross nuclear DNA damage. Adapted from 7.
In order to show that mitochondrially generated H2O2 can freely diffuse to the nucleus we used a nuclear H2O2 sensor (pHyper-nuc) and a single-cell ratiometric cysteine oxidation imaging technique. Together these complementary approaches indicated that the nucleus suffers attack by H2O2 24 h after the initial singlet oxygen-mediated mitochondrial insult. One consequence of this nuclear oxidative damage was replication stress as evidenced by a reduction in bromodeoxyuridine (BrdU) incorporation, an increase in phosphorylation of replication protein A 32 kDa subunit (RPA32), and a reduction of cyclin E protein levels. Analysis of the activation of ataxia telangiectasia mutated (ATM) signaling suggested direct DNA double-strand breaks instead of direct oxidation by H2O2, as evidenced by the phosphorylation of checkpoint kinase 2 (CHK2) and KRAB-associated protein-1 (KAP1) and lack of an oxidative formation of ATM dimer.8 Interestingly, inhibiting ATM kinase sensitized cells treated with MG-2I and light to apoptotic cell death, suggesting a potential therapeutic strategy with this FAP technology. Surprisingly, no nuclear DNA damage could be detected after MG-2I and light treatment in the HEK293 Mito-FAP-expressing cells using a highly sensitive alkaline assay for DNA strand breaks (single- or double-strand breaks). The lack of gross nuclear DNA damage was also supported by a quantitative PCR (qPCR) assay,1 which showed no reduction in amplification of nuclear polymerase β gene. These intriguing results indicated direct activation of ATM in the absence of perceptible DNA strand breaks by traditional DNA damage detection methods.
It has been previously shown that telomeric DNA sequences, TTAGGG, are seven-fold more prone to damage by H2O2 due to the tendency of iron to bind to these sequences and facilitate Fenton chemistry to generate highly reactive hydroxyl radicals (.OH).9 We focused on the analysis of telomeres for double-strand breaks by measuring the colocalization of a peptide nucleic acid (PNA) telomere probe and tumor protein p53-binding protein 1 (53BP1), a marker of DNA double-strand breaks. We observed a two-fold increase in the number of 53BP1 colocalizing with telomeres 48 h after MG-2I and light treatment. Moreover, we observed a significant increase in telomere fragility and telomere signal loss in these cells, Figure 1. Furthermore, addition of the antioxidant N-acetylcysteine (NAC) to cells before and during the MG-2I and light treatment prevented the telomere damage. Finally, cells that lacked functional mitochondrial ETC, through mitochondrial DNA (mtDNA) depletion, did not show telomeric damage after MG-2I and light treatment, indicating that the telomeric damage is a direct result of damage to functional mitochondrial ETC.
In summary, using the Mito-FAP approach, we demonstrated that mitochondrial dysfunction can directly and specifically damage telomeres, without causing general nuclear DNA damage, Figure 1. Our data suggest direct crosstalk between dysfunctional mitochondria and telomeres, with H2O2 as an essential mediator, can lead to the amplification of cellular damage. We have thus established a mechanism underlying telomere dysfunction and mitochondrial initiated ROS, which serves as a basis for understanding pathological conditions such as cancer and aging. Current work with this promising Mito-FAP system in different animal models is underway to further delineate mitochondrial dysfunction in various diseases.
Funding Statement
This work was supported by the National Institute of Environmental Health Sciences [R33ES025606].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Emily Beckwitt for reading and commenting on this manuscript. We would also like to thank our collaborators, Marcel Bruchez, Edward Burton, Patricia Opresko and Simon Watkins, without whom this work would not have been possible.
References
- 1.Yakes FM, Van Houten B.. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94(2):1–3. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saki M, Prakash A.. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic Biol Med. 2017;107:216–227. doi: 10.1016/j.freeradbiomed.2016.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst). 2006;5(2):145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Bulina ME, Lukyanov KA, Britanova OV, Onichtchouk D, Lukyanov S, Chudakov DM. Chromophore-assisted light inactivation (CALI) using the phototoxic fluorescent protein KillerRed. Nat Protoc. 2006;1(2):947–953. doi: 10.1038/nprot.2006.89. [DOI] [PubMed] [Google Scholar]
- 5.Wang FS, Jay DG. Chromophore-assisted laser inactivation (CALI): probing protein function in situ with a high degree of spatial and temporal resolution. Trends Cell Biol. 1996;6:442–445. [DOI] [PubMed] [Google Scholar]
- 6.He J, Wang Y, Missinato MA, Onuoha E, Perkins LA, Watkins SC, St Croix CM, Tsang M, Bruchez MP. A genetically targetable near-infrared photosensitizer. Nat Methods. 2016;13(3):263–268. doi: 10.1038/nmeth.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian W, Kumar N, Roginskaya V, Fouquerel E, Opresko PL, Shiva S, Watkins SC, Kolodieznyi D, Bruchez MP, Van Houten B. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc Natl Acad Sci U S A. 2019;116(37):18435–18444. doi: 10.1073/pnas.1910574116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee J-H, Mand MR, Kao C-H, Zhou Y, Ryu SW, Richards AL, Coon JJ, Paull TT. ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci Signal. 2018;11(512):eaan5598. doi: 10.1126/scisignal.aan5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henle ES, Han Z, Tang N, Rai P, Luo Y, Linn S. Sequence-specific DNA cleavage by Fe2+-mediated fenton reactions has possible biological implications. J Biol Chem. 1999;274(2):962–971. doi: 10.1074/jbc.274.2.962. [DOI] [PubMed] [Google Scholar]